Label: TYSABRI- natalizumab injection


- NDC Code(s): 64406-008-01
- Packager: Biogen Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated October 27, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TYSABRI safely and effectively. See full prescribing information for TYSABRI.
TYSABRI (natalizumab) injection, for intravenous use
Initial U.S. Approval: 2004WARNING: PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
See full prescribing information for complete boxed warning
- TYSABRI increases the risk of progressive multifocal leukoencephalopathy (PML), an opportunistic viral infection of the brain that usually leads to death or severe disability (5.1)
- Risk factors for the development of PML include the presence of anti-JCV antibodies, duration of therapy, and prior use of immunosuppressants. These factors should be considered in the context of expected benefit when initiating and continuing treatment with TYSABRI (5.1)
- Monitor patients, and withhold TYSABRI immediately at the first sign or symptom suggestive of PML (4, 5.1)
- Because of the risk of PML, TYSABRI is available only through a restricted distribution program called the TOUCH® Prescribing Program (5.1, 5.2)
INDICATIONS AND USAGE
TYSABRI is an integrin receptor antagonist indicated for treatment of:
Multiple Sclerosis (MS)
TYSABRI is indicated as monotherapy for the treatment of relapsing forms of multiple sclerosis, to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. TYSABRI increases the risk of PML [See Warnings and Precautions (5.1)]. When initiating and continuing treatment with TYSABRI, physicians should consider whether the expected benefit of TYSABRI is sufficient to offset this risk. (1.1)
Crohn's Disease (CD)
- TYSABRI is indicated for inducing and maintaining clinical response and remission in adult patients with moderately to severely active Crohn's disease with evidence of inflammation who have had an inadequate response to, or are unable to tolerate, conventional CD therapies and inhibitors of TNF-α. (1.2)
Important Limitations:
- In CD, TYSABRI should not be used in combination with immunosuppressants or inhibitors of TNF-α. (1.2)
DOSAGE AND ADMINISTRATION
- 300 mg infused intravenously over one hour, every four weeks. Do not give as an intravenous push or bolus (2.1, 2.2)
- TYSABRI solution must be administered within 48 hours of preparation (2.3)
- Observe patients during all infusions. Post-infusion, for the first 12 infusions, observe patients for one hour after the infusion is complete. For patients who have received 12 infusions without evidence of a hypersensitivity reaction, observe patients post-infusion for the 13th and subsequent infusions according to clinical judgment. (2.4)
- In CD, discontinue in patients that have not experienced therapeutic benefit by 12 weeks of induction therapy, and in patients that cannot discontinue chronic concomitant steroids within six months of starting therapy (2.2)
DOSAGE FORMS AND STRENGTHS
Injection: 300 mg/15 mL (20 mg/mL) solution in a single-dose vial for dilution prior to infusion (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Herpes infections: Life-threatening and fatal cases have occurred with herpes encephalitis and meningitis infections. Blindness has occurred in patients developing acute retinal necrosis. Discontinue TYSABRI if these infections occur and treat appropriately (5.3)
- Hepatotoxicity: Significant liver injury, including liver failure requiring transplant, has occurred. Discontinue TYSABRI in patients with evidence of liver injury (5.4)
- Hypersensitivity reactions: Serious hypersensitivity reactions (e.g., anaphylaxis) have occurred. Permanently discontinue TYSABRI if such a reaction occurs (5.5)
- Immunosuppression/Infections: TYSABRI may increase the risk for certain infections. Monitor patients for development of infections due to increased risk with use of TYSABRI (5.6)
- Thrombocytopenia: TYSABRI may cause thrombocytopenia. Monitor patients for bleeding abnormalities. Discontinue TYSABRI in patients with thrombocytopenia (5.8)
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 10%):
- MS - headache, fatigue, arthralgia, urinary tract infection, lower respiratory tract infection, gastroenteritis, vaginitis, depression, pain in extremity, abdominal discomfort, diarrhea NOS, and rash (6.1)
- CD - headache, upper respiratory tract infections, nausea, and fatigue (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Biogen at 1-800-456-2255 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
1 INDICATIONS AND USAGE
1.1 Multiple Sclerosis (MS)
1.2 Crohn's Disease (CD)
2 DOSAGE AND ADMINISTRATION
2.1 Multiple Sclerosis (MS)
2.2 Crohn's Disease (CD)
2.3 Dilution Instructions
2.4 Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Progressive Multifocal Leukoencephalopathy
5.2 TYSABRI TOUCH® Prescribing Program
5.3 Herpes Infections
5.4 Hepatotoxicity
5.5 Hypersensitivity/Antibody Formation
5.6 Immunosuppression/Infections
5.7 Laboratory Test Abnormalities
5.8 Thrombocytopenia
5.9 Immunizations
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Multiple Sclerosis
14.2 Crohn's Disease
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
TYSABRI increases the risk of progressive multifocal leukoencephalopathy (PML), an opportunistic viral infection of the brain that usually leads to death or severe disability. Risk factors for the development of PML include the presence of anti-JCV antibodies, duration of therapy, and prior use of immunosuppressants. These factors should be considered in the context of expected benefit when initiating and continuing treatment with TYSABRI [see Warnings and Precautions (5.1)].
- Healthcare professionals should monitor patients on TYSABRI for any new sign or symptom that may be suggestive of PML. TYSABRI dosing should be withheld immediately at the first sign or symptom suggestive of PML. For diagnosis, an evaluation that includes a gadolinium-enhanced magnetic resonance imaging (MRI) scan of the brain and, when indicated, cerebrospinal fluid analysis for JC viral DNA are recommended [see Contraindications (4), Warnings and Precautions (5.1)].
- Because of the risk of PML, TYSABRI is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the TOUCH® Prescribing Program [see Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
1.1 Multiple Sclerosis (MS)
TYSABRI is indicated as monotherapy for the treatment of relapsing forms of multiple sclerosis, to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults. TYSABRI increases the risk of PML [see Warnings and Precautions (5.1)]. When initiating and continuing treatment with TYSABRI, physicians should consider whether the expected benefit of TYSABRI is sufficient to offset this risk.
1.2 Crohn's Disease (CD)
TYSABRI is indicated for inducing and maintaining clinical response and remission in adult patients with moderately to severely active Crohn's disease with evidence of inflammation who have had an inadequate response to, or are unable to tolerate, conventional CD therapies and inhibitors of TNF-α. TYSABRI should not be used in combination with immunosuppressants (e.g., 6-mercaptopurine, azathioprine, cyclosporine, or methotrexate) or inhibitors of TNF-α [see Warnings and Precautions (5.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Multiple Sclerosis (MS)
Only prescribers registered in the MS TOUCH® Prescribing Program may prescribe TYSABRI for multiple sclerosis [see Warnings and Precautions (5.2)]. The recommended dose of TYSABRI for multiple sclerosis is 300 mg intravenous infusion over one hour every four weeks.
2.2 Crohn's Disease (CD)
Only prescribers registered in the CD TOUCH® Prescribing Program may prescribe TYSABRI for Crohn's disease [see Warnings and Precautions (5.2)].
The recommended dose of TYSABRI for Crohn's disease is 300 mg intravenous infusion over one hour every four weeks. TYSABRI should not be used with concomitant immunosuppressants (e.g., 6-mercaptopurine, azathioprine, cyclosporine, or methotrexate) or concomitant inhibitors of TNF-α. Aminosalicylates may be continued during treatment with TYSABRI.
If the patient with Crohn's disease has not experienced therapeutic benefit by 12 weeks of induction therapy, discontinue TYSABRI. For patients with Crohn's disease who start TYSABRI while on chronic oral corticosteroids, commence steroid tapering as soon as a therapeutic benefit of TYSABRI has occurred; if the patient with Crohn's disease cannot be tapered off of oral corticosteroids within six months of starting TYSABRI, discontinue TYSABRI. Other than the initial six-month taper, prescribers should consider discontinuing TYSABRI for patients who require additional steroid use that exceeds three months in a calendar year to control their Crohn's disease.
2.3 Dilution Instructions
- Use aseptic technique when preparing TYSABRI solution for intravenous infusion. Each vial is intended for single use only. Discard any unused portion.
- TYSABRI is a colorless, clear to slightly opalescent solution. Inspect the TYSABRI vial for particulate material and discoloration prior to dilution and administration. If visible particulates are observed and/or the liquid in the vial is discolored, the vial must not be used.
- To prepare the diluted solution, withdraw 15 mL of TYSABRI from the vial using a sterile needle and syringe. Inject TYSABRI into 100 mL of 0.9% Sodium Chloride Injection, USP. No other intravenous diluents may be used to prepare the TYSABRI diluted solution.
- Gently invert the TYSABRI diluted solution to mix completely. Do not shake. Inspect the solution visually for particulate material prior to administration.
- The final dosage diluted solution has a concentration of 2.6 mg/mL.
- Following dilution, infuse TYSABRI solution immediately, or refrigerate the diluted solution at 2°C to 8°C, and use within 48 hours. If stored at 2°C to 8°C, allow the diluted solution to warm to room temperature prior to infusion. DO NOT FREEZE.
2.4 Administration Instructions
- Infuse TYSABRI 300 mg in 100 mL 0.9% Sodium Chloride Injection, USP, over approximately one hour (infusion rate approximately 5 mg per minute). Do not administer TYSABRI as an intravenous push or bolus injection. After the infusion is complete, flush with 0.9% Sodium Chloride Injection, USP.
- Observe patients during all infusions. Promptly discontinue the infusion upon the first observation of any signs or symptoms consistent with a hypersensitivity-type reaction [see Warnings and Precautions (5.5)].
