Label: MYCOBUTIN- rifabutin capsule

-
Contains inactivated NDC Code(s)
NDC Code(s): 54868-2841-0, 54868-2841-1, 54868-2841-2, 54868-2841-3, view more54868-2841-4 - Packager: Physicians Total Care, Inc.
- This is a repackaged label.
- Source NDC Code(s): 0013-5301-17
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated June 4, 2010
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
MYCOBUTIN Capsules contain the antimycobacterial agent rifabutin, which is a semisynthetic ansamycin antibiotic derived from rifamycin S. MYCOBUTIN Capsules for oral administration contain 150 mg of rifabutin, USP, per capsule, along with the inactive ingredients microcrystalline cellulose, magnesium stearate, red iron oxide, silica gel, sodium lauryl sulfate, titanium dioxide, and edible white ink.
The chemical name for rifabutin is 1',4-didehydro-1-deoxy-1,4-dihydro-5'-(2-methylpropyl)-1-oxorifamycin XIV (Chemical Abstracts Service, 9th Collective Index) or (9S, 12E, 14S, 15R, 16S, 17R, 18R, 19R, 20S, 21S, 22E, 24Z)-6,16,18,20-tetrahydroxy-1'-isobutyl-14-methoxy-7,9,15,17,19,21,25-heptamethyl-spiro [9,4-(epoxypentadeca[1,11,13]trienimino)-2H-furo[2',3':7,8]naphth[1,2-d] imidazole-2,4'-piperidine]-5,10,26-(3H,9H)-trione-16-acetate. Rifabutin has a molecular formula of C46H62N4O11, a molecular weight of 847.02 and the following structure:
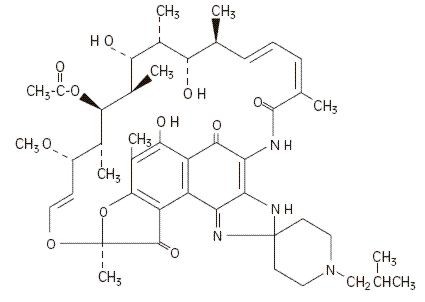
Rifabutin is a red-violet powder soluble in chloroform and methanol, sparingly soluble in ethanol, and very slightly soluble in water (0.19 mg/mL). Its log P value (the base 10 logarithm of the partition coefficient between n-octanol and water) is 3.2 (n-octanol/water).
-
CLINICAL PHARMACOLOGY
Pharmacokinetics
Absorption
Following a single oral dose of 300 mg to nine healthy adult volunteers, rifabutin was readily absorbed from the gastrointestinal tract with mean (±SD) peak plasma levels (Cmax) of 375 (±267) ng/mL (range: 141 to 1033 ng/mL) attained in 3.3 (±0.9) hours (Tmax range: 2 to 4 hours). Absolute bioavailability assessed in five HIV-positive patients, who received both oral and intravenous doses, averaged 20%. Total recovery of radioactivity in the urine indicates that at least 53% of the orally administered rifabutin dose is absorbed from the gastrointestinal tract. The bioavailability of rifabutin from the capsule dosage form, relative to an oral solution, was 85% in 12 healthy adult volunteers. High-fat meals slow the rate without influencing the extent of absorption from the capsule dosage form. Plasma concentrations post-Cmax declined in an apparent biphasic manner. Pharmacokinetic dose-proportionality was established over the 300 to 600 mg dose range in nine healthy adult volunteers (crossover design) and in 16 early symptomatic human immunodeficiency virus (HIV)-positive patients over a 300 to 900 mg dose range.
Distribution
Due to its high lipophilicity, rifabutin demonstrates a high propensity for distribution and intracellular tissue uptake. Following intravenous dosing, estimates of apparent steady-state distribution volume (9.3 ± 1.5 L/kg) in five HIV-positive patients exceeded total body water by approximately 15-fold. Substantially higher intracellular tissue levels than those seen in plasma have been observed in both rat and man. The lung-to-plasma concentration ratio, obtained at 12 hours, was approximately 6.5 in four surgical patients who received an oral dose. Mean rifabutin steady-state trough levels (Cp,minss; 24-hour post-dose) ranged from 50 to 65 ng/mL in HIV-positive patients and in healthy adult volunteers. About 85% of the drug is bound in a concentration-independent manner to plasma proteins over a concentration range of 0.05 to 1 µg/mL. Binding does not appear to be influenced by renal or hepatic dysfunction. Rifabutin was slowly eliminated from plasma in seven healthy adult volunteers, presumably because of distribution-limited elimination, with a mean terminal half-life of 45 (±17) hours (range: 16 to 69 hours). Although the systemic levels of rifabutin following multiple dosing decreased by 38%, its terminal half-life remained unchanged.
Metabolism
Of the five metabolites that have been identified, 25-O-desacetyl and 31-hydroxy are the most predominant, and show a plasma metabolite:parent area under the curve ratio of 0.10 and 0.07, respectively. The former has an activity equal to the parent drug and contributes up to 10% to the total antimicrobial activity.
