Label: PHENYTOIN SODIUM capsule, extended release


-
NDC Code(s):
62756-299-08,
62756-299-13,
62756-299-83,
62756-299-88, view more62756-432-08, 62756-432-13, 62756-432-83, 62756-432-88
- Packager: Sun Pharmaceutical Industries, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated December 14, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EXTENDED PHENYTOIN SODIUM CAPSULES safely and effectively. See full prescribing information for EXTENDED PHENYTOIN SODIUM CAPSULES.
EXTENDED PHENYTOIN SODIUM capsules, for oral use
Initial U.S. Approval: 1953
INDICATIONS AND USAGE
Extended phenytoin sodium capsules are indicated for the treatment of tonic-clonic (grand mal) and psychomotor (temporal lobe) seizures and prevention and treatment of seizures occurring during or following neurosurgery. (1)
DOSAGE AND ADMINISTRATION
- Adult starting dose in patients who have received no previous treatment is one 100 mg extended phenytoin sodium capsule three times a day, with dose adjustments as necessary. For most adults, the satisfactory maintenance dose will be one capsule three to four times a day. An increase, up to two capsules three times a day may be made, if necessary. (2.1)
- Adult once-a-day dose: If seizure control is established with divided doses of three 100 mg extended phenytoin sodium capsules daily, once-a-day dosage with 300 mg extended phenytoin sodium capsules may be considered. (2.1)
- Adult loading dose: reserved for patients in a clinic or hospital setting who require rapid steady-state serum levels and where intravenous administration is not desired. Refer to full prescribing information. (2.1)
- Pediatric starting dose is 5 mg/kg/day in two to three equally divided doses, with dosage adjustments as necessary, up to a maximum of 300 mg daily. Maintenance dosage is 4 to 8 mg/kg/day. (2.2)
- Serum blood level determinations may be necessary for optimal dosage adjustments—the clinically effective serum total concentration is 10 to 20 mcg/mL (unbound phenytoin concentration is 1 to 2 mcg/mL). (2.3)
DOSAGE FORMS AND STRENGTHS
Extended phenytoin sodium capsules are available as 200 mg and 300 mg extended phenytoin sodium capsules. (3) (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Withdrawal Precipitated Seizure: May precipitate status epilepticus. Dose reductions or discontinuation should be done gradually.(5.1)
- Suicidal Behavior and Ideation: Monitor patients for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.(5.2)
- Serious Dermatologic Reactions: Discontinue extended phenytoin sodium capsules at the first sign of a rash, unless the rash is clearly not drug-related. If signs or symptoms suggest SJS/TEN, use of this drug should not be resumed and alternative therapy should be considered. (5.3)
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity: If signs or symptoms of hypersensitivity are present, evaluate the patient immediately. Discontinue if an alternative etiology cannot be established.(5.4)
- Cardiac Effects: Bradycardia and cardiac arrest have been reported.(5.6)
- Angioedema: Discontinue immediately if symptoms of angioedema such as facial, perioral, or upper airway swelling occur. (5.7)
- Hepatic Injury: Cases of acute hepatotoxicity have been reported with extended phenytoin sodium capsules. If this occurs, immediately discontinue.(4,5.8)
- Hematopoietic Complications: If occurs, follow-up observation is indicated and an alternative antiepileptic treatment should be used. (5.9)
ADVERSE REACTIONS
The most common adverse reactions are nervous system reactions, including nystagmus, ataxia, slurred speech, decreased coordination, somnolence, and mental confusion. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Sun Pharmaceutical Industries, Inc. at 1-800-818-4555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2022
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Adult Dosage
2.2 Pediatric Dosage
2.3 Dosage Adjustments
2.4 Switching Between Phenytoin Formulations
2.5 Dosing in Patients with Renal or Hepatic Impairment or Hypoalbuminemia
2.6 Geriatric Dosage
2.7 Dosing during Pregnancy
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Withdrawal Precipitated Seizure, Status Epilepticus
5.2 Suicidal Behavior and Ideation
5.3 Serious Dermatologic Reactions
5.4 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity
5.5 Hypersensitivity
5.6 Cardiac Effects
5.7 Angioedema
5.8 Hepatic Injury
5.9 Hematopoietic Complications
5.10 Effects on Vitamin D and Bone
5.11 Renal or Hepatic Impairment or Hypoalbuminemia
5.12 Exacerbation of Porphyria
5.13 Teratogenicity and Other Harm to the Newborn
5.14 Hyperglycemia
5.15 Serum Phenytoin Levels above Therapeutic Range
6 ADVERSE REACTIONS
7 DRUG INTERACTIONS
7.1 Drugs that Affect Phenytoin Concentrations
7.2 Drugs Affected by Phenytoin
7.3 Hyperammonemia with Concomitant Use of Valproate
7.4 Drug Enteral Feeding/Nutritional Preparations Interaction
7.5 Drug/Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal and/or Hepatic Impairment or Hypoalbuminemia
8.7 Use in Patients with Decreased CYP2C9 Function
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Adult Dosage
Divided daily dosage:
The recommended starting dose for adult patients who have received no previous treatment is one 100 mg extended phenytoin sodium capsules by mouth three times daily. Adjust the dosage to suit individual requirements up to a maximum of two capsules three times a day. For most adults, the satisfactory maintenance dosage will be one capsule three to four times a day.
Once-a-day dosage:
In adults, if seizure control is established with divided doses of three 100 mg extended phenytoin sodium capsules daily, once-a-day dosage with 300 mg of extended phenytoin sodium capsules may be considered. Studies comparing divided doses of 300 mg with a single daily dose of this quantity indicated absorption, peak serum levels, biologic half-life, difference between peak and minimum values, and urinary recovery were equivalent. Once-a-day dosage offers a convenience to the individual patient or to nursing personnel for institutionalized patients and is intended to be used only for patients requiring this amount of drug daily. A major problem in motivating noncompliant patients may also be lessened when the patient can take this drug once a day. However, patients should be cautioned not to miss a dose, inadvertently.
Only extended phenytoin sodium capsules are recommended for once-a-day dosing. Inherent differences in dissolution characteristics and resultant absorption rates of phenytoin due to different manufacturing procedures and/or dosage forms preclude such recommendation for other phenytoin products. When a change in the dosage form or brand is prescribed, careful monitoring of phenytoin serum levels should be carried out.
Loading dose:
Some authorities have advocated use of an oral loading dose of phenytoin in adults who require rapid steady-state serum levels and where intravenous administration is not desirable. This dosing regimen should be reserved for patients in a clinic or hospital setting where phenytoin serum levels can be closely monitored. Patients with a history of renal or liver disease should not receive the oral loading regimen.
Initially, one gram of extended phenytoin sodium capsules are divided into three doses (400 mg, 300 mg, 300 mg) and administered at two-hour intervals.
Normal maintenance dosage is then instituted 24 hours after the loading dose, with frequent serum level determinations.2.2 Pediatric Dosage
The recommended starting dosage for pediatric patients is 5 mg/kg/day by mouth in two or three equally divided doses, with subsequent dosage individualized to a maximum of 300 mg daily in divided doses. A recommended daily maintenance dosage is usually 4 to 8 mg/kg/day in equally divided doses. Children over 6 years and adolescents may require the minimum adult dosage (300 mg/day).