- Post-infusion, for the first 12 infusions, observe patients for one hour after the infusion is complete. For patients who have received 12 infusions without evidence of a hypersensitivity reaction, observe patients post-infusion for the 13th and subsequent infusions according to clinical judgment.
- Use of filtration devices during administration has not been evaluated. Other medications should not be injected into infusion set side ports or mixed with TYSABRI.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- TYSABRI is contraindicated in patients who have or have had progressive multifocal leukoencephalopathy (PML) [see Warnings and Precautions (5.1)].
- TYSABRI is contraindicated in patients who have had a hypersensitivity reaction to TYSABRI. Observed reactions range from urticaria to anaphylaxis [see Warnings and Precautions (5.5)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Progressive Multifocal Leukoencephalopathy
Progressive multifocal leukoencephalopathy (PML), an opportunistic viral infection of the brain caused by the JC virus (JCV) that typically only occurs in patients who are immunocompromised, and that usually leads to death or severe disability, has occurred in patients who have received TYSABRI.
Three factors that are known to increase the risk of PML in TYSABRI-treated patients have been identified:
- The presence of anti-JCV antibodies. Patients who are anti-JCV antibody positive have a higher risk for developing PML.
- Longer treatment duration, especially beyond 2 years.
- Prior treatment with an immunosuppressant (e.g., mitoxantrone, azathioprine, methotrexate, cyclophosphamide, mycophenolate mofetil).
These factors should be considered in the context of expected benefit when initiating and continuing treatment with TYSABRI.
Table 1: Estimated United States Incidence of PML Stratified by Risk Factor Notes: The risk estimates are based on postmarketing data in the United States from approximately 100,000 TYSABRI exposed patients.
The anti-JCV antibody status was determined using an anti-JCV antibody test (ELISA) that has been analytically and clinically validated and is configured with detection and inhibition steps to confirm the presence of JCV-specific antibodies with an analytical false negative rate of 3%.
Anti-JCV Antibody Negative TYSABRI Exposure Anti-JCV Antibody Positive No Prior Immunosuppressant Use Prior Immunosuppressant Use 1/10,000 1-24 months <1/1,000 1/1,000 25-48 months 2/1,000 6/1,000 49-72 months 4/1,000 7/1,000 73-96 months 2/1,000 6/1,000 Retrospective analyses of postmarketing data from various sources, including observational studies and spontaneous reports obtained worldwide, suggest that the risk of developing PML may be associated with relative levels of serum anti-JCV antibody compared to a calibrator as measured by ELISA (often described as an anti-JCV antibody index value).
Ordinarily, patients receiving chronic immunosuppressant or immunomodulatory therapy or who have systemic medical conditions resulting in significantly compromised immune system function should not be treated with TYSABRI. Infection by the JC virus is required for the development of PML. Anti-JCV antibody testing should not be used to diagnose PML. Anti-JCV antibody negative status indicates that antibodies to the JC virus have not been detected. Patients who are anti-JCV antibody negative have a lower risk of PML than those who are positive. Patients who are anti-JCV antibody negative are still at risk for the development of PML due to the potential for a new JCV infection or a false negative test result. The reported rate of seroconversion in patients with MS (changing from anti-JCV antibody negative to positive and remaining positive in subsequent testing) is 3 to 8 percent annually. In addition, some patients' serostatus may change intermittently. Therefore, patients with a negative anti-JCV antibody test result should be retested periodically. For purposes of risk assessment, a patient with a positive anti-JCV antibody test at any time is considered anti-JCV antibody positive regardless of the results of any prior or subsequent anti-JCV antibody testing. When assessed, anti-JCV antibody status should be determined using an analytically and clinically validated immunoassay. After plasma exchange (PLEX), wait at least two weeks to test for anti-JCV antibodies to avoid false negative test results caused by the removal of serum antibodies. After infusion of intravenous immunoglobulin (IVIg), wait at least 6 months (5 half-lives) for the IVIg to clear in order to avoid false positive anti-JCV antibody test results.
Healthcare professionals should monitor patients on TYSABRI for any new sign or symptom suggestive of PML. Symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes. The progression of deficits usually leads to death or severe disability over weeks or months. Withhold TYSABRI dosing immediately and perform an appropriate diagnostic evaluation at the first sign or symptom suggestive of PML.
MRI findings may be apparent before clinical signs or symptoms. Cases of PML, diagnosed based on MRI findings and the detection of JCV DNA in the cerebrospinal fluid in the absence of clinical signs or symptoms specific to PML, have been reported. Many of these patients subsequently became symptomatic with PML. Therefore, monitoring with MRI for signs that may be consistent with PML may be useful, and any suspicious findings should lead to further investigation to allow for an early diagnosis of PML, if present. Consider monitoring patients at high risk for PML more frequently. Lower PML-related mortality and morbidity have been reported following TYSABRI discontinuation in patients with PML who were initially asymptomatic compared to patients with PML who had characteristic clinical signs and symptoms at diagnosis. It is not known whether these differences are due to early detection and discontinuation of TYSABRI or due to differences in disease in these patients.
There are no known interventions that can reliably prevent PML or that can adequately treat PML if it occurs. PML has been reported following discontinuation of TYSABRI in patients who did not have findings suggestive of PML at the time of discontinuation. Patients should continue to be monitored for any new signs or symptoms that may be suggestive of PML for at least six months following discontinuation of TYSABRI.
Because of the risk of PML, TYSABRI is available only under a restricted distribution program, the TOUCH® Prescribing Program.
In multiple sclerosis patients, an MRI scan should be obtained prior to initiating therapy with TYSABRI. This MRI may be helpful in differentiating subsequent multiple sclerosis symptoms from PML.
In Crohn's disease patients, a baseline brain MRI may also be helpful to distinguish pre-existent lesions from newly developed lesions, but brain lesions at baseline that could cause diagnostic difficulty while on TYSABRI therapy are uncommon.
For diagnosis of PML, an evaluation including a gadolinium-enhanced MRI scan of the brain and, when indicated, cerebrospinal fluid analysis for JC viral DNA are recommended. If the initial evaluations for PML are negative but clinical suspicion for PML remains, continue to withhold TYSABRI dosing, and repeat the evaluations.
There are no known interventions that can adequately treat PML if it occurs. Three sessions of PLEX over 5 to 8 days were shown to accelerate TYSABRI clearance in a study of 12 patients with MS who did not have PML, although in the majority of patients, alpha-4 integrin receptor binding remained high. Adverse events which may occur during PLEX include clearance of other medications and volume shifts, which have the potential to lead to hypotension or pulmonary edema. Although PLEX has not been prospectively studied in TYSABRI-treated patients with PML, it has been used in such patients in the postmarketing setting to remove TYSABRI more quickly from the circulation. There is no evidence that PLEX has any benefit in the treatment of opportunistic infections such as PML.
JC virus infection of granule cell neurons in the cerebellum (i.e., JC virus granule cell neuronopathy [JCV GCN]) has been reported in patients treated with TYSABRI. JCV GCN can occur with or without concomitant PML. JCV GCN can cause cerebellar dysfunction (e.g., ataxia, incoordination, apraxia, visual disorders), and neuroimaging can show cerebellar atrophy. For diagnosis of JCV GCN, an evaluation that includes a gadolinium-enhanced MRI scan of the brain and, when indicated, cerebrospinal fluid analysis for JC viral DNA, is recommended. JCV GCN should be managed similarly to PML.
Immune reconstitution inflammatory syndrome (IRIS) has been reported in the majority of TYSABRI treated patients who developed PML and subsequently discontinued TYSABRI. In almost all cases, IRIS occurred after PLEX was used to eliminate circulating TYSABRI. It presents as a clinical decline in the patient's condition after TYSABRI removal (and in some cases after apparent clinical improvement) that may be rapid, can lead to serious neurological complications or death, and is often associated with characteristic changes in the MRI. TYSABRI has not been associated with IRIS in patients discontinuing treatment with TYSABRI for reasons unrelated to PML. In TYSABRI treated patients with PML, IRIS has been reported within days to several weeks after PLEX. Monitoring for development of IRIS and appropriate treatment of the associated inflammation should be undertaken.
5.2 TYSABRI TOUCH® Prescribing Program
TYSABRI is available only through a restricted program under a REMS called the TOUCH® Prescribing Program because of the risk of PML [see Warnings and Precautions (5.1)].
For prescribers and patients, the TOUCH® Prescribing Program has two components: MS TOUCH® (for patients with multiple sclerosis) and CD TOUCH® (for patients with Crohn's disease).
Selected requirements of the TOUCH® Prescribing Program include the following:
- Prescribers must be certified and comply with the following:
- –
- Review the TOUCH® Prescribing Program prescriber educational materials, including the full prescribing information.
- –
- Review, complete, and sign the Prescriber Enrollment Form.
- –
- Educate patients on the benefits and risks of treatment with TYSABRI, ensure that patients receive the Medication Guide, and encourage them to ask questions.
- –
- Review, complete, and sign the Patient Enrollment Form for each patient.
- –
- Evaluate patients three months after the first infusion, six months after the first infusion, every six months thereafter, and for at least six months after discontinuing TYSABRI.
- –
- Determine every six months whether patients should continue on treatment and, if so, authorize treatment for another six months.
- –
- Submit to Biogen the “TYSABRI Patient Status Report and Reauthorization Questionnaire” six months after initiating treatment and every six months thereafter.
- –
- Complete an “Initial Discontinuation Questionnaire” when TYSABRI is discontinued, and a “6-Month Discontinuation Questionnaire” following discontinuation of TYSABRI.