Excretion
A mass-balance study in three healthy adult volunteers with 14C-labeled rifabutin showed that 53% of the oral dose was excreted in the urine, primarily as metabolites. About 30% of the dose is excreted in the feces. Mean systemic clearance (CLs/F) in healthy adult volunteers following a single oral dose was 0.69 (±0.32) L/hr/kg (range: 0.46 to 1.34 L/hr/kg). Renal and biliary clearance of unchanged drug each contribute approximately 5% to CLs/F.
Pharmacokinetics in Special Populations
Geriatric
Compared to healthy volunteers, steady-state kinetics of MYCOBUTIN are more variable in elderly patients (>70 years).
Pediatric
The pharmacokinetics of MYCOBUTIN have not been studied in subjects under 18 years of age.
Renal Insufficiency
The disposition of rifabutin (300 mg) was studied in 18 patients with varying degrees of renal function. Area under plasma concentration time curve (AUC) increased by about 71% in patients with severe renal insufficiency (creatinine clearance below 30 mL/min) compared to patients with creatinine clearance (Crcl) between 61–74 mL/min. In patients with mild to moderate renal insufficiency (Crcl between 30–61 mL/min), the AUC increased by about 41%. A reduction in the dosage of rifabutin is recommended for patients with Crcl< 30 mL/min (see DOSAGE AND ADMINISTRATION).
Drug-Drug Interactions
(see also PRECAUTIONS-Drug Interactions)
Rifabutin induces the enzymes of the cytochrome P450 3A subfamily (CYP3A) and therefore may reduce the plasma concentrations of drugs that are principally metabolized by those enzymes. Rifabutin is also metabolized by CYP3A. Thus, some drugs that inhibit CYP3A may significantly increase plasma concentrations of rifabutin.
Antifungals
Fluconazole
Fluconazole (200 mg/day for 2 weeks) increased the AUC of rifabutin (300 mg/day for 2 weeks) by 82% and Cmax by 88% in 12 HIV-infected patients who were on zidovudine (500 mg/day) maintenance therapy (see PRECAUTIONS-Drug Interactions). Rifabutin did not affect the pharmacokinetics of fluconazole.
Itraconazole
Coadministration of itraconazole (200 mg/day) with rifabutin (300 mg/day) in six HIV-infected patients reduced both the AUC and Cmax of itraconazole by 70% to 75% (see PRECAUTIONS-Drug Interactions).
Antipneumocystis Agents
Dapsone
Rifabutin (300 mg/day) decreased the AUC of dapsone (50 mg/day) in HIV-infected patients (n=16) by about 27% to 40%.
Sulfamethoxazole-trimethoprim
Coadministration of rifabutin (300 mg/day) and sulfamethoxazole-trimethoprim (double strength) in 12 HIV-infected patients decreased the AUC of sulfamethoxazole-trimethoprim by about 15% to 20%. When trimethoprim was given alone, the AUC of trimethoprim was decreased by 14% and the Cmax by 6%.
Sulfamethoxazole-trimethoprim did not alter the pharmacokinetics of rifabutin.
Antiretroviral Agents
Delavirdine
In 7 HIV-infected patients, rifabutin (300 mg/day) decreased delavirdine (400 mg q 8h) AUC by about 80%, Cmax by about 75%, and mean trough plasma concentrations by about 95%. Based on comparisons with historical data, delavirdine appeared to increase the AUC of rifabutin by at least 100% (see PRECAUTIONS-Drug Interactions).
Didanosine
In 12 HIV-infected patients, coadministration of rifabutin (300 or 600 mg/day) and didanosine (167–375 mg BID) did not alter the pharmacokinetics of either drug.
Indinavir
In healthy volunteers, coadministration of indinavir (800 mg q 8h) and rifabutin (300 mg/day) decreased the AUC of indinavir by about 30% and increased the AUC of rifabutin by about 200% (see PRECAUTIONS-Drug Interactions).
Nelfinavir
Coadministration of nelfinavir (750 mg q 8h for 8 days) and rifabutin (300 mg/day for 7–8 days) decreased the AUC and Cmax of nelfinavir by about 32% and 25%, respectively, and increased the AUC and Cmax of rifabutin by about 207% and 146%, respectively (see PRECAUTIONS-Drug Interactions).
Ritonavir
Coadministration of ritonavir (500 mg q 12h) and rifabutin (150 mg/day) increased the AUC and Cmax of rifabutin by more than 400% and 250%, respectively (see PRECAUTIONS-Drug Interactions).
Saquinavir
In 12 HIV-infected patients, rifabutin (300 mg/day) decreased the AUC of saquinavir (600 mg TID) by about 40% (see PRECAUTIONS-Drug Interactions).
Zidovudine
In 16 HIV-infected patients on zidovudine (100 or 200 mg q 4h), rifabutin (300 or 450 mg/day) lowered the Cmax and AUC of zidovudine by about 48% and 32%, respectively. However, zidovudine levels remained within the therapeutic range during coadministration of rifabutin. Zidovudine did not affect the pharmacokinetics of rifabutin.