2.3 Dosage Adjustments
Dosage should be individualized to provide maximum benefit. In some cases, serum blood level determinations may be necessary for optimal dosage adjustments. Trough levels provide information about clinically effective serum level range and confirm patient compliance, and are obtained just prior to the patient’s next scheduled dose. Peak levels indicate an individual’s threshold for emergence of dose-related side effects and are obtained at the time of expected peak concentration. Therapeutic effect without clinical signs of toxicity occurs more often with serum total concentrations between 10 and 20 mcg/mL (unbound phenytoin concentrations between 1 and 2 mcg/mL), although some mild cases of tonic-clonic (grand mal) epilepsy may be controlled with lower serum levels of phenytoin. In patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of unbound phenytoin concentrations may be more relevant [see Dosage and Administration (2.5)].
With recommended dosage, a period of seven to ten days may be required to achieve steady-state blood levels with phenytoin and changes in dosage (increase or decrease) should not be carried out at intervals shorter than seven to ten days.
2.4 Switching Between Phenytoin Formulations
The free acid form of phenytoin is used in phenytoin-125 oral suspension and phenytoin chewable tablets. Extended phenytoin sodium capsules and parenteral phenytoin are formulated with the sodium salt of phenytoin. Because there is approximately an 8% increase in drug content with the free acid form over that of the sodium salt, dosage adjustments and serum level monitoring may be necessary when switching from a product formulated with the free acid to a product formulated with the sodium salt and vice versa.
2.5 Dosing in Patients with Renal or Hepatic Impairment or Hypoalbuminemia
Because the fraction of unbound phenytoin is increased in patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of phenytoin serum levels should be based on the unbound fraction in those patients [see Warnings and Precautions (5.11) and Use in Specific Populations (8.6)].
2.6 Geriatric Dosage
Phenytoin clearance is decreased slightly in elderly patients and lower or less frequent dosing may be required [see Clinical Pharmacology (12.3)].
2.7 Dosing during Pregnancy
Decreased serum concentrations of phenytoin may occur during pregnancy because of altered phenytoin pharmacokinetics. Periodic measurement of serum phenytoin concentrations should be performed during pregnancy, and the extended phenytoin sodium capsules dosage should be adjusted as necessary. Postpartum restoration of the original dosage will probably be indicated [see Use in Specific Populations (8.1)]. Because of potential changes in protein binding during pregnancy, the monitoring of phenytoin serum levels should be based on the unbound fraction.
-
3 DOSAGE FORMS AND STRENGTHS
Extended phenytoin sodium capsules, USP are available as 200 mg or 300 mg of phenytoin sodium, USP
- The 200 mg capsule has a blue transparent cap and a light blue transparent body. The hard-shell gelatin capsule is filled with white to off-white powder. The capsule is printed axially with 299 in black ink on both the cap and the body.
- The 300 mg capsule has a light blue transparent cap and a light blue transparent body. The hard-shell gelatin capsule is filled with white to off-white powder. The capsule is printed axially with 432 in black ink on both the cap and the body.
-
4 CONTRAINDICATIONS
Extended phenytoin sodium capsules are contraindicated in patients with:
- A history of hypersensitivity to phenytoin, its inactive ingredients, or other hydantoins [see Warnings and Precautions (5.5)]. Reactions have included angioedema.
- A history of prior acute hepatotoxicity attributable to phenytoin [see Warnings and Precautions (5.8)].
- Coadministration with delavirdine because of the potential for loss of virologic response and possible resistance to delavirdine or to the class of non-nucleoside reverse transcriptase inhibitors.
-
5 WARNINGS AND PRECAUTIONS
5.1 Withdrawal Precipitated Seizure, Status Epilepticus
Abrupt withdrawal of phenytoin in epileptic patients may precipitate status epilepticus. When, in the judgment of the clinician, the need for dosage reduction, discontinuation, or substitution of alternative anticonvulsant medication arises, this should be done gradually. However, in the event of an allergic or hypersensitivity reaction, more rapid substitution of alternative therapy may be necessary. In this case, alternative therapy should be an anticonvulsant drug not belonging to the hydantoin chemical class.
5.2 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including extended phenytoin sodium capsules, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5 to 100 years) in the clinical trials analyzed.
Table 1 shows absolute and relative risk by indication for all evaluated AEDs.
Table 1 Risk by indication for antiepileptic drugs in the pooled analysis
Indication
Placebo Patients
with Events
Per 1000
Patients
Drug Patients
with Events Per
1000 Patients
Relative Risk:
Incidence of
Events in Drug
Patients/Incidence
in Placebo Patients
Risk Difference:
Additional Drug
Patients with
Events Per 1000
Patients
Epilepsy
Psychiatric
Other
Total
1.0
5.7
1.0
2.4
3.4
8.5
1.8
4.3
3.5
1.5
1.9
1.8
2.4
2.9
0.9
1.9
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing extended phenytoin sodium capsules or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
Patients, their caregivers, and families should be informed that AEDs increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of the signs and symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers.5.3 Serious Dermatologic Reactions
Extended phenytoin sodium capsules can cause severe cutaneous adverse reactions (SCARs), which may be fatal. Reported reactions in phenytoin-treated patients have included toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome (SJS), acute generalized exanthematous pustulosis (AGEP), and Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) [see Warnings and Precautions (5.4)].The onset of symptoms is usually within 28 days, but can occur later. Extended phenytoin sodium capsules should be discontinued at the first sign of a rash, unless the rash is clearly not drug-related. If signs or symptoms suggest a severe cutaneous adverse reaction, use of this drug should not be resumed and alternative therapy should be considered. If a rash occurs, the patient should be evaluated for signs and symptoms of SCARs.
Studies in patients of Chinese ancestry have found a strong association between the risk of developing SJS/TEN and the presence of HLA-B*1502, an inherited allelic variant of the HLA B gene, in patients using carbamazepine. Limited evidence suggests that HLAB* 1502 may be a risk factor for the development of SJS/TEN in patients of Asian ancestry taking other antiepileptic drugs associated with SJS/TEN, including phenytoin. In addition, retrospective, case-control, genome-wide association studies in patients of southeast Asian ancestry have also identified an increased risk of SCARs in carriers of the decreased function CYP2C9*3 variant, which has also been associated with decreased clearance of phenytoin. Consider avoiding extended phenytoin sodium capsules as an alternative to carbamazepine in patients who are positive for HLA-B*1502 or in CYP2C9*3 carriers [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.5)].
The use of HLA-B*1502 or CYP2C9 genotyping has important limitations and must never substitute for appropriate clinical vigilance and patient management. The role of other possible factors in the development of, and morbidity from, SJS/TEN, such as antiepileptic drug (AED) dose, compliance, concomitant medications, comorbidities, and the level of dermatologic monitoring have not been studied.
5.4 Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as Multiorgan hypersensitivity, has been reported in patients taking antiepileptic drugs, including extended phenytoin sodium capsules. Some of these events have been fatal or life-threatening. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy, and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis sometimes resembling an acute viral infection. Eosinophilia is often present. Because this disorder is variable in its expression, other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. Extended phenytoin sodium capsules should be discontinued if an alternative etiology for the signs or symptoms cannot be established.