- –
- Report cases of PML, hospitalizations due to opportunistic infections, and deaths to Biogen at 1-800-456-2255 as soon as possible.
- Patients must be enrolled in the TOUCH® Prescribing Program, read the Medication Guide, understand the risks associated with TYSABRI, and complete and sign the Patient Enrollment Form.
- Pharmacies and infusion centers must be specially certified to dispense or infuse TYSABRI.
5.3 Herpes Infections
Herpes Encephalitis and Meningitis
TYSABRI increases the risk of developing encephalitis and meningitis caused by herpes simplex and varicella zoster viruses. Serious, life-threatening, and sometimes fatal cases have been reported in the postmarketing setting in multiple sclerosis patients receiving TYSABRI. Laboratory confirmation in those cases was based on positive PCR for viral DNA in the cerebrospinal fluid. The duration of treatment with TYSABRI prior to onset ranged from a few months to several years. Monitor patients receiving TYSABRI for signs and symptoms of meningitis and encephalitis. If herpes encephalitis or meningitis occurs, TYSABRI should be discontinued, and appropriate treatment for herpes encephalitis/meningitis should be administered.
Acute Retinal Necrosis
Acute retinal necrosis (ARN) is a fulminant viral infection of the retina caused by the family of herpes viruses (e.g., varicella zoster, herpes simplex virus). A higher risk of ARN has been observed in patients being administered TYSABRI. Patients presenting with eye symptoms, including decreased visual acuity, redness, or eye pain, should be referred for retinal screening for ARN. Some ARN cases occurred in patients with central nervous system (CNS) herpes infections (e.g., herpes meningitis or encephalitis). Serious cases of ARN led to blindness of one or both eyes in some patients. Following clinical diagnosis of ARN, consider discontinuation of TYSABRI. The treatment reported in ARN cases included anti-viral therapy and, in some cases, surgery.
5.4 Hepatotoxicity
Clinically significant liver injury, including acute liver failure requiring transplant, has been reported in patients treated with TYSABRI in the postmarketing setting. Signs of liver injury, including markedly elevated serum hepatic enzymes and elevated total bilirubin, occurred as early as six days after the first dose; signs of liver injury have also been reported for the first time after multiple doses. In some patients, liver injury recurred upon rechallenge, providing evidence that TYSABRI caused the injury. The combination of transaminase elevations and elevated bilirubin without evidence of obstruction is generally recognized as an important predictor of severe liver injury that may lead to death or the need for a liver transplant in some patients.
TYSABRI should be discontinued in patients with jaundice or other evidence of significant liver injury (e.g., laboratory evidence).
5.5 Hypersensitivity/Antibody Formation
Hypersensitivity reactions have occurred in patients receiving TYSABRI, including serious systemic reactions (e.g., anaphylaxis), which occurred at an incidence of <1%. These reactions usually occur within two hours of the start of the infusion. Symptoms associated with these reactions can include urticaria, dizziness, fever, rash, rigors, pruritus, nausea, flushing, hypotension, dyspnea, and chest pain. Generally, these reactions are associated with antibodies to TYSABRI.
If a hypersensitivity reaction occurs, discontinue administration of TYSABRI, and initiate appropriate therapy. Patients who experience a hypersensitivity reaction should not be re-treated with TYSABRI. Hypersensitivity reactions were more frequent in patients with antibodies to TYSABRI compared to patients who did not develop antibodies to TYSABRI in both MS and CD studies. Therefore, the possibility of antibodies to TYSABRI should be considered in patients who have hypersensitivity reactions [see Adverse Reactions (6.2)].
Antibody testing: If the presence of persistent antibodies is suspected, antibody testing should be performed. Antibodies may be detected and confirmed with sequential serum antibody tests. Antibodies detected early in the treatment course (e.g., within the first six months) may be transient and may disappear with continued dosing. It is recommended that testing be repeated three months after an initial positive result to confirm that antibodies are persistent. Prescribers should consider the overall benefits and risks of TYSABRI in a patient with persistent antibodies.
Patients who receive TYSABRI for a short exposure (1 to 2 infusions) followed by an extended period without treatment are at higher risk of developing anti-natalizumab antibodies and/or hypersensitivity reactions on re-exposure, compared to patients who received regularly scheduled treatment. Given that patients with persistent antibodies to TYSABRI experience reduced efficacy, and that hypersensitivity reactions are more common in such patients, consideration should be given to testing for the presence of antibodies in patients who wish to recommence therapy following a dose interruption. Following a period of dose interruption, patients testing negative for antibodies prior to re-dosing have a risk of antibody development with re-treatment that is similar to TYSABRI naïve patients [see Adverse Reactions (6.2)].
5.6 Immunosuppression/Infections
The immune system effects of TYSABRI may increase the risk for infections. In Study MS1 [see Clinical Studies (14.1)], certain types of infections, including pneumonias and urinary tract infections (including serious cases), gastroenteritis, vaginal infections, tooth infections, tonsillitis, and herpes infections, occurred more often in TYSABRI-treated patients than in placebo-treated patients [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. One opportunistic infection, a cryptosporidial gastroenteritis with a prolonged course, was observed in a patient who received TYSABRI in Study MS1.
In Studies MS1 and MS2, an increase in infections was seen in patients concurrently receiving short courses of corticosteroids. However, the increase in infections in TYSABRI-treated patients who received steroids was similar to the increase in placebo-treated patients who received steroids.
In a long-term safety study of patients treated with TYSABRI for multiple sclerosis, opportunistic infections (pulmonary mycobacterium avium intracellulare, aspergilloma, cryptococcal fungemia and meningitis, and Candida pneumonia) have been observed in <1% of TYSABRI-treated patients.
In CD clinical studies, opportunistic infections (pneumocystis carinii pneumonia, pulmonary mycobacterium avium intracellulare, bronchopulmonary aspergillosis, and burkholderia cepacia) have been observed in <1% of TYSABRI-treated patients; some of these patients were receiving concurrent immunosuppressants [see Warnings and Precautions (5.1), Adverse Reactions (6.1)].
In Studies CD1 and CD2, an increase in infections was seen in patients concurrently receiving corticosteroids. However, the increase in infections was similar in placebo-treated and TYSABRI-treated patients who received steroids. Concurrent use of antineoplastic, immunosuppressant, or immunomodulating agents may further increase the risk of infections, including PML and other opportunistic infections, over the risk observed with use of TYSABRI alone [see Warnings and Precautions (5.1), Adverse Reactions (6.1)]. The safety and efficacy of TYSABRI in combination with antineoplastic, immunosuppressant, or immunomodulating agents have not been established. Patients receiving chronic immunosuppressant or immunomodulatory therapy or who have systemic medical conditions resulting in significantly compromised immune system function should not ordinarily be treated with TYSABRI. The risk of PML is also increased in patients who have been treated with an immunosuppressant prior to receiving TYSABRI [see Warnings and Precautions (5.1)].
For patients with Crohn's disease who start TYSABRI while on chronic corticosteroids, commence steroid withdrawal as soon as a therapeutic benefit has occurred. If the patient cannot discontinue systemic corticosteroids within six months, discontinue TYSABRI.
5.7 Laboratory Test Abnormalities
In clinical trials, TYSABRI was observed to induce increases in circulating lymphocytes, monocytes, eosinophils, basophils, and nucleated red blood cells. Observed changes persisted during TYSABRI exposure, but were reversible, returning to baseline levels usually within 16 weeks after the last dose. Elevations of neutrophils were not observed. TYSABRI induces mild decreases in hemoglobin levels (mean decrease of 0.6 g/dL) that are frequently transient.
5.8 Thrombocytopenia
Cases of thrombocytopenia, including immune thrombocytopenic purpura (ITP), have been reported with the use of TYSABRI in the postmarketing setting. Symptoms of thrombocytopenia may include easy bruising, abnormal bleeding, and petechiae. Delay in the diagnosis and treatment of thrombocytopenia may lead to serious and life-threatening sequelae. If thrombocytopenia is suspected, TYSABRI should be discontinued.
Cases of neonatal thrombocytopenia, at times associated with anemia, have been reported in newborns with in utero exposure to TYSABRI [see Use in Specific Populations (8.1)]. A CBC should be obtained in neonates with in utero exposure to TYSABRI.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described below and elsewhere in the labeling:
- Progressive Multifocal Leukoencephalopathy (PML) [see Warnings and Precautions (5.1)]
- Herpes Infections [see Warnings and Precautions (5.3)]
- Hepatotoxicity [see Warnings and Precautions (5.4)]
- Hypersensitivity/Antibody Formation [see Warnings and Precautions (5.5 )]
- Immunosuppression/Infections [see Warnings and Precautions (5.6 )]
- Thrombocytopenia [see Warnings and Precautions (5.8 )]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The most common adverse reactions (incidence ≥ 10%) were headache and fatigue in both the multiple sclerosis (MS) and Crohn's disease (CD) studies. Other common adverse reactions (incidence ≥ 10%) in the MS population were arthralgia, urinary tract infection, lower respiratory tract infection, gastroenteritis, vaginitis, depression, pain in extremity, abdominal discomfort, diarrhea NOS, and rash. Other common adverse reactions (incidence ≥ 10%) in the CD population were upper respiratory tract infections and nausea.
The most frequently reported adverse reactions resulting in clinical intervention (i.e., discontinuation of TYSABRI) in the MS studies were urticaria (1%) and other hypersensitivity reactions (1%), and in the CD studies (Studies CD1 and CD2) were the exacerbation of Crohn's disease (4.2%) and acute hypersensitivity reactions (1.5%) [see Warnings and Precautions (5.5)].
