Label: PREGABALIN capsule

- NDC Code(s): 80425-0123-3
- Packager: Advanced Rx Pharmacy of Tennessee, LLC
- This is a repackaged label.
- Source NDC Code(s): 69238-1313
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: CV
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated November 10, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
1. Indications and Usage Section
1 INDICATIONS AND USAGE
Pregabalin capsules are indicated for:
Management of neuropathic pain associated with diabetic peripheral neuropathy
Management of postherpetic neuralgia
Adjunctive therapy for the treatment of partial-onset seizures in patients 17 years of age and older
Management of fibromyalgia
Management of neuropathic pain associated with spinal cord injuryPediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
-
2. Dosage and Administration Section
2.1 Important Administration Instructions
Pregabalin capsules are given orally with or without food.
When discontinuing pregabalin capsules, taper gradually over a minimum of 1 week [see Warnings and Precautions (5.6)].
Because pregabalin is eliminated primarily by renal excretion, adjust the dose in adult patients with reduced renal function [see Dosage and Administration (2.7)].
2.2 Neuropathic Pain Associated with Diabetic Peripheral Neuropathy in Adults
The maximum recommended dose of pregabalin capsule is 100 mg three times a day (300 mg/day) in patients with creatinine clearance of at least 60 mL/min. Begin dosing at 50 mg three times a day (150 mg/day). The dose may be increased to 300 mg/day within 1 week based on efficacy and tolerability.
Although pregabalin capsule was also studied at 600 mg/day, there is no evidence that this dose confers additional significant benefit and this dose was less well tolerated. In view of the dose-dependent adverse reactions, treatment with doses above 300 mg/day is not recommended [see Adverse Reactions (6.1)].
2.3 Postherpetic Neuralgia in Adults
The recommended dose of pregabalin capsule is 75 mg to 150 mg two times a day, or 50 mg to 100 mg three times a day (150 to 300 mg/day) in patients with creatinine clearance of at least 60 mL/min. Begin dosing at 75 mg two times a day, or 50 mg three times a day (150 mg/day). The dose may be increased to 300 mg/day within 1 week based on efficacy and tolerability.
Patients who do not experience sufficient pain relief following 2 to 4 weeks of treatment with 300 mg/day, and who are able to tolerate pregabalin capsules, may be treated with up to 300 mg two times a day, or 200 mg three times a day (600 mg/day). In view of the dose-dependent adverse reactions and the higher rate of treatment discontinuation due to adverse reactions, reserve dosing above 300 mg/day for those patients who have on-going pain and are tolerating 300 mg daily [see Adverse Reactions (6.1)].
2.4 Adjunctive Therapy for Partial-Onset Seizures in Patients 17 Years of Age and Older
The recommended dosages for adult patients 17 years of age and older is included in Table 1. Administer the total daily dosage orally in two or three divided doses as indicated in Table 1. Based on clinical response and tolerability, dosage may be increased, approximately weekly.
Table 1. Recommended Dosage for Adult Patients 17 Years and Older
Age and Body Weight
Recommended Initial Dosage
Recommended Maximum Dosage
Frequency of Administration Adults (17 years and older)
150 mg/day
600 mg/day
2 or 3 divided doses Both the efficacy and adverse event profiles of pregabalin capsules have been shown to be dose-related.
The effect of dose escalation rate on the tolerability of pregabalin capsules has not been formally studied.
The efficacy of adjunctive pregabalin capsules in patients taking gabapentin has not been evaluated in controlled trials. Consequently, dosing recommendations for the use of pregabalin capsules with gabapentin cannot be offered.
Pediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
2.5 Management of Fibromyalgia in Adults
The recommended dose of pregabalin capsules for fibromyalgia is 300 to 450 mg/day. Begin dosing at 75 mg two times a day (150 mg/day). The dose may be increased to 150 mg two times a day (300 mg/day) within 1 week based on efficacy and tolerability. Patients who do not experience sufficient benefit with 300 mg/day may be further increased to 225 mg two times a day (450 mg/day). Although pregabalin capsule was also studied at 600 mg/day, there is no evidence that this dose confers additional benefit and this dose was less well tolerated. In view of the dose-dependent adverse reactions, treatment with doses above 450 mg/day is not recommended [see Adverse Reactions (6.1)].
2.6 Neuropathic Pain Associated with Spinal Cord Injury in Adults
The recommended dose range of pregabalin capsules for the treatment of neuropathic pain associated with spinal cord injury is 150 to 600 mg/day. The recommended starting dose is 75 mg two times a day (150 mg/day). The dose may be increased to 150 mg two times a day (300 mg/day) within 1 week based on efficacy and tolerability. Patients who do not experience sufficient pain relief after 2 to 3 weeks of treatment with 150 mg two times a day and who tolerate pregabalin capsules may be treated with up to 300 mg two times a day [see Clinical Studies (14.5)].
2.7 Dosing for Adult Patients with Renal Impairment
In view of dose-dependent adverse reactions and since pregabalin is eliminated primarily by renal excretion, adjust the dose in adult patients with reduced renal function. The use of pregabalin capsules in pediatric patients with compromised renal function has not been studied.
Base the dose adjustment in patients with renal impairment on creatinine clearance (CLcr), as indicated in Table 2. To use this dosing table, an estimate of the patient's CLcr in mL/min is needed. CLcr in mL/min may be estimated from serum creatinine (mg/dL) determination using the Cockcroft and Gault equation:
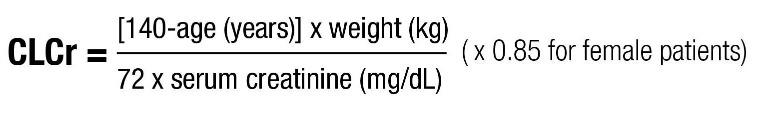
Next, refer to the Dosage and Administration section to determine the recommended total daily dose based on indication, for a patient with normal renal function (CLcr greater than or equal to 60 mL/min). Then refer to Table 2 to determine the corresponding renal adjusted dose.
