Label: TOBRAMYCIN injection, solution


- NDC Code(s): 0409-3578-01, 0409-3578-11
- Packager: Hospira, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated May 10, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
SPL UNCLASSIFIED SECTION
Injection, USP
Multiple Dose
Fliptop VialRx only
To reduce the development of drug-resistant bacteria and maintain the effectiveness of tobramycin and other antibacterial drugs, tobramycin should be used only to treat or prevent infections that are proven or strongly suspected to be caused by bacteria.
-
BOXED WARNING
(What is this?)
WARNINGS
Patients treated with Tobramycin Injection, USP and other aminoglycosides should be under close clinical observation, because these drugs have an inherent potential for causing ototoxicity and nephrotoxicity.
Neurotoxicity, manifested as both auditory and vestibular ototoxicity, can occur. The auditory changes are irreversible, are usually bilateral, and may be partial or total. Eighth-nerve impairment and nephrotoxicity may develop, primarily in patients having pre-existing renal damage and in those with normal renal function to whom aminoglycosides are administered for longer periods or in higher doses than those recommended. Other manifestations of neurotoxicity may include numbness, skin tingling, muscle twitching, and convulsions. The risk of aminoglycoside-induced hearing loss increases with the degree of exposure to either high peak or high trough serum concentrations. Patients who develop cochlear damage may not have symptoms during therapy to warn them of eighth-nerve toxicity, and partial or total irreversible bilateral deafness may continue to develop after the drug has been discontinued. Rarely, nephrotoxicity may not become apparent until the first few days after cessation of therapy. Aminoglycoside-induced nephrotoxicity usually is reversible.
Renal and eighth-nerve function should be closely monitored in patients with known or suspected renal impairment and also in those whose renal function is initially normal but who develop signs of renal dysfunction during therapy. Peak and trough serum concentrations of aminoglycosides should be monitored periodically during therapy to assure adequate levels and to avoid potentially toxic levels. Prolonged serum concentrations above 12 mcg/mL should be avoided. Rising trough levels (above 2 mcg/mL) may indicate tissue accumulation. Such accumulation, excessive peak concentrations, advanced age, and cumulative dose may contribute to ototoxicity and nephrotoxicity (see PRECAUTIONS). Urine should be examined for decreased specific gravity and increased excretion of protein, cells, and casts. Blood urea nitrogen, serum creatinine, and creatinine clearance should be measured periodically. When feasible, it is recommended that serial audiograms be obtained in patients old enough to be tested, particularly high-risk patients. Evidence of impairment of renal, vestibular, or auditory function requires discontinuation of the drug or dosage adjustment.
Tobramycin should be used with caution in premature and neonatal infants because of their renal immaturity and the resulting prolongation of serum half-life of the drug.
Concurrent and sequential use of other neurotoxic and/or nephrotoxic antibiotics, particularly other aminoglycosides (e.g., amikacin, streptomycin, neomycin, kanamycin, gentamicin, and paromomycin), cephaloridine, viomycin, polymyxin B, colistin, cisplatin, and vancomycin, should be avoided. Other factors that may increase patient risk are advanced age and dehydration.
Aminoglycosides should not be given concurrently with potent diuretics, such as ethacrynic acid and furosemide. Some diuretics themselves cause ototoxicity, and intravenously administered diuretics enhance aminoglycoside toxicity by altering antibiotic concentrations in serum and tissue.
Aminoglycosides can cause fetal harm when administered to a pregnant woman (see PRECAUTIONS).
-
DESCRIPTION
Tobramycin Sulfate, a water-soluble antibiotic of the aminoglycoside group, is derived from the actinomycete Streptomyces tenebrarius. Tobramycin Injection, USP is a clear and colorless sterile aqueous solution for parenteral administration.
Each mL contains tobramycin sulfate equivalent to 10 mg (pediatric) or 40 mg tobramycin; sodium metabisulfite added as an antioxidant 3 mg; and edetate disodium added as a stabilizer, 0.1 mg. Contains sulfuric acid and may contain sodium hydroxide for pH adjustment. pH 4.0 (3.0 to 6.5).