Antituberculosis Agents
In studies conducted in healthy volunteers, rifabutin (300 mg) did not alter the pharmacokinetics of ethambutol (n=10) or isoniazid (n=10).
Macrolides
Clarithromycin
In studies conducted in HIV-infected patients, coadministration of rifabutin (300 mg/day) and clarithromycin (500 mg q 12h) decreased the AUC of clarithromycin by about 50% (n=12) and increased the AUC of rifabutin by about 75% (n=14) (see PRECAUTIONS-Drug Interactions).
Other Drugs
Methadone
Rifabutin did not alter the pharmacokinetics of methadone in 24 HIV-infected, methadone-maintained, former intravenous drug users.
Oral contraceptives
In 22 healthy female volunteers receiving an oral contraceptive (35 mcg ethinylestradiol (EE) and 1 mg norethindrone (NE) daily for 21 days), rifabutin decreased EE (AUC) and Cmax by 35% and 20%, respectively, and NE AUC by 46% (see PRECAUTIONS-Drug Interactions).
-
CLINICAL STUDIES
Two randomized, double-blind clinical trials (study 023 and study 027) compared MYCOBUTIN (300 mg/day) to placebo in patients with CDC-defined AIDS and CD4 counts ≤ 200 cells/µL. These studies accrued patients from 2/90 through 2/92. Study 023 enrolled 590 patients, with a median CD4 cell count at study entry of 42 cells/µL (mean 61). Study 027 enrolled 556 patients with a median CD4 cell count at study entry of 40 cells/µL (mean 58).
Endpoints included the following:
- (1)
- MAC bacteremia, defined as at least one blood culture positive for Mycobacterium avium complex (MAC) bacteria.
- (2)
- Clinically significant disseminated MAC disease, defined as MAC bacteremia accompanied by signs or symptoms of serious MAC infection, including one or more of the following: fever, night sweats, rigors, weight loss, worsening anemia, and/or elevations in alkaline phosphatase.
- (3)
- Survival
MAC bacteremia
Participants who received MYCOBUTIN were one-third to one-half as likely to develop MAC bacteremia as were participants who received placebo. These results were statistically significant (study 023: p<0.001; study 027: p = 0.002).
In study 023, the one-year cumulative incidence of MAC bacteremia, on an intent to treat basis, was 9% for patients randomized to MYCOBUTIN and 22% for patients randomized to placebo. In study 027, these rates were 13% and 28% for patients receiving MYCOBUTIN and placebo, respectively.
Most cases of MAC bacteremia (approximately 90% in these studies) occurred among participants whose CD4 count at study entry was ≤100 cells/µL. The median and mean CD4 counts at onset of MAC bacteremia were 13 cells/µL and 24 cells/µL, respectively. These studies did not investigate the optimal time to begin MAC prophylaxis.
Clinically significant disseminated MAC disease
In association with the decreased incidence of bacteremia, patients on MYCOBUTIN showed reductions in the signs and symptoms of disseminated MAC disease, including fever, night sweats, weight loss, fatigue, abdominal pain, anemia, and hepatic dysfunction.
-
MICROBIOLOGY
Mechanism of Action
Rifabutin inhibits DNA-dependent RNA polymerase in susceptible strains of Escherichia coli and Bacillus subtilis but not in mammalian cells. In resistant strains of E. coli, rifabutin, like rifampin, did not inhibit this enzyme. It is not known whether rifabutin inhibits DNA-dependent RNA polymerase in Mycobacterium avium or in M. intracellulare which comprise M. avium complex (MAC).
Susceptibility Testing
In vitro susceptibility testing methods and diagnostic products used for determining minimum inhibitory concentration (MIC) values against M. avium complex (MAC) organisms have not been standardized. Breakpoints to determine whether clinical isolates of MAC and other mycobacterial species are susceptible or resistant to rifabutin have not been established.
In Vitro Studies
Rifabutin has demonstrated in vitro activity against M. avium complex (MAC) organisms isolated from both HIV-positive and HIV-negative people. While gene probe techniques may be used to identify these two organisms, many reported studies did not distinguish between these two species. The vast majority of isolates from MAC-infected, HIV-positive people are M. avium, whereas in HIV-negative people, about 40% of the MAC isolates are M. intracellulare.
Various in vitro methodologies employing broth or solid media, with and without polysorbate 80 (Tween 80), have been used to determine rifabutin MIC values for mycobacterial species. In general, MIC values determined in broth are several fold lower than that observed with methods employing solid media. Utilization of Tween 80 in these assays has been shown to further lower MIC values.
However, MIC values were substantially higher for egg-based compared to agar-based solid media.
Rifabutin activity against 211 MAC isolates from HIV-positive people was evaluated in vitro utilizing a radiometric broth and an agar dilution method. Results showed that 78% and 82% of these isolates had MIC99 values of ≤0.25 µg/mL and ≤1.0 µg/mL, respectively, when evaluated by these two methods. Rifabutin was also shown to be active against phagocytized, M. avium complex in a mouse macrophage cell culture model.
