Label: PRAXBIND- idarucizumab injection

- NDC Code(s): 0597-0197-05
- Packager: Boehringer Ingelheim Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated January 23, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use PRAXBIND safely and effectively. See full prescribing information for PRAXBIND.
PRAXBIND® (idarucizumab) injection, for intravenous use
Initial U.S. Approval: 2015INDICATIONS AND USAGE
PRAXBIND is a humanized monoclonal antibody fragment (Fab) indicated in patients treated with Pradaxa® when reversal of the anticoagulant effects of dabigatran is needed:
- For emergency surgery/urgent procedures
- In life-threatening or uncontrolled bleeding (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 2.5 g/50 mL solution in a single-dose vial (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
- Thromboembolic Risk: Reversing dabigatran therapy exposes patients to the thrombotic risk of their underlying disease. Resume anticoagulant therapy as soon as medically appropriate. (2.4, 5.1)
- Re-elevation of Coagulation Parameters: In patients with elevated coagulation parameters and reappearance of clinically relevant bleeding or requiring a second emergency surgery/urgent procedure, an additional 5 g dose of PRAXBIND may be considered. (5.2)
- Hypersensitivity reactions: Discontinue administration and evaluate. (5.3)
- Risks of Serious Adverse Reactions in Patients with Hereditary Fructose Intolerance due to Sorbitol Excipient: Patients with hereditary fructose intolerance may be at risk of adverse reactions. (5.4)
ADVERSE REACTIONS
- In healthy volunteers, the most frequently reported adverse reactions in ≥5% of subjects treated with idarucizumab was headache. (6.1)
- In patients, the most frequently reported adverse reactions in ≥5% of patients treated with idarucizumab were constipation and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Boehringer Ingelheim Pharmaceuticals, Inc. at 1-800-542-6257 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Preparation
2.3 Administration
2.4 Restarting Antithrombotic Therapy
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Risk
5.2 Re-elevation of Coagulation Parameters
5.3 Hypersensitivity Reactions
5.4 Risks of Serious Adverse Reactions in Patients with Hereditary Fructose Intolerance due to Sorbitol Excipient
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
The recommended dose of PRAXBIND is 5 g, provided as two separate vials each containing 2.5 g/50 mL idarucizumab (see Figure 1). Both vials are packaged together in one carton.
For intravenous use only.
There is limited data to support administration of an additional 5 g of PRAXBIND [see Warnings and Precautions (5.2)].
2.2 Preparation
- Remove both vials (each containing 2.5 g/50 mL idarucizumab) from carton.
- Ensure aseptic handling when preparing the infusion.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- Once solution has been removed from the vial, administration should begin promptly. The solution in vials may be stored at room temperature, 25°C (77°F), but must be used within 6 hours [see How Supplied/Storage and Handling (16.2)].
2.3 Administration
- Do not mix with other medicinal products. Use aseptic technique when administering PRAXBIND.
- Intravenously administer the dose of 5 g (2 vials, each contains 2.5 g) as
- A pre-existing intravenous line may be used for administration of PRAXBIND. The line must be flushed with sterile 0.9% Sodium Chloride Injection, USP solution prior to infusion. No other infusion should be administered in parallel via the same intravenous access.
- PRAXBIND treatment can be used in conjunction with standard supportive measures, which should be considered as medically appropriate [see Clinical Pharmacology (12.2)].
2.4 Restarting Antithrombotic Therapy
Patients being treated with dabigatran therapy have underlying disease states that predispose them to thromboembolic events. Reversing dabigatran therapy exposes patients to the thrombotic risk of their underlying disease. To reduce this risk, resumption of anticoagulant therapy should be considered as soon as medically appropriate.
Idarucizumab is a specific reversal agent for dabigatran, with no impact on the effect of other anticoagulant or antithrombotic therapies.
Pradaxa treatment can be initiated 24 hours after administration of PRAXBIND [see Clinical Pharmacology (12.2)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Thromboembolic Risk
Patients being treated with dabigatran therapy have underlying disease states that predispose them to thromboembolic events. Reversing dabigatran therapy exposes patients to the thrombotic risk of their underlying disease. To reduce this risk, resumption of anticoagulant therapy should be considered as soon as medically appropriate [see Dosage and Administration (2.4)].