5.5 Hypersensitivity
Extended phenytoin sodium capsules and other hydantoins are contraindicated in patients who have experienced phenytoin hypersensitivity [see Contraindications (4) and Warnings and Precautions (5.7)]. Additionally, consider alternatives to structurally similar drugs such as carboxamides (e.g., carbamazepine), barbiturates, succinimides, and oxazolidinediones (e.g., trimethadione) in these same patients. Similarly, if there is a history of hypersensitivity reactions to these structurally similar drugs in the patient or immediate family members, consider alternatives to extended phenytoin sodium capsules.
5.6 Cardiac Effects
Cases of bradycardia and cardiac arrest have been reported in extended phenytoin sodium capsules-treated patients, both at recommended phenytoin doses and levels, and in association with phenytoin toxicity [see Overdosage (10)]. Most of the reports of cardiac arrest occurred in patients with underlying cardiac disease.
5.7 Angioedema
Angioedema has been reported in patients treated with extended phenytoin sodium capsules in the postmarketing setting. Extended phenytoin sodium capsules should be discontinued immediately if symptoms of angioedema, such as facial, perioral, or upper airway swelling occur. Extended phenytoin sodium capsules should be discontinued permanently if a clear alternative etiology for the reaction cannot be established.
5.8 Hepatic Injury
Cases of acute hepatotoxicity, including infrequent cases of acute hepatic failure, have been reported with extended phenytoin sodium capsules. These events may be part of the spectrum of DRESS or may occur in isolation [see Warnings and Precautions (5.4)]. Other common manifestations include jaundice, hepatomegaly, elevated serum transaminase levels, leukocytosis, and eosinophilia. The clinical course of acute phenytoin hepatotoxicity ranges from prompt recovery to fatal outcomes. In these patients with acute hepatotoxicity, extended phenytoin sodium capsules should be immediately discontinued and not readministered.
5.9 Hematopoietic Complications
Hematopoietic complications, some fatal, have occasionally been reported in association with administration of extended phenytoin sodium capsules. These have included thrombocytopenia, leukopenia, granulocytopenia, agranulocytosis, and pancytopenia with or without bone marrow suppression.
There have been a number of reports suggesting a relationship between phenytoin and the development of lymphadenopathy (local or generalized) including benign lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin’s disease. Although a cause and effect relationship has not been established, the occurrence of lymphadenopathy indicates the need to differentiate such a condition from other types of lymph node pathology. Lymph node involvement may occur with or without symptoms and signs of DRESS [see Warnings and Precautions (5.4)].
In all cases of lymphadenopathy, follow-up observation for an extended period is indicated and every effort should be made to achieve seizure control using alternative antiepileptic drugs.5.10 Effects on Vitamin D and Bone
The chronic use of phenytoin in patients with epilepsy has been associated with decreased bone mineral density (osteopenia, osteoporosis, and osteomalacia) and bone fractures. Phenytoin induces hepatic metabolizing enzymes. This may enhance the metabolism of vitamin D and decrease vitamin D levels, which may lead to vitamin D deficiency, hypocalcemia, and hypophosphatemia. Consideration should be given to screening with bone-related laboratory and radiological tests as appropriate and initiating treatment plans according to established guidelines.
5.11 Renal or Hepatic Impairment or Hypoalbuminemia
Because the fraction of unbound phenytoin is increased in patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of phenytoin serum levels should be based on the unbound fraction in those patients.
5.12 Exacerbation of Porphyria
In view of isolated reports associating phenytoin with exacerbation of porphyria, caution should be exercised in using this medication in patients suffering from this disease.
5.13 Teratogenicity and Other Harm to the Newborn
Extended phenytoin sodium capsules may cause fetal harm when administered to a pregnant woman. Prenatal exposure to phenytoin may increase the risks for congenital malformations and other adverse developmental outcomes [see Use in Specific Populations (8.1)].
Increased frequencies of major malformations (such as orofacial clefts and cardiac defects), and abnormalities characteristic of fetal hydantoin syndrome, including dysmorphic skull and facial features, nail and digit hypoplasia, growth abnormalities (including microcephaly), and cognitive deficits, have been reported among children born to epileptic women who took phenytoin alone or in combination with other antiepileptic drugs during pregnancy. There have been several reported cases of malignancies, including neuroblastoma.
A potentially life-threatening bleeding disorder related to decreased levels of vitamin K-dependent clotting factors may occur in newborns exposed to phenytoin in utero. This drug-induced condition can be prevented with vitamin K administration to the mother before delivery and to the neonate after birth.5.14 Hyperglycemia
Hyperglycemia, resulting from the drug’s inhibitory effects on insulin release, has been reported. Phenytoin may also raise the serum glucose level in diabetic patients.
5.15 Serum Phenytoin Levels above Therapeutic Range
Serum levels of phenytoin sustained above the therapeutic range may produce confusional states referred to as “delirium,” “psychosis,” or “encephalopathy,” or rarely irreversible cerebellar dysfunction and/or cerebellar atrophy. Accordingly, at the first sign of acute toxicity, serum levels should be immediately checked. Dose reduction of phenytoin therapy is indicated if serum levels are excessive; if symptoms persist, termination is recommended.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are described elsewhere in the labeling:
- Withdrawal Precipitated Seizure, Status Epilepticus [see Warnings and Precautions (5.1)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.2)]
- Serious Dermatologic Reactions [see Warnings and Precautions (5.3)]
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity [see Warnings and Precautions (5.4)]
- Hypersensitivity [see Warnings and Precautions (5.5)]
- Cardiac Effects [see Warnings and Precautions (5.6)]
- Angioedema [see Warnings and Precautions (5.7)]
- Hepatic Injury [see Warnings and Precautions (5.8)]
- Hematopoietic Complications [see Warnings and Precautions (5.9)]
- Effects on Vitamin D and Bone [see Warnings and Precautions (5.10)]
- Exacerbation of Porphyria [see Warnings and Precautions (5.12)]
- Teratogenicity and Other Harm to the Newborn [see Warnings and Precautions (5.13)]
- Hyperglycemia [see Warnings and Precautions (5.14)]
The following adverse reactions associated with the use of extended phenytoin sodium capsules were identified in clinical studies or postmarketing reports. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Body as a Whole: Allergic reactions in the form of rash and rarely more serious forms and DRESS have been observed, as has angioedema [see Warnings and Precautions (5.3, 5.4, 5.7)]. Anaphylaxis has also been reported.
There have also been reports of coarsening of facial features, systemic lupus erythematosus, periarteritis nodosa, and immunoglobulin abnormalities.
Digestive System: Acute hepatic failure, toxic hepatitis, liver damage, nausea, vomiting, constipation, enlargement of the lips, and gingival hyperplasia.
Hematologic and Lymphatic System: Hematopoietic complications, some fatal, have occasionally been reported in association with administration of phenytoin. These have included thrombocytopenia, leukopenia, granulocytopenia, agranulocytosis, and pancytopenia with or without bone marrow suppression. While macrocytosis and megaloblastic anemia have occurred, these conditions usually respond to folic acid therapy. Lymphadenopathy including benign lymph node hyperplasia, pseudolymphoma, lymphoma, and Hodgkin’s disease have been reported [see Warnings and Precautions (5.9)]. Pure red cell aplasia has also been reported.
Laboratory Test Abnormality: Phenytoin may decrease serum concentrations of thyroid hormone (T4 and T3), sometimes with an accompanying increase in thyroid-stimulating hormone (TSH), but usually in the absence of clinical hypothyroidism. Phenytoin may also produce lower than normal values for dexamethasone or metyrapone tests.