A total of 1617 multiple sclerosis patients in controlled studies received TYSABRI, with a median duration of exposure of 28 months. A total of 1563 patients received TYSABRI in all CD studies for a median exposure of 5 months; of these patients, 33% (n=518) received at least one year of treatment and 19% (n=297) received at least two years of treatment.
Multiple Sclerosis Clinical Studies
The most common serious adverse reactions in Study MS1 [see Clinical Studies (14.1)] with TYSABRI were infections (3.2% versus 2.6% in placebo, including urinary tract infection [0.8% versus 0.3%] and pneumonia [0.6% versus 0%]), acute hypersensitivity reactions (1.1% versus 0.3%, including anaphylaxis/anaphylactoid reaction [0.8% versus 0%]), depression (1.0% versus 1.0%, including suicidal ideation or attempt [0.6% versus 0.3%]), and cholelithiasis (1.0% versus 0.3%). In Study MS2, serious adverse reactions of appendicitis were also more common in patients who received TYSABRI (0.8% versus 0.2% in placebo).
Table 2 enumerates adverse reactions and selected laboratory abnormalities that occurred in Study MS1 at an incidence of at least 1 percentage point higher in TYSABRI-treated patients than was observed in placebo-treated patients.
Table 2: Adverse Reactions in Study MS1 (Monotherapy Study) Adverse Reactions
(Preferred Term)TYSABRI
n=627
%Placebo
n=312
%*Percentage based on female patients only.
** Acute versus other hypersensitivity reactions are defined as occurring within 2 hours post-infusion versus more than 2 hours.
General
Headache
38
33
Fatigue
27
21
Arthralgia
19
14
Chest discomfort
5
3
Other hypersensitivity reactions**
5
2
Acute hypersensitivity reactions**
4
<1
Seasonal allergy
3
2
Rigors
3
<1
Weight increased
2
<1
Weight decreased
2
<1
Infection Urinary tract infection
21
17
Lower respiratory tract infection
17
16
Gastroenteritis
11
9
Vaginitis*
10
6
Tooth infections
9
7
Herpes
8
7
Tonsillitis
7
5
Psychiatric Depression
19
16
Musculoskeletal/Connective Tissue Disorders
Pain in extremity
Muscle cramp
Joint swelling16
5
214
3
1
Gastrointestinal
Abdominal discomfort
Diarrhea NOS
Abnormal liver function test
11
10
5
10
9
4Skin
Rash
Dermatitis
Pruritus
Night sweats
12
7
4
1
9
4
2
0
Menstrual Disorders*
Irregular menstruation
Dysmenorrhea
Amenorrhea
Ovarian cyst
5
3
2
2
4
<1
1
<1
Neurologic Disorders
Vertigo
Somnolence
6
2
5
<1
Renal and Urinary Disorders
Urinary urgency/frequency
Urinary incontinence
9
4
7
3
Injury
Limb injury NOS
Skin laceration
Thermal burn
3
2
1
2
<1
<1In Study MS2, peripheral edema was more common in patients who received TYSABRI (5% versus 1% in placebo).
Crohn's Disease Clinical Studies
The following serious adverse reactions in the induction Studies CD1 and CD2 [see Clinical Studies (14.2)] were reported more commonly with TYSABRI than placebo and occurred at an incidence of at least 0.3%: intestinal obstruction or stenosis (2% vs. 1% in placebo), acute hypersensitivity reactions (0.5% vs. 0%), abdominal adhesions (0.3% vs. 0%), and cholelithiasis (0.3% vs. 0%). Similar serious adverse reactions were seen in the maintenance Study CD3. Table 3 enumerates adverse reactions that occurred in Studies CD1 and CD2 (median exposure of 2.8 months). Table 4 enumerates adverse reactions that occurred in Study CD3 (median exposure of 11.0 months).
Table 3: Adverse Reactions in Studies CD1 and CD2 (Induction Studies) Adverse Reactions*
TYSABRI
n=983
%Placebo
n=431
%* Occurred at an incidence of at least 1% higher in TYSABRI-treated patients than placebo-treated patients.
** Percentage based on female patients only.
General
Headache
Fatigue
Arthralgia
Influenza-like illness
Acute hypersensitivity reactions
Tremor
32
10
8
5
2
1
23
8
6
4
<1
<1
Infection
Upper respiratory tract infection
Vaginal infections**
Viral infection
Urinary tract infection
22
4
3
3
16
2
2
1
Respiratory
Pharyngolaryngeal pain
Cough
6
3
4
<1
Gastrointestinal
Nausea
Dyspepsia
Constipation
Flatulence
Aphthous stomatitis
17
5
4
3
2
15
3
2
2
<1
Skin
Rash
Dry skin
6
1
4
0
Menstrual Disorder
Dysmenorrhea**
2
<1
Table 4: Adverse Reactions in Study CD3 (Maintenance Study) Adverse Reactions*
TYSABRI
n=214
%Placebo
n=214
%* Occurred at an incidence of at least 2% higher in TYSABRI-treated patients than placebo-treated patients.
** Percentage based on female patients only.
General
Headache
Influenza-like illness
Peripheral edema
Toothache
37
11
6
4
31
6
3
<1
Infection
Influenza
Sinusitis
Vaginal infections**
Viral infection
12
8
8
7
5
4
<1
3
Respiratory
Cough
7
5
Gastrointestinal
Lower abdominal pain
4
2
Musculoskeletal and Connective Tissue
Back pain
12
8
Menstrual Disorder
Dysmenorrhea**
6
3
Infections
Progressive Multifocal Leukoencephalopathy (PML) occurred in three patients who received TYSABRI in clinical trials [see Warnings and Precautions (5.1)]. Two cases of PML were observed in the 1869 patients with multiple sclerosis who were treated for a median of 120 weeks. These two patients had received TYSABRI in addition to interferon beta-1a [see Warnings and Precautions (5.1)]. The third case occurred after eight doses in one of the 1043 patients with Crohn's disease who were evaluated for PML. In the postmarketing setting, additional cases of PML have been reported in TYSABRI-treated multiple sclerosis and Crohn's disease patients who were not receiving concomitant immunomodulatory therapy.
In Studies MS1 and MS2 [see Clinical Studies (14.1)], the rate of any type of infection was approximately 1.5 per patient-year in both TYSABRI-treated patients and placebo-treated patients. The infections were predominately upper respiratory tract infections, influenza, and urinary tract infections. In Study MS1, the incidence of serious infection was approximately 3% in TYSABRI-treated patients and placebo-treated patients. Most patients did not interrupt treatment with TYSABRI during infections. The only opportunistic infection in the multiple sclerosis clinical trials was a case of cryptosporidial gastroenteritis with a prolonged course.
In Studies CD1 and CD2 [see Clinical Studies (14.2)], the rate of any type of infection was 1.7 per patient-year in TYSABRI-treated patients and 1.4 per patient-year in placebo-treated patients. In Study CD3, the incidence of any type of infection was 1.7 per patient-year in TYSABRI-treated patients and was similar in placebo-treated patients. The most common infections were nasopharyngitis, upper respiratory tract infection, and influenza. The majority of patients did not interrupt TYSABRI therapy during infections, and recovery occurred with appropriate treatment. Concurrent use of TYSABRI in CD clinical trials with chronic steroids and/or methotrexate, 6-MP, and azathioprine did not result in an increase in overall infections compared to TYSABRI alone; however, the concomitant use of such agents could lead to an increased risk of serious infections.
In Studies CD1 and CD2, the incidence of serious infection was approximately 2.1% in both TYSABRI-treated patients and placebo-treated patients. In Study CD3, the incidence of serious infection was approximately 3.3% in TYSABRI-treated patients and approximately 2.8% in placebo-treated patients.
In clinical studies for CD, opportunistic infections (pneumocystis carinii pneumonia, pulmonary mycobacterium avium intracellulare, bronchopulmonary aspergillosis, and burkholderia cepacia) have been observed in <1% of TYSABRI-treated patients; some of these patients were receiving concurrent immunosuppressants [see Warnings and Precautions (5.6)]. Two serious non-bacterial meningitides occurred in TYSABRI-treated patients compared to none in placebo-treated patients.
Infusion-related Reactions
An infusion-related reaction was defined in clinical trials as any adverse event occurring within two hours of the start of an infusion. In MS clinical trials, approximately 24% of TYSABRI-treated multiple sclerosis patients experienced an infusion-related reaction, compared to 18% of placebo-treated patients. In the controlled CD clinical trials, infusion-related reactions occurred in approximately 11% of patients treated with TYSABRI compared to 7% of placebo-treated patients. Reactions more common in the TYSABRI-treated MS patients compared to the placebo-treated MS patients included headache, dizziness, fatigue, urticaria, pruritus, and rigors. Acute urticaria was observed in approximately 2% of patients. Other hypersensitivity reactions were observed in 1% of patients receiving TYSABRI. Serious systemic hypersensitivity infusion reactions occurred in <1% of patients [see Warnings and Precautions (5.5)]. All patients recovered with treatment and/or discontinuation of the infusion.
Infusion-related reactions that were more common in CD patients receiving TYSABRI than those receiving placebo included headache, nausea, urticaria, pruritus, and flushing. Serious infusion reactions occurred in Studies CD1, CD2, and CD3 at an incidence of <1% in TYSABRI-treated patients.