(For example: A patient initiating pregabalin therapy for postherpetic neuralgia with normal renal function (CLcr greater than or equal to 60 mL/min), receives a total daily dose of 150 mg/day pregabalin. Therefore, a renal impaired patient with a CLcr of 50 mL/min would receive a total daily dose of 75 mg/day pregabalin administered in two or three divided doses.)
For patients undergoing hemodialysis, adjust the pregabalin daily dose based on renal function. In addition to the daily dose adjustment, administer a supplemental dose immediately following every 4-hour hemodialysis treatment (see Table 2).
Table 2. Pregabalin Dosage Adjustment Based on Renal Function
Next, refer to the Dosage and Administration section to determine the recommended total daily dose based on indication, for a patient with normal renal function (CLcr greater than or equal to 60 mL/min). Then refer to Table 2 to determine the corresponding renal adjusted dose.
(For example: A patient initiating pregabalin therapy for postherpetic neuralgia with normal renal function (CLcr greater than or equal to 60 mL/min), receives a total daily dose of 150 mg/day pregabalin. Therefore, a renal impaired patient with a CLcr of 50 mL/min would receive a total daily dose of 75 mg/day pregabalin administered in two or three divided doses.)
For patients undergoing hemodialysis, adjust the pregabalin daily dose based on renal function. In addition to the daily dose adjustment, administer a supplemental dose immediately following every 4-hour hemodialysis treatment (see Table 2).
Table 2. Pregabalin Dosage Adjustment Based on Renal Function
Creatinine Clearance (CLcr)
(mL/min)
Total Pregabalin Daily Dose
(mg/day)*
Dose Regimen
Greater than or equal to 60
150
300
450
600
BID or TID
30 to 60
75
150
225
300
BID or TID
15 to 30
25 to 50
75
100 to 150
150
QD or BID
Less than 15
25
25 to 50
50 to 75
75
QD
Supplementary dosage following hemodialysis (mg)†
Patients on the 25 mg QD regimen: take one supplemental dose of 25 mg or 50 mg
Patients on the 25 to 50 mg QD regimen: take one supplemental dose of 50 mg or 75 mg
Patients on the 50 to 75 mg QD regimen: take one supplemental dose of 75 mg or 100 mg
Patients on the 75 mg QD regimen: take one supplemental dose of 100 mg or 150 mg
TID= Three divided doses; BID = Two divided doses; QD = Single daily dose.
*Total daily dose (mg/day) should be divided as indicated by dose regimen to provide mg/dose.
†Supplementary dose is a single additional dose.
-
3. Dosage Forms and Strengths
Pregabalin Capsules are available in 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 225 mg, and 300 mg strengths [see Description (11) and How Supplied/Storage and Handling (16)].
Pregabalin capsules, 25 mg are supplied as white, hard gelatin capsule printed with black ink “AN” on cap & “1310” on body.
Pregabalin capsules, 50 mg are supplied as white, hard gelatin capsule printed with black ink “AN” on cap & “1311” on body.
Pregabalin capsules, 75 mg are supplied as white/orange, hard gelatin capsule printed with black ink “AN” on cap & “1312” on body.
Pregabalin capsules, 100 mg are supplied as orange, hard gelatin capsule printed with black ink “AN” on cap & “1313” on body.
Pregabalin capsules, 150 mg are supplied as white, hard gelatin capsule printed with black ink “AN” on cap & “1314” on body.
Pregabalin capsules, 200 mg are supplied as light orange, hard gelatin capsule printed with black ink “AN” on cap & “1315” on body.
Pregabalin capsules, 225 mg are supplied as white/light orange, hard gelatin capsule printed with black ink “AN” on cap & “1316” on body.
Pregabalin capsules, 300 mg are supplied as white/orange, hard gelatin capsule printed with black ink “AN” on cap & “1317” on body. - 4. Contraindications
-
5. Warnings and Precautions
p-value, vs. placebo5.1 Angioedema
There have been postmarketing reports of angioedema in patients during initial and chronic treatment with pregabalin. Specific symptoms included swelling of the face, mouth (tongue, lips, and gums), and neck (throat and larynx). There were reports of life-threatening angioedema with respiratory compromise requiring emergency treatment. Discontinue pregabalin immediately in patients with these symptoms.Exercise caution when prescribing pregabalin to patients who have had a previous episode of angioedema. In addition, patients who are taking other drugs associated with angioedema (e.g., angiotensin converting enzyme inhibitors [ACE-inhibitors]) may be at increased risk of developing angioedema.
5.2 Hypersensitivity
There have been postmarketing reports of hypersensitivity in patients shortly after initiation of treatment with pregabalin. Adverse reactions included skin redness, blisters, hives, rash, dyspnea, and wheezing. Discontinue pregabalin immediately in patients with these symptoms.5.3 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including pregabalin, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Monitor patients treated with any AED for any indication for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5 to 100 years) in the clinical trials analyzed.
Table 3 shows absolute and relative risk by indication for all evaluated AEDs.