Phenytoin may cause increased serum levels of glucose [see Warnings and Precautions (5.14)], alkaline phosphatase, and gamma glutamyl transpeptidase (GGT).
Nervous System: The most common adverse reactions encountered with phenytoin therapy are nervous system reactions and are usually dose-related. Reactions include nystagmus, ataxia, slurred speech, decreased coordination, somnolence, and mental confusion. Dizziness, vertigo, insomnia, transient nervousness, motor twitchings, paresthesias, and headaches have also been observed. There have also been rare reports of phenytoin-induced dyskinesias, including chorea, dystonia, tremor and asterixis, similar to those induced by phenothiazine and other neuroleptic drugs. Cerebellar atrophy has been reported, and appears more likely in settings of elevated phenytoin levels and/or long-term phenytoin use [see Warnings and Precautions (5.15)].
A predominantly sensory peripheral polyneuropathy has been observed in patients receiving long-term phenytoin therapy.
Skin and Appendages: Dermatological manifestations sometimes accompanied by fever have included scarlatiniform or morbilliform rashes. A morbilliform rash (measles-like) is the most common; other types of dermatitis are seen more rarely. Other more serious forms which may be fatal have included bullous, exfoliative or purpuric dermatitis, acute generalized exanthematous pustulosis, Stevens-Johnson syndrome, and toxic epidermal necrolysis [see Warnings and Precautions (5.3)]. There have also been reports of hypertrichosis and urticaria.
Special Senses: Altered taste sensation including metallic taste.
Urogenital: Peyronie’s disease
-
7 DRUG INTERACTIONS
Phenytoin is extensively bound to plasma proteins and is prone to competitive displacement. Phenytoin is primarily metabolized by the hepatic cytochrome P450 enzyme CYP2C9 and to a lesser extent by CYP2C19, and is particularly susceptible to inhibitory drug interactions because it is subject to saturable metabolism. Inhibition of metabolism may produce significant increases in circulating phenytoin concentrations and enhance the risk of drug toxicity. Monitoring of phenytoin serum levels is recommended when a drug interaction is suspected.
Phenytoin is a potent inducer of hepatic drug-metabolizing enzymes.
7.1 Drugs that Affect Phenytoin Concentrations
Table 2 includes commonly occurring drug interactions that affect phenytoin concentrations. However, this list is not intended to be inclusive or comprehensive. Individual prescribing information from relevant drugs should be consulted. The addition or withdrawal of these agents in patients on phenytoin therapy may require an adjustment of the phenytoin dose to achieve optimal clinical outcome.
Table 2: Drugs That Affect Phenytoin Concentrations
Interacting Agent
Examples
Drugs that may increase phenytoin serum levels
Antiepileptic drugs
Ethosuximide, felbamate, oxcarbazepine, methsuximide,
topiramate
Azoles
Fluconazole, ketoconazole, itraconazole, miconazole,
voriconazole
Antineoplastic agents
Capecitabine, fluorouracil
Antidepressants
Fluoxetine, fluvoxamine, sertraline
Gastric acid reducing
agents
H2 antagonists (cimetidine), omeprazole
Sulfonamides
Sulfamethizole, sulfaphenazole, sulfadiazine, sulfamethoxazoletrimethoprim
Other
Acute alcohol intake, amiodarone, chloramphenicol,
chlordiazepoxide, disulfiram, estrogen, fluvastatin, isoniazid,
methylphenidate, phenothiazines, salicylates, ticlopidine,
tolbutamide, trazodone, warfarin
Drugs that may decrease phenytoin serum levels
Antacidsa
Calcium carbonate, aluminum hydroxide, magnesium hydroxide
Prevention or Management: Phenytoin and antacids should
not be taken at the same time of day
Antineoplastic agents
usually in
combination
Bleomycin, carboplatin, cisplatin, doxorubicin, methotrexate
Antiviral agents
Fosamprenavir, nelfinavir, ritonavir
Antiepileptic drugs
Carbamazepine, vigabatrin
Other
Chronic alcohol abuse, diazepam, diazoxide, folic acid,
reserpine, rifampin, St. John’s wortb, sucralfate, theophylline
Drugs that may either increase or decrease phenytoin serum levels
Antiepileptic drugs
Phenobarbital, valproate sodiumc, valproic acidc
a Antacids may affect absorption of phenytoin.
b The induction potency of St. John’s wort may vary widely based on preparation.C Valproate sodium and valproic acid are similar medications. The term valproate has been used to represent these medications.
7.2 Drugs Affected by Phenytoin
Table 3 includes commonly occurring drug interactions affected by phenytoin. However, this list is not intended to be inclusive or comprehensive. Individual drug package inserts should be consulted. The addition or withdrawal of phenytoin during concomitant therapy with these agents may require adjustment of the dose of these agents to achieve optimal clinical outcome.
Table 3: Drugs Affected by Phenytoin
Interacting Agent
Examples
Drugs whose efficacy is impaired by phenytoin
Azoles
Fluconazole, ketoconazole, itraconazole, posaconazole,
voriconazole
Antineoplastic agents
Irinotecan, paclitaxel, teniposide
Delavirdine
Phenytoin can substantially reduce the concentrations of
delavirdine. This can lead to loss of virologic response and
possible resistance [see Contraindications (4)].
Neuromuscular
blocking agents
Cisatracurium, pancuronium, rocuronium and vecuronium:
resistance to the neuromuscular blocking action of the
nondepolarizing neuromuscular blocking agents has occurred in
patients chronically administered phenytoin. Whether or not
phenytoin has the same effect on other non-depolarizing agents
is unknown.
Prevention or Management: Patients should be monitored
closely for more rapid recovery from neuromuscular blockade
than expected, and infusion rate requirements may be higher.
Warfarin
Increased and decreased PT/INR responses have been reported
when phenytoin is coadministered with warfarin
Other
Corticosteroids, doxycycline, estrogens, furosemide, oral
contraceptives, paroxetine, quinidine, rifampin, sertraline,
theophylline, and vitamin D
Drugs whose level is decreased by phenytoin
Anticoagulants
Apixaban, dabigatran, edoxaban, rivaroxaban
Antiepileptic drugsa
Carbamazepine, felbamate, lamotrigine, topiramate,
oxcarbazepine, lacosamide
Antilipidemic agents
Atorvastatin, fluvastatin, simvastatin
Antiplatelets
Ticagrelor
Antiviral agents
Efavirenz, lopinavir/ritonavir, indinavir, nelfinavir, ritonavir,
saquinavir
Fosamprenavir: phenytoin when given with fosamprenavir alone
may decrease the concentration of amprenavir, the active
metabolite. Phenytoin when given with the combination of
fosamprenavir and ritonavir may increase the concentration of
amprenavir
Calcium channel
blockers
Nifedipine, nimodipine, nisoldipine, verapamil
Other
Albendazole (decreases active metabolite), chlorpropamide,
clozapine, cyclosporine, digoxin, disopyramide, folic acid, methadone, mexiletine, praziquantel, quetiapine
a The effect of phenytoin on phenobarbital, valproic acid and sodium valproate serum levels is unpredictable
7.3 Hyperammonemia with Concomitant Use of Valproate
Concomitant administration of phenytoin and valproate has been associated with an increased risk of valproate-associated hyperammonemia. Patients treated concomitantly with these two drugs should be monitored for signs and symptoms of hyperammonemia.