MS and CD patients who became persistently positive for antibodies to TYSABRI were more likely to have an infusion-related reaction than those who were antibody-negative.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to natalizumab in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
Patients in Study MS1 [see Clinical Studies (14.1)] were tested for antibodies to natalizumab every 12 weeks. The assays used were unable to detect low to moderate levels of antibodies to natalizumab. Approximately 9% of patients receiving TYSABRI developed detectable antibodies at least once during treatment. Approximately 6% of patients had positive antibodies on more than one occasion. Approximately 82% of patients who became persistently antibody-positive developed detectable antibodies by 12 weeks. Anti-natalizumab antibodies were neutralizing in vitro.
The presence of anti-natalizumab antibodies was correlated with a reduction in serum natalizumab levels. In Study MS1, the Week 12 pre-infusion mean natalizumab serum concentration in antibody-negative patients was 15 mcg/mL compared to 1.3 mcg/mL in antibody-positive patients. Persistent antibody-positivity resulted in a substantial decrease in the effectiveness of TYSABRI. The risk of increased disability and the annualized relapse rate were similar in persistently antibody-positive TYSABRI-treated patients and patients who received placebo. A similar phenomenon was also observed in Study MS2.
Infusion-related reactions that were most often associated with persistent antibody-positivity included urticaria, rigors, nausea, vomiting, headache, flushing, dizziness, pruritus, tremor, feeling cold, and pyrexia. Additional adverse reactions more common in persistently antibody-positive patients included myalgia, hypertension, dyspnea, anxiety, and tachycardia.
Patients in CD studies [see Clinical Studies (14.2)] were first tested for antibodies at Week 12, and in a substantial proportion of patients, this was the only test performed given the 12-week duration of placebo-controlled studies. Approximately 10% of patients were found to have anti-natalizumab antibodies on at least one occasion. Five percent (5%) of patients had positive antibodies on more than one occasion. Persistent antibodies resulted in reduced efficacy and an increase in infusion-related reactions with symptoms that include urticaria, pruritus, nausea, flushing, and dyspnea.
The long-term immunogenicity of TYSABRI and the effects of low to moderate levels of antibody to natalizumab are unknown [see Warnings and Precautions (5.5), Adverse Reactions (6.1)].
6.3 Postmarketing Experience
The following adverse reactions have been identified during post approval use of TYSABRI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood disorders: hemolytic anemia, thrombocytopenia (including immune thrombocytopenic purpura).
-
7 DRUG INTERACTIONS
Because of the potential for increased risk of PML and other infections, Crohn's disease patients receiving TYSABRI should not be treated with concomitant immunosuppressants (e.g., 6-mercaptopurine, azathioprine, cyclosporine, or methotrexate) or inhibitors of TNF-α, and corticosteroids should be tapered in those patients with Crohn's disease who are on chronic corticosteroids when they start TYSABRI therapy [see Indications and Usage (1.2), Warnings and Precautions (5.1, 5.6)]. Ordinarily, MS patients receiving chronic immunosuppressant or immunomodulatory therapy should not be treated with TYSABRI [see Indications and Usage (1.1), Warnings and Precautions (5.1, 5.6)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the risk of major birth defects, miscarriage, or other adverse maternal outcomes associated with the use of TYSABRI in pregnant women. Adverse fetal outcomes of neonatal thrombocytopenia, at times associated with anemia, have been reported (see Clinical Considerations). In animal studies, administration of natalizumab during pregnancy produced fetal immunologic and hematologic effects in monkeys at doses similar to the human dose and reduced offspring survival in guinea pigs at doses greater than the human dose. These doses were not maternally toxic but produced the expected pharmacological effects in maternal animals [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Cases of neonatal thrombocytopenia, at times associated with anemia, in infants born to women exposed to TYSABRI during pregnancy were reported in the post-marketing setting [see Warnings and Precautions (5.8)]. Therefore, a CBC should be obtained in neonates who were exposed to TYSABRI in utero.
Data
Animal Data
In developmental toxicity studies conducted in guinea pigs and monkeys, at natalizumab doses up to 30 mg/kg (7 times the recommended human dose based on body weight [mg/kg]), transplacental transfer and in utero exposure of the embryo/fetus was demonstrated in both species.
In a study in which pregnant guinea pigs were administered natalizumab (0, 3, 10, or 30 mg/kg) by intravenous (IV) infusion on alternate days throughout organogenesis (gestation days [GD] 4-30), no effects on embryofetal development were observed.
When pregnant monkeys were administered natalizumab (0, 3, 10, or 30 mg/kg) by IV infusion on alternative days throughout organogenesis (GDs 20-70), serum levels in fetuses at delivery were approximately 35% of maternal serum natalizumab levels. There were no effects on embryofetal development; however, natalizumab-related immunological and hematologic changes were observed in the fetuses at the two highest doses. These changes included decreases in lymphocytes (CD3+ and CD20+), changes in lymphocyte subpopulation percentages, mild anemia, reduced platelet count, increased spleen weights, and reduced liver and thymus weights associated with increased splenic extramedullary hematopoiesis, thymic atrophy, and decreased hepatic hematopoiesis.
In a study in which monkeys were exposed to natalizumab during pregnancy (IV infusion of 30 mg/kg) on alternate days from GD20 to GD70 or GD20 to term, abortions were increased approximately 2-fold compared to controls. In offspring born to mothers administered natalizumab on alternate days from GD20 until delivery, hematologic effects (decreased lymphocyte and platelet counts) were also observed. These effects were reversed upon clearance of natalizumab. There was no evidence of anemia in these offspring. Offspring exposed in utero and during lactation had a normal immune response to challenge with a T-cell dependent antigen.
In a study in which pregnant guinea pigs were exposed to natalizumab (30 mg/kg IV) on alternate dates during GDs 30-64, a reduction in pup survival was observed.
8.2 Lactation
Risk Summary
Natalizumab has been detected in human milk. There are no data on the effects of this exposure on the breastfed infant or the effects of the drug on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TYSABRI and any potential adverse effects on the breastfed infant from TYSABRI or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness in pediatric patients with multiple sclerosis or Crohn's disease below the age of 18 years have not been established. TYSABRI is not indicated for use in pediatric patients.
8.5 Geriatric Use
Clinical studies of TYSABRI did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently than younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Natalizumab is a recombinant humanized IgG4κ monoclonal antibody produced in murine myeloma cells. Natalizumab contains human framework regions and the complementarity-determining regions of a murine antibody that binds to α4-integrin. The molecular weight of natalizumab is 149 kilodaltons.
TYSABRI (natalizumab) injection is supplied as a sterile, colorless, and clear to slightly opalescent solution for intravenous infusion. Each 15 mL of solution contains 300 mg natalizumab; sodium chloride, USP (123 mg); sodium phosphate, monobasic, monohydrate, USP (17 mg); sodium phosphate, dibasic, heptahydrate, USP (7.24 mg); polysorbate 80, USP/NF (3 mg), in Water for Injection, USP at pH 6.1.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Natalizumab binds to the α4-subunit of α4β1 and α4β7 integrins expressed on the surface of all leukocytes except neutrophils, and inhibits the α4-mediated adhesion of leukocytes to their counter-receptor(s). The receptors for the α4 family of integrins include vascular cell adhesion molecule-1 (VCAM-1), which is expressed on activated vascular endothelium, and mucosal addressin cell adhesion molecule-1 (MAdCAM-1) present on vascular endothelial cells of the gastrointestinal tract. Disruption of these molecular interactions prevents transmigration of leukocytes across the endothelium into inflamed parenchymal tissue. In vitro, anti-α4-integrin antibodies also block α4-mediated cell binding to ligands such as osteopontin and an alternatively spliced domain of fibronectin, connecting segment-1 (CS-1). In vivo, natalizumab may further act to inhibit the interaction of α4-expressing leukocytes with their ligand(s) in the extracellular matrix and on parenchymal cells, thereby inhibiting further recruitment and inflammatory activity of activated immune cells.
The specific mechanism(s) by which TYSABRI exerts its effects in multiple sclerosis and Crohn's disease have not been fully defined.
In multiple sclerosis, lesions are believed to occur when activated inflammatory cells, including T-lymphocytes, cross the blood-brain barrier (BBB). Leukocyte migration across the BBB involves interaction between adhesion molecules on inflammatory cells and their counter-receptors present on endothelial cells of the vessel wall. The clinical effect of natalizumab in multiple sclerosis may be secondary to blockade of the molecular interaction of α4β1-integrin expressed by inflammatory cells with VCAM-1 on vascular endothelial cells, and with CS-1 and/or osteopontin expressed by parenchymal cells in the brain. Data from an experimental autoimmune encephalitis animal model of multiple sclerosis demonstrate reduction of leukocyte migration into brain parenchyma and reduction of plaque formation detected by magnetic resonance imaging (MRI) following repeated administration of natalizumab. The clinical significance of these animal data is unknown.
In Crohn's disease, the interaction of the α4β7 integrin with the endothelial receptor MAdCAM-1 has been implicated as an important contributor to the chronic inflammation that is a hallmark of the disease. MAdCAM-1 is mainly expressed on gut endothelial cells and plays a critical role in the homing of T lymphocytes to gut lymph tissue found in Peyer's patches. MAdCAM-1 expression has been found to be increased at active sites of inflammation in patients with CD, which suggests it may play a role in the recruitment of leukocytes to the mucosa and contribute to the inflammatory response characteristic of CD. The clinical effect of natalizumab in CD may therefore be secondary to blockade of the molecular interaction of the α4ß7-integrin receptor with MAdCAM-1 expressed on the venular endothelium at inflammatory foci. VCAM-1 expression has been found to be upregulated on colonic endothelial cells in a mouse model of IBD and appears to play a role in leukocyte recruitment to sites of inflammation. The role of VCAM-1 in CD, however, is not clear.