Tobramycin sulfate is O-3-amino-3-deoxy-α-D-glucopyranosyl-(1→4)-O-[2,6-diamino-2,3,6-trideoxy-α-D-ribo-hexopyranosyl-(1→6)]-2-deoxy-L-streptamine sulfate (2:5)(salt) and has the chemical formula (C18H37N5O9)2 • 5H2SO4. The molecular weight is 1,425.45. The structural formula for tobramycin is as follows:
-
CLINICAL PHARMACOLOGY
Tobramycin is rapidly absorbed following intramuscular administration. Peak serum concentrations of tobramycin occur between 30 and 90 minutes after intramuscular administration.
Following an intramuscular dose of 1 mg/kg of body weight, maximum serum concentrations reach about 4 mcg/mL, and measurable levels persist for as long as 8 hours. Therapeutic serum levels are generally considered to range from 4 to 6 mcg/mL. When tobramycin injection is administered by intravenous infusion over a 1-hour period, the serum concentrations are similar to those obtained by intramuscular administration. Tobramycin is poorly absorbed from the gastrointestinal tract.
In patients with normal renal function, except neonates, tobramycin administered every 8 hours does not accumulate in the serum. However, in those patients with reduced renal function and in neonates, the serum concentration of the antibiotic is usually higher and can be measured for longer periods of time than in normal adults. Dosage for such patients must, therefore, be adjusted accordingly (see DOSAGE AND ADMINISTRATION).
Following parenteral administration, little, if any, metabolic transformation occurs, and tobramycin is eliminated almost exclusively by glomerular filtration. Renal clearance is similar to that of endogenous creatinine. Ultrafiltration studies demonstrate that practically no serum protein binding occurs. In patients with normal renal function, up to 84% of the dose is recoverable from the urine in 8 hours and up to 93% in 24 hours.
Peak urine concentrations ranging from 75 to 100 mcg/mL have been observed following the intramuscular injection of a single dose of 1 mg/kg. After several days of treatment, the amount of tobramycin excreted in the urine approaches the daily dose administered. When renal function is impaired, excretion of tobramycin is slowed, and accumulation of the drug may cause toxic blood levels.
The serum half-life in normal individuals is 2 hours. An inverse relationship exists between serum half-life and creatinine clearance, and the dosage schedule should be adjusted according to the degree of renal impairment (see DOSAGE AND ADMINISTRATION). In patients undergoing dialysis, 25% to 70% of the administered dose may be removed, depending on the duration and type of dialysis.
Tobramycin can be detected in tissues and body fluids after parenteral administration. Concentrations in bile and stools ordinarily have been low, which suggests minimum biliary excretion. Tobramycin has appeared in low concentration in the cerebrospinal fluid following parenteral administration, and concentrations are dependent on dose, rate of penetration, and degree of meningeal inflammation. It has also been found in sputum, peritoneal fluid, synovial fluid, and abscess fluids, and it crosses the placental membranes. Concentrations in the renal cortex are several times higher than the usual serum levels.
Probenecid does not affect the renal tubular transport of tobramycin.
Microbiology
Tobramycin is an aminoglycoside antibiotic with activity against Gram-positive and Gram-negative bacteria.
Mechanism of Action
Tobramycin acts by inhibiting synthesis of protein in bacterial cells. In vitro tests demonstrate that tobramycin is bactericidal.
Resistance
Cross-resistance between aminoglycosides may occur.
Interactions With Other Antimicrobials
Although most strains of enterococci demonstrate in vitro resistance, some strains in this group are susceptible. In vitro studies have shown that an aminoglycoside combined with an antibiotic that interferes with cell-wall synthesis affects some enterococcal strains synergistically. The combination of penicillin G and tobramycin results in a synergistic bactericidal effect in vitro against certain strains of Enterococcus faecalis. However, this combination is not synergistic against other closely related organisms, e.g. Enterococcus faecium. Speciation of enterococci alone cannot be used to predict susceptibility. Susceptibility testing and tests for antibiotic synergism are emphasized.
