Label: EMPAVELI- pegcetacoplan injection, solution


- NDC Code(s): 73606-010-01
- Packager: Apellis Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated July 30, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use EMPAVELI safely and effectively. See full prescribing information for EMPAVELI.
EMPAVELI® (pegcetacoplan) injection, for subcutaneous use
Initial U.S. Approval: 2021WARNING: SERIOUS INFECTIONS CAUSED BY ENCAPSULATED BACTERIA
See full prescribing information for complete boxed warning.
EMPAVELI increases the risk of serious and life-threatening infections caused by encapsulated bacteria including Streptococcus pneumoniae, Neisseria meningitidis and Haemophilus influenzae type B.
- Complete or update vaccination for encapsulated bacteria at least 2 weeks prior to the first dose of EMPAVELI, unless the risks of delaying EMPAVELI outweigh the risks of developing a serious infection. Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for vaccinations against encapsulated bacteria in patients receiving a complement inhibitor. (5.1)
- Patients receiving EMPAVELI are at increased risk for invasive disease caused by encapsulated bacteria, even if they develop antibodies following vaccination. Monitor patients for early signs and symptoms of serious infections and evaluate immediately if infection is suspected. (5.1)
EMPAVELI is available only through a restricted program called EMPAVELI REMS.
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
EMPAVELI is a complement inhibitor indicated:
DOSAGE AND ADMINISTRATION
PNH (2.2)
Recommended dosage is 1,080 mg administered subcutaneously twice weekly.
C3G or Primary IC-MPGN (2.3)
- Recommended dosage for adults is 1,080 mg administered subcutaneously twice weekly.
- Recommended dosage for pediatric patients is dependent upon patient weight. See full prescribing information for the recommended dosage in patients with C3G or IC-MPGN. (2.2)
- EMPAVELI can be administered via a commercially available pump or with EMPAVELI Injector. (2.4)
- See Full Prescribing Information for instructions on preparation and administration. (2.2, 2.3, 2.4)
DOSAGE FORMS AND STRENGTHS
- Injection: 1,080 mg/20 mL (54 mg/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Serious infections caused by encapsulated bacteria. (5.1)
- Infusion-Related Reactions: Monitor patients for infusion-related reactions and institute appropriate medical management as needed. (5.3)
- Interference with Laboratory Tests: Use of silica reagents in coagulation panels may result in artificially prolonged activated partial thromboplastin time (aPTT). (5.5)
ADVERSE REACTIONS
- The most common adverse reactions in patients with PNH (incidence ≥10%) were injection site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, pain in extremity, hypokalemia, fatigue, viral infection, cough, arthralgia, dizziness, headache, and rash. (6.1)
- The most common adverse reactions in patients with C3G or primary IC-MPGN (incidence ≥10%) were infusion site reactions, pyrexia, nasopharyngitis, influenza, cough, and nausea. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Apellis Pharmaceuticals, Inc. at 1-833-866-3346 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 7/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS INFECTIONS CAUSED BY ENCAPSULATED BACTERIA
1 INDICATIONS AND USAGE
1.1 Paroxysmal Nocturnal Hemoglobinuria
1.2 C3 glomerulopathy or primary immune-complex membranoproliferative glomerulonephritis
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination and Prophylaxis
2.2 Recommended Dosage - PNH
2.3 Recommended Dosage - C3G or Primary IC-MPGN
2.4 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections Caused by Encapsulated Bacteria
5.2 EMPAVELI REMS
5.3 Infusion-Related Reactions
5.4 Monitoring PNH Manifestations after Discontinuation of EMPAVELI
5.5 Interference with Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Paroxysmal Nocturnal Hemoglobinuria
14.2 C3 Glomerulopathy (C3G) or Primary Immune-Complex Membranoproliferative Glomerulonephritis (IC-MPGN)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS INFECTIONS CAUSED BY ENCAPSULATED BACTERIA
EMPAVELI, a complement inhibitor, increases the risk of serious infections, especially those caused by encapsulated bacteria, such as Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B [see Warnings and Precautions (5.1)]. Life-threatening and fatal infections with encapsulated bacteria have occurred in patients treated with complement inhibitors. These infections may become rapidly life-threatening or fatal if not recognized and treated early.
- Complete or update vaccination for encapsulated bacteria at least 2 weeks prior to the first dose of EMPAVELI, unless the risks of delaying therapy with EMPAVELI outweigh the risk of developing a serious infection. Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for vaccinations against encapsulated bacteria in patients receiving a complement inhibitor. See Warnings and Precautions (5.1) for additional guidance on the management of the risk of serious infections caused by encapsulated bacteria.
- Patients receiving EMPAVELI are at increased risk for invasive disease caused by encapsulated bacteria, even if they develop antibodies following vaccination. Monitor patients for early signs and symptoms of serious infections and evaluate immediately if infection is suspected.
Because of the risk of serious infections caused by encapsulated bacteria, EMPAVELI is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the EMPAVELI REMS [see Warnings and Precautions (5.2)].
-
1 INDICATIONS AND USAGE
1.1 Paroxysmal Nocturnal Hemoglobinuria
EMPAVELI® is indicated for the treatment of adult patients with paroxysmal nocturnal hemoglobinuria (PNH).
1.2 C3 glomerulopathy or primary immune-complex membranoproliferative glomerulonephritis
EMPAVELI® is indicated for the treatment of adult and pediatric patients aged 12 years and older with C3 glomerulopathy (C3G) or primary immune-complex membranoproliferative glomerulonephritis (IC-MPGN), to reduce proteinuria.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination and Prophylaxis
Vaccinate patients against encapsulated bacteria, including Streptococcus pneumoniae and Neisseria meningitidis (serogroups A, C, W, Y and B), according to current ACIP recommendations at least 2 weeks prior to initiation of EMPAVELI therapy [see Warnings and Precautions (5.1)].
If urgent EMPAVELI therapy is indicated in a patient who is not up to date with vaccines for Streptococcus pneumoniae and Neisseria meningitidis, according to ACIP recommendations, provide the patient with antibacterial drug prophylaxis and administer these vaccines as soon as possible.
Healthcare professionals who prescribe EMPAVELI must enroll in the REMS for EMPAVELI [see Warnings and Precautions (5.2)].
2.2 Recommended Dosage - PNH
The recommended dose of EMPAVELI is 1,080 mg administered subcutaneously twice weekly [see Dosage and Administration (2.4)].
Dosage for patients switching to EMPAVELI from C5 inhibitors
To reduce the risk of hemolysis with abrupt treatment discontinuation:
- For patients switching from eculizumab, initiate EMPAVELI while continuing eculizumab at its current dose. After 4 weeks, discontinue eculizumab before continuing on monotherapy with EMPAVELI.
- For patients switching from ravulizumab, initiate EMPAVELI no more than 4 weeks after the last dose of ravulizumab.
2.3 Recommended Dosage - C3G or Primary IC-MPGN
For adults (18 years and older)
The recommended dose of EMPAVELI is 1,080 mg (20 mL) administered subcutaneously twice weekly [see Dosage and Administration (2.4)].
For pediatric patients (12 years to less than 18 years of age)
For pediatric patients 12 years of age and older, administer EMPAVELI dose and volume based upon body weight, according to the schedule in Table 1. Administer the recommended dose of EMPAVELI subcutaneously twice weekly [see Dosage and Administration (2.4)] .
Table 1: Dosing recommendation in C3G or primary IC-MPGN Patient Body Weight First dose
(infusion volume)Second dose
(infusion volume)Maintenance dose
(infusion volume)50 kg or higher 1,080 mg (20 mL) 1,080 mg (20 mL) 1,080 mg twice weekly (20 mL) 35 kg to less than 50 kg 648 mg (12 mL) 810 mg (15 mL) 810 mg twice weekly (15 mL) Less than 35 kg 540 mg (10 mL) 540 mg (10 mL) 648 mg twice weekly (12 mL) 2.4 Administration
EMPAVELI is for subcutaneous administration using:
- A commercially available infusion pump with a reservoir of at least 20 mL OR
- EMPAVELI Injector, a single-use, disposable on body injector
EMPAVELI is intended for use under the guidance of a healthcare professional. Patients may self-administer or caregivers may administer EMPAVELI after proper training by a healthcare professional on how to prepare and administer EMPAVELI.
Follow the steps below and use aseptic technique to prepare and administer EMPAVELI, either by an infusion pump or EMPAVELI Injector:
- Prior to use‚ allow EMPAVELI to reach room temperature 20°C to 25°C (68°F to 77°F) for approximately 30 minutes. Keep the vial in the carton until ready for use to protect from light.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. EMPAVELI is a clear, colorless to slightly yellowish solution. Do not use if the liquid looks cloudy, contains particles, or is dark yellow.
- Discard any unused portion of EMPAVELI.
Preparation with Infusion Pump
- Refer to the EMPAVELI Instructions for Use and the infusion pump manufacturer's instructions for full preparation and administration information.
- Use a needleless transfer device (such as a vial adapter) or a transfer needle to fill the syringe.
- Rotate infusion sites (i.e., abdomen, thighs, hips, upper arms) from one infusion to the next. Do not infuse where the skin is tender, bruised, red, or hard. Avoid infusing into tattoos, scars, or stretch marks.
- If multi-infusion sets are needed, ensure the infusion sites are at least 3 inches apart.
- The typical infusion time is approximately 30 minutes (if using two infusion sites) or approximately 60 minutes (if using one infusion site).
Preparation with EMPAVELI Injector
- Refer to the EMPAVELI Injector Instructions for Use, which comes with the device.
- Use a needleless transfer device (such as a vial adapter).
- EMPAVELI Injector is for abdominal subcutaneous use only. Rotate the site of each subcutaneous administration. Do not inject where the skin is tender, bruised, red, or hard. Avoid injecting into tattoos, scars, or stretch marks.
- Injection time is approximately 30 to 60 minutes.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
EMPAVELI is contraindicated:
- in patients with hypersensitivity to pegcetacoplan or to any of the excipients [see Warnings and Precautions (5.3)].
- for initiation in patients with unresolved serious infection caused by encapsulated bacteria including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections Caused by Encapsulated Bacteria
EMPAVELI, a complement inhibitor, increases a patient's susceptibility to serious, life-threatening, or fatal infections caused by encapsulated bacteria including Streptococcus pneumoniae, Neisseria meningitidis (caused by any serogroup, including non-groupable strains), and Haemophilus influenzae type B. Life-threatening and fatal infections with encapsulated bacteria have occurred in both vaccinated and unvaccinated patients treated with complement inhibitors. The initation of EMPAVELI treatment is contraindicated in patients with unresolved serious infection caused by encapsulated bacteria.
Complete or update vaccination against encapsulated bacteria at least 2 weeks prior to administration of the first dose of EMPAVELI, according to the most current ACIP recommendations for patients receiving a complement inhibitor. Revaccinate patients in accordance with ACIP recommendations considering the duration of therapy with EMPAVELI. Note that, ACIP recommends an administration schedule in patients receiving complement inhibitors that differs from the administration schedule in the vaccine prescribing information. If urgent EMPAVELI therapy is indicated in a patient who is not up to date with vaccines against encapsulated bacteria according to ACIP recommendations, provide the patient with antibacterial drug prophylaxis and administer these vaccines as soon as possible. Various durations and regimens of antibacterial drug prophylaxis have been considered, but the optimal durations and drug regimens for prophylaxis and their efficacy have not been studied in unvaccinated or vaccinated patients receiving complement inhibitors, including EMPAVELI. The benefits and risks of treatment with EMPAVELI, as well as the benefits and risks of antibacterial drug prophylaxis in unvaccinated or vaccinated patients, must be considered against the known risks for serious infections caused by encapsulated bacteria.
Vaccination does not eliminate the risk of serious encapsulated bacterial infections, despite development of antibodies following vaccination. Closely monitor patients for early signs and symptoms of serious infection and evaluate patients immediately if an infection is suspected. Inform patients of these signs and symptoms and instruct patients to seek immediate medical care if these signs and symptoms occur. Promptly treat known infections. Serious infection may become rapidly life-threatening or fatal if not recognized and treated early. Consider interruption of EMPAVELI in patients who are undergoing treatment for serious infections.
EMPAVELI is available only through a restricted program under a REMS [see Warnings and Precautions (5.2)].
5.2 EMPAVELI REMS
EMPAVELI is available only through a restricted program under a REMS called EMPAVELI REMS, because of the risk of serious infections caused by encapsulated bacteria [see Warnings and Precautions (5.1)].
Notable requirements of the EMPAVELI REMS include the following:
- Prescribers must enroll in the REMS.
- Prescribers must counsel patients about the risk of serious infections caused by encapsulated bacteria.
- Prescribers must provide the patients with the REMS educational materials.
- Prescribers must assess patient vaccination status for encapsulated bacteria and vaccinate if needed according to current ACIP recommendations two weeks prior to the first dose of EMPAVELI.
- Prescribers must provide a prescription for antibacterial drug prophylaxis if treatment must be started urgently, and the patient is not up to date with vaccinations against encapsulated bacteria according to current ACIP recommendations at least two weeks prior to the first dose of EMPAVELI.