Table 3. Risk by Indication for Antiepileptic Drugs in the Pooled Analysis
Indication
Placebo Patients with Events Per 1,000 Patients
Drug Patients with Events Per 1,000 Patients
Relative Risk: Incidence of Events in Drug Patients/Incidence in Placebo Patients
Risk Difference: Additional Drug Patients with Events Per 1,000 Patients
Epilepsy
1.0
3.4
3.5
2.4
Psychiatric
5.7
8.5
1.5
2.9
Other
1.0
1.8
1.9
0.9
Total
2.4
4.3
1.8
1.9
The relative risk for suicidal thoughts or behavior was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
Anyone considering prescribing pregabalin or any other AED must balance the risk of suicidal thoughts or behavior with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
5.4 Respiratory Depression
There is evidence from case reports, human studies, and animal studies associating pregabalin with serious, life-threatening, or fatal respiratory depression when co-administered with central nervous system (CNS) depressants, including opioids, or in the setting of underlying respiratory impairment. When the decision is made to co-prescribe pregabalin with another CNS depressant, particularly an opioid, or to prescribe pregabalin to patients with underlying respiratory impairment, monitor patients for symptoms of respiratory depression and sedation, and consider initiating pregabalin at a low dose. The management of respiratory depression may include close observation, supportive measures, and reduction or withdrawal of CNS depressants (including pregabalin).
There is more limited evidence from case reports, animal studies, and human studies associating pregabalin with serious respiratory depression, without co-administered CNS depressants or without underlying respiratory impairment.
5.5 Dizziness and Somnolence
Pregabalin may cause dizziness and somnolence. Inform patients that pregabalin-related dizziness and somnolence may impair their ability to perform tasks such as driving or operating machinery [see Patient Counseling Information (17)].
In the pregabalin controlled trials in adult patients, dizziness was experienced by 30% of pregabalin-treated patients compared to 8% of placebo-treated patients; somnolence was experienced by 23% of pregabalin-treated patients compared to 8% of placebo-treated patients. Dizziness and somnolence generally began shortly after the initiation of pregabalin therapy and occurred more frequently at higher doses. Dizziness and somnolence were the adverse reactions most frequently leading to withdrawal (4% each) from controlled studies. In pregabalin-treated patients reporting these adverse reactions in short-term, controlled studies, dizziness persisted until the last dose in 30% and somnolence persisted until the last dose in 42% of patients [see Drug Interactions (7)].
Pediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
5.6 Increased Risk of Adverse Reactions with Abrupt or Rapid Discontinuation
As with all antiepileptic drugs (AEDs), withdraw pregabalin gradually to minimize the potential of increased seizure frequency in patients with seizure disorders.
Following abrupt or rapid discontinuation of pregabalin, some patients reported symptoms including insomnia, nausea, headache, anxiety, hyperhidrosis, and diarrhea.
If pregabalin is discontinued, taper the drug gradually over a minimum of 1 week rather than discontinue the drug abruptly.
5.7 Peripheral Edema
Pregabalin treatment may cause peripheral edema. In short-term trials of patients without clinically significant heart or peripheral vascular disease, there was no apparent association between peripheral edema and cardiovascular complications such as hypertension or congestive heart failure. Peripheral edema was not associated with laboratory changes suggestive of deterioration in renal or hepatic function.
In controlled clinical trials in adult patients, the incidence of peripheral edema was 6% in the pregabalin group compared with 2% in the placebo group. In controlled clinical trials, 0.5% of pregabalin patients and 0.2% placebo patients withdrew due to peripheral edema.
Higher frequencies of weight gain and peripheral edema were observed in patients taking both pregabalin and a thiazolidinedione antidiabetic agent compared to patients taking either drug alone. The majority of patients using thiazolidinedione antidiabetic agents in the overall safety database were participants in studies of pain associated with diabetic peripheral neuropathy. In this population, peripheral edema was reported in 3% (2/60) of patients who were using thiazolidinedione antidiabetic agents only, 8% (69/859) of patients who were treated with pregabalin only, and 19% (23/120) of patients who were on both pregabalin and thiazolidinedione antidiabetic agents. Similarly, weight gain was reported in 0% (0/60) of patients on thiazolidinediones only; 4% (35/859) of patients on pregabalin only; and 7.5% (9/120) of patients on both drugs.
As the thiazolidinedione class of antidiabetic drugs can cause weight gain and/or fluid retention, possibly exacerbating or leading to heart failure, exercise caution when co-administering pregabalin and these agents.
Because there are limited data on congestive heart failure patients with New York Heart Association (NYHA) Class III or IV cardiac status, exercise caution when using pregabalin in these patients.
5.8 Weight Gain
Pregabalin treatment may cause weight gain. In pregabalin controlled clinical trials in adult patients of up to 14 weeks, a gain of 7% or more over baseline weight was observed in 9% of pregabalin-treated patients and 2% of placebo-treated patients. Few patients treated with pregabalin (0.3%) withdrew from controlled trials due to weight gain. Pregabalin associated weight gain was related to dose and duration of exposure, but did not appear to be associated with baseline BMI, gender, or age. Weight gain was not limited to patients with edema [see WARNINGS AND PRECAUTIONS (5.7)].
Although weight gain was not associated with clinically important changes in blood pressure in short-term controlled studies, the long-term cardiovascular effects of pregabalin-associated weight gain are unknown.
Among diabetic patients, pregabalin-treated patients gained an average of 1.6 kg (range: -16 to 16 kg), compared to an average 0.3 kg (range: -10 to 9 kg) weight gain in placebo patients. In a cohort of 333 diabetic patients who received pregabalin for at least 2 years, the average weight gain was 5.2 kg.
While the effects of pregabalin-associated weight gain on glycemic control have not been systematically assessed, in controlled and longer-term open label clinical trials with diabetic patients, pregabalin treatment did not appear to be associated with loss of glycemic control (as measured by HbA1C).