Antimicrobial Activity
Tobramycin has been shown to be active against most strains of the following organisms both in vitro and in clinical infections: (see INDICATIONS AND USAGE)
Aerobic and facultative Gram-positive microorganisms
Staphylococcus aureus
Aerobic and facultative Gram-negative microorganisms
Citrobacter sp Pseudomonas aeruginosa
Enterobacter sp Proteus mirabilis
Escherichia coli Proteus vulgaris
Klebsiella sp Providencia sp
Morganella morganii Serratia sp
Aminoglycosides have a low order of activity against most Gram-positive organisms, including Streptococcus pyogenes, Streptococcus pneumoniae, and enterococci.
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
-
INDICATIONS AND USAGE
To reduce the development of drug-resistant bacteria and maintain the effectiveness of tobramycin and other antibacterial drugs, tobramycin should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
Tobramycin is indicated for the treatment of serious bacterial infections caused by susceptible strains of the designated microorganisms in the diseases listed below:
Septicemia in the pediatric patient and adult caused by P. aeruginosa, E. coli, and Klebsiella sp.
Lower respiratory tract infections caused by P. aeruginosa, Klebsiella sp, Enterobacter sp, Serratia sp, E. coli, and S. aureus (penicillinase and non-penicillinase-producing strains).
Serious central-nervous-system infections (meningitis) caused by susceptible organisms.
Intra-abdominal infections, including peritonitis, caused by E. coli, Klebsiella sp, and Enterobacter sp.
Skin, bone, and skin-structure infections caused by P. aeruginosa, Proteus sp, E. coli, Klebsiella sp, Enterobacter sp, and S. aureus.
Complicated and recurrent urinary tract infections caused by P. aeruginosa, Proteus sp (indole-positive and indole-negative), E. coli, Klebsiella sp, Enterobacter sp, Serratia sp, S. aureus, Providencia sp, and Citrobacter sp.
Aminoglycosides, including tobramycin, are not indicated in uncomplicated initial episodes of urinary tract infections unless the causative organisms are not susceptible to antibiotics having less potential toxicity. Tobramycin may be considered in serious staphylococcal infections when penicillin or other potentially less toxic drugs are contraindicated and when bacterial susceptibility testing and clinical judgment indicate its use.
Bacterial cultures should be obtained prior to and during treatment to isolate and identify etiologic organisms and to test their susceptibility to tobramycin. If susceptibility tests show that the causative organisms are resistant to tobramycin, other appropriate therapy should be instituted. In patients in whom a serious life-threatening gram-negative infection is suspected, including those in whom concurrent therapy with a penicillin or cephalosporin and an aminoglycoside may be indicated, treatment with tobramycin sulfate may be initiated before the results of susceptibility studies are obtained. The decision to continue therapy with tobramycin should be based on the results of susceptibility studies, the severity of the infection, and the important additional concepts discussed in the WARNINGS box above.
- CONTRAINDICATIONS
-
WARNINGS
See WARNINGS box above.
Tobramycin Injection, USP contains sodium metabisulfite, a sulfite that may cause allergic-type reactions, including anaphylactic symptoms and life-threatening or less severe asthmatic episodes, in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in nonasthmatic people.
Serious allergic reactions including anaphylaxis and dermatologic reactions including exfoliative dermatitis, toxic epidermal necrolysis, erythema multiforme, and Stevens-Johnson Syndrome have been reported rarely in patients on tobramycin therapy. Although rare, fatalities have been reported (see CONTRAINDICATIONS).
If an allergic reaction occurs, the drug should be discontinued and appropriate therapy instituted.