- Pharmacies that dispense EMPAVELI must be certified in the EMPAVELI REMS and must verify prescribers are certified.
- Patients must receive counseling from the prescriber about the need to receive vaccinations against encapsulated bacteria per ACIP recommendations, the need to take antibiotics as directed by the prescriber, and the signs and symptoms of serious infections.
- Patients must be instructed to carry the Patient Safety Card with them at all times during and for 2 months following treatment discontinuation with EMPAVELI.
Further information is available at www.empavelirems.com or 1-888-343-7073
5.3 Infusion-Related Reactions
Systemic hypersensitivity reactions (e.g., facial swelling, rash, urticaria, pyrexia) have occurred in patients treated with EMPAVELI, which may resolve after treatment with antihistamines. Cases of anaphylaxis leading to treatment discontinuation have been reported. If a severe hypersensitivity reaction (including anaphylaxis) occurs, discontinue EMPAVELI infusion immediately, institute appropriate treatment, per standard of care, and monitor until signs and symptoms are resolved.
5.4 Monitoring PNH Manifestations after Discontinuation of EMPAVELI
After discontinuing treatment with EMPAVELI, closely monitor for signs and symptoms of hemolysis, identified by elevated LDH levels along with sudden decrease in PNH clone size or hemoglobin, or reappearance of symptoms such as fatigue, hemoglobinuria, abdominal pain, dyspnea, major adverse vascular events (including thrombosis), dysphagia, or erectile dysfunction. Monitor any patient who discontinues EMPAVELI for at least 8 weeks to detect hemolysis and other reactions. If hemolysis, including elevated LDH, occurs after discontinuation of EMPAVELI, consider restarting treatment with EMPAVELI.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
- Serious Infections Caused by Encapsulated Bacteria [see Warnings and Precautions (5.1)]
- Infusion-Related Reactions [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Paroxysmal Nocturnal Hemoglobinuria
Study in Complement-Inhibitor Experienced Adult Patients with PNH (Study APL2-302)
The data described below reflect the exposure in 80 adult patients with PNH who received EMPAVELI (n=41) or eculizumab (n=39) at the recommended dosing regimens for 16 weeks. Serious adverse reactions were reported in 7 (17%) patients with PNH receiving EMPAVELI. The most common serious adverse reaction in patients treated with EMPAVELI was infections (5%). The most common adverse reactions (≥10%) with EMPAVELI were injection-site reactions, infections, diarrhea, abdominal pain, respiratory tract infection, viral infection, and fatigue.
Table 2 describes the adverse reactions that occurred in ≥5% of patients treated with EMPAVELI in Study APL2-302.
Table 2: Adverse Reactions Reported in ≥5% of Patients Treated with EMPAVELI in Study APL2-302 Adverse Reaction EMPAVELI
(N=41)
n (%)Eculizumab
(N=39)
n (%)- *
- Grouped terms
- †
- Term includes injection-site erythema, injection-site reaction, injection-site swelling, injection-site induration, injection-site bruising, injection-site pain, injection-site pruritus, vaccination site reaction, administration site swelling, injection-site hemorrhage, injection -site edema, injection-site warmth, administration site pain, application site pain, injection-site mass, injection-site rash, vaccination site pain
General disorders and administration site conditions Injection-site reaction*† 16 (39) 2 (5) Fatigue* 5 (12) 9 (23) Chest pain* 3 (7) 1 (3) Infections and infestations Infections* 12 (29) 10 (26) Respiratory tract infection* 6 (15) 5 (13) Viral Infection* 5 (12) 3 (8) Gastrointestinal disorders Diarrhea 9 (22) 1 (3) Abdominal pain* 8 (20) 4 (10) Musculoskeletal disorders Back pain* 3 (7) 4 (10) Nervous system disorders Headache 3 (7) 9 (23) Vascular disorders Systemic hypertension* 3 (7) 1 (3) Clinically relevant adverse reactions in less than 5% of patients include:
- Intestinal ischemia
- Biliary sepsis
- Hypersensitivity pneumonitis
After the randomized control period, 77 patients continued the study, and all were treated with EMPAVELI monotherapy at the recommended dosing regimen for up to 48 weeks. Serious adverse reactions were reported in 18 patients (23%). Additional adverse reactions reported in >5% of patients treated with EMPAVELI during the open-label part of the study compared to the randomized controlled part in Table 1 were cough (12%), arthralgia (8%), oropharyngeal pain (8%), pyrexia (8%), pain in extremity (7%), thrombocytopenia (7%), abdominal distension (5%), acute kidney injury (5%), anxiety (5%), and myalgia (5%). One patient (1%) died due to COVID-19 infection.
Study in Complement-Inhibitor Naïve Adult Patients with PNH (Study APL2-308)
The data described below reflect the exposure in adult patients with PNH who received EMPAVELI (n=46) or the control arm (supportive care excluding complement inhibitors) (n=18) in Study APL2-308 [see Clinical Studies (14.1)]. One patient (2%) who received EMPAVELI died due to septic shock. Serious adverse reactions were reported in 6 (13%) patients with PNH receiving EMPAVELI. The most common adverse reaction (≥10%) in patients treated with EMPAVELI were injection site reactions, infections, viral infection, pain in extremity, hypokalemia, arthralgia, dizziness, abdominal pain, rash, and headache.
Table 3 describes the adverse reactions that occurred in ≥5% of patients treated with EMPAVELI in Study APL2‑308.
Table 3: Adverse Reactions Reported in ≥5% of Patients Treated with EMPAVELI in Study APL2-308 Adverse Reaction EMPAVELI
(N=46)
n (%)Control Arm*
(N=18)
n (%)Exposure Adjusted Rate (per 100 pt yrs) Exposure Adjusted Rate (per 100 pt yrs) EMPAVELI (N=46) group includes patients who received EMPAVELI at any point during the study, including patients randomized to EMPAVELI (N=35) and patients randomized to the control arm and crossed over to EMPAVELI treatment (N=11). General disorders and administration site conditions Injection-site reaction†‡ 12 (26)
420
0Pyrexia 4(9)
140
0Peripheral edema† 3 (7)
110
0Infections and Infestations Infections† 9 (20)
324 (22)
74Viral infection† 6 (13)
212 (11)
37Musculoskeletal and connective tissue disorders Pain in extremity 6 (13)
210
0Arthralgia 5 (11)
180
0Musculoskeletal pain 3 (7)
110
0Metabolism and nutrition disorders Hypokalemia 6 (13)
212 (11)
37Nervous system disorders Dizziness 5 (11)
180
0Headache 5 (11)
180
0Somnolence 3 (7)
110
0Gastrointestinal disorders Abdominal pain† 5 (11)
181 (6)
18Skin and subcutaneous tissue disorders Rash† 5(11)
180
0Ecchymosis 3 (7)
110
0Erythema 3 (7)
110
0Blood and lymphatic system disorders Thrombocytopenia 3 (7)
111 (6)
18Respiratory, thoracic and mediastinal disorders Cough† 4 (9)
140
0Epistaxis 3 (7)
110
0Investigations Blood creatinine increased 3 (7)
110
0C3 Glomerulopathy or Primary IC-MPGN
Study in Adult and Pediatric Patients 12 Years of age and older with C3G or primary IC-MPGN (Study APL2-C3G-310)
The data described below reflects the exposure in adult (n=35) and pediatric patients 12 years of age and older (n=28) with native kidney C3G (n=46), native kidney primary IC-MPGN (n=12), or recurrent C3G following kidney transplant (n=5) who received EMPAVELI at the recommended dosing regimens during the 26-week placebo-controlled period of APL2-C3G-310. Serious adverse reactions due to viral infections resulting in hospitalizations occurred in 2 (3%) patients with C3G or primary IC-MPGN receiving EMPAVELI and 1 (2%) patient on placebo. One patient (2%) on EMPAVELI with native kidney C3G died because of respiratory failure due to COVID-19 pneumonia; there were no deaths in the placebo arm.
Table 4 describes the adverse reactions that were reported in ≥5% of patients (adults and pediatric patients 12 years of age and older) treated with EMPAVELI and at a greater incidence than placebo in APL2-C3G-310. Adverse reactions in pediatric patients were similar to those seen in adults.
The placebo-controlled period of APL2-C3G-310 was followed by a 26-week open-label period. During the open-label period, one patient with native kidney C3G had a serious adverse event of pneumonia secondary to Streptococcus pneumoniae, and one patient with recurrent C3G following kidney transplant developed herpes zoster meningoencephalitis while on concomitant immunosuppression, leading to treatment discontinuation.
Table 4: Adverse Reactions Reported in ≥5% of Patients (adult and pediatric) Treated with EMPAVELI and Greater than Placebo in Study APL2-C3G-310 Adverse Reaction EMPAVELI
(N=63)
n (%)Placebo
(N=61)
n (%)- *
- Term includes the following reactions at the infusion site: erythema, pruritus, swelling, bruising, induration, pain, hemorrhage, discomfort, oedema, rash, and hypoaesthesia.
General disorders and administration site conditions Infusion-site reactions* 16 (25) 14 (23) Pyrexia 12 (19) 6 (10) Fatigue 4 (6) 1 (2) Infections and infestations Nasopharyngitis 11 (18) 7 (12) Influenza 7 (11) 3 (5) Gastrointestinal disorders Nausea 6 (10) 4 (7) Respiratory, thoracic and mediastinal disorders Cough 6 (10) 1 (2) Study in Adult recurrent C3G or primary IC-MPGN following kidney transplant (Study APL2-C3G-204)
In a study in 13 adults with recurrent C3G or primary IC-MPGN after kidney transplant (NCT#04572854), one patient with primary IC-MPGN experienced a serious adverse event of Pneumocystis jirovecii pneumonia while on EMPAVELI and concurrent immunosuppressive medications.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of EMPAVELI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to EMPAVELI exposure.
- Anaphylaxis and urticaria [see Warnings and Precautions (5.3)]
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are insufficient data on EMPAVELI use in pregnant women to inform a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. There are risks to the mother and fetus associated with untreated PNH in pregnancy (see Clinical Considerations). The use of EMPAVELI may be considered following an assessment of the risks and benefits.
Treatment of pregnant cynomolgus monkeys with pegcetacoplan at a subcutaneous dose of 28 mg/kg/day (2.9 times human exposure based on AUC) from the gestation period through parturition resulted in a statistically significant increase in abortions or stillbirths compared to controls (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of major birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriages in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or fetal/neonatal risk
PNH in pregnancy is associated with adverse maternal outcomes, including worsening cytopenias, thrombotic events, infections, bleeding, miscarriages and increased maternal mortality, and adverse fetal outcomes, including fetal death and premature delivery.
Data
Animal Data
Animal reproduction studies with pegcetacoplan were conducted in cynomolgus monkeys. Pegcetacoplan treatment of pregnant cynomolgus monkeys at a subcutaneous dose of 28 mg/kg/day (2.9 times human exposure based on AUC) from the gestation period through parturition resulted in a statistically significant increase in abortions and stillbirths compared to controls. No increase in abortions or stillbirths occurred at a dose of 7 mg/kg/day (1.3 times human exposure based on AUC). No maternal toxicity or teratogenic effects were observed in offspring delivered at term. No developmental effects were observed in infants up to 6 months postpartum. Systemic exposure to pegcetacoplan of less than 1% of maternal levels was detected in fetuses from monkeys treated with 28 mg/kg/day from the period of organogenesis through the second trimester.
8.2 Lactation
Risk Summary
It is not known whether pegcetacoplan is secreted in human milk or whether there is potential for absorption and harm to the infant. There are no data on the effects of pegcetacoplan on milk production. Pegcetacoplan is present in milk of lactating monkeys (see Animal Data). Since many medicinal products are secreted into human milk, and because of the potential for serious adverse reaction in a breastfeeding child, breastfeeding should be discontinued during treatment and for 40 days after the last dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
EMPAVELI may cause embryo-fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)]. Pregnancy testing is recommended for females of reproductive potential prior to treatment with EMPAVELI. Advise female patients of reproductive potential to use effective contraception during treatment with EMPAVELI and for 40 days after the last dose.
8.4 Pediatric Use
The safety and effectiveness of EMPAVELI for the treatment of C3G or primary IC-MPGN have been established in pediatric patients aged 12 years and older. Use of EMPAVELI for this indication is supported by evidence from an adequate and well controlled trial that enrolled 55 pediatric patients aged 12 years and older [see Adverse Reactions (6.1), and Clinical Studies (14.2)]. The safety and effectiveness of EMPAVELI in pediatric patients less than 12 years of age with C3G or IC-MPGN have not been established.
Safety and effectiveness of EMPAVELI in pediatric patients with PNH have not been established.