5.9 Tumorigenic Potential
In standard preclinical in vivo lifetime carcinogenicity studies of pregabalin, an unexpectedly high incidence of hemangiosarcoma was identified in two different strains of mice [see NONCLINICAL TOXICOLOGY (13.1)]. The clinical significance of this finding is unknown. Clinical experience during pregabalin’s premarketing development provides no direct means to assess its potential for inducing tumors in humans.
In clinical studies across various patient populations, comprising 6,396 patient-years of exposure in patients greater than 12 years of age, new or worsening-preexisting tumors were reported in 57 patients. Without knowledge of the background incidence and recurrence in similar populations not treated with pregabalin, it is impossible to know whether the incidence seen in these cohorts is or is not affected by treatment.
5.10 Ophthalmological Effects
In controlled studies in adult patients, a higher proportion of patients treated with pregabalin reported blurred vision (7%) than did patients treated with placebo (2%), which resolved in a majority of cases with continued dosing. Less than 1% of patients discontinued pregabalin treatment due to vision-related events (primarily blurred vision).
Prospectively planned ophthalmologic testing, including visual acuity testing, formal visual field testing and dilated funduscopic examination, was performed in over 3,600 patients. In these patients, visual acuity was reduced in 7% of patients treated with pregabalin, and 5% of placebo-treated patients. Visual field changes were detected in 13% of pregabalin-treated, and 12% of placebo-treated patients. Funduscopic changes were observed in 2% of pregabalin-treated and 2% of placebo-treated patients.
Although the clinical significance of the ophthalmologic findings is unknown, inform patients to notify their physician if changes in vision occur. If visual disturbance persists, consider further assessment. Consider more frequent assessment for patients who are already routinely monitored for ocular conditions [see PATIENT COUNSELING INFORMATION (17)].
5.11 Creatine Kinase Elevations
Pregabalin treatment was associated with creatine kinase elevations. Mean changes in creatine kinase from baseline to the maximum value were 60 U/L for pregabalin-treated patients and 28 U/L for the placebo patients. In all controlled trials in adult patients across multiple patient populations, 1.5% of patients on pregabalin and 0.7% of placebo patients had a value of creatine kinase at least three times the upper limit of normal. Three pregabalin-treated subjects had events reported as rhabdomyolysis in premarketing clinical trials. The relationship between these myopathy events and pregabalin is not completely understood because the cases had documented factors that may have caused or contributed to these events. Instruct patients to promptly report unexplained muscle pain, tenderness, or weakness, particularly if these muscle symptoms are accompanied by malaise or fever. Discontinue treatment with pregabalin if myopathy is diagnosed or suspected or if markedly elevated creatine kinase levels occur.
5.12 Decreased Platelet Count
Pregabalin treatment was associated with a decrease in platelet count. Pregabalin-treated subjects experienced a mean maximal decrease in platelet count of 20 × 103/μL, compared to 11 × 103/μL in placebo patients. In the overall database of controlled trials in adult patients, 2% of placebo patients and 3% of pregabalin patients experienced a potentially clinically significant decrease in platelets, defined as 20% below baseline value and less than 150 × 103/μL. A single pregabalin-treated subject developed severe thrombocytopenia with a platelet count less than 20 × 103/ μL. In randomized controlled trials, pregabalin was not associated with an increase in bleeding-related adverse reactions.
5.13 PR Interval Prolongation
Pregabalin treatment was associated with PR interval prolongation. In analyses of clinical trial ECG data in adult patients, the mean PR interval increase was 3 to 6 msec at pregabalin doses greater than or equal to 300 mg/day. This mean change difference was not associated with an increased risk of PR increase greater than or equal to 25% from baseline, an increased percentage of subjects with on-treatment PR greater than 200 msec, or an increased risk of adverse reactions of second or third degree AV block.
Subgroup analyses did not identify an increased risk of PR prolongation in patients with baseline PR prolongation or in patients taking other PR prolonging medications. However, these analyses cannot be considered definitive because of the limited number of patients in these categories.
-
6. Adverse Reactions
The following serious adverse reactions are described elsewhere in the labeling:
- Angioedema [see WARNINGS AND PRECAUTIONS (5.1)]
- Hypersensitivity [see WARNINGS AND PRECAUTIONS (5.2)]
- Suicidal Behavior and Ideation [see WARNINGS AND PRECAUTIONS (5.3)]
- Respiratory Depression [see WARNINGS AND PRECAUTIONS (5.4)]
- Dizziness and Somnolence [see WARNINGS AND PRECAUTIONS (5.5)]
- Increased Risk of Adverse Reactions with Abrupt or Rapid Discontinuation [see WARNINGS AND PRECAUTIONS (5.6)]
- Peripheral Edema [see WARNINGS AND PRECAUTIONS (5.7)]
- Weight Gain [see WARNINGS AND PRECAUTIONS (5.8)]
- Tumorigenic Potential [see WARNINGS AND PRECAUTIONS (5.9)]
- Ophthalmological Effects [see WARNINGS AND PRECAUTIONS (5.10)]
- Creatine Kinase Elevations [see WARNINGS AND PRECAUTIONS (5.11)]
- Decreased Platelet Count [see WARNINGS AND PRECAUTIONS (5.12)]
- PR Interval Prolongation [see WARNINGS AND PRECAUTIONS (5.13)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In all controlled and uncontrolled trials across various patient populations during the premarketing development of pregabalin, more than 10,000 patients have received pregabalin. Approximately 5,000 patients were treated for 6 months or more, over 3,100 patients were treated for 1 year or longer, and over 1,400 patients were treated for at least 2 years.