7.4 Drug Enteral Feeding/Nutritional Preparations Interaction
Literature reports suggest that patients who have received enteral feeding preparations and/or related nutritional supplements have lower than expected phenytoin serum levels. It is therefore suggested that phenytoin not be administered concomitantly with an enteral feeding preparation. More frequent serum phenytoin level monitoring may be necessary in these patients.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antiepileptic drugs (AEDs), such as extended phenytoin sodium capsules, during pregnancy.
Physicians are advised to recommend that pregnant patients taking extended phenytoin sodium capsules enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. This can be done by calling the tollfree number 1-888-233-2334, and must be done by patients themselves.
Information on the registry can also be found at the website http://www.aedpregnancyregistry.org/
Risk Summary
In humans, prenatal exposure to phenytoin may increase the risks for congenital malformations and other adverse developmental outcomes. Prenatal phenytoin exposure is associated with an increased incidence of major malformations, including orofacial clefts and cardiac defects. In addition, the fetal hydantoin syndrome, a pattern of abnormalities including dysmorphic skull and facial features, nail and digit hypoplasia, growth abnormalities (including microcephaly), and cognitive deficits has been reported among children born to epileptic women who took phenytoin alone or in combination with other antiepileptic drugs during pregnancy [see Data]. There have been several reported cases of malignancies, including neuroblastoma, in children whose mothers received phenytoin during pregnancy.
Administration of phenytoin to pregnant animals resulted in an increased incidence of fetal malformations and other manifestations of developmental toxicity (including embryofetal death, growth impairment, and behavioral abnormalities) in multiple species at clinically relevant doses [see Data].
In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Clinical Considerations
Disease-associated maternal risk
An increase in seizure frequency may occur during pregnancy because of altered phenytoin pharmacokinetics. Periodic measurement of serum phenytoin concentrations may be valuable in the management of pregnant women as a guide to appropriate adjustment of dosage [see Dosage and Administration (2.3, 2.7)]. However, postpartum restoration of the original dosage will probably be indicated [see Clinical Pharmacology (12.3)].
Fetal/Neonatal Adverse Reactions
A potentially life-threatening bleeding disorder related to decreased levels of vitamin K-dependent clotting factors may occur in newborns exposed to phenytoin in utero. This drug-induced condition can be prevented with vitamin K administration to the mother before delivery and to the neonate after birth.
Data
Human Data
Meta-analyses using data from published observational studies and registries have estimated an approximately 2.4-fold increased risk for any major malformation in children with prenatal phenytoin exposure compared to controls. An increased risk of heart defects, facial clefts, and digital hypoplasia has been reported. The fetal hydantoin syndrome is a pattern of congenital anomalies including craniofacial anomalies, nail and digital hypoplasia, prenatal-onset growth deficiency, and neurodevelopmental deficiencies.
Animal Data
Administration of phenytoin to pregnant rats, rabbits, and mice during organogenesis resulted in embryofetal death, fetal malformations, and decreased fetal growth.
Malformations (including craniofacial, cardiovascular, neural, limb, and digit abnormalities) were observed in rats, rabbits, and mice at doses as low as 100, 75, and 12.5 mg/kg, respectively.
8.2 Lactation
Risk Summary
Phenytoin is secreted in human milk. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for extended phenytoin sodium capsules and any potential adverse effects on the breastfed infant from extended phenytoin sodium capsules or from the underlying maternal condition.
8.4 Pediatric Use
Initially, 5 mg/kg/day in two or three equally divided doses, with subsequent dosage individualized to a maximum of 300 mg daily. A recommended daily maintenance dosage is usually 4 to 8 mg/kg. Children over 6 years and adolescents may require the minimum adult dosage (300 mg/day) [see Dosage and Administration (2.2)].
8.5 Geriatric Use
Phenytoin clearance tends to decrease with increasing age [see Clinical Pharmacology (12.3)]. Lower or less frequent dosing may be required [see Dosage and Administration (2.6)].
8.6 Renal and/or Hepatic Impairment or Hypoalbuminemia
The liver is the chief site of biotransformation of phenytoin; patients with impaired liver function, elderly patients, or those who are gravely ill may show early signs of toxicity.
Because the fraction of unbound phenytoin is increased in patients with renal or hepatic disease, or in those with hypoalbuminemia, the monitoring of phenytoin serum levels should be based on the unbound fraction in those patients.
8.7 Use in Patients with Decreased CYP2C9 Function
Patients who are intermediate or poor metabolizers of CYP2C9 substrates (e.g., *1/*3, *2/*2, *3/*3) may exhibit increased phenytoin serum concentrations compared to patients who are normal metabolizers (e.g., *1/*1). Thus, patients who are known to be intermediate or poor metabolizers may ultimately require lower doses of phenytoin to maintain similar steady-state concentrations compared to normal metabolizers. If early signs of dose-related central nervous system (CNS) toxicity develop, serum concentrations should be checked immediately [see Clinical Pharmacology (12.5)].
-
10 OVERDOSAGE
The lethal dose in pediatric patients is not known. The lethal dose in adults is estimated to be 2 to 5 grams. The initial symptoms are nystagmus, ataxia, and dysarthria. Other signs are tremor, hyperreflexia, lethargy, slurred speech, blurred vision, nausea, and vomiting. The patient may become comatose and hypotensive. Bradycardia and cardiac arrest have been reported [see Warnings and Precautions (5.6)]. Death is caused by respiratory and circulatory depression.
There are marked variations among individuals with respect to phenytoin serum levels where toxicity may occur. Nystagmus, on lateral gaze, usually appears at 20 mcg/mL, ataxia at 30 mcg/mL; dysarthria and lethargy appear when the serum concentration is over 40 mcg/mL, but as high a concentration as 50 mcg/mL has been reported without evidence of toxicity. As much as 25 times the therapeutic dose has been taken to result in a serum concentration over 100 mcg/mL with complete recovery. Irreversible cerebellar dysfunction and atrophy have been reported.
Treatment: Treatment is nonspecific since there is no known antidote.
The adequacy of the respiratory and circulatory systems should be carefully observed and appropriate supportive measures employed. Hemodialysis can be considered since phenytoin is not completely bound to plasma proteins. Total exchange transfusion has been used in the treatment of severe intoxication in pediatric patients.
In acute overdosage the possibility of other CNS depressants, including alcohol, should be borne in mind.
-
11 DESCRIPTION
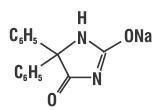
Phenytoin sodium is related to the barbiturates in chemical structure, but has a five-membered ring. The chemical name is 5,5- Diphenylhydantoin sodium salt, having the following structural formula:
Each extended phenytoin sodium capsule, USP, for oral administration contains 200 mg or 300 mg of phenytoin sodium, USP. Each capsule also contains the following inactive ingredients: magnesium stearate, lactitol monohydrate, sodium lauryl sulfate, and talc. In addition, each of the empty gelatin capsule contains the following: gelatin, FD&C Blue # 1, FD&C Red # 40, D&C Red # 28 (200 mg) and FD&C Red # 3 (200 mg). The imprinting ink contains shellac, dehydrated alcohol, butyl alcohol, propylene glycol, strong ammonia solution, black iron oxide and potassium hydroxide. Product in vivo performance is characterized by a slow and extended rate of absorption with peak blood concentrations expected in 4 to 12 hours as contrasted to prompt phenytoin sodium capsules, USP, with a rapid rate of absorption with peak blood concentration expected in 1½ to 3 hours.