5.2 Re-elevation of Coagulation Parameters
In a limited number of patients in the clinical program, between 12 and 24 hours after administration of 5 g idarucizumab, elevated coagulation parameters (e.g., activated partial thromboplastin time (aPTT) or ecarin clotting time (ECT)) have been observed [see Dosage and Administration (2.1)].
If reappearance of clinically relevant bleeding together with elevated coagulation parameters is observed after administration of 5 g PRAXBIND, administration of an additional 5 g dose of PRAXBIND may be considered. Similarly, patients who require a second emergency surgery/urgent procedure and have elevated coagulation parameters may receive an additional 5 g dose of PRAXBIND.
The safety and effectiveness of repeat treatment with PRAXBIND have not been established [see Clinical Pharmacology (12.2)].
5.3 Hypersensitivity Reactions
There is insufficient clinical experience with PRAXBIND in patients to evaluate risk of hypersensitivity to idarucizumab. In clinical studies adverse events possibly indicative of hypersensitivity reactions where a possible relationship could not be excluded were reported [see Adverse Reactions (6.1)]. The risk of using PRAXBIND in patients with known hypersensitivity (e.g., anaphylactoid reaction) to idarucizumab or to any of the excipients needs to be weighed cautiously against the potential benefit of such an emergency treatment. If an anaphylactic reaction or other serious allergic reaction occurs, immediately discontinue administration of PRAXBIND and institute appropriate treatment.
5.4 Risks of Serious Adverse Reactions in Patients with Hereditary Fructose Intolerance due to Sorbitol Excipient
In patients with the condition of hereditary fructose intolerance who have received parenteral administration of sorbitol, serious adverse reactions, including fatal reactions, have been reported. Reactions have included hypoglycemia, hypophosphatemia, metabolic acidosis, increase in uric acid, acute liver failure with breakdown of excretory and synthetic function.
The recommended dose of PRAXBIND contains 4 g sorbitol as an excipient. When prescribing PRAXBIND to patients with hereditary fructose intolerance, consider the combined daily metabolic load of sorbitol/fructose from all sources, including PRAXBIND and other drugs containing sorbitol. The minimum amount of sorbitol at which serious adverse reactions may occur in these patients is not known.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described in more detail elsewhere in the labeling:
- Thromboembolic Risk [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Risks of Serious Adverse Reactions in Patients with Hereditary Fructose Intolerance due to Sorbitol Excipient [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
In three healthy volunteer clinical trials, 224 subjects were treated with idarucizumab. In these trials, during the treatment period, the overall frequency of adverse events was similar between idarucizumab-treated subjects (55/224, 25%) and placebo-treated subjects (26/105, 25%). Among those subjects treated with idarucizumab, adverse reactions reported in ≥5% of subjects was headache (12/224, 5%).
In the RE-VERSE-AD® (RE-VERSal Effects of idarucizumab on Active Dabigatran) trial, a total of 503 dabigatran-treated patients were administered idarucizumab either because they required an emergency surgery or urgent procedure, or because they presented with life-threatening or uncontrolled bleeding [see Clinical Studies (14)]. The adverse reactions reported in ≥5% of patients were constipation (33/503, 7%) and nausea (23/503, 5%). Of the 503 dabigatran-treated patients in the entire study period, 101 patients died, 19 within the first day after idarucizumab dosing; each of these deaths could be attributed either as a complication of the index event or associated with co-morbidities.
Thromboembolic Events
In the RE-VERSE-AD trial, 33 of 503 patients reported thrombotic events, 11 patients within 5 days after treatment with idarucizumab and 22 patients 6 days or more after treatment with idarucizumab. Most of these patients were not on antithrombotic therapy at the time of the event, and in each of these cases, the thrombotic event could be attributed to the underlying medical condition of the patient [see Warnings and Precautions (5.1)].
Hypersensitivity
Pyrexia, bronchospasm, hyperventilation, rash, and pruritus have been reported in clinical trials with idarucizumab [see Warnings and Precautions (5.3)].