Adverse Reactions Most Commonly Leading to Discontinuation in All Premarketing Controlled Clinical Studies
In premarketing controlled trials of all adult populations combined, 14% of patients treated with pregabalin and 7% of patients treated with placebo discontinued prematurely due to adverse reactions. In the pregabalin treatment group, the adverse reactions most frequently leading to discontinuation were dizziness (4%) and somnolence (4%). In the placebo group, 1% of patients withdrew due to dizziness and less than 1% withdrew due to somnolence. Other adverse reactions that led to discontinuation from controlled trials more frequently in the pregabalin group compared to the placebo group were ataxia, confusion, asthenia, thinking abnormal, blurred vision, incoordination, and peripheral edema (1% each).
Most Common Adverse Reactions in All Controlled Clinical Studies in Adults
In premarketing controlled trials of all adult patient populations combined (including DPN, PHN, and adult patients with partial-onset seizures), dizziness, somnolence, dry mouth, edema, blurred vision, weight gain, and "thinking abnormal" (primarily difficulty with concentration/attention) were more commonly reported by subjects treated with pregabalin than by subjects treated with placebo (greater than or equal to 5% and twice the rate of that seen in placebo).
Controlled Studies with Neuropathic Pain Associated with Diabetic Peripheral Neuropathy
Adverse Reactions Leading to Discontinuation
In clinical trials in adults with neuropathic pain associated with diabetic peripheral neuropathy, 9% of patients treated with pregabalin and 4% of patients treated with placebo discontinued prematurely due to adverse reactions. In the pregabalin treatment group, the most common reasons for discontinuation due to adverse reactions were dizziness (3%) and somnolence (2%). In comparison, less than 1% of placebo patients withdrew due to dizziness and somnolence. Other reasons for discontinuation from the trials, occurring with greater frequency in the pregabalin group than in the placebo group, were asthenia, confusion, and peripheral edema. Each of these events led to withdrawal in approximately 1% of patients.
Most Common Adverse Reactions
Table 4 lists all adverse reactions, regardless of causality, occurring in greater than or equal to 1% of patients with neuropathic pain associated with diabetic neuropathy in the combined pregabalin group for which the incidence was greater in this combined pregabalin group than in the placebo group. A majority of pregabalin-treated patients in clinical studies had adverse reactions with a maximum intensity of "mild" or "moderate”.
Table 4. Adverse Reaction Incidence in Controlled Trials in Neuropathic Pain Associated with Diabetic Peripheral Neuropathy
Body system Preferred term 75 mg/day [N=77]
%150 mg/day [N=212]
%300 mg/day [N=321]
%600 mg/day [N=369]
%All PGB* [N=979]
%Placebo [N=459]
%Body as a whole Asthenia 4 2 4 7 5 2 Accidental injury 5 2 2 6 4 3 Back pain 0 2 1 2 2 0 Chest pain 4 1 1 2 2 1 Face edema 0 1 1 2 1 0 Digestive system Dry mouth 3 2 5 7 5 1 Constipation 0 2 4 6 4 2 Flatulence 3 0 2 3 2 1 Metabolic and nutritional disorders Peripheral edema 4 6 9 12 9 2 Weight gain 0 4 4 6 4 0 Edema 0 2 4 2 2 0 Hypoglycemia 1 3 2 1 2 1 Nervous system Dizziness 8 9 23 29 21 5 Somnolence 4 6 13 16 12 3 Neuropathy 9 2 2 5 4 3 Ataxia 6 1 2 4 3 1 Vertigo 1 2 2 4 3 1 Confusion 0 1 2 3 2 1 Euphoria 0 0 3 2 2 0 Incoordination 1 0 2 2 2 0 Thinking abnormal† 1 0 1 3 2 0 Tremor 1 1 1 2 1 0 Abnormal gait 1 0 1 3 1 0 Amnesia 3 1 0 2 1 0 Nervousness 0 1 1 1 1 0 Respiratory system Dyspnea 3 0 2 2 2 1 Special senses Blurry vision‡ 3 1 3 6 4 2 Abnormal vision 1 0 1 1 1 0 * PGB: pregabalin
†Thinking abnormal primarily consists of events related to difficulty with concentration/attention but also includes events related to cognition and language problems and slowed thinking.
‡Investigator term; summary level term is amblyopiaControlled Studies in Postherpetic Neuralgia
Adverse Reactions Leading to Discontinuation
In clinical trials in adults with postherpetic neuralgia, 14% of patients treated with pregabalin and 7% of patients treated with placebo discontinued prematurely due to adverse reactions. In the pregabalin treatment group, the most common reasons for discontinuation due to adverse reactions were dizziness (4%) and somnolence (3%). In comparison, less than 1% of placebo patients withdrew due to dizziness and somnolence. Other reasons for discontinuation from the trials, occurring in greater frequency in the pregabalin group than in the placebo group, were confusion (2%), as well as peripheral edema, asthenia, ataxia, and abnormal gait (1% each).
Most Common Adverse Reactions
Table 5 lists all adverse reactions, regardless of causality, occurring in greater than or equal to 1% of patients with neuropathic pain associated with postherpetic neuralgia in the combined pregabalin group for which the incidence was greater in this combined pregabalin group than in the placebo group. In addition, an event is included, even if the incidence in the all pregabalin group is not greater than in the placebo group, if the incidence of the event in the 600 mg/day group is more than twice that in the placebo group. A majority of pregabalin-treated patients in clinical studies had adverse reactions with a maximum intensity of "mild" or "moderate". Overall, 12.4% of all pregabalin-treated patients and 9.0% of all placebo-treated patients had at least one severe event while 8% of pregabalin-treated patients and 4.3% of placebo-treated patients had at least one severe treatment-related adverse event.