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including Tobramycin, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
Risk of Ototoxicity Due to Mitochondrial DNA Variants
Cases of ototoxicity with aminoglycosides have been observed in patients with certain variants in the mitochondrially encoded 12S rRNA gene (MT-RNR1), particularly the m.1555A>G variant. Ototoxicity occurred in some patients even when their aminoglycoside serum levels were within the recommended range. Mitochondrial DNA variants are present in less than 1% of the general US population, and the proportion of the variant carriers who may develop ototoxicity as well as the severity of ototoxicity is unknown. In case of known maternal history of ototoxicity due to aminoglycoside use or a known mitochondrial DNA variant in the patient, consider alternative treatments other than aminoglycosides unless the increased risk of permanent hearing loss is outweighed by the severity of infection and lack of safe and effective alternative therapies.
-
PRECAUTIONS
Serum and urine specimens for examination should be collected during therapy, as recommended in the WARNINGS box. Serum calcium, magnesium, and sodium should be monitored.
Peak and trough serum levels should be measured periodically during therapy. Prolonged concentrations above 12 mcg/mL should be avoided. Rising trough levels (above 2 mcg/mL) may indicate tissue accumulation. Such accumulation, advanced age, and cumulative dosage may contribute to ototoxicity and nephrotoxicity. It is particularly important to monitor serum levels closely in patients with known renal impairment.
A useful guideline would be to perform serum level assays after 2 or 3 doses, so that the dosage could be adjusted if necessary, and also at 3- to 4-day intervals during therapy. In the event of changing renal function, more frequent serum levels should be obtained and the dosage or dosage interval adjusted according to the guidelines provided in the DOSAGE AND ADMINISTRATION section.
In order to measure the peak level, a serum sample should be drawn about 30 minutes following intravenous infusion or 1 hour after an intramuscular injection. Trough levels are measured by obtaining serum samples at 8 hours or just prior to the next dose of tobramycin. These suggested time intervals are intended only as guidelines and may vary according to institutional practices. It is important, however, that there be consistency within the individual patient program unless computerized pharmacokinetic dosing programs are available in the institution. These serum-level assays may be especially useful for monitoring the treatment of severely ill patients with changing renal function or of those infected with less sensitive organisms or those receiving maximum dosage.
Neuromuscular blockade and respiratory paralysis have been reported in cats receiving very high doses of tobramycin (40 mg/kg). The possibility of prolonged or secondary apnea should be considered if tobramycin is administered to anesthetized patients who are also receiving neuromuscular blocking agents, such as succinylcholine, tubocurarine, or decamethonium, or to patients receiving massive transfusions of citrated blood. If neuromuscular blockade occurs, it may be reversed by the administration of calcium salts.
Cross-allergenicity among aminoglycosides has been demonstrated.
In patients with extensive burns or cystic fibrosis, altered pharmacokinetics may result in reduced serum concentrations of aminoglycosides. In such patients treated with tobramycin, measurement of serum concentration is especially important as a basis for determination of appropriate dosage.
Elderly patients may have reduced renal function that may not be evident in the results of routine screening tests, such as BUN or serum creatinine. A creatinine clearance determination may be more useful. Monitoring of renal function during treatment with aminoglycosides is particularly important in such patients.
An increased incidence of nephrotoxicity has been reported following concomitant administration of aminoglycoside antibiotics and cephalosporins.
Aminoglycosides should be used with caution in patients with muscular disorders, such as myasthenia gravis or parkinsonism, since these drugs may aggravate muscle weakness because of their potential curare-like effect on neuromuscular function.
Aminoglycosides may be absorbed in significant quantities from body surfaces after local irrigation or application and may cause neurotoxicity and nephrotoxicity.
Aminoglycosides have not been approved for intraocular and/or subconjunctival use. Physicians are advised that macular necrosis has been reported following administration of aminoglycosides, including tobramycin, by these routes.
See WARNINGS box regarding concurrent use of potent diuretics and concurrent and sequential use of other neurotoxic or nephrotoxic drugs.