6.2 Immunogenicity
As with all proteins there is a potential for immunogenicity with idarucizumab. Detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications and underlying disease. For these reasons, comparison of the incidence of antibodies to idarucizumab in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Using an electro-chemiluminescence (ECL) based assay, plasma samples from 283 subjects (224 treated with idarucizumab) in phase I trials and 501 patients were tested for antibodies cross-reacting with idarucizumab. Pre-existing antibodies with cross-reactivity to idarucizumab were detected in approximately 12% (33/283) of the subjects and 4% (19/501) of patients. The majority of pre-existing antibodies were shown to have low titers. No impact on the pharmacokinetics or the reversal effect of idarucizumab or hypersensitivity reactions were observed. Treatment-emergent possibly persisting anti-idarucizumab antibodies with low titers were observed in 4% (10/224) of the subjects and 2% (8/501) of patients treated with idarucizumab. Nine patients were re-dosed with idarucizumab. All nine patients were re-dosed within 6 days after the first idarucizumab dose. None of these patients re-dosed with idarucizumab tested positive for anti-idarucizumab antibodies.
The epitope specificity of antibodies to idarucizumab was characterized using probe molecules. For pre-existing antibodies in patients, 95% (18/19) had specificity for the C-terminus, a region of idarucizumab to which dabigatran does not bind. For treatment emergent antibodies in patients, 67% (6/9) had specificity for the C-terminus, 22% (2/9) had specificity for the variable region, and 11% (1/9) had mixed specificity.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on PRAXBIND use in pregnant women to inform a drug-associated risk of major birth defects and miscarriage. Animal reproductive and development studies have not been conducted with idarucizumab. It is also not known whether PRAXBIND can cause fetal harm when administered to a pregnant woman or can affect reproduction capacity. PRAXBIND should be given to a pregnant woman only if clearly needed.
The background risk of major birth defects and miscarriage for the indicated population is unknown. Adverse outcomes in pregnancy occur regardless of the health of the mother or the use of medications. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
8.2 Lactation
Risk Summary
There are no data on the effects of PRAXBIND on the breastfed child or on milk production.
It is not known whether idarucizumab is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when PRAXBIND is administered to a nursing woman.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for PRAXBIND and any potential adverse effects on the breastfed child from PRAXBIND or from the underlying maternal condition.
8.5 Geriatric Use
A total of 454 (90%) patients treated with idarucizumab in the case series trial were 65 years of age and older, and 318 (63%) were 75 years of age and older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
-
11 DESCRIPTION
Idarucizumab is a humanized monoclonal antibody fragment (Fab) derived from an IgG1 isotype molecule, whose target is the direct thrombin inhibitor dabigatran. Using recombinant expression technology, idarucizumab is produced in a well characterized recombinant (mammalian) CHO cell line and is purified using standard technology. Idarucizumab is composed of a light chain of 219 amino acids and a heavy chain fragment of 225 amino acids, covalently linked together by one disulfide bond between cysteine 225 of the heavy chain fragment and cysteine 219 of the light chain, and has an estimated molecular mass of approximately 47,766 Daltons.
PRAXBIND (idarucizumab) is a sterile, preservative-free, colorless to slightly yellow, clear to slightly opalescent solution for intravenous administration. PRAXBIND (idarucizumab) is supplied in 2 single-dose vials, each containing 2.5 g of idarucizumab in 50 mL formulated as a buffered, isotonic, solution containing acetic acid glacial (10.05 mg), polysorbate 20 (10 mg), sodium acetate (88.82 mg), sorbitol (2004.20 mg), and water for injection with an osmolality of 270-330 mOsm/kg and a pH of 5.3-5.7.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Idarucizumab is a specific reversal agent for dabigatran. It is a humanized monoclonal antibody fragment (Fab) that binds to dabigatran and its acylglucuronide metabolites with higher affinity than the binding affinity of dabigatran to thrombin, neutralizing their anticoagulant effect.
12.2 Pharmacodynamics
In healthy subjects aged 45 to 64 years, the plasma concentrations of unbound dabigatran were reduced to below the lower limit of quantification immediately after the administration of 5 g idarucizumab. Subjects' diluted thrombin time (dTT), ECT, aPTT, thrombin time (TT), and activated clotting time (ACT) parameters returned to baseline levels (see Figure 4 and Figure 5). This reduction of dabigatran plasma concentration was observed over the entire observation period of at least 24 hours. Similar findings were also observed in elderly subjects (aged 65 to 80 years) as well as subjects with mild and moderate renal impairment [see Clinical Pharmacology (12.3)].