Extended phenytoin sodium capsules, USP 200 mg and 300 mg meet USP Dissolution Test 3.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism by which phenytoin exerts its therapeutic effect has not been established but is thought to involve the voltage-dependent blockade of membrane sodium channels resulting in a reduction in sustained high-frequency neuronal discharges.
12.3 Pharmacokinetics
Absorption
For extended phenytoin sodium capsules, peak serum levels occur 4 to 12 hours after administration. Steady-state therapeutic levels are achieved at least 7 to 10 days (5–7 half-lives) after initiation of therapy with recommended doses of 300 mg/day. When serum level determinations are necessary, they should be obtained at least 5–7 half-lives after treatment initiation, dosage change, or addition or subtraction of another drug to the regimen so that equilibrium or steady-state will have been achieved.
Distribution
Phenytoin is extensively bound to serum plasma proteins.
Elimination
The plasma half-life in man after oral administration of phenytoin averages 22 hours, with a range of 7 to 42 hours.
Metabolism
Phenytoin is primarily metabolized by the hepatic cytochrome P450 enzyme CYP2C9 and to a lesser extent by CYP2C19. Because phenytoin is hydroxylated in the liver by an enzyme system which is saturable at high serum levels, small incremental doses may increase the half-life and produce very substantial increases in serum levels, when these are in the upper range. The steady-state level may be disproportionately increased, with resultant intoxication, from an increase in dosage of 10% or more.
In most patients maintained at a steady dosage, stable phenytoin serum levels are achieved. There may be wide interpatient variability in phenytoin serum levels with equivalent dosages. Patients with unusually low levels may be noncompliant or hypermetabolizers of phenytoin. Unusually high levels result from liver disease, variant CYP2C9 and CYP2C19 alleles, or drug interactions which result in metabolic interference. The patient with large variations in phenytoin serum levels, despite standard doses, presents a difficult clinical problem. Serum level determinations in such patients may be particularly helpful. As phenytoin is highly protein bound, free phenytoin levels may be altered in patients whose protein binding characteristics differ from normal.
Excretion
Most of the drug is excreted in the bile as inactive metabolites which are then reabsorbed from the intestinal tract and excreted in the urine. Urinary excretion of phenytoin and its metabolites occurs partly with glomerular filtration but, more importantly, by tubular secretion.
Specific Populations
Age: Geriatric Population:
Phenytoin clearance tends to decrease with increasing age (20% less in patients over 70 years of age relative to that in patients 20 to 30 years of age). Since phenytoin clearance is decreased slightly in elderly patients, lower or less frequent dosing may be required [see Dosage and Administration (2.6)].
Sex/Race:
Gender and race have no significant impact on phenytoin pharmacokinetics.
Renal or Hepatic Impairment:
Increased fraction of unbound phenytoin in patients with renal or hepatic disease, or in those with hypoalbuminemia has been reported.
Pregnancy:
It has been reported in the literature that the plasma clearance of phenytoin generally increased during pregnancy, reached a peak in the third trimester and returned to the level of pre-pregnancy after few weeks or months of delivery.
Drug Interaction Studies
Phenytoin is primarily metabolized by the hepatic cytochrome P450 enzyme CYP2C9 and to a lesser extent by CYP2C19.
Phenytoin is a potent inducer of hepatic drug-metabolizing enzymes [see Drug Interactions (7.1, 7.2)].12.5 Pharmacogenomics
CYP2C9 activity is decreased in individuals with genetic variants such as the CYP2C9*2 and CYP2C9*3 alleles. Carriers of variant alleles, resulting in intermediate (e.g., *1/*3, *2/*2) or poor metabolism (e.g., *2/*3, *3/*3) have decreased clearance of phenytoin. Other decreased or nonfunctional CYP2C9 alleles may also result in decreased clearance of phenytoin (e.g., *5, *6, *8, *11).
The prevalence of the CYP2C9 poor metabolizer phenotype is approximately 2-3% in the White population, 0.5-4% in the Asian population, and <1% in the African American population. The CYP2C9 intermediate phenotype prevalence is approximately 35% in the White population, 24% in the African American population, and 15-36% in the Asian population [see Warnings and Precautions (5.3) and Use in Specific Populations (8.7)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis [see Warnings and Precautions (5.9)]
In carcinogenicity studies, phenytoin was administered in the diet to mice (10, 25, or 45 mg/kg/day) and rats (25, 50, or 100 mg/kg/day) for 2 years. The incidences of hepatocellular tumors were increased in male and female mice at the highest dose. No increases in tumor incidence were observed in rats. The highest doses tested in these studies were associated with peak serum phenytoin levels below human therapeutic concentrations.
In carcinogenicity studies reported in the literature, phenytoin was administered in the diet for 2 years at doses up to 600 ppm (approximately 160 mg/kg/day) to mice and up to 2400 ppm (approximately 120 mg/kg/day) to rats. The incidences of hepatocellular tumors were increased in female mice at all but the lowest dose tested. No increases in tumor incidence were observed in rats.
Mutagenesis
Phenytoin was negative in the Ames test and in the in vitro clastogenicity assay in Chinese hamster ovary (CHO) cells.
In studies reported in the literature, phenytoin was negative in the in vitro mouse lymphoma assay and the in vivo micronucleus assay in mouse. Phenytoin was clastogenic in the in vitro sister chromatid exchange assay in CHO cells.
Fertility
Phenytoin has not been adequately assessed for effects on male or female fertility.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Extended Phenytoin Sodium Capsules, USP are available containing 200 mg or 300 mg of phenytoin sodium, USP.
The 200 mg capsule has a blue transparent cap and a light blue transparent body. The hard-shell gelatin capsule is filled with white to off-white powder. The capsule is printed axially with 299 in black ink on both the cap and the body.
They are available as follows:
Bottle of 30 with child-resistant cap...................NDC 62756-299-83
Bottle of 100 with child-resistant cap.................NDC 62756-299-88
Bottle of 100.........NDC 62756-299-08
Bottle of 500.........NDC 62756-299-13
The 300 mg capsule has a light blue transparent cap and a light blue transparent body. The hard-shell gelatin capsule is filled with white to off-white powder. The capsule is printed axially with 432 in black ink on both the cap and the body.
They are available as follows:
Bottle of 30 with child-resistant cap...................NDC 62756-432-83
Bottle of 100 with child-resistant cap.................NDC 62756-432-88
Bottle of 100.........NDC 62756-432-08
Bottle of 500.........NDC 62756-432-13
-
17 PATIENT COUNSELING INFORMATION
Advise patients to read the FDA-approved patient labeling (Medication Guide).
Administration Information
Advise patients taking phenytoin of the importance of adhering strictly to the prescribed dosage regimen, and of informing the physician of any clinical condition in which it is not possible to take the drug orally as prescribed, e.g., surgery, etc.
Advise patients not to use capsules which are discolored.
Withdrawal of Antiepileptic Drugs
Advise patients not to discontinue use of extended phenytoin sodium capsules without consulting with their healthcare provider. Extended phenytoin sodium capsules should normally be gradually withdrawn to reduce the potential for increased seizure frequency and status epilepticus [see Warnings and Precautions (5.1)].