The inactivation of tobramycin and other aminoglycosides by β-lactam-type antibiotics (penicillins or cephalosporins) has been demonstrated in vitro and in patients with severe renal impairment. Such inactivation has not been found in patients with normal renal function who have been given the drugs by separate routes of administration.
Therapy with tobramycin may result in overgrowth of nonsusceptible organisms. If overgrowth of nonsusceptible organisms occurs, appropriate therapy should be initiated.
General: Prescribing tobramycin in the absence of a proven or strongly suspected bacterial infection or a prophylactic indication is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria.
Pregnancy: Aminoglycosides can cause fetal harm when administered to a pregnant woman. Aminoglycoside antibiotics cross the placenta, and there have been several reports of total irreversible bilateral congenital deafness in children whose mothers received streptomycin during pregnancy. Serious side effects to mother, fetus, or newborn have not been reported in the treatment of pregnant women with other aminoglycosides. If tobramycin is used during pregnancy or if the patient becomes pregnant while taking tobramycin, she should be apprised of the potential hazard to the fetus.
Information for Patients: Patients should be counseled that antibacterial drugs including tobramycin should only be used to treat bacterial infections. They do not treat viral infections (e.g., the common cold). When tobramycin is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by tobramycin or other antibacterial drugs in the future.
Diarrhea is a common problem caused by antibiotics, which usually ends when the antibiotic is discontinued. Sometimes after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
Geriatric Use: Elderly patients may be at a higher risk of developing nephrotoxicity and ototoxicity while receiving tobramycin (see WARNINGS, PRECAUTIONS and OVERDOSAGE). Other factors that may contribute to nephrotoxicity and ototoxicity are rising trough levels, excessive peak concentrations, dehydration, concomitant use of other neurotoxic or nephrotoxic drugs, and cumulative dose. Peak and trough serum levels should be measured periodically during therapy to assure adequate levels and to avoid potentially toxic levels (see WARNINGS and PRECAUTIONS).
Tobramycin is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Dose reduction is required for patients with impaired renal function (see DOSAGE AND ADMINISTRATION). Elderly patients may have reduced renal function that may not be evident in the results of routine screening tests, such as BUN or serum creatinine. A creatinine clearance determination may be more useful. Monitoring of renal function during treatment with aminoglycosides is particularly important in the elderly (see PRECAUTIONS).
Tobramycin 80 mg/2 mL vials contain 1.4 mg (0.06 mEq) of sodium.
-
ADVERSE REACTIONS
Neurotoxicity: Adverse effects on both the vestibular and auditory branches of the eighth nerve have been noted, especially in patients receiving high doses or prolonged therapy, in those given previous courses of therapy with an ototoxin, and in cases of dehydration. Symptoms include dizziness, vertigo, tinnitus, roaring in the ears, and hearing loss. Hearing loss is usually irreversible and is manifested initially by diminution of high-tone acuity. Tobramycin and gentamicin closely parallel each other in regard to ototoxic potential.
Nephrotoxicity: Renal function changes, as shown by rising BUN, NPN, and serum creatinine and by oliguria, cylindruria, and increased proteinuria, have been reported, especially in patients with a history of renal impairment who are treated for longer periods or with higher doses than those recommended. Adverse renal effects can occur in patients with initially normal renal function.
Clinical studies and studies in experimental animals have been conducted to compare the nephrotoxic potential of tobramycin and gentamicin. In some of the clinical studies and in the animal studies, tobramycin caused nephrotoxicity significantly less frequently than gentamicin. In some other clinical studies, no significant difference in the incidence of nephrotoxicity between tobramycin and gentamicin was found.
Other reported adverse reactions possibly related to tobramycin include anemia, granulocytopenia, and thrombocytopenia; and fever, rash, exfoliative dermatitis, itching, urticaria, nausea, vomiting, diarrhea, headache, lethargy, pain at the injection site, mental confusion, and disorientation. Laboratory abnormalities possibly related to tobramycin include increased serum transaminases (AST, ALT); increased serum LDH and bilirubin; decreased serum calcium, magnesium, sodium, and potassium; and leukopenia, leukocytosis, and eosinophilia.