-
11 DESCRIPTION
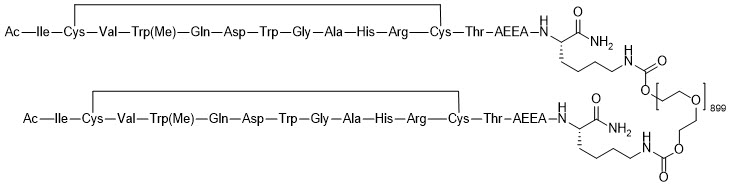
EMPAVELI contains pegcetacoplan, a complement inhibitor. Pegcetacoplan is a symmetrical molecule comprised of two identical pentadecapeptides covalently bound to the ends of a linear 40-kiloDalton (kDa) PEG molecule. The peptide portions of pegcetacoplan contain 1-methyl-L-tryptophan (Trp(Me)) in position 4 and amino(ethoxyethoxy)acetic acid (AEEA) in position 14.
The molecular weight of pegcetacoplan is approximately 43.5 kDa. The molecular formula is C1970H3848N50O947S4. The structure of pegcetacoplan is shown below.
EMPAVELI injection is a sterile, clear, colorless to slightly yellowish aqueous solution for subcutaneous use and is supplied in a 20-mL single-dose vial. Each 1 mL of solution contains 54 mg of pegcetacoplan, 41 mg of sorbitol, 0.384 mg of glacial acetic acid, 0.490 mg of sodium acetate trihydrate, and Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for adjustment to a target pH of 5.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Pegcetacoplan binds to complement protein C3 and its activation fragment C3b, thereby regulating the cleavage of C3 and the generation of downstream effectors of complement activation.
In PNH, extravascular hemolysis (EVH) is facilitated by C3b opsonization while intravascular hemolysis (IVH) is mediated by the downstream membrane attack complex (MAC). Pegcetacoplan acts proximally in the complement cascade controlling both C3b-mediated EVH and terminal complement-mediated IVH.
In C3G and primary IC-MPGN, complement dysregulation and overactivation causes deposition of C3 fragments in glomeruli, which contributes to the pathogenesis of C3G and is thought to contribute to the pathogenesis of IC-MPGN. Pegcetacoplan binds C3 and its activation fragment C3b, therefore inhibiting C3 activation, decreasing C3 glomerular fragment deposition, and decreasing C5 convertase activity and subsequent assembly of C5b-9.
12.2 Pharmacodynamics
In patients with PNH administered multiple doses of pegcetacoplan, the mean C3 concentration increased from 94 mg/dL at baseline to 380 mg/dL at Week 16 and sustained through Week 48 (Study APL2-302). In study APL2-308, the mean C3 concentration increased from 95 mg/dL at baseline to 356 mg/dL at Week 26 [see Clinical Studies (14.1)].
The percentage of PNH Type II + III RBCs increased from 66.2% at baseline to 93.9% at Week 16 and sustained through Week 48 (Study APL2-302). In Study APL2-308, the mean percentage of PNH Type II + III RBCs increased from 42.4% at baseline to 90% at Week 26.
The mean percentage of PNH Type II + III RBCs with C3 deposition decreased from 17.8% at baseline to 0.2% at Week 16 and sustained through Week 48 (Study APL2-302). In Study APL2-308, the mean percentage of PNH Type II + III RBCs with C3 deposition decreased from 2.85% at baseline to 0.09% at Week 26.
In patients with C3G or primary IC-MPGN administered pegcetacoplan SC infusion twice weekly, mean (SD) serum C3 levels increased from 62 (48) mg/dL at baseline to 371 (120) mg/dL at Week 26 compared to no change with placebo. Mean (SD) plasma soluble C5b-9 levels decreased from 903 (698) ng/mL at baseline to 290 (249) ng/mL at Week 26 with EMPAVELI compared to no change with placebo. Of patients with evaluable kidney biopsies (n=69), 74% of patients on pegcetacoplan had a decrease in C3 complement staining by at least 2 orders of magnitude from baseline to Week 26 compared to 12% on placebo.
12.3 Pharmacokinetics
In patients with PNH, the serum pegcetacoplan concentrations achieved steady-state approximately 4 to 6 weeks following the first dose. The exposure of pegcetacoplan increased proportionally over a dose range from 45 to 1,440 mg (0.04 to 1.33 times the approved recommended dose). The mean (CV%) trough serum concentration observed at Week 16 was 706 (15.1%) mcg/mL and sustained through Week 48 (Study APL2-302). In Study APL2-308, mean (CV%) trough serum concentration was 744 (25.5%) mcg/mL at Week 26.
In patients with C3G or primary IC-MPGN, serum pegcetacoplan concentrations reached steady state approximately 4 to 8 weeks following twice weekly SC infusion. The mean (CV%) trough serum concentrations ranged between 716 (31%) and 766 (23%) mcg/mL from Week 4 to Week 26.
Absorption
The median Tmax of pegcetacoplan is between 108 and 144 hours (4.5 to 6 days) after a single dose.
Distribution
The mean (CV%) volume of distribution of pegcetacoplan is approximately 3.98 L (32%) in patients with PNH.
The estimated mean (CV%) volume of distribution of pegcetacoplan is approximately 4.79 L (38%) and 4.58 L (29%) in patients with C3G or primary IC-MPGN, respectively.
Elimination
The estimated mean (CV%) of clearance (CL) is 0.36 L/day (30%) and median effective half-life of elimination (t1/2) is 8.6 days in patients with PNH.
The estimated mean (CV%) of clearance (CL) is 0.32 L/day (54%) and median t1/2 is 10.2 days in patients with C3G. The estimated mean (CV%) of clearance (CL) is 0.3 L/day (51%) and median t1/2 is 10.8 days in patients with primary IC-MPGN.
Specific Populations
There were no clinically significant differences on the pharmacokinetics of pegcetacoplan based on age (19 to 81 years old), sex, race (Asian vs. non‑Asian), renal impairment, and hepatic function as evaluated by total bilirubin (0.06‑8.8 mg/dL), albumin (1.8‑5.5 g/dL), aspartate aminotransferase (6‑302 IU/L), or alanine aminotransferase (4‑209 IU/L).
In patients with C3G or primary IC-MPGN, there were no clinically significant differences in pharmacokinetics of pegcetacoplan based on age (12 to 74 years old), diagnosis (C3G vs primary IC-MPGN), and urine protein-to-creatinine ratio (UPCR) (133-13300 mg/g).
12.6 Immunogenicity
PNH
There is insufficient information to characterize the anti-drug antibody response to EMPAVELI and the effects of anti-drug antibodies on pharmacokinetics, pharmacodynamics, safety, or effectiveness of pegcetacoplan products in PNH patients.
C3G or primary IC-MPGN
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the study described below with the incidence of anti-drug antibodies in other studies.
During the 26-week placebo-controlled period in Study APL2-C3G-310, 14 of 62 participants (23%) with C3G or primary IC-MPGN randomized to pegcetacoplan developed anti–pegcetacoplan peptide antibodies. Two of 62 participants (3%) developed anti–pegcetacoplan peptide neutralizing antibodies. There was no identified clinically significant effect of anti-drug antibodies on pharmacokinetics, pharmacodynamics, safety, or effectiveness of pegcetacoplan over the treatment duration of 26 weeks.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal carcinogenicity studies of pegcetacoplan have not been conducted.
Pegcetacoplan was not mutagenic when tested in an in vitro bacterial reverse mutation (Ames) and was not genotoxic in an in vitro assay in human TK6 cells or in an in vivo micronucleus assay in mice.
Effects of pegcetacoplan on fertility have not been studied in animals. There were no microscopic abnormalities in male or female reproductive organs in toxicity studies in rabbits and monkeys.
13.2 Animal Toxicology and/or Pharmacology
In toxicology studies in rabbits and cynomolgus monkeys, epithelial vacuolation and infiltrates of vacuolated macrophages were observed in multiple tissues, including the renal tubules, following daily subcutaneous doses of pegcetacoplan up to 7 times the human dose. These findings are attributable to uptake of the PEG moieties of pegcetacoplan. Renal degeneration was observed microscopically in rabbits at exposures (Cmax and AUC) less than those for the human dose, and in monkeys at exposures approximately 2.7-fold those for the human dose. The clinical significance of these findings is uncertain.
-
14 CLINICAL STUDIES
14.1 Paroxysmal Nocturnal Hemoglobinuria
The efficacy and safety of EMPAVELI in patients with PNH were assessed in two open-label, randomized-controlled Phase 3 studies: Study APL2-302 (NCT03500549) and Study APL2-308 (NCT04085601). All patients who completed the studies were eligible to enroll in a separate long-term extension study.
In both studies, patients were vaccinated against Streptococcus pneumoniae, Neisseria meningitidis types A, C, W, Y, and B, and Haemophilus influenzae type B (Hib), either within 2 years prior to Day 1 or within 2 weeks after starting treatment with EMPAVELI. Patients vaccinated after initiation of treatment with EMPAVELI received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination. In addition, prophylactic antibiotic therapy was administered at the discretion of the investigator in accordance with local treatment guidelines for patients with PNH receiving treatment with a complement inhibitor.
A dose of 1,080 mg twice weekly was used for patients randomized to the EMPAVELI group of each study. If required, the dose of EMPAVELI could be adjusted to 1,080 mg every 3 days. EMPAVELI was administered as a subcutaneous infusion; the infusion time was approximately 20 to 40 minutes.
Study in Complement-Inhibitor Experienced Adult Patients with PNH (Study APL2-302)
The study enrolled patients with PNH who had been treated with a stable dose of eculizumab for at least the previous 3 months and with Hb levels less than 10.5 g/dL.
Eligible patients entered a 4-week run-in period during which they received EMPAVELI 1,080 mg subcutaneously twice weekly in addition to their current dose of eculizumab. Patients were then randomized in a 1:1 ratio to receive either 1,080 mg of EMPAVELI twice weekly or their current dose of eculizumab through the duration of the 16-week randomized controlled period (RCP).
Randomization was stratified based on the number of packed red blood cell (PRBC) transfusions within the 12 months prior to Day -28 (<4; ≥4) and platelet count at screening (<100,000/mm3; ≥100,000/mm3). Following completion of the RCP, all patients entered a 32-week open-label period (OLP) and received monotherapy with EMPAVELI. Patients initially randomized to eculizumab entered a second 4-week run-in period during which they received EMPAVELI in addition to eculizumab before continuing on to receive EMPAVELI monotherapy. All patients who completed the 48-week period were eligible to enroll in a separate long-term extension study.
A total of 80 patients were randomized to receive treatment, 41 to EMPAVELI and 39 to eculizumab. Demographics and baseline disease characteristics were generally well balanced between treatment groups (see Table 5). The median times from PNH diagnosis to Day -28 were 6 and 9.7 years, respectively, for EMPAVELI and eculizumab. The baseline mean total PNH RBC clone sizes (Type III) were 47% for EMPAVELI and 50% for eculizumab. Twenty-nine percent and 23% of patients had a history of major adverse vascular events, and 37% and 26% had a history of thrombosis for patients receiving EMPAVELI or eculizumab, respectively. Within 28 days prior to the first dose of EMPAVELI or eculizumab, respectively, 34% and 31% of patients used anti-thrombotic agents (anti-platelet and/or anticoagulants). During Study APL2-302, 37% and 36% of patients on EMPAVELI and eculizumab, respectively, used antithrombotic agents. A total of 38 patients in the group treated with EMPAVELI and 39 patients in the eculizumab group completed the 16-week RCP and continued into the 32-week OLP. Because of adverse reactions of hemolysis, 3 patients were discontinued from the EMPAVELI group during the RCP. Two out of 41 patients in the EMPAVELI group needed the dose adjustment to 1,080 mg every 3 days.
Table 5: Patient Baseline Demographics and Characteristics in Study APL2-302 Parameter Statistics EMPAVELI
(N=41)Eculizumab
(N=39)Age (years) Mean (SD) 50.2 (16.3) 47.3 (15.8) Sex Female n (%) 27 (65.9) 22 (56.4) Race Asian n (%) 5 (12.2) 7 (17.9) Black or African American n (%) 2 (4.9) 0 White n (%) 24 (58.5) 25 (64.1) Other n (%) 0 1 (2.6) Not reported n (%) 10 (24.4) 6 (15.4) Ethnicity Hispanic or Latino n (%) 2 (4.9) 1 (2.6) Not Hispanic or Latino n (%) 29 (70.7) 32 (82.1) Not reported n (%) 10 (24.4) 6 (15.4) Hemoglobin level (g/dL) Mean (SD) 8.7 (1.1) 8.7 (0.9) Absolute reticulocyte count
(109 cells/L)Mean (SD) 218 (75) 216 (69.1) LDH level (U/L) Mean (SD) 257.5 (97.7) 308.6 (284.8) Number of transfusions in last 12 months prior to Day -28 Mean (SD) 6.1 (7.3) 6.9 (7.7) <4 n (%) 20 (48.8) 16 (41) ≥4 n (%) 21 (51.2) 23 (59) The efficacy of EMPAVELI was based on change from baseline to Week 16 (during RCP) in hemoglobin level. Baseline was defined as the average of measurements recorded prior to taking the first dose of EMPAVELI. Supportive efficacy data included transfusion avoidance, defined as the proportion of patients who did not require a transfusion during the RCP, and change from baseline to Week 16 in absolute reticulocyte count (ARC).