In a limited number of patients, re-distribution of dabigatran from the periphery to plasma led to re-elevation of dTT, ECT, aPTT, and TT [see Warnings and Precautions (5.2)].
Re-dosing with 2.5 g idarucizumab in 6 healthy subjects aged 45-64 years at 2 months after first infusion revealed no differences in safety and no indication of allergic reactions [see Clinical Pharmacology (12.3)].
No changes in the pharmacokinetics or pharmacodynamics of dabigatran were noted upon re-initiation 24 hours after the administration of idarucizumab [see Dosage and Administration (2.4)].
Figure 4 Plasma-Levels of Unbound Dabigatran in the Representative Group of Healthy Subjects (Administration of Idarucizumab or Placebo at 0 h)
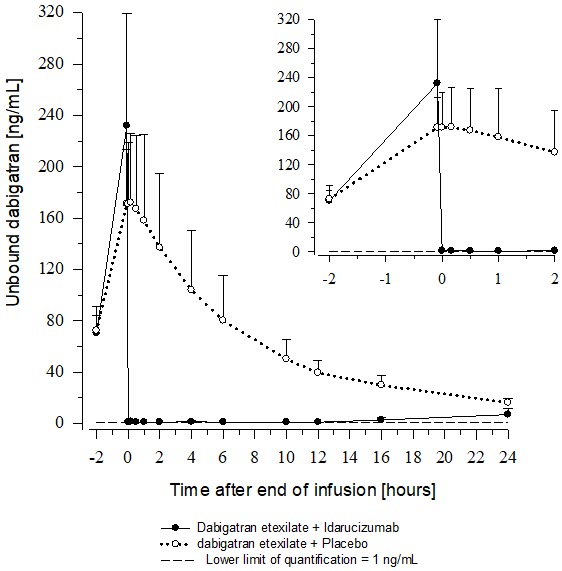
Figure 5 Change of ECT from Baseline in the Representative Group of Healthy Subjects (Administration of Idarucizumab or Placebo at 0 h)
Thrombin Generation Parameters
Idarucizumab alone has shown no procoagulant effect measured as endogenous thrombin potential (ETP).
Cardiac Electrophysiology
Clinical trials with idarucizumab in healthy subjects measured heart rate and electrocardiogram (ECG) parameters (waveform morphology, P wave duration, and PR, QRS, QT, and QTc intervals). There were no clinically relevant abnormal findings related to ECG.
Drug Interactions
In vitro Assessment of Drug Interactions
In vitro data suggest that the inhibition of dabigatran by idarucizumab is not affected by coagulation factor concentrates [3- or 4-factor prothrombin complex concentrates (PCCs), activated PCC, or recombinant Factor VIIa].
Assessment of Drug Interactions in Animal Studies
The potential effect of the binding of idarucizumab to dabigatran in the presence of volume replacement agents (e.g., crystalloids, colloids, and retransfusion of washed red blood cells) was investigated in swine. The results of this study suggest that neutralization of dabigatran anticoagulant activity is not influenced by 50% hemodilution with routinely used volume replacement strategies.
12.3 Pharmacokinetics
There were no obvious differences in the idarucizumab plasma concentration time profiles when idarucizumab was administered alone or after pretreatment with dabigatran. A dose-dependent increase in the fraction of unchanged idarucizumab excreted in urine was observed.
Distribution
Idarucizumab exhibited multiphasic disposition kinetics and limited extravascular distribution. Following the intravenous infusion of a 5 g dose, the geometric mean volume of distribution at steady state (Vss) was 8.9 L (geometric coefficient of variation (gCV 24.8%)).
Elimination
Idarucizumab was rapidly eliminated with a total clearance of 47.0 mL/min (gCV 18.4%), an initial half-life of 47 minutes (gCV 11.4%), and a terminal half-life of 10.3 h (gCV 18.9%). After intravenous administration of 5 g idarucizumab, 32.1% (gCV 60.0%) of the dose was recovered in urine within a collection period of 6 hours and less than 1% in the following 18 hours. The remaining part of the dose is assumed to be eliminated via protein catabolism, mainly in the kidney.
Metabolism
Several pathways have been described that may contribute to the metabolism of antibodies. All of these pathways involve biodegradation of the antibody to smaller molecules, i.e., small peptides or amino acids which are then reabsorbed and incorporated in the general protein synthesis.