Rifabutin has in vitro activity against many strains of Mycobacterium tuberculosis. In one study, utilizing the radiometric broth method, each of 17 and 20 rifampin-naive clinical isolates tested from the United States and Taiwan, respectively, were shown to be susceptible to rifabutin concentrations of ≤0.125 µg/mL.
Cross-resistance between rifampin and rifabutin is commonly observed with M. tuberculosis and M. avium complex isolates. Isolates of M. tuberculosis resistant to rifampin are likely to be resistant to rifabutin. Rifampicin and rifabutin MIC99 values against 523 isolates of M. avium complex were determined utilizing the agar dilution method (Ref. Heifets, Leonid B. and Iseman, Michael D. 1985. Determination of in vitro susceptibility of Mycobacteria to Ansamycin. Am. Rev. Respir. Dis. 132 (3):710–711).
SUSCEPTIBILITY OF M. AVIUM COMPLEX STRAINS TO RIFAMPIN AND RIFABUTIN % of Strains Susceptible/Resistant to
Different Concentrations of Rifabutin (µg/mL)Susceptibility to Rifampin
(µg/mL)Number of Strains Susceptible to 0.5 Resistant to 0.5 only Resistant to 1.0 Resistant to 2.0 Susceptible to 1.0 30 100.0 0.0 0.0 0.0 Resistant to 1.0 only 163 88.3 11.7 0.0 0.0 Resistant to 5.0 105 38.0 57.1 2.9 2.0 Resistant to 10.0 225 20.0 50.2 19.6 10.2 TOTAL 523 49.5 36.7 9.0 4.8 Rifabutin in vitro MIC99 values of ≤0.5 µg/mL, determined by the agar dilution method, for M. kansasii, M. gordonae and M. marinum have been reported; however, the clinical significance of these results is unknown.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
MYCOBUTIN Capsules must not be administered for MAC prophylaxis to patients with active tuberculosis. Tuberculosis in HIV-positive patients is common and may present with atypical or extrapulmonary findings. Patients are likely to have a nonreactive purified protein derivative (PPD) despite active disease. In addition to chest X-ray and sputum culture, the following studies may be useful in the diagnosis of tuberculosis in the HIV-positive patient: blood culture, urine culture, or biopsy of a suspicious lymph node.
When MYCOBUTIN is used concomitantly with clarithromycin for MAC treatment, a decreased dose of MYCOBUTIN is recommended due to the increase in plasma concentrations of MYCOBUTIN (see PRECAUTIONS-Drug Interactions). Due to the possible occurrence of uveitis, patients should also be carefully monitored when MYCOBUTIN is given in combination with clarithromycin (or other macrolides) and/or fluconazole (and related compounds). If uveitis is suspected, the patient should be referred to an ophthalmologist and, if considered necessary, treatment with MYCOBUTIN should be suspended (see also ADVERSE REACTIONS).
Patients who develop complaints consistent with active tuberculosis while on prophylaxis with MYCOBUTIN should be evaluated immediately, so that those with active disease may be given an effective combination regimen of anti-tuberculosis medications. Administration of MYCOBUTIN as a single agent to patients with active tuberculosis is likely to lead to the development of tuberculosis that is resistant both to MYCOBUTIN and to rifampin.
There is no evidence that MYCOBUTIN is effective prophylaxis against M. tuberculosis. Patients requiring prophylaxis against both M. tuberculosis and Mycobacterium avium complex may be given isoniazid and MYCOBUTIN concurrently.
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, including MYCOBUTIN (rifabutin capsules, USP), and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibiotic use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibiotic use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibiotic treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
In accordance with the commonly accepted criteria for the treatment of mycobacterial infections, MYCOBUTIN should always be given in combination with other anti-mycobacterial drugs not belonging to the family of rifamycins.
For patients with severe liver insufficiency a dose reduction should be considered. Mild hepatic impairment does not require a dose modification.
Severe renal impairment (creatinine clearance below 30 mL/min) requires a dosage reduction of 50%. Mild to moderate renal impairment does not require any dosage adjustment.
Protease inhibitors act as substrates or inhibitors of CYP450 IIIA4 mediated metabolism. Therefore, due to significant drug-drug interactions between protease inhibitors and rifabutin, their concomitant use should be based on the overall assessment of the patient and a patient-specific drug profile (see PRECAUTIONS-Drug Interactions). For further recommendations regarding protease inhibitors, please refer to current, official product monographs or contact the specific manufacturer.
-
PRECAUTIONS
General
Because treatment with MYCOBUTIN Capsules may be associated with neutropenia, and more rarely thrombocytopenia, physicians should consider obtaining hematologic studies periodically in patients receiving prophylaxis with MYCOBUTIN.
Information for Patients
Patients should be advised of the signs and symptoms of both MAC and tuberculosis, and should be instructed to consult their physicians if they develop new complaints consistent with either of these diseases. In addition, since MYCOBUTIN may rarely be associated with myositis and uveitis, patients should be advised to notify their physicians if they develop signs or symptoms suggesting either of these disorders.