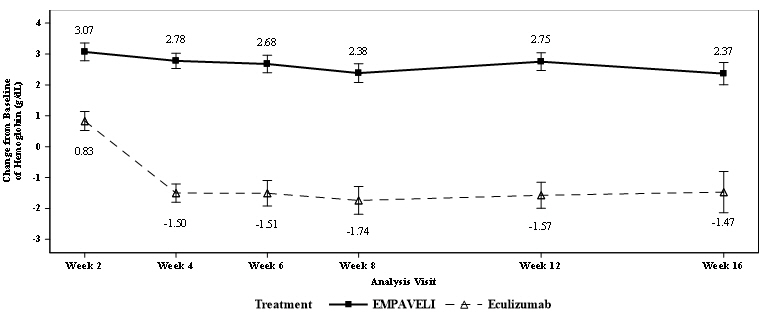
EMPAVELI was superior to eculizumab for the change from baseline in hemoglobin level at Week 16 (p<0.0001). The adjusted mean change from baseline in hemoglobin level was 2.37 g/dL in the group treated with EMPAVELI versus -1.47 g/dL in the eculizumab group (Figure 1), demonstrating an adjusted mean increase of 3.84 g/dL with EMPAVELI compared to eculizumab at Week 16 (95% CI, 2.33-5.34).
- *
- Treatment effect estimates from a mixed model are shown. The mixed model contained the categorical effects of treatment, visit, treatment by visit interaction, and stratification factors (transfusion history and platelet count at screening), and the continuous covariate of baseline value.
Figure 1: Adjusted Mean (± SE) Change from Baseline to Week 16 in Hemoglobin (g/dL) in Study APL2-302*
Non-inferiority was demonstrated in the endpoints of transfusion avoidance and change from baseline in ARC at Week 16.
The adjusted means, treatment differences, and confidence intervals (CIs) for additional efficacy results are shown in Table 6.
Table 6: Additional Efficacy Results at Week 16 in Study APL2-302 EMPAVELI
(N=41)Eculizumab
(N=39)Difference
(95% CI)Transfusion avoidance, n (%) 35 (85%) 6 (15%) 63%*
(48%, 77%)Change from baseline in ARC (109 cells/L), LS† mean (SE)‡ -136 (6.5) 28 (11.9) -164
(-189.9, -137.3)Efficacy was generally similar across subgroups based on sex, race, and age.
All 77 patients who completed the RCP entered the 32- week OLP, during which all patients received EMPAVELI, resulting in a total exposure of up to 48 weeks. Between Week 16 and Week 48, 10 patients discontinued the study, all due to adverse reactions, and thirteen patients had a dose adjustment to 1,080 mg every three days. The efficacy results at Week 48 were generally consistent with those at Week 16.
Study in Complement-Inhibitor Naïve Adult Patients with PNH (Study APL2-308)
Study APL2-308 enrolled patients with PNH who had not been treated with any complement inhibitor within 3 months prior to enrollment and with Hb levels less than the lower limit of normal (LLN). Eligible patients were randomized in a 2:1 ratio to receive EMPAVELI or supportive care [excluding complement inhibitors (e.g., transfusions, corticosteroids, supplements such as iron, folate, and vitamin B12), hereafter referred to as the control arm] through the duration of the 26-week treatment period. Randomization was stratified based on the number of packed red blood cell (PRBC) transfusions within the 12 months prior to Day -28 (<4; ≥4). At any point during the study, a patient assigned to the control arm treatment group who had Hb levels ≥2 g/dL below baseline or presented with a PNH associated thromboembolic event was offered cross-over to EMPAVELI for the remainder of the study.
A total of 53 patients were randomized, 35 to EMPAVELI and 18 to the control arm. Demographics and baseline disease characteristics were generally well balanced between treatment groups (see Table 7). The mean times from PNH diagnosis to Day 1 were 5.7 and 5.5 years, respectively, for EMPAVELI and the control arm. The baseline mean total PNH RBC clone sizes (Type III) were 31% for EMPAVELI and 28% for the control arm. In the EMPAVELI group, 2.9% of patients had a history of major adverse vascular events. Two patients (5.7%) in the EMPAVELI group and 3 patients (16.7%) in the control arm group had a history of at least 1 type of thrombosis. Within 28 days prior to the first dose of EMPAVELI or the control arm, respectively, 17.1% and 27.8% of patients used anti-thrombotic agents (anti-platelet and/or anticoagulants). During Study APL2-308, 8.6% and 0% of patients on EMPAVELI and the control arm, respectively, used antithrombotic agents. Eleven of 18 patients randomized to the control transitioned to cross-over therapy with EMPAVELI due to a decreased Hb level ≥2 g/dL below baseline. Three patients treated with EMPAVELI required dose adjustment to 1,080 mg every 3 days. Three patients (5.7%; two patients in the EMPAVELI group and one patient in the control arm group) discontinued the study, none due to an adverse reaction.
Table 7: Patient Baseline Demographics and Characteristics in Study APL2-308 Parameter Statistics EMPAVELI
(N=35)Control Arm* (N=18) - *
- Control Arm = supportive care (excluding complement inhibitors)
Age (years) Mean (SD) 42.2 (12.7) 49.1 (15.6) Sex Female n (%) 16 (45.7) 8 (44.4) Race American Indian or Alaska n (%) 9 (25.7) 2 (11.1) Native Asian n (%) 23 (65.7) 16 (88.9) Black or African American n (%) 2 (5.7) 0 Other n (%) 1 (2.9) 0 Ethnicity Hispanic or Latino n (%) 12 (34.3) 2 (11.1) Not Hispanic or Latino n (%) 23 (65.7) 16 (88.9) Hemoglobin level (g/dL) Mean (SD) 9.4 (1.4) 8.7 (0.8) Absolute reticulocyte count (109 cells/L) Mean (SD) 230.2 (81) 180.3 (109.1) LDH level (U/L) Mean (SD) 2151 (909.4) 1945.9 (1003.7) Number of transfusions in last 12 months prior to Day -28 Mean (SD) 3.9 (4.4) 5.1 (5) <4 n (%) 21 (60) 8 (44.4) ≥4 n (%) 14 (40) 10 (55.6) The efficacy of EMPAVELI was based on the percentage of patients achieving hemoglobin stabilization, defined as avoidance of a >1 g/dL decrease in hemoglobin levels from baseline in the absence of transfusion, and the change from baseline in LDH level. Supportive efficacy data included change from baseline in absolute reticulocyte count (ARC), change from baseline in hemoglobin, and transfusion avoidance, defined as the proportion of patients who did not require a transfusion through Week 26. Baseline was defined as the average of measurements recorded prior to taking the first dose of EMPAVELI or prior to randomization to the control arm treatment group.
Efficacy results are shown in Table 8 below.
Table 8: Efficacy Results During the 26-Week Study in Study APL2-308 EMPAVELI
(N=35)Control Arm* Difference
(95% CI)(N=18) p-value Data collected after cross-over from the control arm is excluded in analyses. - *
- Control Arm = supportive care (excluding complement inhibitors)
- †
- Patients who crossed over from the control arm group to the EMPAVELI group, withdrew from the study, or were lost to follow up are considered as failing to achieve the criteria.
- ‡
- p-value is obtained by stratified Cochran-Mantel-Haenszel test.
- §
- The post baseline missing values (including the values after cross-over from the control arm) are imputed using a multiple imputation method.
- ¶
- LS = Least square
- #
- SE = Standard error
Hemoglobin Stabilization†
(n, %)30 (85.7%) 0 (0%) 73% (57%, 89%)
p<0.0001‡Change from Baseline in LDH§ (LS¶ Mean CFB, SE#) -1870 (101) -400 (313) -1470 (-2113.4, -827.3)
p<0.0001Change from baseline in ARC§
(LS¶ Mean CFB, SE#)-123 (9.2) -19 (25.2) -103 (-158.9, -48.7)
p = 0.0002Change from baseline in Hb§
(LS¶ Mean CFB, SE#)2.9 (0.38) 0.3 (0.76) 2.7 (0.99, 4.35)
p = 0.0019Transfusion Avoidance†
(n, %)32 (91%) 1 (6%) 72% (56%, 89%)
p<0.0001‡14.2 C3 Glomerulopathy (C3G) or Primary Immune-Complex Membranoproliferative Glomerulonephritis (IC-MPGN)
The efficacy of EMPAVELI in reducing proteinuria in adult and pediatric patients aged 12 years and older with native kidney C3G, native kidney IC-MPGN, or recurrent C3G following kidney transplant was demonstrated in Study APL2-C3G-310. Safety and effectiveness of EMPVAELI in patients with recurrent IC-MPGN following kidney transplant have not been established.
APL2-C3G-310 is a randomized, double-blind, placebo-controlled study that included 124 adult and pediatric patients aged 12 years and older and weighing at least 30 kg with biopsy-proven, native kidney or post-transplant recurrent C3G, or native kidney primary IC-MPGN, eGFR ≥30 mL/min/1.73 m2, proteinuria ≥1 g/day, and urine protein-to-creatinine ratio (UPCR) ≥1 g/g (NTC 05067127). For at least 12 weeks before randomization and throughout the 26-week placebo-controlled period, patients were required to be on stable and optimized doses of angiotensin-converting enzyme inhibitors, angiotensin receptor blockers and/or sodium-glucose cotransporter-2 (SGLT2) inhibitors. Immunosuppressant medication doses (e.g., steroids no higher than 20 mg daily, mycophenolate mofetil, tacrolimus) had to be stable for at least 12 weeks before randomization and throughout the 26-week placebo-controlled period.
Patients were randomized (1:1) to EMPAVELI or placebo, administered twice weekly as a subcutaneous (SC) infusion for 26 weeks. Randomization was stratified by post-transplant recurrence and by kidney biopsy obtained within 28 weeks of screening. Adults and pediatric patients weighing 50 kg or more received EMPAVELI 1,080 mg (20 mL) twice weekly. Pediatric patients weighing 35 kg to less than 50 kg received 648 mg (12 mL) for the first infusion and 810 mg (15 mL) for each infusion thereafter. Pediatric patients weighing 30 kg to less than 35 kg received EMPAVELI 540 mg (10 mL) for the first 2 infusions and 648 mg (12 mL) twice weekly thereafter.
Patients were vaccinated against Streptococcus pneumoniae, Neisseria meningitidis types A, C, W, Y, and B, and Haemophilus influenzae type B (Hib) at least 14 days prior to randomization, unless documented evidence existed that participants received the recommended vaccinations or were non-responders to vaccination.
At baseline, the mean age was 26 years (range 12 to 74 years); 57% were female, 73% White, 15% Asian, 1% Black or African American, and 11% others. The study population included 55 pediatric patients 12 years to less than 18 years of age; mean age 14.7 years (28 randomized to EMPAVELI and 27 to placebo). Disease type was reasonably balanced between the treatment groups. Overall, 88 patients (71%) had native kidney C3G, 27 patients (22%) had native kidney primary IC-MPGN, and 8 patients (6%) had post-kidney transplant recurrent C3G. At baseline, mean UPCR from triplicate first morning urine (FMU) collections was 3.1 g/g and 2.5 g/g in the pegcetacoplan and placebo groups, respectively; mean eGFR (mL/min/1.73 m2) was 79 and 87 in the pegcetacoplan and placebo groups, respectively; and mean baseline serum albumin was approximately 3.4 g/dL in both treatment groups.
Approximately 91% of patients were treated with angiotensin-converting enzyme inhibitors (ACEi) or angiotensin II receptor blockers (ARB), 72% with immunosuppressants (e.g., mycophenolate mofetil, tacrolimus), 40% with systemic corticosteroids, and 11% with sodium-glucose co-transporter 2 (SGLT2) inhibitors. Use of each of these medications was balanced between the treatment groups.
The primary efficacy endpoint was the log-transformed ratio of UPCR (sampled from first morning urine collections) at Week 26 compared to baseline. At Week 26, the geometric mean UPCR ratio relative to baseline was 0.33 (95% CI: 0.25, 0.43) and 1.03 (95% CI: 0.91, 1.16) in the EMPAVELI and placebo groups, respectively, resulting in a 68% reduction in UPCR from baseline in the EMPAVELI group compared to placebo (p<0.0001). The treatment effect was consistent across all subgroups including disease type, age, transplant status (C3G), sex, race, baseline disease characteristics (eGFR and UPCR), and immunosuppressant use.
Figure 2 shows the geometric mean UPCR ratio compared to baseline over time.
Abbreviations: FMU = first-morning spot urine;
UPCR = urine protein-to-creatinine ratio.Figure 2: Geometric Mean of UPCR Ratio Compared to Baseline Over 26 Weeks of Treatment - Study APL2-C3G-310 
During the 26-week placebo-controlled period, 49% of patients in the EMPAVELI group achieved a composite renal endpoint defined as a ≥50% reduction in UPCR and stable eGFR (≤15% reduction from baseline) compared with 3% of patients in the placebo group (odds ratio [95% CI] of 27 [6, 124], p<0.0001). Sixty percent of patients in the EMPAVELI group achieved a 50% or greater reduction in UPCR from baseline to Week 26 compared with 5% in the placebo arm, and 68% of patients in the EMPAVELI group had a stable eGFR (≤15% reduction from baseline to Week 26) compared with 59% in the placebo group. Over the first 6 months of treatment, EMPAVELI reduced the loss of kidney function compared to placebo (Figure 3). The efficacy of EMPAVELI in pediatric patients 12 years of age and older was similar to adults.