12.2 Pharmacodynamics
TYSABRI administration increases the number of circulating leukocytes (including lymphocytes, monocytes, basophils, and eosinophils) due to inhibition of transmigration out of the vascular space. TYSABRI does not affect the absolute count of circulating neutrophils [see Warnings and Precautions (5.7)].
12.3 Pharmacokinetics
Multiple Sclerosis (MS) Patients:
In patients with MS, following the repeat intravenous administration of a 300 mg dose of TYSABRI, the mean ± SD maximum observed serum concentration was 110 ± 52 mcg/mL. Mean average steady-state trough concentrations ranged from 23 mcg/mL to 29 mcg/mL. The observed time to steady-state was approximately 24 weeks after every four weeks of dosing. The mean ± SD half-life, volume of distribution, and clearance of natalizumab were 11 ± 4 days, 5.7 ± 1.9 L, and 16 ± 5 mL/hour, respectively.
The effects of covariates such as body weight, age, gender, and presence of anti-natalizumab antibodies on natalizumab pharmacokinetics were investigated in a population pharmacokinetic study (n=2195). Natalizumab clearance increased with body weight in a less than proportional manner such that a 43% increase in body weight resulted in a 32% increase in clearance. The presence of persistent anti-natalizumab antibodies increased natalizumab clearance approximately 3-fold [see Adverse Reactions (6.2)].
Crohn's Disease (CD) Patients:
In patients with CD, following the repeat intravenous administration of a 300 mg dose of TYSABRI, the mean ± SD maximum observed serum concentration was 101 ± 34 mcg/mL. The mean ± SD average steady-state trough concentration was 10 ± 9 mcg/mL. The estimated time to steady-state was approximately 16 to 24 weeks after every four weeks of dosing. The mean ± SD half-life, volume of distribution, and clearance of natalizumab were 10 ± 7 days, 5.2 ± 2.8 L, and 22 ± 22 mL/hour, respectively.
The effects of total body weight, age, gender, race, selected hematology and serum chemistry measures, co-administered medications (infliximab, immunosuppressants, or steroids), and the presence of anti-natalizumab antibodies were investigated in a population pharmacokinetic analysis (n=1156). The presence of anti-natalizumab antibodies was observed to increase natalizumab clearance [see Adverse Reactions (6.2)].
Pharmacokinetics of natalizumab in patients with renal or hepatic insufficiency have not been studied.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No clastogenic or mutagenic effects of natalizumab were observed in the Ames test or in vitro chromosomal aberration assay in human lymphocytes. Natalizumab showed no effects in in vitro assays of α4-integrin positive human tumor line proliferation/cytotoxicity. Xenograft transplantation models in SCID and nude mice with two α4-integrin positive human tumor lines (leukemia, melanoma) demonstrated no increase in tumor growth rates or metastasis resulting from natalizumab treatment.
In male guinea pigs administered natalizumab (0, 3, 10, or 30 mg/kg) by intravenous (IV) infusion on alternate days from 28 days prior to and continuing throughout mating (to untreated females), no effects on fertility were observed. The highest dose tested is 6 times the recommended human dose (RHD) (300 mg) on a body weight (mg/kg) basis.
In a separate study in female guinea pigs (mated with untreated males), natalizumab (0, 3, 10, or 30 mg/kg), administered by IV infusion on alternate days from gestation day (GD) 30 of the first pregnancy through GD 30 of the second pregnancy, resulted in a decrease in pregnancy rate and number of implantations at 30 mg/kg. (Fertility parameters were assessed for the second pregnancy.) The no-effect dose for effects on female fertility (10 mg/kg) is 2 times the RHD on a body weight basis.
-
14 CLINICAL STUDIES
14.1 Multiple Sclerosis
TYSABRI was evaluated in two randomized, double-blind, placebo-controlled trials in patients with multiple sclerosis. Both studies enrolled patients who experienced at least one clinical relapse during the prior year and had a Kurtzke Expanded Disability Status Scale (EDSS) score between 0 and 5.0. Results for each study are shown in Table 5 and Table 6. Median time on study drug was 120 weeks in each study. In both studies, neurological evaluations were performed every 12 weeks and at times of suspected relapse. Magnetic resonance imaging evaluations for T1-weighted gadolinium (Gd)-enhancing lesions and T2-hyperintense lesions were performed annually.
Study MS1 enrolled patients who had not received any interferon-beta or glatiramer acetate for at least the previous 6 months; approximately 94% had never been treated with these agents. Median age was 37, with a median disease duration of 5 years. Patients were randomized in a 2:1 ratio to receive TYSABRI 300 mg intravenous infusion (n=627) or placebo (n=315) every 4 weeks for up to 28 months (30 infusions).
Study MS2 enrolled patients who had experienced one or more relapses while on treatment with AVONEX® (Interferon beta-1a) 30 mcg intramuscularly (IM) once weekly during the year prior to study entry. Median age was 39, with a median disease duration of 7 years. Patients were evenly randomized to receive TYSABRI 300 mg (n=589) or placebo (n=582) every 4 weeks for up to 28 months (30 infusions). All patients continued to receive AVONEX 30 mcg IM once weekly. The efficacy of TYSABRI alone was not compared with the efficacy of TYSABRI plus AVONEX.
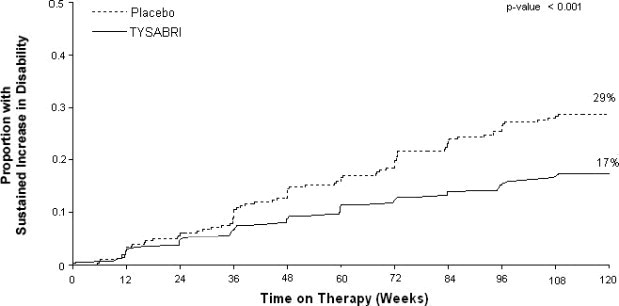
The primary endpoint at 2 years was time to onset of sustained increase in disability, defined as an increase of at least 1 point on the EDSS from baseline EDSS ≥ 1.0 that was sustained for 12 weeks, or at least a 1.5 point increase on the EDSS from baseline EDSS=0 that was sustained for 12 weeks. Time to onset of sustained increase in disability was longer in TYSABRI-treated patients than in placebo-treated patients in Studies MS1 (Figure 1) and MS2. The proportion of patients with increased disability and the annualized relapse rate were also lower in TYSABRI-treated patients than in placebo-treated patients in Studies MS1 and MS2 (Table 5 and Table 6).
Table 5: Clinical and MRI Endpoints in Study MS1 (Monotherapy Study) at 2 Years TYSABRI
n=627Placebo
n=315All analyses were intent-to-treat. For each endpoint, p<0.001. Determination of p-values: Increase in disability by Cox proportional hazards model adjusted for baseline EDSS and age; relapse rate by Poisson regression adjusting for baseline relapse rate, EDSS, presence of Gd-enhancing lesions, age; percentage relapse-free by logistic regression adjusting for baseline relapse rate; and lesion number by ordinal logistic regression adjusting for baseline lesion number.
Annualized relapse rate is calculated as the number of relapses for each subject divided by the number of years followed in the study for that subject. The value reported is the mean across all subjects.
*Values do not total 100% due to rounding.
CLINICAL ENDPOINTS Percentage with sustained increase in disability 17% 29%
Relative Risk Reduction 42% (95% CI 23%, 57%) Annualized relapse rate 0.22 0.67 Relative reduction (percentage) 67% Percentage of patients remaining relapse-free 67% 41% MRI ENDPOINTS New or newly enlarging T2-hyperintense lesions
Median
0.0 5.0 Percentage of patients with*:
0 lesions
57% 15% 1 lesion
17% 10% 2 lesions
8% 8% 3 or more lesions 18% 68% Gd-enhancing lesions
Median
0.0 0.0 Percentage of patients with:
0 lesions
97% 72% 1 lesion
2% 12% 2 or more lesions 1% 16% Table 6: Clinical and MRI Endpoints in Study MS2 (Add-On Study) at 2 Years TYSABRI
plus AVONEX
n= 589Placebo
plus AVONEX
n=582All analyses were intent-to-treat. For disability accumulation p=0.024, for all other endpoints, p<0.001. Determination of p-values: Increase in disability by Cox proportional hazards model adjusted for baseline EDSS; relapse rate by Poisson regression adjusting for baseline relapse rate, EDSS, presence of Gd-enhancing lesions, age; percentage relapse-free by logistic regression adjusting for baseline relapse rate; and lesion number by ordinal logistic regression adjusting for baseline lesion number.
Annualized relapse rate is calculated as the number of relapses for each subject divided by the number of years followed in the study for that subject. The value reported is the mean across all subjects.
*Values do not total 100% due to rounding.
CLINICAL ENDPOINTS Percentage with sustained increase in disability 23% 29%
Relative Risk Reduction 24% (95% CI 4%, 39%) Annualized relapse rate 0.33 0.75 Relative reduction (percentage) 56% Percentage of patients remaining relapse-free 54% 32% MRI ENDPOINTS New or newly enlarging T2-hyperintense lesions
Median
0.0 3.0 Percentage of patients with*:
0 lesions
67% 30% 1 lesion
13% 9% 2 lesions
7% 10% 3 or more lesions 14% 50% Gd-enhancing lesions
Median
0.0 0.0 Percentage of patients with*:
0 lesions
96% 75% 1 lesion
2% 12% 2 or more lesions 1% 14% Figure 1: Time to Increase in Disability Sustained for 12 Weeks in Study MS1
14.2 Crohn's Disease
The safety and efficacy of TYSABRI were evaluated in three randomized, double-blind, placebo-controlled clinical trials in 1414 adult patients with moderately to severely active Crohn's disease (Crohn's Disease Activity Index [CDAI] ≥220 and ≤450) [see References (15)]. Concomitant inhibitors of TNF-α were not permitted. Concomitant stable doses of aminosalicylates, corticosteroids, and/or immunosuppressants (e.g., 6-mercatopurine, azathioprine, or methotrexate) were permitted, and 89% of patients continued to receive at least one of these medications. Although permitted in the clinical trials, combination therapy with immunosuppressants is not recommended [see Indications and Usage (1.2)]. Overall, approximately two-thirds of patients were not taking concomitant immunosuppressants, and approximately one-third of patients were taking neither concomitant immunosuppressants nor concomitant corticosteroids.