Table 5. Adverse Reaction Incidence in Controlled Trials in Neuropathic Pain Associated with Postherpetic Neuralgia Body system Preferred term
Body system Preferred term 75 mg/d [N=84]
%150 mg/d [N=302]
%300 mg/d [N=312]
%600 mg/d [N=154]
%All PGB* [N=852]
%Placebo [N=398]
%Body as a whole Infection 14 8 6 3 7 4 Headache 5 9 5 8 7 5 Pain 5 4 5 5 5 4 Accidental injury 4 3 3 5 3 2 Flu syndrome 1 2 2 1 2 1 Face edema 0 2 1 3 2 1 Digestive system Dry mouth 7 7 6 15 8 3 Constipation 4 5 5 5 5 2 Flatulence 2 1 2 3 2 1 Vomiting 1 1 3 3 2 1 Metabolic and nutritional disorders Peripheral edema 0 8 16 16 12 4 Weight gain 1 2 5 7 4 0 Edema 0 1 2 6 2 1 Musculoskeletal system Myasthenia 1 1 1 1 1 0 Nervous system Dizziness 11 18 31 37 26 9 Somnolence 8 12 18 25 16 5 Ataxia 1 2 5 9 5 1 Abnormal gait 0 2 4 8 4 1 Confusion 1 2 3 7 3 0 Thinking abnormal† 0 2 1 6 2 2 Incoordination 2 2 1 3 2 0 Amnesia 0 1 1 4 2 0 Speech disorder 0 0 1 3 1 0 Respiratory system Bronchitis 0 1 1 3 1 1 Special senses Blurry vision‡ 1 5 5 9 5 3 Diplopia 0 2 2 4 2 0 Abnormal vision 0 1 2 5 2 0 Eye Disorder 0 1 1 2 1 0 Urogenital System Urinary Incontinence 0 1 1 2 1 0 * PGB: pregabalin
†Thinking abnormal primarily consists of events related to difficulty with concentration/attention but also includes events related to cognition and language problems and slowed thinking.
‡ Investigator term; summary level term is amblyopiaAdverse Reactions Leading to DiscontinuationControlled Studies of Adjunctive Therapy for Partial-Onset Seizures in Adult Patients
Approximately 15% of patients receiving pregabalin and 6% of patients receiving placebo in trials of adjunctive therapy for partial-onset seizures discontinued prematurely due to adverse reactions. In the pregabalin treatment group, the adverse reactions most frequently leading to discontinuation were dizziness (6%), ataxia (4%), and somnolence (3%). In comparison, less than 1% of patients in the placebo group withdrew due to each of these events. Other adverse reactions that led to discontinuation of at least 1% of patients in the pregabalin group and at least twice as frequently compared to the placebo group were asthenia, diplopia, blurred vision, thinking abnormal, nausea, tremor, vertigo, headache, and confusion (which each led to withdrawal in 2% or less of patients).
Most Common Adverse Reactions
Table 6 lists all dose-related adverse reactions occurring in at least 2% of all pregabalin-treated patients. Dose-relatedness was defined as the incidence of the adverse event in the 600 mg/day group was at least 2% greater than the rate in both the placebo and 150 mg/day groups. In these studies, 758 patients received pregabalin and 294 patients received placebo for up to 12 weeks. A majority of pregabalin-treated patients in clinical studies had adverse reactions with a maximum intensity of "mild" or "moderate”.
Table 6. Dose-related Adverse Reaction Incidence in Controlled Trials of Adjunctive Therapy for Partial-Onset Seizures in Adult Patients
Body System Preferred Term 150 mg/d
[N = 185]
%300 mg/d
[N = 90]
%600 mg/d
[N = 395]
%All PGB*
[N = 670]†
%Placebo
[N = 294]
%Body as a Whole Accidental Injury 7 11 10 9 5 Pain 3 2 5 4 3 Digestive System Increased Appetite 2 3 6 5 1 Dry Mouth 1 2 6 4 1 Constipation 1 1 7 4 2 Metabolic and Nutritional Disorders Weight Gain 5 7 16 12 1 Peripheral Edema 3 3 6 5 2 Nervous System Dizziness 18 31 38 32 11 Somnolence 11 18 28 22 11 Ataxia 6 10 20 15 4 Tremor 3 7 11 8 4 Thinking Abnormal‡ 4 8 9 8 2 Amnesia 3 2 6 5 2 Speech Disorder 1 2 7 5 1 Incoordination 1 3 6 4 1 Abnormal Gait 1 3 5 4 0 Twitching 0 4 5 4 1 Confusion 1 2 5 4 2 Myoclonus 1 0 4 2 0 Special Senses Blurred Vision§ 5 8 12 10 4 Diplopia 5 7 12 9 4 Abnormal Vision 3 1 5 4 1 * PGB: pregabalin
† Excludes patients who received the 50 mg dose in Study E1.
‡ Thinking abnormal primarily consists of events related to difficulty with concentration/attention but also includes events related to cognition and language problems and slowed thinking.
§ Investigator term; summary level term is amblyopiaControlled Studies with Fibromyalgia
Adverse Reactions Leading to Discontinuation
In clinical trials of patients with fibromyalgia, 19% of patients treated with pregabalin (150 to 600 mg/day) and 10% of patients treated with placebo discontinued prematurely due to adverse reactions. In the pregabalin treatment group, the most common reasons for discontinuation due to adverse reactions were dizziness (6%) and somnolence (3%). In comparison, less than 1% of placebo-treated patients withdrew due to dizziness and somnolence. Other reasons for discontinuation from the trials, occurring with greater frequency in the pregabalin treatment group than in the placebo treatment group, were fatigue, headache, balance disorder, and weight increased. Each of these adverse reactions led to withdrawal in approximately 1% of patients.