To report SUSPECTED ADVERSE EVENTS, contact FDA at 1-800-FDA-1088 or www.fda.gov.
-
OVERDOSAGE
Signs and Symptoms: The severity of the signs and symptoms following a tobramycin overdose are dependent on the dose administered, the patient's renal function, state of hydration, and age and whether or not other medications with similar toxicities are being administered concurrently. Toxicity may occur in patients treated more than 10 days, in adults given more than 5 mg/kg/day, pediatric patients given more than 7.5 mg/kg/day, or patients with reduced renal function whose dose has not been appropriately adjusted.
Nephrotoxicity following the parenteral administration of an aminoglycoside is most closely related to the area under the curve of the serum concentration versus time graph. Nephrotoxicity is more likely if trough blood concentrations fail to fall below 2 mcg/mL and is also proportional to the average blood concentration. Patients who are elderly, have abnormal renal function, are receiving other nephrotoxic drugs, or are volume depleted are at greater risk for developing acute tubular necrosis. Auditory and vestibular toxicity has been associated with aminoglycoside overdose; these toxicities occur in patients treated longer than 10 days, in patients with abnormal renal function, in dehydrated patients, or in patients receiving medications with additive auditory toxicities. These patients may not have signs or symptoms or may experience dizziness, tinnitus, vertigo, and a loss of high-tone acuity, as ototoxicity progresses. Ototoxicity signs and symptoms may not begin to occur until long after the drug has been discontinued.
Neuromuscular blockade or respiratory paralysis may occur following administration of many aminoglycosides. Neuromuscular blockade, respiratory failure, and prolonged respiratory paralysis may occur more commonly in patients with myasthenia gravis or Parkinson's disease. Prolonged respiratory paralysis may also occur in patients receiving decamethonium, tubocurarine, or succinylcholine. If neuromuscular blockade occurs, it may be reversed by the administration of calcium salts but mechanical assistance may be necessary.
If tobramycin were ingested, toxicity would be less likely because aminoglycosides are poorly absorbed from an intact gastrointestinal tract.
Treatment: In all cases of suspected overdosage, call your Regional Poison Control Center to obtain the most up-to-date information about the treatment of overdose. This recommendation is made because, in general, information regarding the treatment of overdose may change more rapidly than the package insert. In managing overdosage, consider the possibility of multiple drug overdoses, interaction among drugs, and unusual drug kinetics in your patient.
The initial intervention in a tobramycin overdose is to establish an airway and ensure oxygenation and ventilation. Resuscitative measures should be initiated promptly if respiratory paralysis occurs.
Patients that have received an overdose of tobramycin and have normal renal function should be adequately hydrated to maintain a urine output of 3 to 5 mL/kg/hr. Fluid balance, creatinine clearance, and tobramycin plasma levels should be carefully monitored until the serum tobramycin level falls below 2 mcg/mL.
Patients in whom the elimination half-life is greater than 2 hours or whose renal function is abnormal may require more aggressive therapy. In such patients, hemodialysis may be beneficial.
-
DOSAGE AND ADMINISTRATION
Tobramycin Injection, USP may be given intramuscularly or intravenously. Recommended dosages are the same for both routes. The patient's pretreatment body weight should be obtained for calculation of correct dosage. It is desirable to measure both peak and trough serum concentrations (see WARNINGS box and PRECAUTIONS).
Administration for Patients with Normal Renal Function
Adults with Serious Infections: 3 mg/kg/day in 3 equal doses every 8 hours (see Table 1).
Adults with Life-Threatening Infections: Up to 5 mg/kg/day may be administered in 3 or 4 equal doses (see Table 1). The dosage should be reduced to 3 mg/kg/day as soon as clinically indicated. To prevent increased toxicity due to excessive blood levels, dosage should not exceed 5 mg/kg/day unless serum levels are monitored (see WARNINGS box and PRECAUTIONS).