Induction of clinical response (defined as ≥70-point decrease in CDAI from baseline) was evaluated in two studies. In Study CD1, 896 patients were randomized 4:1 to receive three monthly infusions of either 300 mg TYSABRI or placebo. Clinical results were assessed at Week 10, and patients with incomplete information were considered as not having a clinical response. At Week 10, 56% of the 717 patients receiving TYSABRI were in response compared to 49% of the 179 patients receiving placebo (treatment effect: 7%; 95% confidence interval (CI): [-1%, 16%]; p=0.067). In a post hoc analysis of the subset of 653 patients with elevated baseline C-reactive protein (CRP), indicative of active inflammation, 57% of TYSABRI patients were in response compared to 45% of those receiving placebo (treatment effect: 12%; 95% CI: [3%, 22%]; nominal p=0.01).
In the second induction trial, Study CD2, only patients with elevated serum C-reactive Protein (CRP) were studied. A total of 509 patients were randomized 1:1 to receive three monthly infusions of either 300 mg TYSABRI or placebo. In Study CD2, in contrast to Study CD1, clinical response and clinical remission (defined as CDAI score <150) were required to be met at both Weeks 8 and 12, rather than at a single time-point; patients with incomplete information were considered as not having a response (Table 7).
Table 7: Induction of Clinical Response and Remission in Study CD2 *p <0.005
Response is defined as a ≥70-point reduction in CDAI score from baseline.
Remission is defined as CDAI <150.
TYSABRI
n=259Placebo
n=250Treatment Difference
(95% CI)Clinical Response at:
Week 8
Week 12
Both Weeks 8 & 12*
56%
60%
48%
40%
44%
32%
16% (8%, 26%)
16% (7%, 25%)
16% (7%, 24%)Clinical Remission at:
Week 8
Week 12
Both Weeks 8 & 12*
32%
37%
26%
21%
25%
16%
11% (3%, 19%)
12% (4%, 21%)
10% (3%, 18%)In studies CD1 and CD2, for subgroups defined by prior use of, or by inadequate response to prior therapies (i.e., corticosteroids, immunosuppressants, and inhibitors of TNF-α), the treatment effect was generally similar to that seen in the whole study population. In the subgroup of patients that were taking neither concomitant immunosuppressants nor concomitant corticosteroids, the treatment effect was generally similar to that seen in the whole study population. Patients with inadequate response to inhibitors of TNF-α appeared to have lower clinical response and lower clinical remission in both the treatment and placebo groups. For patients in Study CD2 with an inadequate response to prior treatment with inhibitors of TNF-α, clinical response at both Weeks 8 and 12 was seen in 38% of those randomized to TYSABRI, and clinical remission at both Weeks 8 and 12 was seen in 17%.
Maintenance therapy was evaluated in Study CD3. In this study, 331 patients from Study CD1 that had had a clinical response to TYSABRI at both Weeks 10 and 12 were re-randomized 1:1 to treatment with continuing monthly infusions of either 300 mg TYSABRI or placebo.
Maintenance of response was assessed by the proportion of patients who did not lose clinical response at any study visit for an additional 6 and 12 months of treatment (i.e., Month 9 and Month 15 after initial treatment with TYSABRI). The study also assessed the proportion of patients who did not lose clinical remission at any study visit within the subset of those who were in remission at study entry. Requiring maintenance of response or remission at each visit, as opposed to just at Month 9 or Month 15, may result in lower proportions meeting endpoint criteria, and may make a comparison of these results with those of other products used to treat Crohn's disease misleading (Table 8).
Table 8: Maintenance of Clinical Response and Remission in Study CD3 *p<0.005
†Number of patients included for analysis of “through” Month 9 and Month 15 includes only those in remission upon entry into Study CD3.
Response is defined as CDAI <220 and a ≥70-point reduction in CDAI score compared to Baseline from Study CD1.
Remission is defined as CDAI <150.
TYSABRI Placebo
Treatment Difference
(95% CI)
Clinical Response through:
Month 9*
Month 15n=164
61%
54%n=167
29%
20%
32% (21%, 43%)
34% (23%, 44%)
Clinical Remission through:
Month 9*
Month 15n=128†
45%
40%n=118†
26%
15%
19% (6%, 31%)
25% (13%, 36%)For subgroups in study CD3 defined by prior use of, or by inadequate response to prior therapies (i.e., corticosteroids, immunosuppressants, and inhibitors of TNF-α), the treatment effect was generally similar to that seen in the whole study population. In the subgroup of patients that were taking neither concomitant immunosuppressants nor concomitant corticosteroids, the treatment effect was generally similar to that seen in the whole study population. Patients with inadequate response to inhibitors of TNF-α appeared to have lower maintenance of clinical response and lower maintenance of clinical remission in both the treatment and placebo groups. For patients in study CD3 with an inadequate response to prior treatment with inhibitors of TNF-α, maintenance of clinical response through Month 9 was seen in 52% of those randomized to TYSABRI, and maintenance of clinical remission through Month 9 was seen in 30%.
Given the requirement to discontinue chronic steroids it is important to note that in the subgroup of patients (n=65) who were receiving corticosteroid medication at baseline, responded to TYSABRI in Study CD1, and were re-randomized to TYSABRI in Study CD3, approximately two-thirds were able to discontinue steroids within 10 weeks of initiating a steroid taper.
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
TYSABRI (natalizumab) injection, a sterile, preservative-free, colorless and clear to slightly opalescent solution for dilution prior to intravenous infusion, is supplied as one 300 mg/15 mL (20 mg/mL) single-dose vial per carton (NDC 64406-008-01).
TYSABRI is available only through registered infusion centers participating in the TOUCH® Prescribing Program. To locate these infusion centers, contact Biogen at 1-800-456-2255.
TYSABRI single-dose vials must be refrigerated between 2°C to 8°C (36°F to 46°F). Do not use beyond the expiration date stamped on the carton and vial label. DO NOT SHAKE OR FREEZE. Protect from light.
Store diluted TYSABRI solution refrigerated at 2°C to 8°C (36°F to 46°F) [see Dosage and Administration (2.3)]
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
General Counseling Information
Counsel patients to understand the risks and benefits of TYSABRI before an initial prescription is written. The patient may be educated by either the enrolled prescriber or a healthcare provider under that prescriber's direction. INSTRUCT PATIENTS USING TYSABRI TO:
- Read the Medication Guide before starting TYSABRI and before each TYSABRI infusion.
- Promptly report any new or continuously worsening symptoms that persist over several days to their prescriber [see Warnings and Precautions (5.1)].
- Inform all of their physicians that they are receiving TYSABRI.
- Plan to see their prescriber three months after the first infusion, six months after the first infusion, every six months thereafter, and for at least six months after discontinuing TYSABRI.
Progressive Multifocal Leukoencephalopathy
Inform patients that Progressive Multifocal Leukoencephalopathy (PML) has occurred in patients who received TYSABRI. Instruct the patient of the importance of contacting their doctor if they develop any symptoms suggestive of PML. Instruct the patient that typical symptoms associated with PML are diverse, progress over days to weeks, and include progressive weakness on one side of the body or clumsiness of limbs, disturbance of vision, and changes in thinking, memory, and orientation leading to confusion and personality changes. Instruct the patient that the progression of deficits usually leads to death or severe disability over weeks or months.
Instruct patients to continue to look for new signs and symptoms suggestive of PML for approximately 6 months following discontinuation of TYSABRI [see Warnings and Precautions (5.1)].
TYSABRI TOUCH® Prescribing Program
Advise the patient that TYSABRI is only available through a restricted program called the TOUCH® Prescribing Program. Inform the patient of the following requirements:
Patients must read the Medication Guide and sign the Patient Enrollment Form. Advise patients that TYSABRI is available only from certified pharmacies and infusion centers participating in the program [see Warnings and Precautions (5.2)].
Herpes Infections
Inform patients that TYSABRI increases the risk of developing encephalitis and meningitis, which could be fatal, and acute retinal necrosis, which could lead to blindness, caused by the family of herpes viruses (e.g., herpes simplex and varicella zoster viruses). Instruct patients to immediately report any possible symptoms of encephalitis and meningitis (such as fever, headache, and confusion) or acute retinal necrosis (such as decreased visual acuity, eye redness, or eye pain) [see Warnings and Precautions (5.3)].
Hepatotoxicity
Inform patients that TYSABRI may cause liver injury. Instruct patients treated with TYSABRI to report promptly any symptoms that may indicate liver injury, including fatigue, anorexia, right upper abdominal discomfort, dark urine, or jaundice [see Warnings and Precautions (5.4)].
Hypersensitivity Reactions
Instruct patients to report immediately if they experience symptoms consistent with a hypersensitivity reaction (e.g., urticaria with or without associated symptoms) during or following an infusion of TYSABRI [see Warnings and Precautions (5.5)].