Urine, feces, saliva, sputum, perspiration, tears, and skin may be colored brown-orange with rifabutin and some of its metabolites. Soft contact lenses may be permanently stained. Patients to be treated with MYCOBUTIN should be made aware of these possibilities.
Diarrhea is a common problem caused by antibiotics which usually ends when the antibiotic is discontinued. Sometimes, after starting treatment with antibiotics, patients can develop watery and bloody stools (with or without stomach cramps and fever) even as late as two or more months after having taken the last dose of the antibiotic. If this occurs, patients should contact their physician as soon as possible.
There is no reason to believe that MYCOBUTIN has any adverse effect on the ability to drive and/or use machines.
Drug Interactions
Multiple dosing of rifabutin has been associated with induction of hepatic metabolic enzymes of the CYP450 IIIA subfamily. Rifabutin's predominant metabolite (25-desacetyl rifabutin; LM 565), may also contribute to this effect. Metabolic induction due to rifabutin is likely to produce a decrease in circulating levels of concomitantly administered drugs (especially those metabolized by the CYP450 IIIA pathway). Kinetic data suggest that enzymatic induction by rifabutin is complete within 5 days and is dose-independent over the 300 to 600 mg dose-range. Similarly, concomitant medications that competitively inhibit the CYP450 IIIA activity may increase circulating levels of rifabutin.
Malabsorption
Gastric pH alteration due to progressing HIV disease has been linked with malabsorption of some drugs used in HIV-positive patients (e.g., rifampin, isoniazid). Drug serum concentration data from AIDS patients with varying disease severity (based on CD4+ counts) suggest that rifabutin absorption is not influenced by progressing HIV disease.
Effects on Other Drugs
Rifabutin induces CYP3A enzymes and therefore may reduce the plasma concentrations of drugs metabolized by those enzymes. This effect may reduce the efficacy of standard doses of such drugs, which include itraconazole, clarithromycin, and saquinavir (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions).
Effects on Rifabutin
Some drugs that inhibit CYP3A may significantly increase the plasma concentration of rifabutin. Because high plasma levels of rifabutin may increase the risk of adverse reactions, carefully monitor patients receiving coadministration of such drugs, which include fluconazole and clarithromycin (see CLINICAL PHARMACOLOGY-Drug-Drug Interactions). In some cases, the dosage of MYCOBUTIN may need to be reduced when it is coadministered with such a drug (see below).
The following table summarizes the results and magnitude of the pertinent drug interactions assessed with rifabutin. The clinical relevance of these interactions and subsequent dose modifications should be judged in light of the population studied, severity of the disease, patient's drug profile, and the likely impact on the risk/benefit ratio.
Rifabutin Interaction Studies* Coadministered Drugs Effect on Rifabutin Effect on Coadministered Drug Comments ANTIVIRALS Amprenavir 2.9-fold ↑ AUC, 2.2-fold ↑ Cmax No significant change in kinetics. A 50% reduction in the rifabutin dose is recommended when combined with amprenavir. Increased monitoring for adverse reactions is warranted. Delavirdine ND Oral clearance ↑ 5-fold resulting in significantly lower mean trough plasma concentrations (18±15 to 1.0±0.7 µM) Study conducted in HIV-1 infected patients Rifabutin is not recommended for patients dosed with delavirdine mesylate 400 mg q8h. Didanosine No significant change in kinetics. No significant change in kinetics at steady state. Fosamprenavir/ritonavir 64% ↑ AUC † 35% ↑ AUC and 36% ↑ Cmax, no effect Ctrough (amprenavir) Dosage reduction of rifabutin by at least 75% (to 150 mg every other day or three times per week) is recommended when combined with fosamprenavir Indinavir 204% ↑ in AUC 32%↓ in AUC Lopinavir/ritonavir 5.7-fold ↑ AUC, 3.4 fold ↑ Cmax† No significant change in lopinavir kinetics. Dosage reduction of rifabutin by at least 75% of the usual dose of 300 mg/day is recommended (i.e., a maximum dose of 150 mg every other day or three times per week). Increased monitoring for adverse reactions is warranted. Further dosage reduction of rifabutin may be necessary. Saquinavir ND 40% ↓ in AUC Ritonavir 4 fold increase in AUC, 2.5 fold increase in Cmax ND In the presence of ritonavir the subsequent risk of side effects, including uveitis may be increased . If a protease inhibitor is required in a patient treated with rifabutin, agents other than ritonavir should be considered. Tipranavir/ritonavir[133] 2.9-fold ↑ AUC, 1.7-fold ↑ Cmax No significant change in tipranavir kinetics. Therapeutic drug monitoring of rifabutin is recommended. Zidovudine No significant change in kinetics. Approximately 32%↓ in Cmax and AUC A large controlled clinical study has shown that these changes are of no clinical relevance. ANTIFUNGALS ANTIFUNGALS 82% ↑ in AUC No significant change in steady-state plasma concentrations Itraconazole ND 70% to 75% ↓ in Cmax and AUC One case report suggests a kinetic interaction resulting in an increase in serum rifabutin levels and a risk for developing uveitis in the