Table 1. Dosage Schedule Guide for Tobramycin Injection, USP in Adults with Normal Renal Function (Dosage at 8-Hour Intervals) For
Patient
WeighingUsual Dose for
Serious InfectionsMaximum Dose for Life-
Threatening Infections
(Reduce as soon as possible)1 mg/kg q8h
(Total, 3 mg/kg/day)1.66 mg/kg q8h
(Total, 5 mg/kg/day)mg/dose
mL/dose*
mg/dose
mL/dose*
kg
lb
q8h
q8h
120
264
120 mg
3 mL
200 mg
5 mL
115
253
115 mg
2.9 mL
191 mg
4.75 mL
110
242
110 mg
2.75 mL
183 mg
4.5 mL
105
231
105 mg
2.6 mL
175 mg
4.4 mL
100
220
100 mg
2.5 mL
166 mg
4.2 mL
95
209
95 mg
2.4 mL
158 mg
4 mL
90
198
90 mg
2.25 mL
150 mg
3.75 mL
85
187
85 mg
2.1 mL
141 mg
3.5 mL
80
176
80 mg
2 mL
133 mg
3.3 mL
75
165
75 mg
1.9 mL
125 mg
3.1 mL
70
154
70 mg
1.75 mL
116 mg
2.9 mL
65
143
65 mg
1.6 mL
108 mg
2.7 mL
60
132
60 mg
1.5 mL
100 mg
2.5 mL
55
121
55 mg
1.4 mL
91 mg
2.25 mL
50
110
50 mg
1.25 mL
83 mg
2.1 mL
45
99
45 mg
1.1 mL
75 mg
1.9 mL
40
88
40 mg
1 mL
66 mg
1.6 mL
* Applicable to all product forms except Tobramycin Injection, USP, 10 mg/mL (Pediatric).
Pediatric Patients (Greater Than 1 Week of Age):
6 to 7.5 mg/kg/day in 3 or 4 equally divided doses (2 to 2.5 mg/kg every 8 hours or 1.5 to 1.89 mg/kg every 6 hours).
Premature or Full-Term Neonates 1 Week of Age or Less: Up to 4 mg/kg/day may be administered in 2 equal doses every 12 hours.
It is desirable to limit treatment to a short term. The usual duration of treatment is 7 to 10 days. A longer course of therapy may be necessary in difficult and complicated infections. In such cases, monitoring of renal, auditory, and vestibular functions is advised, because neurotoxicity is more likely to occur when treatment is extended longer than 10 days.
Dosage in Patients with Cystic Fibrosis
In patients with cystic fibrosis, altered pharmacokinetics may result in reduced serum concentration of aminoglycosides. Measurement of tobramycin serum concentration during treatment is especially important as a basis for determining appropriate dose. In patients with severe cystic fibrosis, an initial dosing regimen of 10 mg/kg/day in 4 equally divided doses is recommended. This dosing regimen is suggested only as a guide. The serum levels of tobramycin should be measured directly during treatment due to a wide interpatient variability.
Administration for Patients with Impaired Renal Function
Whenever possible, serum tobramycin concentrations should be monitored during therapy.
Following a loading dose of 1 mg/kg, subsequent dosage in these patients must be adjusted, either with reduced doses administered at 8-hour intervals or with normal doses given at prolonged intervals. Both of these methods are suggested as guides to be used when serum levels of tobramycin cannot be measured directly. They are based on either the creatinine clearance or the serum creatinine of the patient, because these values correlate with the half-life of tobramycin. The dosage schedules derived from either method should be used in conjunction with careful clinical and laboratory observations of the patient and should be modified as necessary. Neither method should be used when dialysis is being performed.