Suicidal Ideation and Behavior
Counsel patients, their caregivers, and families that AEDs, including extended phenytoin sodium capsules, may increase the risk of suicidal thoughts and behavior and advise them of the need to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Behaviors of concern should be reported immediately to healthcare providers [see Warnings and Precautions (5.2)].
Serious Dermatologic ReactionsAdvise patients of the early signs and symptoms of severe cutaneous adverse reactions and to report any occurrence immediately to a physician [see Warnings and Precautions (5.3)].
Potential Signs of Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) and Other Systemic Reactions
Advise patients of the early toxic signs and symptoms of potential hematologic, dermatologic, hypersensitivity, or hepatic reactions. These symptoms may include, but are not limited to, fever, sore throat, rash, ulcers in the mouth, easy bruising, lymphadenopathy, facial swelling, and petechial or purpuric hemorrhage, and in the case of liver reactions, anorexia, nausea/vomiting, or jaundice. Advise the patient that, because these signs and symptoms may signal a serious reaction, that they must report any occurrence immediately to a physician. In addition, advise the patient that these signs and symptoms should be reported even if mild or when occurring after extended use [see Warnings and Precautions (5.3, 5.4, 5.5, 5.8, 5.9)].
Cardiac Effects
Counsel patients that cases of bradycardia and cardiac arrest have been reported, both at recommended phenytoin doses and levels, and in association with phenytoin toxicity. Patients should report cardiac signs or symptoms to their healthcare provider [see Warnings and Precautions (5.6) and Overdosage (10)].
AngioedemaAdvise patients to discontinue extended phenytoin sodium capsules and seek immediate medical care if they develop signs or symptoms of angioedema, such as facial, perioral, or upper airway swelling [see Warnings and Precautions (5.7)].
Effects of Alcohol Use and Other Drugs and Over-the-Counter Drug Interactions
Caution patients against the use of other drugs or alcoholic beverages without first seeking their physician’s advice [Drug Interactions (7.1, 7.2)].
Inform patients that certain over-the-counter medications (e.g., antacids, cimetidine, and omeprazole), vitamins (e.g., folic acid), and herbal supplements (e.g., St. John’s wort) can alter their phenytoin levels.
Hyperglycemia
Advise patients that extended phenytoin sodium capsules may cause an increase in blood glucose levels [see Warnings and Precautions (5.14)].
Gingival Hyperplasia
Advise patients of the importance of good dental hygiene in order to minimize the development of gingival hyperplasia and its complications.
Neurologic Effects
Counsel patients that extended phenytoin sodium capsules may cause dizziness, gait disturbance, decreased coordination and somnolence. Advise patients taking extended phenytoin sodium capsules not to drive, operate complex machinery, or engage in other hazardous activities until they have become accustomed to any such effects associated with extended phenytoin sodium capsules.
Use in Pregnancy
Inform pregnant women and women of childbearing potential that use of extended phenytoin sodium capsules during pregnancy can cause fetal harm, including an increased risk for cleft lip and/or cleft palate (oral clefts), cardiac defects, dysmorphic skull and facial features, nail and digit hypoplasia, growth abnormalities (including microcephaly), and cognitive deficits. When appropriate, counsel pregnant women and women of childbearing potential about alternative therapeutic options. Advise women of childbearing potential who are not planning a pregnancy to use effective contraception while using extended phenytoin sodium capsules, keeping in mind that there is a potential for decreased hormonal contraceptive efficacy [see Drug Interactions (7.2)].
Instruct patients to notify their physician if they become pregnant or intend to become pregnant during therapy, and to notify their physician if they are breastfeeding or intend to breastfeed during therapy [see Use in Specific Populations (8.1, 8.2)].
Encourage patients to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy [see Use in Specific Populations (8.1)].
-
MEDICATION GUIDE
Dispense with Medication Guide available at: https://www.sunpharma.com/usa/products
MEDICATION GUIDE
Extended Phenytoin (FEN-i-toyn) Sodium Capsules, USP
What is the most important information I should know about extended phenytoin sodium capsules?
1. Do not stop taking extended phenytoin sodium capsules without first talking to your healthcare provider.
- Stopping extended phenytoin sodium capsules suddenly can cause serious problems.
- Stopping a seizure medicine suddenly can cause you to have seizures more often or seizures that will not stop (status epilepticus).
- Thoughts about suicide or dying
- New or worse anxiety
- Trouble sleeping (insomnia)
- Acting on dangerous impulses
- Attempts to commit suicide
- Feeling agitated or restless
- New or worse irritability
- An extreme increase in activity and talking (mania)
- New or worse depression
- Panic attacks
- Acting aggressive, being angry, or violent
- Other unusual changes in behavior or mood
Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions?
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
3. Extended phenytoin sodium capsules can cause a type of serious allergic reaction that may affect different parts of the body such as your liver, kidneys, blood, heart, skin or other parts of your body. These can be very serious and cause death. Call your healthcare provider right away if you have any or all of these symptoms:
- Fever
- Sore throat
- Not wanting to eat (anorexia)
- Rash
- Sores in your mouth
- Nausea
- Swollen lymph glands
- Bruise easily
- Vomiting
- Swelling of your face, eye, lips, or tongue
- Purple or red spots on your skin
- Yellowing of the skin and the white part of your eyes (jaundice)
- Trouble swallowing or Breathing
- Increase infections
Call your healthcare provider even if the symptoms are mild or if you have been taking extended phenytoin sodium capsules for an extended period of time. These symptoms can be a sign of a serious allergic reaction.
4. Extended phenytoin sodium capsules can cause problems with your heart, including a slow heartbeat. Let your healthcare provider know right away if you have any of these symptoms:
- dizziness
- tiredness
- feeling like your heart is beating slowly or skipping beats
- chest pain
What is extended phenytoin sodium capsule?
Extended phenytoin sodium capsule is a prescription medicine used to treat certain types of seizures called tonic-clonic (grand mal) and psychomotor (temporal lobe) seizures.
Do not take extended phenytoin sodium capsules if you:
- Are allergic to phenytoin or any of the ingredients in extended phenytoin sodium capsules. See the end of this leaflet for a complete list of ingredients in extended phenytoin sodium capsules.
- Have had an allergic reaction to CEREBYX* (fosphenytoin), PEGANONE* (ethotoin), or MESANTOIN* (mephenytoin).
- Have had liver problems from taking phenytoin.
- Take delavirdine.
Before taking extended phenytoin sodium capsules, tell your healthcare provider about all of your medical conditions, including if you:
- Have or have had depression, mood problems, or suicidal thoughts or behavior
- Have had an allergic reaction to a medicine similar to extended phenytoin sodium capsules called carboxamides, barbiturates, succinimides, and oxazolidinediones
- Have or had liver or kidney problems
- Have or had an enzyme problem called porphyria
- Have or had high blood sugar (hyperglycemia)
- Drink alcohol
- Are pregnant or plan to become pregnant. Extended phenytoin sodium capsules may harm your unborn baby.
- If you take extended phenytoin sodium capsules during pregnancy, your baby is at risk for serious birth defects.
- If you become pregnant while taking extended phenytoin sodium capsules, the level of extended phenytoin sodium capsules in your blood may decrease, causing your seizures to become worse. Your healthcare provider may change your dose of extended phenytoin sodium capsules.
- If you take extended phenytoin sodium capsules during pregnancy, your baby is also at risk for bleeding problems right after birth. Your healthcare provider may give you and your baby medicine to prevent this.