Specific Populations
Age, Sex, Race and Body Weight
Age, sex, race (Caucasian vs Asian) and body weight had no clinically important effect on systemic exposure of idarucizumab based on population pharmacokinetic analyses which included data from 220 volunteers and 486 patients.
Patients with Renal Impairment
Idarucizumab has been studied in 12 subjects with mild renal impairment (creatinine clearance [CrCl] 60 to <90 mL/min, by Cockcroft-Gault equation) and 6 subjects with moderate impairment (CrCl30 to <60 mL/min). Compared to healthy subjects, the total clearance was reduced, leading to an increase in idarucizumab's area under the curve (AUC) by 43.5% and 83.5% in mild and moderate renal impairment, respectively. No dose adjustment is required in renally impaired patients.
Based on pharmacokinetic data from 347 patients with varying degrees of renal function (median CrCl 21 to 99 mL/min) it is estimated that mean idarucizumab exposure (AUC0–24h) increased by 38%, 90%, and 146% in patients with mild renal impairment (CrCl 50 to <80 mL/min), moderate renal impairment (CrCl 30 to <50 mL/min) and severe renal impairment (CrCl 0 to <30 mL/min), respectively. Since dabigatran is also excreted primarily via the kidneys, increases in the exposure to dabigatran are also seen with worsening renal function.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
The safety and effectiveness of PRAXBIND was investigated in three randomized placebo-controlled healthy volunteer trials, Trials 1321.1, 1321.2 and 1321.5 (NCT01688830, NCT01955720, NCT02028780), and in RE-VERSE-AD (RE-VERSal Effects of idarucizumab on Active Dabigatran) trial (NCT02104947), a single cohort case series trial with dabigatran-treated patients who have life-threatening or uncontrolled bleeding, or who require emergency surgery or urgent procedure.
Healthy Volunteers
Trials 1321.1, 1321.2 and 1321.5 in a total of 283 subjects assessed the safety, dose-response, and effect of idarucizumab on reducing unbound dabigatran and coagulation parameters. Of the 283 subjects, 224 received at least one dose of idarucizumab. These trials included 19 females and 30 subjects aged 65 years or older (overall mean age 36 years).
The tables below summarize the idarucizumab effect on coagulation parameters dTT, aPTT, ECT, TT, and ACT over time for 14 subjects treated in Trial 1321.2. Fourteen subjects received dabigatran 220 mg orally twice daily for three days and an additional single 220 mg dose of dabigatran on day four, two hours before receiving idarucizumab. Idarucizumab was administered as one 5 g intravenous infusion over five minutes. Table 1 shows the results of the idarucizumab treatment group and Table 2 shows the results of the placebo treatment group.
Table 1 Change in Coagulation Parameters in 14 Dabigatran-exposed Subjects Treated with 5 g Idarucizumab Clotting Assay
(Mean and Standard Deviation)Pre-Idarucizumab
(N=14)End of infusion of Idarucizumab
(N=14)24 hours after Idarucizumab
(N=14)dTT [s] 66.6 (12.0) 32.1 (1.38) 33.0 (1.69) aPTT [s] 67.8 (14.5) 29.2 (4.74) 31.9 (5.71) ECT [s] 122 (42.2) 34.7 (1.92) 38.8 (2.86) TT [s] 127 (62.6) 12.5 (0.786) 19.3 (5.14) ACT [s] 236 (47.6) 116 (7.71) 140 (10.1) Table 2 Change in Coagulation Parameters in 14 Dabigatran-exposed Subjects Treated with Placebo Clotting Assay
(Mean and Standard Deviation)Pre-Placebo
(N=14)End of infusion of Placebo
(N=14)24 hours after Placebo
(N=14)dTT [s] 64.7 (9.82) 65.3 (12.1) 36.1 (2.48) aPTT [s] 65.2 (14.0) 66.5 (13.2) 37.0 (7.10) ECT [s] 117 (29.8) 122 (32.9) 44.7 (5.39) TT [s] 132 (35.4) 147 (46.7) 39.5 (11.8) ACT [s] 219 (44.7) 216 (50.5) 148 (15.1) The effect of idarucizumab on reducing unbound dabigatran in healthy volunteers is summarized in section 12.2, Pharmacodynamics.