Abbreviations: LS = least square LS Mean (95% CI) of difference between EMPAVELI and placebo at Week 26 is 6.31 (0.50, 12.12) mL/min/1.73m2. Figure 3: LS Mean of eGFR Compared to Baseline Over 26 Weeks of Treatment - Study APL2-C3G-310 
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
EMPAVELI injection is a clear, colorless to slightly yellowish aqueous solution for subcutaneous infusion supplied as 1,080 mg/20 mL (54 mg/mL) solution in 20-mL single-dose vials.
EMPAVELI is available in 20-mL single-dose vials individually packaged in cartons that are supplied in 8-count convenience cartons. NDC 73606-010-01.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Dosing
For weight-based dosing, instruct caregivers and patients on the proper techniques for preparing, storing, measuring, and administering EMPAVELI via EMPAVELI Injector or commercially available infusion pump.
Serious Infections Caused by Encapsulated Bacteria
Advise patients of the risk of serious infection. Inform patients of the need to complete or update their vaccinations against encapsulated bacteria at least 2 weeks prior to receiving the first dose of EMPAVELI or receive antibacterial drug prophylaxis if EMPAVELI treatment must be initiated immediately and they have not been previously vaccinated. Inform the patient that they are required to be revaccinated according to current ACIP recommendations for encapsulated bacteria while on EMPAVELI therapy [see Warnings and Precautions (5.1)].
Inform patients that vaccination may not prevent serious infection and strongly advise patients to seek immediate medical attention if these signs or symptoms occur. These signs and symptoms include the following:
- fever with or without shivers or the chills
- fever with chest pain and cough
- fever with breathlessness/fast breathing
- fever with high heart rate
- headache and a fever
- headache with a stiff neck or stiff back
- fever and a rash
- confusion
- headache with nausea or vomiting
- body aches with flu-like symptoms
- clammy skin
- eyes sensitive to light
Inform patients that they will be given a Patient Safety Card for EMPAVELI that they should carry with them at all times. This card describes symptoms which, if experienced, should prompt the patient to seek immediate medical evaluation.
EMPAVELI REMS
EMPAVELI is available only through a restricted program called EMPAVELI REMS [see Warnings and Precautions (5.2)].
Inform the patient of the following notable requirements:
- Patients must receive counseling about the risk of serious infections caused by encapsulated bacteria.
- Patients must receive written educational materials about this risk.
- Patients must be instructed to carry the Patient Safety Card with them at all times during and for 2 months following treatment with EMPAVELI.
- Patients must be instructed to complete or update vaccinations against encapsulated bacteria per ACIP recommendations as directed by the prescriber prior to treatment with EMPAVELI.
- Patients must receive antibiotics as directed by the prescriber if they are not up to date with vaccinations against encapsulated bacteria and have to start EMPAVELI right away.
Anaphylaxis and infusion-related reactions
Advise patients of the risk of anaphylaxis and infusion-related reactions. Inform patients that anaphylaxis is life-threatening and strongly advise patients to seek immediate medical attention if these signs or symptoms occur. These signs and symptoms include the following:
- difficulty breathing including shortness of breath and wheezing
- swollen tongue or throat
- feeling faint
- rapid heart rate
- skin reactions, including hives and itching
- nausea or vomiting
- confusion and anxiety
- dizziness or fainting
Discontinuation
Inform patients with PNH that they may develop hemolysis due to PNH when EMPAVELI is discontinued and that they will be monitored by their healthcare professional for at least 8 weeks following discontinuation of EMPAVELI.
Inform patients who discontinue EMPAVELI to keep the Patient Safety Card with them for 2 months after the last dose of EMPAVELI, because the increased risk of serious infection persists for several weeks following discontinuation of EMPAVELI.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 7/2025 MEDICATION GUIDE
EMPAVELI® (em-puh-vel-ee)
(pegcetacoplan)
injection, for subcutaneous useWhat is the most important information I should know about EMPAVELI?
EMPAVELI is a medicine that affects your immune system. EMPAVELI may lower the ability of your immune system to fight infections.-
EMPAVELI increases your chance of getting serious infections caused by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B. These serious infections may quickly become life-threatening or cause death if not recognized and treated early.
- You must complete or be up to date with the vaccines against Streptococcus pneumoniae and Neisseria meningitidis at least 2 weeks before your first dose of EMPAVELI.
- If you have not completed your vaccines and EMPAVELI must be started right away, you should receive the required vaccines as soon as possible.
- If you have not been vaccinated and EMPAVELI must be started right away, you should also receive antibiotics to take for as long as your healthcare provider tells you.
- If you have been vaccinated against these bacteria in the past, you might need additional vaccines before starting EMPAVELI. Your healthcare provider will decide if you need additional vaccines.
- Vaccines do not prevent all infections caused by encapsulated bacteria. Call your healthcare provider or get emergency medical care right away if you get any of these signs and symptoms of a serious infection:
- fever with or without shivers or the chills
- fever with chest pain and cough
- fever with high heart rate
- headache and fever
- confusion
- clammy skin
- fever and a rash
- fever with breathlessness or fast breathing
- headache with nausea or vomiting
- headache with a stiff neck or stiff back
- body aches with flu-like symptoms
- eyes sensitive to light
Your healthcare provider will give you a Patient Safety Card about the risk of serious infections. Carry it with you at all times during treatment and for 2 months after your last dose of EMPAVELI. Your risk of serious infections may continue for several weeks after your last dose of EMPAVELI. It is important to show this card to any healthcare provider who treats you. This will help them diagnose and treat you quickly.
EMPAVELI is only available through a program called the EMPAVELI Risk Evaluation and Mitigation Strategy (REMS). Before you can take EMPAVELI, your healthcare provider must:- enroll in the EMPAVELI REMS program
- counsel you about the risk of serious infections caused by certain bacteria
- give you information about the symptoms of serious infections
- make sure that you are vaccinated against serious infections caused by encapsulated bacteria and that you receive antibiotics if you need to start EMPAVELI right away and you are not up to date on your vaccines
- give you a Patient Safety Card about your risk of serious infections, as discussed above
- For more information about side effects, see "What are the possible side effects of EMPAVELI?"
What is EMPAVELI?
EMPAVELI is a prescription medicine used to:- treat adults with a disease called paroxysmal nocturnal hemoglobinuria (PNH).
- treat adults and children 12 years of age and older with a kidney disease called complement 3 glomerulopathy (C3G) or primary immune-complex membranoproliferative glomerulonephritis (IC-MPGN), to reduce levels of protein in the urine (proteinuria).
It is not known if EMPAVELI is safe and effective in children under 12 years of age with C3G or primary IC-MPGN.Do not take EMPAVELI if you: - are allergic to pegcetacoplan or any of the ingredients in EMPAVELI. See the end of this Medication Guide for a complete list of ingredients in EMPAVELI.
- have a serious infection caused by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, or Haemophilus influenzae type B when you are starting EMPAVELI treatment.
Before you take EMPAVELI, tell your healthcare provider about all of your medical conditions, including if you: - have an infection or fever.
- are pregnant or plan to become pregnant. EMPAVELI may harm your unborn baby.
Females who are able to become pregnant:- should have a pregnancy test before starting treatment with EMPAVELI.
- use an effective method of birth control (contraception) during treatment with EMPAVELI and for 40 days after the last dose.
- are breastfeeding or plan to breastfeed. It is not known if EMPAVELI passes into your breast milk. You should not breastfeed during treatment with EMPAVELI and for 40 days after the last dose.
Know the medicines you take and the vaccines you receive. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.How should I take EMPAVELI? - See the detailed Instructions for Use that comes with your EMPAVELI for information about how to prepare and infuse your dose of EMPAVELI with your infusion pump.
- See the detailed Instructions for Use that comes with your EMPAVELI Injector for information about how to prepare and inject your dose of EMPAVELI with your EMPAVELI Injector.
- Your healthcare provider should show you how to prepare and administer EMPAVELI before you use it for the first time.
- Use EMPAVELI exactly as your healthcare provider tells you. Do not use more or less than your healthcare provider tells you to.
- If you miss a dose of EMPAVELI, take the missed dose as soon as possible. Take your next dose at your regularly scheduled time.
- EMPAVELI is given under the skin (subcutaneously) 2 times each week. If there is an increase in your LDH, an enzyme in your blood, your healthcare provider may tell you to take EMPAVELI every 3 days.
- If you are changing treatment from eculizumab to EMPAVELI, you should continue eculizumab for 4 weeks after your first dose of EMPAVELI. After 4 weeks, you should stop treatment with eculizumab.
- If you are changing treatment from ravulizumab to EMPAVELI, you should take your starting dose of EMPAVELI no more than 4 weeks after your last dose of ravulizumab.
-
If you have PNH and you stop taking EMPAVELI, your healthcare provider will need to monitor you closely for at least 8 weeks after stopping EMPAVELI. Stopping treatment with EMPAVELI may cause a breakdown of red blood cells due to PNH.
Symptoms or problems that can happen due to red blood cell breakdown include:
- decreased hemoglobin level in your blood
- blood in your urine
- shortness of breath
- trouble swallowing
- tiredness
- pain in the stomach (abdomen)
- blood clots
- erectile dysfunction (ED)
For adults and children 12 years of age and older with C3G or primary IC-MPGN: - EMPAVELI is given under the skin (subcutaneously) 2 times each week.
What are the possible side effects of EMPAVELI?
EMPAVELI can cause serious side effects including:- See "What is the most important information I should know about EMPAVELI?"
- Allergic reactions. Allergic reactions can happen during your EMPAVELI infusion and can be life-threatening. Stop your EMPAVELI infusion and get emergency medical care right away if you get any of these symptoms during your EMPAVELI infusion:
- chest pain
- trouble breathing or shortness of breath
- wheezing
- swelling of your face, tongue, or throat
- feel dizzy or faint or pass out
- fast heart rate
- nausea or vomiting
- feel confused or anxious
- skin reactions, including rash, hives, and itching
The most common side effects in adults with PNH treated with EMPAVELI include: - injection-site reactions
- infections
- diarrhea
- pain in the stomach (abdomen)
- respiratory tract infection
- pain in the arms, hands, legs or feet
- low potassium in blood
- tiredness
- viral infection
- cough
- joint pain
- dizziness
- headache
- rash
The most common side effects in adults and children 12 years of age and older with C3G or primary IC-MPGN treated with EMPAVELI include: - injection-site reactions
- fever
- common cold
- flu
- cough
- nausea
Tell your healthcare provider about any side effect that bothers you or that does not go away. These are not all of the possible side effects of EMPAVELI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store EMPAVELI? - Store vials of EMPAVELI in the refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not use EMPAVELI past the expiration date stamped on the carton.
General information about the safe and effective use of EMPAVELI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use EMPAVELI for a condition for which it was not prescribed. Do not give EMPAVELI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about EMPAVELI that is written for health professionals.What are the ingredients in EMPAVELI?
Active ingredient: pegcetacoplan
Inactive ingredients: sorbitol, glacial acetic acid, sodium acetate trihydrate, Water for Injection USP. EMPAVELI may also contain sodium hydroxide and/or additional glacial acetic acid for pH adjustment.
Manufactured for:
Apellis Pharmaceuticals, Inc. 100 Fifth Avenue Waltham, MA 02451
For patent information: www.apellis.com/productpatent
Copyright © 2025 Apellis Pharmaceuticals, Inc. All rights reserved.
EMPAVELI is a registered trademark of Apellis Pharmaceuticals, Inc.
For more information, go to www.EMPAVELI.com or call 1-866-692-7527.
EMP-MG-28Jul2025-7.0 -
EMPAVELI increases your chance of getting serious infections caused by encapsulated bacteria, including Streptococcus pneumoniae, Neisseria meningitidis, and Haemophilus influenzae type B. These serious infections may quickly become life-threatening or cause death if not recognized and treated early.
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
EMPAVELI® (em-puh-vel-ee)
(pegcetacoplan)
injection, for subcutaneous use
infusion pumpImportant Information
This Instructions for Use is for the infusion pump only. If using EMPAVELI Injector, follow the Instructions for Use that comes with the EMPAVELI Injector.
Read this Instructions for Use before you start using EMPAVELI with an infusion pump and each time you get a refill as there may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. Your healthcare provider should show you or your caregiver how to infuse EMPAVELI the right way before you use it for the first time. Ask your healthcare provider about any instructions you do not understand.
How should I store EMPAVELI?
- Store vials of EMPAVELI in the refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not use EMPAVELI past the expiration date stamped on the carton.
Keep EMPAVELI and all medicines out of the reach of children.
Step 1 Prepare for infusion
Before you start:- Find a well-lit, flat work surface area, like a table.