Most Common Adverse Reactions
Table 9 lists all adverse reactions, regardless of causality, occurring in greater than or equal to 2% of patients with fibromyalgia in the ‘all pregabalin’ treatment group for which the incidence was greater than in the placebo treatment group. A majority of pregabalin-treated patients in clinical studies experienced adverse reactions with a maximum intensity of "mild" or "moderate".
Table 9. Adverse Reaction Incidence in Controlled Trials in Fibromyalgia
System Organ Class Preferred term 150 mg/d [N=132]
%300 mg/d [N=502]
%450 mg/d [N=505]
%600 mg/d [N=378]
%All PGB* [N=1517]
%Placebo [N=505]
%Ear and Labyrinth Disorders Vertigo 2 2 2 1 2 0 Eye Disorders Vision blurred 8 7 7 12 8 1 Gastrointestinal Disorders Dry mouth 7 6 9 9 8 2 Constipation 4 4 7 10 7 2 Vomiting 2 3 3 2 3 2 Flatulence 1 1 2 2 2 1 Abdominal distension 2 2 2 2 2 1 General Disorders and Administrative Site Conditions Fatigue 5 7 6 8 7 4 Edema peripheral 5 5 6 9 6 2 Chest pain 2 1 1 2 2 1 Feeling abnormal 1 3 2 2 2 0 Edema 1 2 1 2 2 1 Feeling drunk 1 2 1 2 2 0 Infections and Infestations Sinusitis 4 5 7 5 5 4 Investigations Weight increased 8 10 10 14 11 2 Metabolism and Nutrition Disorders Increased appetite 4 3 5 7 5 1 Fluid retention 2 3 3 2 2 1 Musculoskeletal and Connective Tissue Disorders Arthralgia 4 3 3 6 4 2 Muscle spasms 2 4 4 4 4 2 Back pain 2 3 4 3 3 3 Nervous System Disorders Dizziness 23 31 43 45 38 9 Somnolence 13 18 22 22 20 4 Headache 11 12 14 10 12 12 Disturbance in attention 4 4 6 6 5 1 Balance disorder 2 3 6 9 5 0 Memory impairment 1 3 4 4 3 0 Coordination abnormal 2 1 2 2 2 1 Hypoesthesia 2 2 3 2 2 1 Lethargy 2 2 1 2 2 0 Tremor 0 1 3 2 2 0 Psychiatric Disorders Euphoric Mood 2 5 6 7 6 1 Confusional state 0 2 3 4 3 0 Anxiety 2 2 2 2 2 1 Disorientation 1 0 2 1 2 0 Depression 2 2 2 2 2 2 Respiratory, Thoracic and Mediastinal Disorders Pharyngolaryngeal pain 2 1 3 3 2 2 * PGB: pregabalin
Adverse Reactions Leading to DiscontinuationControlled Studies in Neuropathic Pain Associated with Spinal Cord Injury
In clinical trials of adults with neuropathic pain associated with spinal cord injury, 13% of patients treated with pregabalin and 10% of patients treated with placebo discontinued prematurely due to adverse reactions. In the pregabalin treatment group, the most common reasons for discontinuation due to adverse reactions were somnolence (3%) and edema (2%). In comparison, none of the placebo-treated patients withdrew due to somnolence and edema. Other reasons for discontinuation from the trials, occurring with greater frequency in the pregabalin treatment group than in the placebo treatment group, were fatigue and balance disorder. Each of these adverse reactions led to withdrawal in less than 2% of patients.
Most Common Adverse Reactions
Table 10 lists all adverse reactions, regardless of causality, occurring in greater than or equal to 2% of patients for which the incidence was greater than in the placebo treatment group with neuropathic pain associated with spinal cord injury in the controlled trials. A majority of pregabalin-treated patients in clinical studies experienced adverse reactions with a maximum intensity of "mild" or "moderate".
Table 10. Adverse Reaction Incidence in Controlled Trials in Neuropathic Pain Associated with Spinal Cord Injury
System Organ Class
Preferred termPGB* (N=182) Placebo (N=174) % % Ear and labryrinth disorders Vertigo 2.7 1.1 Eye disorders Vision blurred 6.6 1.1 Gastrointestinal disorders Dry mouth 11.0 2.9 Constipation 8.2 5.7 Nausea 4.9 4.0 Vomiting 2.7 1.1 General disorders and administration site conditions Fatigue 11.0 4.0 Edema peripheral 10.4 5.2 Edema 8.2 1.1 Pain 3.3 1.1 Infections and infestations Nasopharyngitis 8.2 4.6 Investigations Weight increased 3.3 1.1 Blood creatine phosphokinase increased 2.7 0 Musculoskeletal and connective tissue disorders Muscular weakness 4.9 1.7 Pain in extremity 3.3 2.3 Neck pain 2.7 1.1 Back pain 2.2 1.7 Joint swelling 2.2 0 Nervous system disorders Somnolence 35.7 11.5 Dizziness 20.9 6.9 Disturbance in attention 3.8 0 Memory impairment 3.3 1.1 Paresthesia 2.2 0.6 Psychiatric disorders Insomnia 3.8 2.9 Euphoric mood 2.2 0.6 Renal and urinary disorders Urinary incontinence 2.7 1.1 Skin and subcutaneous tissue disorders Decubitus ulcer 2.7 1.1 Vascular disorders Hypertension 2.2 1.1 Hypotension 2.2 0 Other Adverse Reactions Observed During the Clinical Studies of Pregabalin
Following is a list of treatment-emergent adverse reactions reported by patients treated with pregabalin during all clinical trials. The listing does not include those events already listed in the previous tables or elsewhere in labeling, those events for which a drug cause was remote, those events which were so general as to be uninformative, and those events reported only once which did not have a substantial probability of being acutely life-threatening.