presence of itraconazole. Posaconazole 31%↑ Cmax, 72%↑ AUC 43%↓ Cmax, 49%↓ AUC If the drugs are co-administered, patients should be monitored for adverse events associated with rifabutin administration. Voriconazole 195%↑ Cmax, 331%↑ AUC ‡ Rifabutin (300 mg once daily) decreased the Cmax and AUC of voriconazole at 200 mg twice daily by 69% and 78%, respectively. During co-administration with rifabutin, the Cmax and AUC of voriconazole at 350 mg twice daily were 96% and 68% of the levels when administered alone at 200 mg twice daily. At a voriconazole dose of 400 mg twice daily Cmax and AUC were 104% and 87% higher, respectively, compared with voriconazole alone at 200 mg twice daily. If the benefit outweighs the risk, rifabutin may be coadministered with voriconazole if the maintenance dose of voriconazole is increased to 5 mg/kg intravenously every 12 hours or from 200 mg to 350 mg orally, every 12 hours (100 mg to 200 mg orally, every 12 hours in patients less than 40 kg). Careful monitoring of full blood counts and adverse events to rifabutin (e.g. uveitis) is recommended when rifabutin is coadministered with voriconazole ANTI-PCP (Pneumocystis carinii pneumonia) Dapsone ND Approximately 27% to 40% ↓ in AUC Study conducted in HIV infected patients (rapid and slow acetylators). Sulfamethoxazole-Trimethoprim No significant change in Cmax and AUC Approximately 15% to 20% ↓ in AUC In another study, only trimethoprim (not sulfamethoxazole) had 14% ↓ in AUC and 6%↓ in Cmax but were not considered clinically significant. ANTI-MAC (Mycobacterium avium intracellulare complex) Azithromycin No PK interaction No PK interaction Clarithromycin Approximately 77% ↑ in AUC Approximately 50%↓ in AUC Study conducted in HIV infected patients. Dose of rifabutin should be adjusted in the presence of clarithromycin ANTI-TB (Tuberculosis) Ethambutol ND No significant change in AUC or Cmax Isoniazid ND Pharmacokinetics not affected Pyrazinamide ND ND Study data being evaluated. OTHER Methadone ND No significant effect No apparent effect of rifabutin on either peak levels of methadone or systemic exposure based upon AUC. Rifabutin kinetics not evaluated. Ethinylestradiol ND 35%↓ AUC
20%↓ CmaxPatients should be advised to use other methods of contraception. Norethindrone ND 46%↓ AUC Patients should be advised to use other methods of contraception. Tacrolimus ND ND Authors report that rifabutin decreases tacrolimus trough blood levels. Theophylline ND No significant change in AUC or Cmax compared with baseline. Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies were conducted with rifabutin in mice and in rats. Rifabutin was not carcinogenic in mice at doses up to 180 mg/kg/day, or approximately 36 times the recommended human daily dose. Rifabutin was not carcinogenic in the rat at doses up to 60 mg/kg/day, about 12 times the recommended human dose.
Rifabutin was not mutagenic in the bacterial mutation assay (Ames Test) using both rifabutin-susceptible and resistant strains. Rifabutin was not mutagenic in Schizosaccharomyces pombe P1 and was not genotoxic in V-79 Chinese hamster cells, human lymphocytes in vitro, or mouse bone marrow cells in vivo.
Fertility was impaired in male rats given 160 mg/kg (32 times the recommended human daily dose).
Pregnancy
There are no adequate and well-controlled studies in pregnant or breastfeeding women.
Pregnancy Category B
Reproduction studies have been carried out in rats and rabbits given rifabutin using dose levels up to 200 mg/kg (40 times the recommended human daily dose). No teratogenicity was observed in either species. In rats, given 200 mg/kg/day, there was a decrease in fetal viability. In rats, at 40 mg/kg/day (8 times the recommended human daily dose), rifabutin caused an increase in fetal skeletal variants. In rabbits, at 80 mg/kg/day (16 times the recommended human daily dose), rifabutin caused maternotoxicity and increase in fetal skeletal anomalies. There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, rifabutin should be used in pregnant women only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether rifabutin is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness of rifabutin for prophylaxis of MAC in children have not been established. Limited safety data are available from treatment use in 22 HIV-positive children with MAC who received MYCOBUTIN in combination with at least two other antimycobacterials for periods from 1 to 183 weeks. Mean doses (mg/kg) for these children were: 18.5 (range 15.0 to 25.0) for infants one year of age, 8.6 (range 4.4 to 18.8) for children 2 to 10 years of age, and 4.0 (range 2.8 to 5.4) for adolescents 14 to 16 years of age. There is no evidence that doses greater than 5 mg/kg daily are useful. Adverse experiences were similar to those observed in the adult population, and included leukopenia, neutropenia, and rash. In addition, corneal deposits have been observed in some patients during routine ophthalmologic surveillance of HIV-positive pediatric patients receiving MYCOBUTIN as part of a multiple-drug regimen for MAC prophylaxis. These are tiny, almost transparent, asymptomatic peripheral and central corneal deposits which do not impair vision. Doses of MYCOBUTIN may be administered mixed with foods such as applesauce.