- All women of child-bearing age should talk to their healthcare provider about using other possible treatments instead of extended phenytoin sodium capsules.
- If you are of childbearing age and are not planning on getting pregnant, you should use effective birth control (contraception) while taking extended phenytoin sodium capsules.
-
Pregnancy Registry: If you become pregnant while taking extended phenytoin sodium capsules, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334. The purpose of this registry is to collect information about the safety of antiepileptic drugs during pregnancy.
- Are breastfeeding or plan to breastfeed. Phenytoin sodium can pass into breast milk. You and your healthcare provider should decide if you will take extended phenytoin sodium capsules while you are breastfeeding.
Taking extended phenytoin sodium capsules with certain other medicines can cause side effects or affect how well they work. Do not start or stop other medicines without talking to your healthcare provider.
Know the medicines you take. Keep a list of them and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take extended phenytoin sodium capsules?
- Take extended phenytoin sodium capsules exactly as your healthcare provider tells you.
- Your healthcare provider will tell you how many extended phenytoin sodium capsules to take and when to take it.
- Your healthcare provider may change your dose if needed. Do not change your dose of extended phenytoin sodium capsules without talking to your healthcare provider.
- If your healthcare provider has prescribed phenytoin oral suspension, ask your pharmacist for a medicine dropper or medicine cup to help you measure the correct amount of extended phenytoin sodium capsules. Do not use a household teaspoon. Ask your pharmacist for instructions on how to use the measuring device the right way.
- Do not stop taking extended phenytoin sodium capsules without first talking to your healthcare provider. Stopping extended phenytoin sodium capsules suddenly can cause serious problems.
What should I avoid while taking extended phenytoin sodium capsules?
- Do not drink alcohol while you take extended phenytoin sodium capsules without first talking to your healthcare provider. Drinking alcohol while taking extended phenytoin sodium capsules may change your blood levels of phenytoin sodium which can cause serious problems.
- Do not drive, operate heavy machinery, or do other dangerous activities until you know how extended phenytoin sodium capsules affects you. Extended phenytoin sodium capsules can slow your thinking and motor skills.
What are the possible side effects of extended phenytoin sodium capsules?
See “What is the most important information I should know about extended phenytoin sodium capsules?”
Extended phenytoin sodium capsules may cause other serious side effects including:
- Liver problems.
- Low blood count which could increase your chance of getting infections, bruising, bleeding and increased fatigue
- Softening of your bones (osteopenia, osteoporosis, and osteomalacia) can cause your bones to break (fractures).
- High blood sugar (hyperglycemia).
- High levels of extended phenytoin sodium capsules in your blood that could cause confusion also known as delirium, psychosis or a more serious condition that affects how your brain works (encephalopathy).
Call your healthcare provider right away, if you have any of the symptoms listed above.
The most common side effects of extended phenytoin sodium capsules include:
- Irregular movement of the eye (nystagmus)
- Slurred speech
- Drowsiness (somnolence)
- Problems with movement and balance (ataxia)
- Decrease in coordination
- Confusion
These are not all of the possible side effects of extended phenytoin sodium capsules.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store extended phenytoin sodium capsules?
- Store at 25°C (77°F); excursions permitted to 15° to 30°C (59° to 86°F).
- Store in tight, light-resistant containers.
- Protect from light and moisture.
- Extended phenytoin sodium capsules come in a child-resistant package.
Keep extended phenytoin sodium capsules and all medicines out of the reach of children.
General information about the safe and effective use of extended phenytoin sodium capsules.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use extended phenytoin sodium capsules for a condition for which it was not prescribed. Do not give extended phenytoin sodium capsules to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about extended phenytoin sodium capsules that is written for health professionals.
What are the ingredients in extended phenytoin sodium capsules?
Extended phenytoin sodium capsules 200 mg and 300 mg:
Active ingredient: phenytoin sodium
Inactive ingredients: lactitol monohydrate, sodium lauryl sulfate, talc and magnesium stearate. The capsule shell contains gelatin, FD&C Blue # 1, FD&C Red # 40, D&C Red # 28 (200 mg) and FD&C Red # 3 (200 mg) and black printing ink, which contains black iron oxide, shellac, dehydrated alcohol, butyl alcohol, propylene glycol, strong ammonia solution, and potassium hydroxide.
For more information call at 1-800-818-4555.
*All trademarks are the properties of their respective owners.
Dispense with Medication Guide available at: https://www.sunpharma.com/usa/products
Distributed by:
Sun Pharmaceutical Industries, Inc.
Cranbury, NJ 08512
Manufactured by:
Sun Pharmaceutical Industries Ltd.
Halol-Baroda Highway,
Halol-389 350, Gujarat, India.
5237839
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised. 12/2022
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-300 mg
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL-200 mg
-
INGREDIENTS AND APPEARANCE
PHENYTOIN SODIUM
phenytoin sodium capsule, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:62756-299 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PHENYTOIN SODIUM (UNII: 4182431BJH) (PHENYTOIN - UNII:6158TKW0C5) PHENYTOIN SODIUM 200 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) LACTITOL MONOHYDRATE (UNII: UH2K6W1Y64) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TALC (UNII: 7SEV7J4R1U) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C RED NO. 40 (UNII: WZB9127XOA) D&C RED NO. 28 (UNII: 767IP0Y5NH) ALCOHOL (UNII: 3K9958V90M) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) SHELLAC (UNII: 46N107B71O) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) FD&C RED NO. 3 (UNII: PN2ZH5LOQY) Product Characteristics Color BLUE (blue transparent) , BLUE (light blue transparent) Score no score Shape CAPSULE Size 19mm Flavor Imprint Code 299 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:62756-299-83 30 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 2 NDC:62756-299-88 100 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 3 NDC:62756-299-08 100 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 4 NDC:62756-299-13 500 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA040731 06/30/2008 PHENYTOIN SODIUM
phenytoin sodium capsule, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:62756-432 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PHENYTOIN SODIUM (UNII: 4182431BJH) (PHENYTOIN - UNII:6158TKW0C5) PHENYTOIN SODIUM 300 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) LACTITOL MONOHYDRATE (UNII: UH2K6W1Y64) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TALC (UNII: 7SEV7J4R1U) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C RED NO. 40 (UNII: WZB9127XOA) ALCOHOL (UNII: 3K9958V90M) BUTYL ALCOHOL (UNII: 8PJ61P6TS3) PROPYLENE GLYCOL (UNII: 6DC9Q167V3) AMMONIA (UNII: 5138Q19F1X) FERROSOFERRIC OXIDE (UNII: XM0M87F357) POTASSIUM HYDROXIDE (UNII: WZH3C48M4T) SHELLAC (UNII: 46N107B71O) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) Product Characteristics Color BLUE (light blue transparent) , BLUE (light blue transparent) Score no score Shape CAPSULE Size 22mm Flavor Imprint Code 432 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:62756-432-83 30 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 2 NDC:62756-432-88 100 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 3 NDC:62756-432-08 100 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 4 NDC:62756-432-13 500 in 1 BOTTLE; Type 0: Not a Combination Product 06/30/2008 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA040731 06/30/2008 Labeler - Sun Pharmaceutical Industries, Inc. (146974886) Establishment Name Address ID/FEI Business Operations Sun Pharmaceutical Industries Limited 725959238 ANALYSIS(62756-299, 62756-432) , MANUFACTURE(62756-299, 62756-432)