Reduced Dosage at 8-hour Intervals
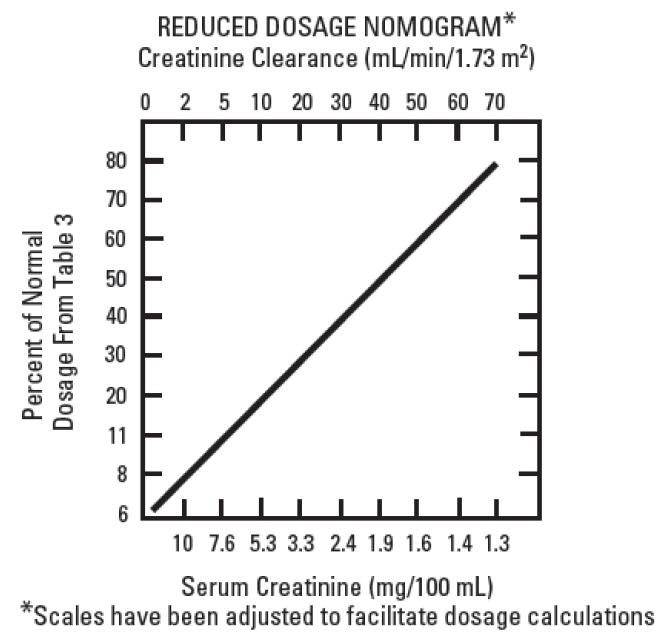
When the creatinine clearance rate is 70 mL or less per minute or when the serum creatinine value is known, the amount of the reduced dose can be determined by multiplying the normal dose from Table 1 by the percent of normal dose from the accompanying nomogram.
An alternate rough guide for determining reduced dosage at 8-hour intervals (for patients whose steady-state serum creatinine values are known) is to divide the normally recommended dose by the patient's serum creatinine.
Normal Dosage at Prolonged Intervals
If the creatinine clearance rate is not available and the patient's condition is stable, a dosage frequency in hours for the dosage given in Table 1 can be determined by multiplying the patient's serum creatinine by 6.
Dosage in Obese Patients
The appropriate dose may be calculated by using the patient's estimated lean body weight plus 40% of the excess as the basic weight on which to figure mg/kg.
Intramuscular Administration
Tobramycin Injection, USP may be administered by withdrawing the appropriate dose directly from a vial. Tobramycin Sulfate in 0.9% Sodium Chloride is not intended for intramuscular administration.
Intravenous Administration
For intravenous administration, the usual volume of diluent (0.9% Sodium Chloride Injection or 5% Dextrose Injection) is 50 to 100 mL for adult doses. For pediatric patients, the volume of diluent should be proportionately less than for adults. The diluted solution usually should be infused over a period of 20 to 60 minutes. Infusion periods of less than 20 minutes are not recommended because peak serum levels may exceed 12 mcg/mL (see WARNINGS box).
Tobramycin Injection, USP should not be physically premixed with other drugs but should be administered separately according to the recommended dose and route.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
- HOW SUPPLIED
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 80 mg/2 mL Vial Label
- PRINCIPAL DISPLAY PANEL - 80 mg/2 mL Vial Tray
-
INGREDIENTS AND APPEARANCE
TOBRAMYCIN
tobramycin injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0409-3578 Route of Administration INTRAMUSCULAR, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TOBRAMYCIN SULFATE (UNII: HJT0RXD7JK) (TOBRAMYCIN - UNII:VZ8RRZ51VK) TOBRAMYCIN 40 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM METABISULFITE (UNII: 4VON5FNS3C) 3 mg in 1 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 0.1 mg in 1 mL SULFURIC ACID (UNII: O40UQP6WCF) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0409-3578-01 25 in 1 TRAY 05/05/2005 1 NDC:0409-3578-11 2 mL in 1 VIAL, MULTI-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA063111 05/05/2005 Labeler - Hospira, Inc. (141588017) Establishment Name Address ID/FEI Business Operations Hospira, Inc. 093132819 ANALYSIS(0409-3578) , MANUFACTURE(0409-3578) , PACK(0409-3578) , LABEL(0409-3578) Establishment Name Address ID/FEI Business Operations Hospira, Inc. 827731089 ANALYSIS(0409-3578)