Geriatric Use
Clinical studies of MYCOBUTIN did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy (see CLINICAL PHARMACOLOGY).
-
ADVERSE REACTIONS
MYCOBUTIN Capsules were generally well tolerated in the controlled clinical trials. Discontinuation of therapy due to an adverse event was required in 16% of patients receiving MYCOBUTIN, compared to 8% of patients receiving placebo in these trials. Primary reasons for discontinuation of MYCOBUTIN were rash (4% of treated patients), gastrointestinal intolerance (3%), and neutropenia (2%).
The following table enumerates adverse experiences that occurred at a frequency of 1% or greater, among the patients treated with MYCOBUTIN in studies 023 and 027.
CLINICAL ADVERSE EXPERIENCES REPORTED IN ≥1% OF PATIENTS TREATED WITH MYCOBUTIN ADVERSE EVENT MYCOBUTIN
(n = 566) %PLACEBO
(n = 580) %BODY AS A WHOLE Abdominal Pain 4 3 Asthenia 1 1 Chest Pain 1 1 Fever 2 1 Headache 3 5 Pain 1 2 DIGESTIVE SYSTEM Anorexia 2 2 Diarrhea 3 3 Dyspepsia 3 1 Eructation 3 1 Flatulence 2 1 Nausea 6 5 Nausea and Vomiting 3 2 Vomiting 1 1 MUSCULOSKELETAL SYSTEM Myalgia 2 1 NERVOUS SYSTEM Insomnia 1 1 SKIN AND APPENDAGES Rash 11 8 SPECIAL SENSES Taste Perversion 3 1 UROGENITAL SYSTEM Discolored Urine 30 6 CLINICAL ADVERSE EVENTS REPORTED IN <1% OF PATIENTS WHO RECEIVED MYCOBUTIN
Considering data from the 023 and 027 pivotal trials, and from other clinical studies, MYCOBUTIN appears to be a likely cause of the following adverse events which occurred in less than 1% of treated patients: flu-like syndrome, hepatitis, hemolysis, arthralgia, myositis, chest pressure or pain with dyspnea, and skin discoloration.
The following adverse events have occurred in more than one patient receiving MYCOBUTIN, but an etiologic role has not been established: seizure, paresthesia, aphasia, confusion, and non-specific T wave changes on electrocardiogram.
When MYCOBUTIN was administered at doses from 1050 mg/day to 2400 mg/day, generalized arthralgia and uveitis were reported. These adverse experiences abated when MYCOBUTIN was discontinued.
The following table enumerates the changes in laboratory values that were considered as laboratory abnormalities in studies 023 and 027.
PERCENTAGE OF PATIENTS WITH LABORATORY ABNORMALITIES LABORATORY ABNORMALITIES MYCOBUTIN
(n = 566) %PLACEBO
(n = 580) %INCLUDES GRADE 3 OR 4 TOXICITIES AS SPECIFIED: Chemistry: Increased Alkaline Phosphatase * < 1 3 Increased SGOT † 7 12 Increased SGPT † 9 11 Hematology: Anemia ‡ 6 7 Eosinophilia 1 1 Leukopenia § 17 16 Neutropenia ¶ 25 20 Thrombocytopenia # 5 4 The incidence of neutropenia in patients treated with MYCOBUTIN was significantly greater than in patients treated with placebo (p = 0.03). Although thrombocytopenia was not significantly more common among patients treated with MYCOBUTIN in these trials, MYCOBUTIN has been clearly linked to thrombocytopenia in rare cases. One patient in study 023 developed thrombotic thrombocytopenic purpura, which was attributed to MYCOBUTIN.
Uveitis is rare when MYCOBUTIN is used as a single agent at 300 mg/day for prophylaxis of MAC in HIV-infected persons, even with the concomitant use of fluconazole and/or macrolide antibiotics. However, if higher doses of MYCOBUTIN are administered in combination with these agents, the incidence of uveitis is higher.
Patients who developed uveitis had mild to severe symptoms that resolved after treatment with corticosteroids and/or mydriatic eye drops; in some severe cases, however, resolution of symptoms occurred after several weeks.
When uveitis occurs, temporary discontinuance of MYCOBUTIN and ophthalmologic evaluation are recommended. In most mild cases, MYCOBUTIN may be restarted; however, if signs or symptoms recur, use of MYCOBUTIN should be discontinued (Morbidity and Mortality Weekly Report, September 9, 1994).
Adverse reactions identified through clinical trials or post-marketing surveillance by system organ class (SOC) are listed below.