- Remove a single vial carton from the refrigerator. Keep the vial in the carton at room temperature 68°F to 77°F (20°C to 25°C) and allow it to warm up for about 30 minutes.
- Do not try to speed up the warming process.
Gather your supplies (See Figure A): - Infusion pump and manufacturer's instructions (not shown)
- Compatible syringe for your infusion pump
- Transfer needle OR
- Needleless transfer device to draw up the medicine from the vial
- Infusion set (not shown; varies according to device manufacturer's instructions)
- Infusion tubing
- Sharps container
- Alcohol wipes
- Gauze and tape, or transparent dressing
Figure A: Supplies 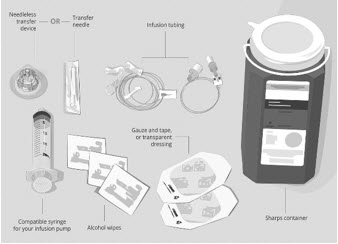
Clean your work surface well using an alcohol wipe. Wash your hands well with soap and water. Dry your hands. Step 2 Check the vial and liquid - Remove the vial from the carton. Carefully look at the liquid in the vial of EMPAVELI.
- EMPAVELI is a clear, colorless to slightly yellowish liquid. Check for particles or color changes (See Figure B).
- The liquid looks cloudy, contains particles, or is dark yellow.
- The protective flip cap is missing or damaged.
- The expiration date on the label has passed.
Figure B 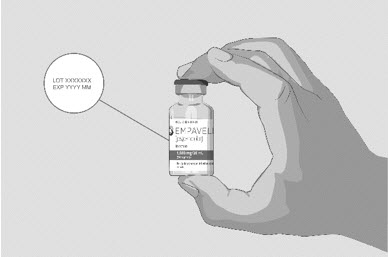
Step 3 Prepare and fill syringe - Remove the protective flip cap from the top of the vial to show the middle part of the gray rubber stopper of the EMPAVELI vial (See Figure C). Throw away the protective flip cap.
- Clean the gray rubber stopper with a new alcohol wipe and allow the gray rubber stopper to dry for at least 30 seconds.
Do not touch the exposed gray rubber stopper after wiping.
Figure C 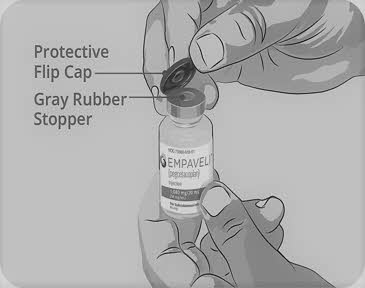
Option 1: If using a needleless transfer device (such as a vial adapter), follow the instructions provided by the device manufacturer.
OR
Option 2: If transfer is done using a transfer needle and a syringe, follow the instructions below:- Attach a sterile transfer needle to a sterile syringe.
- Pull back the plunger to the 20-mL mark to fill the syringe with air (See Figure D).
- Push the air-filled syringe with transfer needle attached down through the center of the vial gray rubber stopper.
- The tip of the transfer needle should not be in the solution to avoid creating air bubble(s) (See Figure E).
- Gently push the air from the syringe into the vial. This will inject the air from the syringe into the vial.
Figure D
Figure E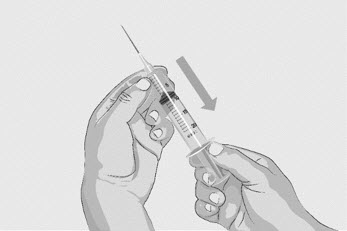
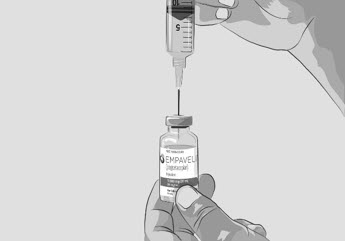
- Turn the vial upside down and insert the transfer needle in the EMPAVELI solution (See Figure F).
Figure F 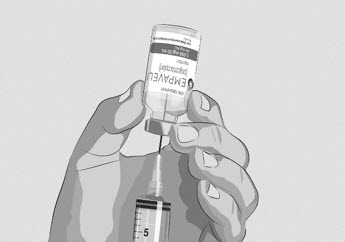
- With the transfer needle tip in the EMPAVELI solution, slowly pull the plunger back to fill the syringe with your prescribed dose (See Figure G). Your first dose, second dose, and maintenance dose may be different.
- Double check that you have withdrawn your prescribed dose.
- Remove the filled syringe with EMPAVELI and the transfer needle from the vial.
- Throw away the vial with the needleless transfer device attached and any remaining EMPAVELI solution into the household trash.
Figure G 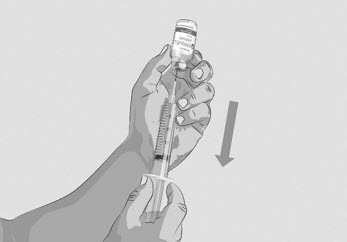
- Remove the transfer needle by using 1 hand to slide the needle into the needle cap and scoop upwards to cover the needle (See Figure H).
Figure H 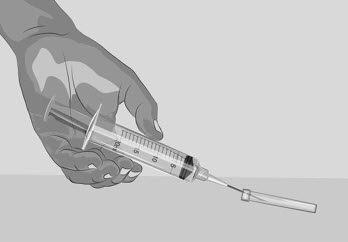
- After the needle is covered, push the needle cap down towards the syringe to fully attach it with 1 hand to prevent an accidental stick with the needle (See Figure I).
Figure I 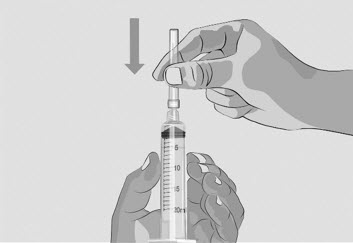
- Twist off and remove the transfer needle (See Figure J).
Figure J 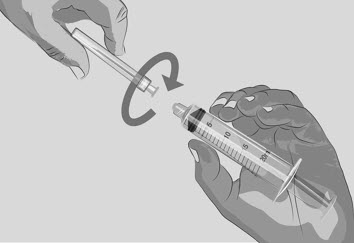
Step 4 Prepare infusion pump and tubing - Gather the infusion pump supplies and follow the device manufacturer's instructions to prepare the pump and tubing.
Step 5 Prepare the infusion site(s) - Select an area on your stomach (abdomen), thighs, hips, or upper arms for the infusion(s) (See Figure K).
Avoid the following infusion areas:- Do not infuse into areas where the skin is tender, bruised, red, or hard.
- Avoid infusing into tattoos, scars, or stretch marks.
Figure K 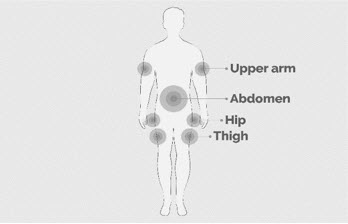
- Use a different site(s) from the last time you infused EMPAVELI. If there are multiple infusion sites, they should be at least 3 inches apart. Change (rotate) infusion sites in between each infusion (See Figure L).
Figure L 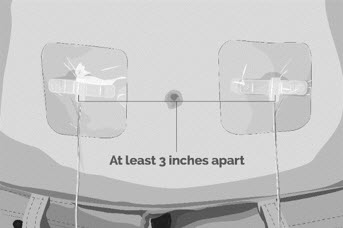
- Clean the skin at each infusion site(s) with a new alcohol wipe, starting at the center of each infusion site and working outward in a circular motion (See Figure M).
- Let the skin dry.
Figure M 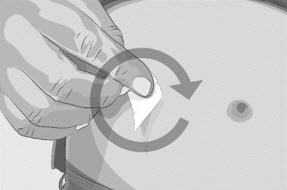
Step 6 Insert and secure the infusion needle(s) - Pinch the skin between your thumb and forefinger around the infusion site (where you plan to insert the needle).
- Insert the needle into the skin (See Figure N).
Figure N 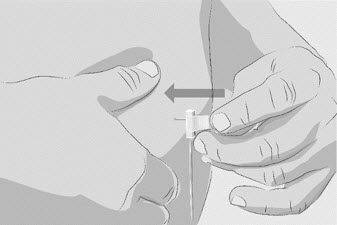
- Secure the needle(s) using gauze and tape or a transparent dressing placed over the infusion site(s) (See Figure O).
Figure O 
Step 7 Start infusion - Follow the device manufacturer's instructions to start the infusion.
- Start the infusion right away after drawing EMPAVELI into the syringe.
- EMPAVELI infusion takes about 30 minutes (if using 2 infusion sites) or about 60 minutes (if using 1 infusion site) to complete.
Step 8 Complete infusion - Follow the device manufacturer's instructions to complete the infusion.
Step 9 Record infusion - Record your treatment as directed by your healthcare provider.
Step 10 Clean up - After the infusion is complete, remove the dressing and slowly take out the needle(s). Cover the infusion site with a new dressing.
- Remove the infusion set from the pump and throw it away into the sharps container (See Figure P).
- Clean and store the infusion pump according to the device manufacturer's instructions.
Step 11 Dispose of (throw away) used needles and syringes and EMPAVELI infusion tubing. - Put the used needles, syringes, and EMPAVELI infusion tubing in an FDA-cleared sharps disposal container right away after use (See Figure P).
- Do not dispose of (throw away) the used needles, syringes, and EMPAVELI infusion tubing in your household trash.
- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
- For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal.
- Do not throw away your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
Figure P 
Call 1-866-692-7527 to speak with an Apellis representative.
Manufactured for:
Apellis Pharmaceuticals, Inc. 100 Fifth Avenue Waltham, MA 02451
Copyright © 2025 Apellis Pharmaceuticals, Inc. All rights reserved.
EMPAVELI is a registered trademark of Apellis Pharmaceuticals, Inc.This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised 7/2025EMP-IFU-28Jul2025-5.0
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE
EMPAVELI® Injector
(pegcetacoplan)
injection, for subcutaneous use
Single-use on-body injectorImportant Information
This Instructions for Use is for the EMPAVELI Injector only.Read this Instructions for Use before you start using the Injector and each time you get a refill as there may be new information. The EMPAVELI Injector is placed on your body to give medicine under the skin. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. Your healthcare provider should show you or your caregiver how to inject EMPAVELI the right way before you use it for the first time. It is important that you do not try to give yourself or someone else the injection unless you have received training from your healthcare provider. Ask your healthcare provider about any instructions you do not understand.
If you have questions, concerns, or need of help, please call ApellisAssist® at 1-866-MY-APL-ASSIST (1-866-692-7527).
SIDE 1:
Filling the Syringe
Start Here
Complete these instructions on how to prepare EMPAVELI before completing EMPAVELI Injector administration instructions on the back of this page.How should I store EMPAVELI?
- Store vials of EMPAVELI in the refrigerator between 36°F to 46°F (2°C to 8°C) in the original carton to protect from light.
- Do not use EMPAVELI past the expiration date stamped on the carton.
Keep EMPAVELI, EMPAVELI Injector, and all medicines out of the reach of children.
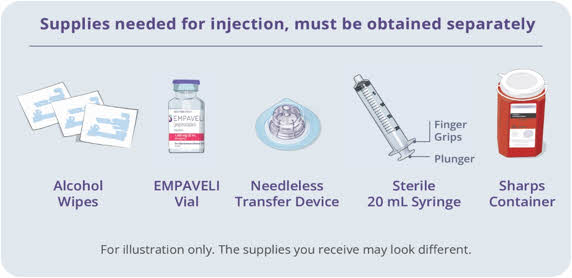
Prepare for injection 1 Before you start: - Find a well-lit, flat work surface area, like a table.
- Remove a single vial carton of EMPAVELI from the refrigerator. Keep the vial in the carton at room temperature 68°F to 77°F (20°C to 25°C) and allow it to warm up for about 30 minutes.
 Do not try to speed up the warming process.
Do not try to speed up the warming process.
2 - Wash your hands well with soap and water.
- Dry your hands.
Check the vial and liquid 3
- Remove the vial from the carton. Carefully look at the liquid in the vial of EMPAVELI.
- EMPAVELI is a clear, colorless to slightly yellowish liquid. Check for particles or color changes.
Do not use and call ApellisAssist if:- —
- The liquid looks cloudy, contains particles, or is dark yellow.
- —
- The protective flip cap is missing from the top of the vial or damaged.
- —
- The expiration date on the label has passed.
4
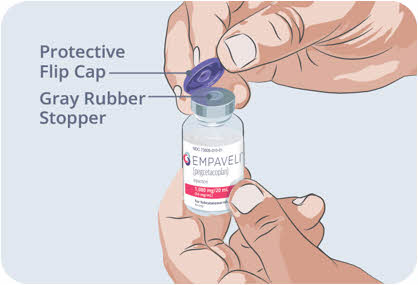
- Flip up to remove the protective flip cap from the top of the vial to show the exposed middle part of the gray rubber stopper of the EMPAVELI vial.
- Throw away the protective flip cap.