Immunosuppression/Infections
Inform patients that TYSABRI may lower the ability of their immune system to fight infections. Instruct the patient of the importance of contacting their doctor if they develop any symptoms of infection [see Warnings and Precautions (5.6)].
Thrombocytopenia
Inform patients that TYSABRI may cause a low platelet count, which can cause severe bleeding that may be life-threatening. Instruct patients to report any symptoms that may indicate thrombocytopenia, such as easy bruising, prolonged bleeding from cuts, petechiae, abnormally heavy menstrual periods, or bleeding from the nose or gums that is new [see Warnings and Precuations (5.8)].
Pregnancy
Instruct patients that if they become pregnant or plan to become pregnant while taking TYSABRI they should inform their healthcare provider [see Use in Specific Populations (8.1)].
I61061-30
TYSABRI (natalizumab)
Manufactured by:
Biogen Inc.
Cambridge, MA 02142 USA
US License No. 1697© 2015-2023 Biogen Inc. All rights reserved.
U.S. Patent Numbers: 5,840,299; 6,602,503 -
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised 04/2023 MEDICATION GUIDE
TYSABRI® (tie-SA-bree)
(natalizumab)
injection, for intravenous useRead this Medication Guide before you start receiving TYSABRI and before you receive each dose. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or your treatment. What is the most important information I should know about TYSABRI?
-
TYSABRI increases your chance (risk) of getting a rare brain infection that usually leads to death or severe disability. This infection is called progressive multifocal leukoencephalopathy (PML). If PML happens, it usually happens in people with weakened immune systems.
- There is no known treatment, prevention, or cure for PML.
- Your chance of getting PML may be higher if you are also being treated with other medicines that can weaken your immune system, including other treatments for Multiple Sclerosis (MS) and Crohn's disease (CD). You should not take certain medicines that weaken your immune system at the same time you are taking TYSABRI. Even if you use TYSABRI alone to treat your MS or CD, you can still get PML.
-
Your risk of getting PML is higher if you:
- have been infected by the John Cunningham Virus (JCV). JCV is a common virus that is harmless in most people but can cause PML in people who have weakened immune systems, such as people taking TYSABRI. Most people who are infected by JCV do not know it or do not have any symptoms. This infection usually happens in childhood. Before you start receiving TYSABRI or during your treatment, your doctor may do a blood test to check if you have been infected by JCV.
- have received TYSABRI for a long time, especially longer than 2 years
- have received certain medicines that can weaken your immune system before you start receiving TYSABRI
-
While you receive TYSABRI, and for 6 months after you stop receiving TYSABRI, it is important that you call your doctor right away if you have any new or worsening medical problems that have lasted several days.
These may be new or sudden and include problems with:
- thinking
- balance
- eyesight
- weakness on 1 side of your body
- strength
- using your arms and legs
Tell all your doctors that you are receiving TYSABRI.
-
Because of your risk of getting PML while you receive TYSABRI, TYSABRI is available only through a restricted distribution program called the TOUCH®Prescribing Program. To receive TYSABRI, you must talk to your doctor and understand the risks and benefits of TYSABRI and agree to follow all of the instructions in the TOUCH® Prescribing Program.
-
TYSABRI is only:
- prescribed by doctors who are enrolled in the TOUCH® Prescribing Program
- given by an infusion nurse from an infusion center that is enrolled in the TOUCH® Prescribing Program
- given to people who are enrolled in the TOUCH® Prescribing Program
-
Before you receive TYSABRI, your doctor will:
- explain the TOUCH® Prescribing Program to you
- have you sign the TOUCH® Patient Enrollment Form
-
TYSABRI is only:
What is TYSABRI?
TYSABRI is a prescription medicine used to treat adults with:
- relapsing forms of Multiple Sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease and active secondary progressive disease. TYSABRI increases the risk of PML. When starting and continuing treatment with TYSABRI, it is important that you discuss with your doctor whether the expected benefit of TYSABRI is enough to outweigh this risk. See “What is the most important information I should know about TYSABRI?”
- moderate to severe Crohn's disease (CD). TYSABRI is used:
- to reduce signs and symptoms of CD
- in people who have not been helped enough by, or cannot use the usual CD medicines and medicines called tumor necrosis factor (TNF) inhibitors.
- It is not known if TYSABRI is safe and effective in children under 18 years of age.
Who should not receive TYSABRI?
Do not receive TYSABRI if you:
- have PML
- are allergic to natalizumab or any of the ingredients in TYSABRI. See the end of this Medication Guide for a complete list of ingredients in TYSABRI.
What should I tell my doctor before receiving each dose of TYSABRI?
Before you receive TYSABRI, tell your doctor if you:
- have medical conditions that can weaken your immune system, including:
- HIV infection or AIDS
- leukemia or lymphoma
- an organ transplant
- other medical conditions that can weaken your immune system
- have any new or worsening medical problems that have lasted several days. These may be new or sudden and include problems with:
- thinking
- strength
- eyesight
- weakness on 1 side of your body
- balance
- using your arms and legs
- have had hives, itching or trouble breathing during or after receiving a dose of TYSABRI
- have a fever or infection (including shingles or any unusually long lasting infection)
- are pregnant or plan to become pregnant. TYSABRI may cause low platelets, and in some cases also low red blood cells (anemia), in your newborn baby if you take TYSABRI while you are pregnant. It is not known if TYSABRI can cause birth defects.
- are breastfeeding or plan to breastfeed. TYSABRI can pass into your breast milk. It is not known if the TYSABRI that passes into your breast milk can harm your baby. Talk to your doctor about the best way to feed your baby while you receive TYSABRI.
Know the medicines you take. Keep a list of them to show your doctor and pharmacist when you get a new medicine.How should I receive TYSABRI?
- TYSABRI is given 1 time every 4 weeks through a needle placed in your vein (IV infusion). Each infusion will last about 1 hour.
- Before each TYSABRI dose you will be asked questions to make sure TYSABRI is still right for you.
What are the possible side effects of TYSABRI?
TYSABRI may cause serious side effects, including:
- See “What is the most important information I should know about TYSABRI?”
- Herpes Infections. TYSABRI may increase your risk of getting an infection of the brain or the covering of your brain and spinal cord (encephalitis or meningitis) caused by herpes viruses that may lead to death. Call your doctor right away if you have sudden fever, severe headache, or if you feel confused after receiving TYSABRI. Herpes infections of the eye, causing blindness in some patients, have also occurred. Call your doctor right away if you have changes in vision, eye redness, or eye pain.
-
Liver damage. Symptoms of liver damage can include:
- yellowing of the skin and eyes (jaundice)
- unusual darkening of the urine
- nausea
- feeling tired or weak
- vomiting
Call your doctor right away if you have symptoms of liver damage. Your doctor can do blood tests to check for liver damage.
-
Allergic reactions, including serious allergic reactions. Symptoms of an allergic reaction can include:
- hives
- dizziness
- nausea
- itching
- wheezing
- flushing of skin
- trouble breathing
- chills
- low blood pressure
- chest pain
- rash
Serious allergic reactions usually happen within 2 hours of the start of your infusion, but they can happen at any time after you receive TYSABRI.
Tell your doctor right away if you have any symptom of an allergic reaction, even if it happens after your infusion. You may need treatment if you are having an allergic reaction.- Infections. TYSABRI may increase your chance of getting an unusual or serious infection because TYSABRI can weaken your immune system. You have a higher risk of getting infections if you also take other medicines that can weaken your immune system.
-
Low platelet counts. TYSABRI may cause the number of platelets in your blood to be reduced. Call your healthcare provider if you have any of the following symptoms:
- easy bruising
- heavier menstrual periods than are normal
- bleeding from your gums or nose that is new or takes longer than usual to stop
- bleeding from a cut that is hard to stop
- small scattered red spots on your skin that are red, pink, or purple
- headache
- lung infection
- vaginitis
- stomach area pain
- feeling tired
- depression
- rash
- urinary tract infection
- pain in your arm and legs
- nose and throat infections
- joint pain
- diarrhea
- nausea
Tell your doctor about any side effect that bothers you or that does not go away.
These are not all the possible side effects of TYSABRI. Ask your doctor for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of TYSABRI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide.
This Medication Guide summarizes the most important information about TYSABRI. If you would like more information, talk with your doctor. You can ask your pharmacist or doctor for information about TYSABRI that is written for healthcare professionals.
For more information, go to www.TYSABRI.com or call 1-800-456-2255.What are the ingredients in TYSABRI?
Active ingredient: natalizumab
Inactive Ingredients: sodium chloride, sodium phosphate, monobasic, monohydrate; sodium phosphate, dibasic, heptahydrate; polysorbate 80, and water for injection
Manufactured by: Biogen Inc.; Cambridge, MA 02142 USA -
TYSABRI increases your chance (risk) of getting a rare brain infection that usually leads to death or severe disability. This infection is called progressive multifocal leukoencephalopathy (PML). If PML happens, it usually happens in people with weakened immune systems.
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
TYSABRI
natalizumab injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64406-008 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength natalizumab (UNII: 3JB47N2Q2P) (natalizumab - UNII:3JB47N2Q2P) natalizumab 300 mg in 15 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) sodium phosphate, monobasic, monohydrate (UNII: 593YOG76RN) sodium phosphate, dibasic, heptahydrate (UNII: 70WT22SF4B) polysorbate 80 (UNII: 6OZP39ZG8H) water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64406-008-01 1 in 1 CARTON 11/23/2004 1 15 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125104 11/23/2004 Labeler - Biogen Inc. (121376230)