RE-VERSE-AD Patient Experience
In RE-VERSE-AD, 5 g idarucizumab was administered to patients treated with dabigatran who presented with dabigatran-related life-threatening or uncontrolled bleeding (Group A) or who required emergency surgery or urgent procedures (Group B). The primary endpoint was the maximum percentage reversal of the pharmacodynamic anticoagulant effect of dabigatran within 4 hours after the administration of idarucizumab, based on central laboratory determination of dTT or ECT.
RE-VERSE-AD included data for 503 patients: 301 patients with serious bleeding (Group A) and 202 requiring an urgent procedure (Group B). Approximately half of the patients in each group were male. The median age was 78 years and the median creatinine clearance was 53 mL/min. Approximately 62% of patients in Group A and 62% of patients in Group B had been treated with dabigatran 110 mg BID.
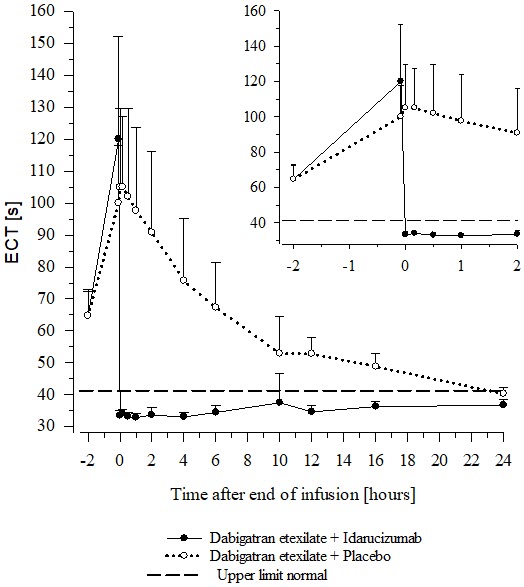
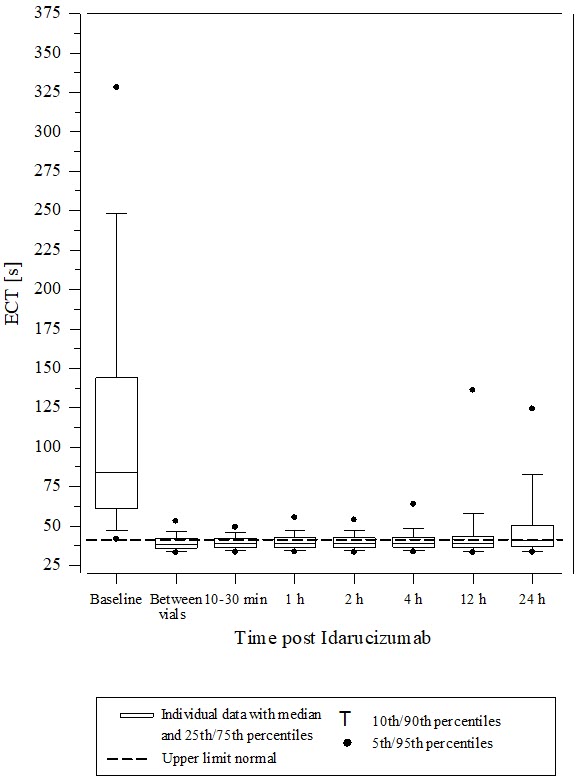
Reversal was evaluable only for those patients showing prolonged coagulation times prior to idarucizumab treatment. Among the evaluable patients, the median maximum reversal of the pharmacodynamic anticoagulant effect of dabigatran as measured by ECT, aPTT or dTT in the first 4 hours after administration of 5 g idarucizumab was 100%, with most patients achieving complete reversal as measured by ECT (82%), aPTT (93%) or dTT (99%). Reversal of the pharmacodynamics effects was evident immediately after administration. Results for Groups A and B were similar. In a limited number of patients, between 12 and 24 hours after administration of 5 g idarucizumab, elevated coagulation parameters (e.g., aPTT or ECT) have been observed. Eight patients were treated with more than 5 g idarucizumab due to rebleeding, a second emergency procedure and/or bleeding after an emergency surgical procedure. ECT measures over the 24-hour observation time are shown in Figure 6.