5
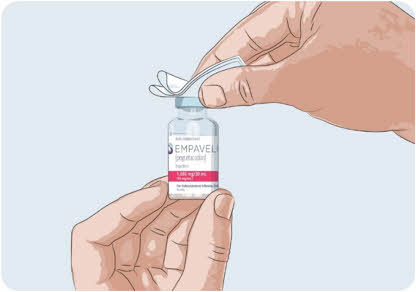
- Clean the gray rubber stopper on the top of the EMPAVELI vial with a new alcohol wipe.
- Allow the gray rubber stopper to dry for at least 30 seconds.
Do not touch the exposed gray rubber stopper after wiping.Prepare and fill the syringe with EMPAVELI using a needleless transfer device (such as a vial adapter)
6
Always follow the Instructions for Use provided by the needleless transfer device's manufacturer (as they may differ from the following steps). Do not remove the needleless transfer device from the blister package. Do not touch the spike or the inside of the needleless transfer device. Do not use the needleless transfer device if it comes out or is dropped out of the package. Do not use the needleless transfer device if the package is opened.7
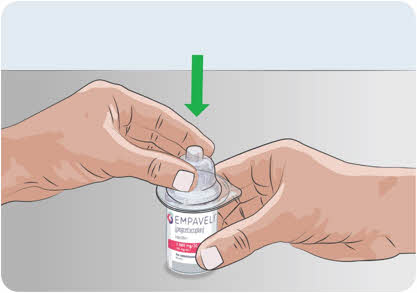
- Remove the cover of the needleless transfer device package.
- Place the vial on a clean, flat surface and hold the vial by the base with one hand.
- Using the outside of the blister package to firmly hold the needleless transfer device, push needleless transfer device straight down onto the vial top until it snaps securely into place.
8
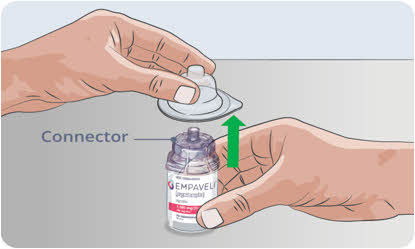
- Remove the blister package from the needleless transfer device and throw the blister package away.
Do not touch the connector at the top of the transfer device.9
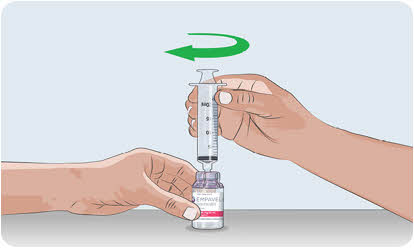
- Remove the syringe from its packaging.
Do not touch the tip of the syringe.- Attach the syringe to the needleless transfer device by twisting the tip of the syringe to the right (clockwise) onto the top of the needleless transfer device.
10
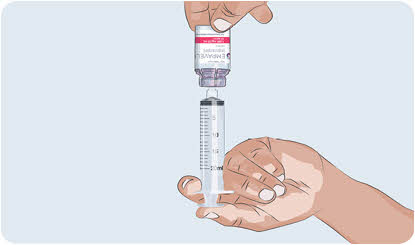
- Turn the EMPAVELI vial upside down.
11
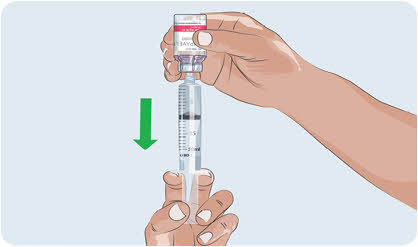
- Slowly pull the syringe plunger down to about the 5 mL mark to partially fill the syringe.
- Remove air from the syringe by gently pushing on the plunger.
12
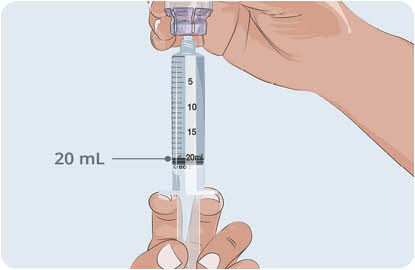
-
Double check your prescribed dose.
- —
- Your first dose, second dose and maintenance dose may be different.
- Slowly pull the syringe plunger down to withdraw your prescribed dose of EMPAVELI to the correct marking amount (mL) of medicine that matches the dose your healthcare provider prescribed. Your dose may be different than the example shown.
- Double check that you have withdrawn your prescribed dose. Your dose cannot be changed after it is filled in the EMPAVELI injector.
13
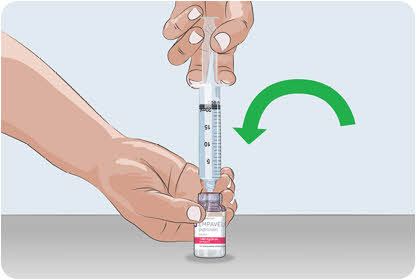
- While holding the EMPAVELI vial and syringe, turn the EMPAVELI vial and filled syringe upright and place the bottom of the EMPAVELI vial on a flat surface.
14
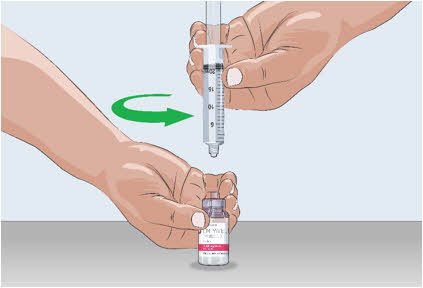
- Remove the filled syringe from the needleless transfer device with one hand while holding the EMPAVELI vial with the other hand and twisting the filled syringe to the left (counterclockwise).
15
- Place the syringe on a clean, flat surface while you prepare the EMPAVELI Injector. The syringe will not leak when set down.
Do not touch the tip of the filled syringe.16
 Do not remove the needleless transfer device from the vial.
Do not remove the needleless transfer device from the vial.- Throw away the vial with the needleless transfer device attached and any remaining EMPAVELI solution into the household trash.
SIDE 2:
Injector AdministrationComplete these instructions for administering the EMPAVELI Injector after completing syringe filling instructions on the front of this page. 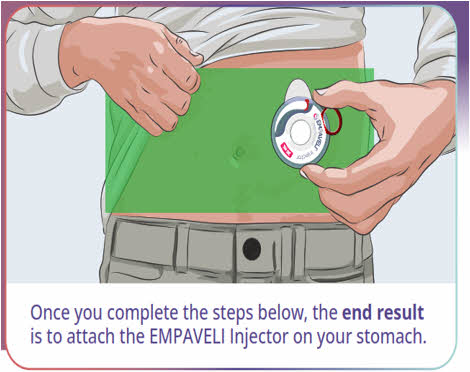

Important information for administration with EMPAVELI Injector
General use: - Do not use EMPAVELI Injector if tamper-proof label has been broken.
- Do not use EMPAVELI Injector if you have a skin condition on your stomach (injection site).
- Do not use if you dropped EMPAVELI Injector.
- Do not use if the sealed plastic tray is open or damaged.
- Do not use if the expiration date on the box has passed.
- Do not use if you have an acrylic allergy. Tell your healthcare provider if you are allergic to acrylic.
- Do not reuse EMPAVELI Injector.
- Do not store the filled EMPAVELI Injector.
- Wear loose clothes so that they do not get in the way of the EMPAVELI Injector.
- Do not store the EMPAVELI Injector in direct sunlight. If the EMPAVELI Injector is stored in direct sunlight, do not use it and call ApellisAssist at 1-866-MY-APL-ASSIST (1-866-692-7527).
Using EMPAVELI Injector: - Do not apply EMPAVELI Injector along the belt line or on areas where the injector will be affected by folds in the skin.
- Do not touch the white adhesive on the bottom of EMPAVELI Injector before attaching to stomach.
- Do not let your clothes touch the clean site.
- Do not remove the Red Safety Tab until EMPAVELI Injector is attached to body.
- Do not remove EMPAVELI Injector from the skin until the button pops out.
- Do not throw away (dispose of) the EMPAVELI Injector into household trash. See the section "Remove and Dispose of EMPAVELI Injector" for information on how to dispose of the EMPAVELI Injector.
- Choose an injection site at least 1 inch from the edge of your belly button and the edge of the EMPAVELI Injector, and 1 inch from last injection site.
- Use the EMPAVELI Injector on stomach only.
During injection: - Do not remove EMPAVELI Injector from the skin during injection.
- Do not bathe, shower, exercise, use hot tubs, whirlpools, or saunas. Avoid getting your stomach wet. The EMPAVELI Injector is not waterproof. Water or sweat may loosen EMPAVELI Injector from skin.
- Do not sleep or bathe during injection.
- Avoid intense physical activity.
- Do not bump or knock the EMPAVELI Injector.
- Do not bump the EMPAVELI Injector Button.
- Do not use anything to hold the EMPAVELI Injector in place.
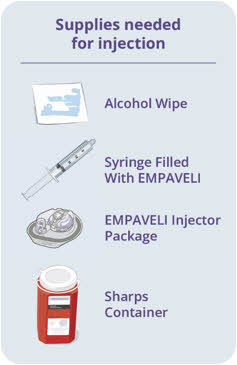
Fill injector with EMPAVELI liquid 17
Peel back the cover and remove the clear packaging insert.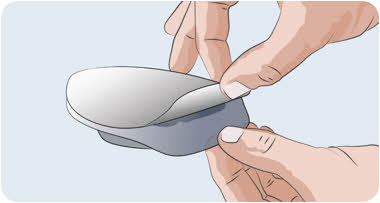
Remove the EMPAVELI Injector and the surrounding Filling Base from the packaging. Place it on a clean, flat surface.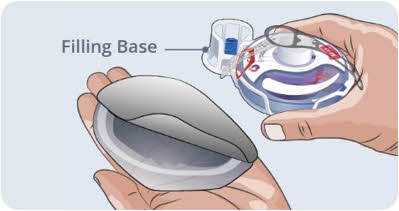
18
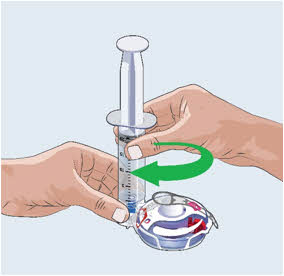
- Pick up the syringe filled with EMPAVELI.
- Twist the filled syringe tip to the right (clockwise) into the Fill Port until it is tight.
19
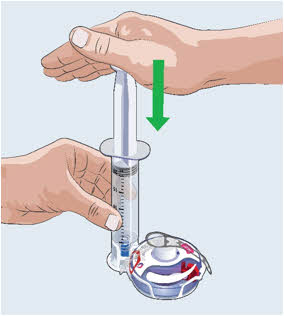
- Firmly push the syringe plunger down.
- The syringe plunger may be hard to push.
- Watch the Fill Gauge move as EMPAVELI is pushed into the injector.

20
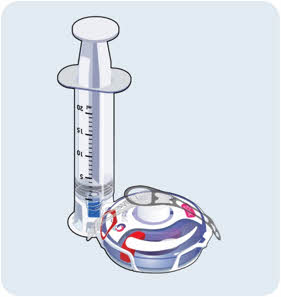
- Make sure the syringe is empty. If needed, press firmly down on the syringe plunger again.
Do not remove syringe from Filling Base. After the EMPAVELI Injector is filled, continue with the preparation and injection. Do not store filled EMPAVELI Injector.Attach EMPAVELI Injector to stomach 21
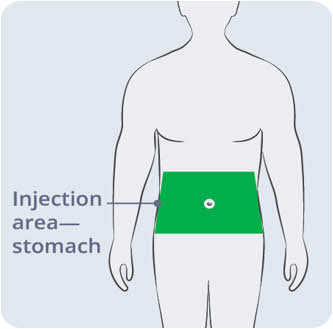
- Select an area on your stomach to place the EMPAVELI Injector.
Use the EMPAVELI Injector on your stomach only.- Choose an injection site at least:
- 1 inch from the edge of your belly button and the edge of the EMPAVELI Injector.
and - 1 inch from your last injection site.
- 1 inch from the edge of your belly button and the edge of the EMPAVELI Injector.
Avoid an injection site that is tender, bruised, red, hard, irritated, scarred, tattooed, or has stretch marks. Do not apply the EMPAVELI Injector along the belt line or on areas where the EMPAVELI injector will be affected by folds in the skin. Wear loose clothes so that they do not get in the way of the EMPAVELI Injector.22
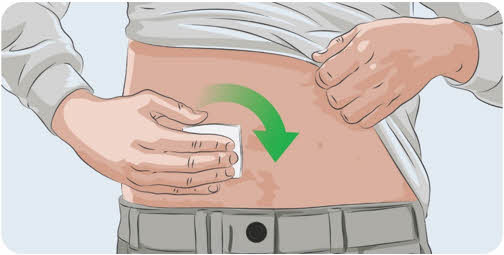
- Clean the injection site with an alcohol wipe.
- Allow the injection site to dry for at least 30 seconds.