Blood and lymphatic system disorders: Pancytopenia, white blood cell disorders (including agranulocytosis, leukopenia, lymphopenia, granulocytopenia, neutropenia, white blood cell count decreased, neutrophil count decreased), thrombocytopenia, platelet count decreased, anemia.
Immune system disorders: Shock, hypersensitivity, bronchospasm, rash, eosinophilia.
Eye disorders: Uveitis, corneal deposits.
Gastrointestinal disorders: Clostridium difficile colitis , nausea, vomiting.
Hepato-biliary disorders: Jaundice, hepatic enzyme increased.
Skin and subcutaneous tissue disorders: Skin discoloration.
Musculoskeletal and connective tissue disorders: Arthralgia, myalgia.
General disorders and administration site conditions: Pyrexia.
Pyrexia, rash and rarely other hypersensitivity reactions such as eosinophilia, bronchospasm and shock might occur, as has been seen with other antibiotics. A limited number of skin discoloration has been reported. Mild to severe, reversible uveitis has been reported less frequently when MYCOBUTIN is used at 300 mg as monotherapy in MAC prophylaxis versus MYCOBUTIN in combination with clarithromycin for MAC treatment (see also WARNINGS). Corneal deposits have been reported during routine ophthalmologic surveillance of some HIV-positive pediatric patients receiving MYCOBUTIN as part of a multiple drug regimen for MAC prophylaxis. The deposits are tiny, almost transparent, asymptomatic peripheral and central corneal deposits, and do not impair vision.
-
ANIMAL TOXICOLOGY
Liver abnormalities (increased bilirubin and liver weight) occurred in all species tested, in rats at doses 5 times, in monkeys at doses 8 times, and in mice at doses 6 times the recommended human daily dose. Testicular atrophy occurred in baboons at doses 4 times the recommended human dose, and in rats at doses 40 times the recommended human daily dose.
-
OVERDOSAGE
No information is available on accidental overdosage in humans.
Treatment
While there is no experience in the treatment of overdose with MYCOBUTIN Capsules, clinical experience with rifamycins suggests that gastric lavage to evacuate gastric contents (within a few hours of overdose), followed by instillation of an activated charcoal slurry into the stomach, may help absorb any remaining drug from the gastrointestinal tract.
Rifabutin is 85% protein bound and distributed extensively into tissues (Vss:8 to 9 L/kg). It is not primarily excreted via the urinary route (less than 10% as unchanged drug); therefore, neither hemodialysis nor forced diuresis is expected to enhance the systemic elimination of unchanged rifabutin from the body in a patient with an overdose of MYCOBUTIN.
-
DOSAGE AND ADMINISTRATION
It is recommended that MYCOBUTIN Capsules be administered at a dose of 300 mg once daily. For those patients with propensity to nausea, vomiting, or other gastrointestinal upset, administration of MYCOBUTIN at doses of 150 mg twice daily taken with food may be useful. For patients with severe renal impairment (creatinine clearance less than 30 mL/min), the dose of MYCOBUTIN should be reduced by 50%. No dosage adjustment is required for patients with mild to moderate renal impairment. Reduction of the dose of MYCOBUTIN may also be needed for patients receiving concomitant treatment with certain other drugs (see PRECAUTIONS-Drug Interactions).
-
HOW SUPPLIED
MYCOBUTIN Capsules (rifabutin capsules, USP) are supplied as hard gelatin capsules having an opaque red-brown cap and body, imprinted with MYCOBUTIN/PHARMACIA & UPJOHN in white ink, each containing 150 mg of rifabutin, USP.
MYCOBUTIN is available as follows:
Bottles of 10
NDC 54868-2841-3
Bottles of 30
NDC 54868-2841-4
Bottles of 50
NDC 54868-2841-0
Bottles of 60
NDC 54868-2841-1
Bottles of 100
NDC 54868-2841-2
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 150 mg Capsule Label
-
INGREDIENTS AND APPEARANCE
MYCOBUTIN
rifabutin capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54868-2841(NDC:0013-5301-17) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIFABUTIN (UNII: 1W306TDA6S) (RIFABUTIN - UNII:1W306TDA6S) RIFABUTIN 150 mg Inactive Ingredients Ingredient Name Strength CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) MAGNESIUM STEARATE (UNII: 70097M6I30) FERRIC OXIDE RED (UNII: 1K09F3G675) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color red (Red-Brown) Score no score Shape CAPSULE Size 21mm Flavor Imprint Code MYCOBUTIN;PHARMACIA;UPJOHN Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54868-2841-0 50 in 1 BOTTLE, PLASTIC 2 NDC:54868-2841-1 60 in 1 BOTTLE, PLASTIC 3 NDC:54868-2841-2 100 in 1 BOTTLE 4 NDC:54868-2841-3 10 in 1 BOTTLE, PLASTIC 5 NDC:54868-2841-4 30 in 1 BOTTLE, PLASTIC Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA050689 03/15/1995 Labeler - Physicians Total Care, Inc. (194123980) Establishment Name Address ID/FEI Business Operations Physicians Total Care, Inc. 194123980 relabel, repack