Figure 6 Change of ECT from Baseline in Dabigatran-exposed Patients (n=487)
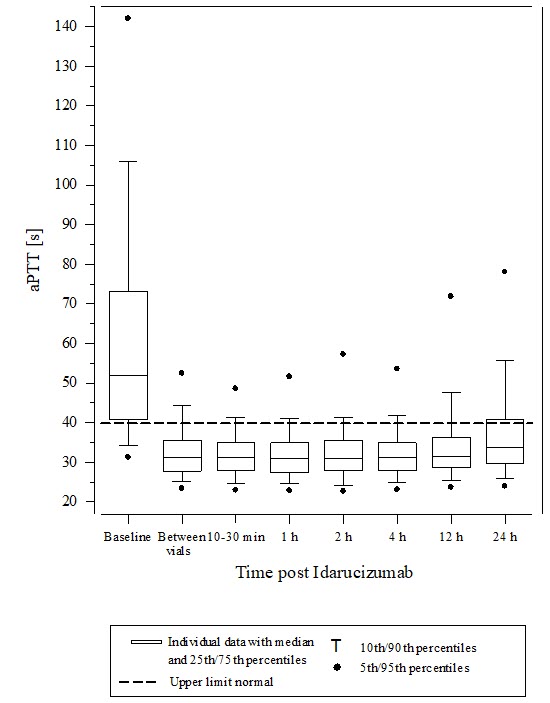
Activated partial thromboplastin time (aPTT) showed similar results to ECT (see Figure 7).
Figure 7 Change of aPTT from Baseline in Dabigatran-exposed Patients (n=486)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
- PRAXBIND is a sterile, preservative-free, colorless to slightly yellow, clear to slightly opalescent solution supplied as 2 single-dose vials each containing 2.5 g/50 mL of idarucizumab.
- NDC number 0597-0197-05: Carton containing two 2.5 g/50 mL vials.
16.2 Storage and Handling
- Store PRAXBIND vials in the refrigerator at 2°C to 8°C (36°F to 46°F) in the original carton to protect from light. Do not freeze. Do not shake.
- PRAXBIND vials may be stored at room temperature, 25°C (77°F), for up to 48 hours in the original carton to protect from light.
- PRAXBIND vials may be stored at room temperature, 25°C (77°F), out of the carton and exposed to light but must be used within 6 hours [see Dosage and Administration (2.2)].
-
17 PATIENT COUNSELING INFORMATION
Thromboembolic Risk
Inform patients that reversing dabigatran therapy exposes them to the thromboembolic risk of their underlying disease. To reduce this risk, resumption of anticoagulant therapy should be considered as soon as the patient is sufficiently stable [see Warnings and Precautions (5.1)].
Recurrence of Bleeding
Inform patients to get immediate medical attention for any signs or symptoms of bleeding [see Warnings and Precautions (5.2)].
Hypersensitivity Reactions
Inform patients of signs and symptoms of allergic hypersensitivity reactions such as anaphylactoid reactions that may be experienced during or after injection of PRAXBIND [see Warnings and Precautions (5.3)].
Risk of Serious Adverse Reactions in Patients with Hereditary Fructose Intolerance due to Sorbitol Excipient
Inform patients with hereditary fructose intolerance (HFI) that PRAXBIND contains sorbitol. Parenteral administration of sorbitol in patients who have HFI has been associated with reports of hypoglycemia, hypophosphatemia, metabolic acidosis, increase in uric acid, acute liver failure with breakdown of excretory and synthetic function, and death and may occur during or after injection of PRAXBIND [see Warnings and Precautions (5.4)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 50 mL Vial Label
-
INGREDIENTS AND APPEARANCE
PRAXBIND
idarucizumab injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0597-0197 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IDARUCIZUMAB (UNII: 97RWB5S1U6) (IDARUCIZUMAB - UNII:97RWB5S1U6) IDARUCIZUMAB 50 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0597-0197-05 2 in 1 CARTON 10/21/2015 1 50 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761025 10/21/2015 Labeler - Boehringer Ingelheim Pharmaceuticals, Inc. (603175944) Registrant - Boehringer Ingelheim Pharmaceuticals, Inc. (603175944) Establishment Name Address ID/FEI Business Operations Boehringer Ingelheim Pharma GmbH and Co. KG 340700520 PACK(0597-0197) , API MANUFACTURE(0597-0197) , LABEL(0597-0197) , ANALYSIS(0597-0197) , MANUFACTURE(0597-0197)