Do not let your clothes touch the clean injection site.23
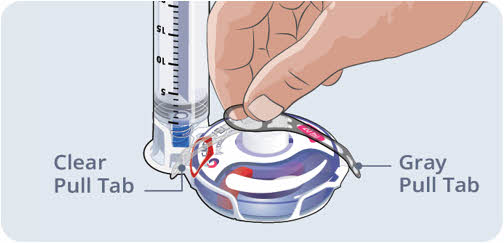
- Hold the Gray Pull Tab and pull. Allow both the Gray and Clear Pull Tabs to fall to the side.
- Both tabs may fall to the side or come off completely.
Do not remove the Red Safety Tab until the EMPAVELI Injector is attached to the body.24
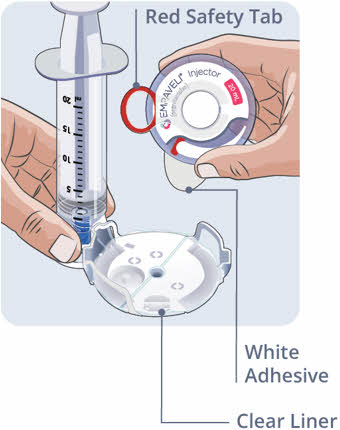
- Hold the sides of the EMPAVELI Injector and pull it straight up to remove it from the Filling Base.
Do not touch the adhesive on the bottom of the EMPAVELI Injector or fold the adhesive onto itself.- The White Adhesive will stay attached to the EMPAVELI Injector and the Clear Liner will stay attached to the Filling Base.
Do not remove the Red Safety Tab until the EMPAVELI Injector is attached to your body. Ensure the injection site has been cleaned before attaching EMPAVELI Injector.25
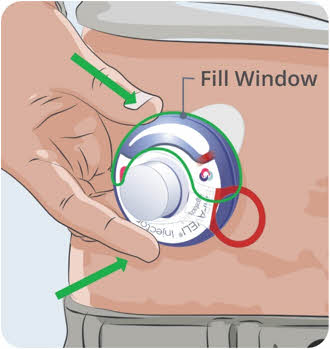
- Position the EMPAVELI Injector so that the Fill Window is pointed up toward your face.
- Press firmly on the clear portion of the EMPAVELI Injector to attach to stomach.
Do not use anything to hold the EMPAVELI Injector in place.Start Injection 26
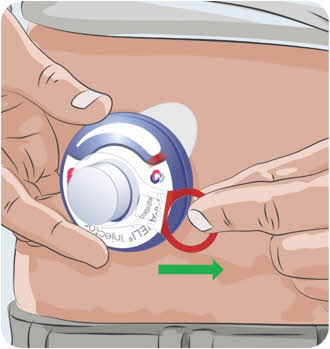
- Hold the EMPAVELI Injector with 1 hand. Use the other hand to pull the Red Safety Tab off.
- The EMPAVELI injection will not start until the Red Safety Tab is removed.
27
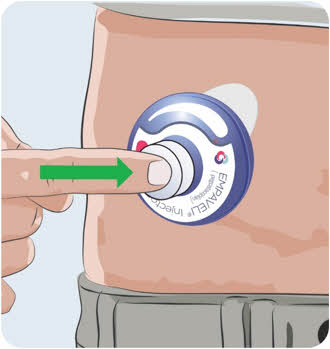
- Right away, Press the button in firmly until it stays in place to start the EMPAVELI injection.
Pushing the Button in will insert the needle into your skin. You may feel the needle go into your skin.- Light daily activities can be done during the EMPAVELI injection.
Be careful not to bump or knock the EMPAVELI Injector or button during the EMPAVELI injection. Keep your stomach dry. Avoid intense physical activity. Do not sleep or bathe during your EMPAVELI injection.28
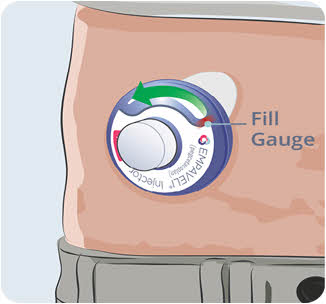
- Your EMPAVELI injection will continue as long as the button is pushed in. It may take about 30 to 60 minutes to complete.
- To track progress, watch the Fill Gauge move across Fill Window toward empty. It may take some time to move and may move slowly.
Do not remove the EMPAVELI Injector until the button pops out. If the button does not pop out after 2 hours (120 minutes), refer to Questions and Answers.If the EMPAVELI Injector falls off of your body, refer to Questions and Answers. Caution: Holding down the button will stop the flow of medicine. Injection will begin again when the button is released.If you have an allergic reaction to the adhesive, call your healthcare provider right away.29
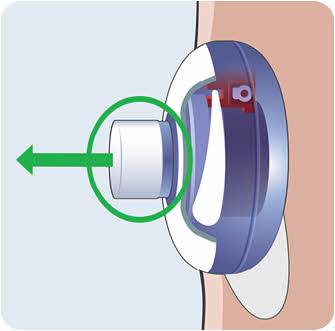
- When the button pops out, the EMPAVELI injection is done. The needle will be pulled out of the skin and back into the EMPAVELI Injector.
The button popping out is the only way to know if the EMPAVELI injection is complete. Do not remove the EMPAVELI Injector until the Button pops out.Remove and dispose of EMPAVELI Injector 30
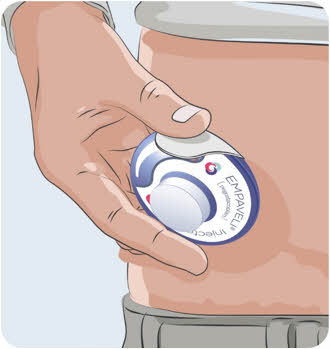
- Use your thumb to lift the Adhesive Tab. Hold the Adhesive Tab against the EMPAVELI Injector.
- Slowly peel the EMPAVELI Injector away from your skin.
31

- Put your used EMPAVELI Injector in an FDA-cleared sharps disposal container right away after use.
Do not throw away the EMPAVELI Injector in the household trash.The Filling Base with the syringe attached, alcohol wipe, and packaging may be placed in your household trash.- If you do not have an FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
- For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http:/www.fda.gov/safesharpsdisposal.
- Do not throw away (dispose of) your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
- Keep the used EMPAVELI Injector and sharps disposal container out of the reach of children.
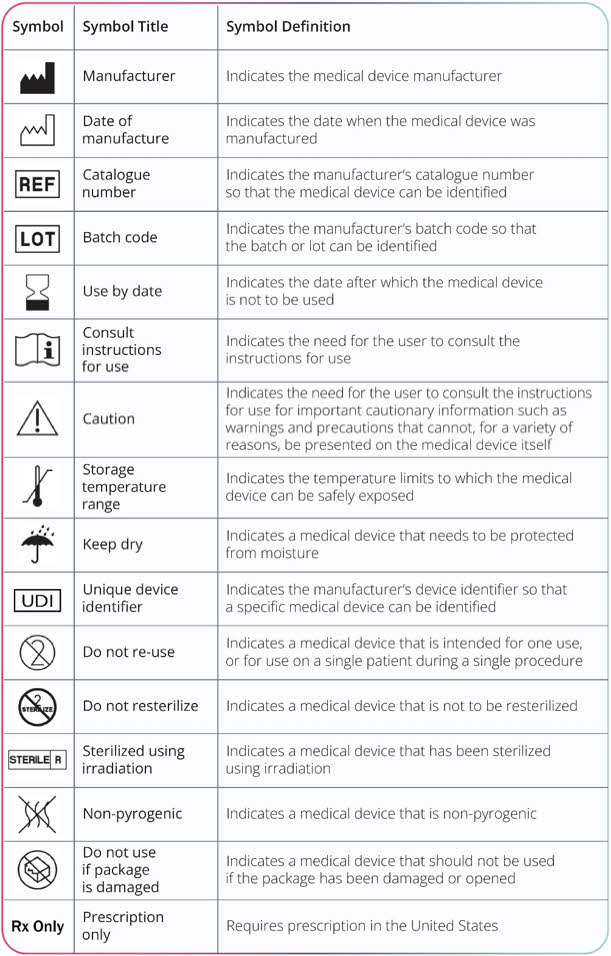
How to store the EMPAVELI Injector - Keep the EMPAVELI Injector in unopened tray inside the original box.
- Do not open the tray until ready for EMPAVELI injection.
- Store the EMPAVELI Injector unit in clean, dry area away from heat and sunlight, at a temperature between 36°F to 86°F (2°C to 30°C).
- Use the EMPAVELI Injector where the temperature is between 41°F to 104°F (5°C to 40°C).
Questions and answers
Can I use more than 1 syringe to fill the EMPAVELI Injector?
No, use only 1 syringe per EMPAVELI Injector.
What should I do if the syringe plunger will not push down to fill the EMPAVELI Injector?
You must firmly press down on the plunger to fill the EMPAVELI Injector. It will feel like there is resistance.
Can I remove the EMPAVELI Injector from my stomach and put it on later to finish injection?
No. The EMPAVELI Injector cannot be reattached. If you take it off, you may not get your full dose.
How long should the injection take?
The injection time is about 30 to 60 minutes.
After 2 hours (120 minutes):- If the button has not popped out, press and hold the button while you remove the EMPAVELI Injector from your skin. Do not touch the bottom of the EMPAVELI Injector as the needle will be exposed. Set the EMPAVELI Injector aside
- If you forgot to start the injection, do not push the button. Remove the EMPAVELI Injector and set it aside.
What if the Button will not push in and lock? Call ApellisAssist at 1-866-MY-APL-ASSIST (1-866-692-7527) right away after putting your device aside.
Make sure that you have taken off the Red Safety Tab. If the Red Safety Tab is removed, make sure you have tried to push the Button in all the way. If you still cannot push the button all the way in, then the EMPAVELI Injector is damaged. Remove your EMPAVELI Injector and set aside. Open a new EMPAVELI Injector and start over. Call ApellisAssist at 1-866-MY-APL-ASSIST (1-866-692-7527).
What if the EMPAVELI Injector falls off of my body?
If the EMPAVELI Injector falls off of your body, pick it up carefully. Do not touch the needle or any medicine that may be on the EMPAVELI Injector. Set the EMPAVELI Injector aside and out of the reach of children. Call ApellisAssist at 1-866-MY-APLASSIST (1-866-692-7527) right away.
Is it normal for skin to be bumpy or irritated during an injection?
No. Your body may be sensitive to the adhesive on the EMPAVELI Injector or to the medicine. Call your healthcare provider right away.
Is it normal for skin to be red after an injection?
Your skin may be slightly red after adhesive removal. If the redness does not go away after 1 to 2 days, call your healthcare provider.Manufactured for:
Apellis Pharmaceuticals, Inc. 100 Fifth Avenue Waltham, MA 02451Manufactured by:
Enable Injections, Inc. 2863 E. Sharon Road Cincinnati, OH 45421, USA
10130600 Rev 04Patent: EnableInjections.com/patent
Copyright © 2025 Apellis Pharmaceuticals, Inc. All rights reserved.
APELLIS, APELLISASSIST, EMPAVELI, and their respective logos are registered trademarks of Apellis Pharmaceuticals, Inc.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 7/2025EMP INJ-IFU-28Jul2025-2.0
- PRINCIPAL DISPLAY PANEL - 1,080 mg/20 mL Vial Carton
- PRINCIPAL DISPLAY PANEL - 20 mL Injector Carton
-
INGREDIENTS AND APPEARANCE
EMPAVELI
pegcetacoplan injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:73606-010 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Pegcetacoplan (UNII: TO3JYR3BOU) (Pegcetacoplan - UNII:TO3JYR3BOU) Pegcetacoplan 1080 mg in 20 mL Inactive Ingredients Ingredient Name Strength SORBITOL (UNII: 506T60A25R) 820 mg in 20 mL ACETIC ACID (UNII: Q40Q9N063P) SODIUM ACETATE (UNII: 4550K0SC9B) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Product Characteristics Color WHITE Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:73606-010-01 1 in 1 CARTON 05/14/2021 1 20 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA215014 05/14/2021 Labeler - Apellis Pharmaceuticals, Inc. (961959629) Establishment Name Address ID/FEI Business Operations Juzen Chemical Corporation 691036974 API MANUFACTURE(73606-010) Establishment Name Address ID/FEI Business Operations Bachem AG 482220311 API MANUFACTURE(73606-010) Establishment Name Address ID/FEI Business Operations Eurofins Lancaster Laboratories, LLC 069777290 ANALYSIS(73606-010) Establishment Name Address ID/FEI Business Operations Eurofins Advantar Laboratories, LLC 849636258 ANALYSIS(73606-010) Establishment Name Address ID/FEI Business Operations Bora Pharmaceuticals Injectables Inc. 119352788 MANUFACTURE(73606-010) Establishment Name Address ID/FEI Business Operations Nelson Laboratories, LLC 151663234 ANALYSIS(73606-010) Establishment Name Address ID/FEI Business Operations AndersonBrecon Inc 053217022 PACK(73606-010) Establishment Name Address ID/FEI Business Operations Cenexi HSC 268155718 ANALYSIS(73606-010) , MANUFACTURE(73606-010) , PACK(73606-010) , STERILIZE(73606-010)