Events are categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse reactions are those occurring on one or more occasions in at least 1/100 patients; infrequent adverse reactions are those occurring in 1/100 to 1/1,000 patients; rare reactions are those occurring in fewer than 1/1,000 patients. Events of major clinical importance are described in the Warnings and Precautions section (5).
Body as a Whole – Frequent: Abdominal pain, Allergic reaction, Fever, Infrequent: Abscess, Cellulitis, Chills, Malaise, Neck rigidity, Overdose, Pelvic pain, Photosensitivity reaction, Rare: Anaphylactoid reaction, Ascites, Granuloma, Hangover effect, Intentional Injury, Retroperitoneal Fibrosis, Shock
Cardiovascular System – Infrequent: Deep thrombophlebitis, Heart failure, Hypotension, Postural hypotension, Retinal vascular disorder, Syncope; Rare: ST Depressed, Ventricular Fibrillation
Digestive System – Frequent: Gastroenteritis, Increased appetite; Infrequent: Cholecystitis, Cholelithiasis, Colitis, Dysphagia, Esophagitis, Gastritis, Gastrointestinal hemorrhage, Melena, Mouth ulceration, Pancreatitis, Rectal hemorrhage, Tongue edema; Rare: Aphthous stomatitis, Esophageal Ulcer, Periodontal abscess
Hemic and Lymphatic System – Frequent: Ecchymosis; Infrequent: Anemia, Eosinophilia, Hypochromic anemia, Leukocytosis, Leukopenia, Lymphadenopathy, Thrombocytopenia; Rare: Myelofibrosis, Polycythemia, Prothrombin decreased, Purpura, Thrombocythemia, Alanine aminotransferase increased, Aspartate aminotransferase increased
Metabolic and Nutritional Disorders – Rare: Glucose Tolerance Decreased, Urate Crystalluria
Musculoskeletal System – Frequent: Arthralgia, Leg cramps, Myalgia, Myasthenia; Infrequent: Arthrosis; Rare: Chondrodystrophy, Generalized Spasm
Nervous System – Frequent: Anxiety, Depersonalization, Hypertonia, Hypoesthesia, Libido decreased, Nystagmus, Paresthesia, Sedation, Stupor, Twitching; Infrequent: Abnormal dreams, Agitation, Apathy, Aphasia, Circumoral paresthesia, Dysarthria, Hallucinations, Hostility, Hyperalgesia, Hyperesthesia, Hyperkinesia, Hypokinesia, Hypotonia, Libido increased, Myoclonus, Neuralgia; Rare: Addiction, Cerebellar syndrome, Cogwheel rigidity, Coma, Delirium, Delusions, Dysautonomia, Dyskinesia, Dystonia, Encephalopathy, Extrapyramidal syndrome, Guillain-Barré syndrome, Hypalgesia, Intracranial hypertension, Manic reaction, Paranoid reaction, Peripheral neuritis, Personality disorder, Psychotic depression, Schizophrenic reaction, Sleep disorder, Torticollis, Trismus
Respiratory System – Rare: Apnea, Atelectasis, Bronchiolitis, Hiccup, Laryngismus, Lung edema, Lung fibrosis, Yawn
Skin and Appendages – Frequent: Pruritus, Infrequent: Alopecia, Dry skin, Eczema, Hirsutism, Skin ulcer, Urticaria, Vesiculobullous rash; Rare: Angioedema, Exfoliative dermatitis, Lichenoid dermatitis, Melanosis, Nail Disorder, Petechial rash, Purpuric rash, Pustular rash, Skin atrophy, Skin necrosis, Skin nodule, Stevens-Johnson syndrome, Subcutaneous nodule
Special senses – Frequent: Conjunctivitis, Diplopia, Otitis media, Tinnitus; Infrequent: Abnormality of accommodation, Blepharitis, Dry eyes, Eye hemorrhage, Hyperacusis, Photophobia, Retinal edema, Taste loss, Taste perversion; Rare: Anisocoria, Blindness, Corneal ulcer, Exophthalmos, Extraocular palsy, Iritis, Keratitis, Keratoconjunctivitis, Miosis, Mydriasis, Night blindness, Ophthalmoplegia, Optic atrophy, Papilledema, Parosmia, Ptosis, Uveitis
Urogenital System – Frequent: Anorgasmia, Impotence, Urinary frequency, Urinary incontinence; Infrequent: Abnormal ejaculation, Albuminuria, Amenorrhea, Dysmenorrhea, Dysuria, Hematuria, Kidney calculus, Leukorrhea, Menorrhagia, Metrorrhagia, Nephritis, Oliguria, Urinary retention, Urine abnormality; Rare: Acute kidney failure, Balanitis, Bladder Neoplasm, Cervicitis, Dyspareunia, Epididymitis, Female lactation, Glomerulitis, Ovarian disorder, Pyelonephritis
Comparison of Gender and Race
The overall adverse event profile of pregabalin was similar between women and men. There are insufficient data to support a statement regarding the distribution of adverse experience reports by race.
Pediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of pregabalin. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Nervous System Disorders – Headache
Gastrointestinal Disorders – Nausea, Diarrhea
Reproductive System and Breast Disorders – Gynecomastia, Breast Enlargement
Skin and subcutaneous tissue disorders – Bullous pemphigoid
There are postmarketing reports of life-threatening or fatal respiratory depression in patients taking pregabalin with opioids or other CNS depressants, or in the setting of underlying respiratory impairment.
In addition, there are postmarketing reports of events related to reduced lower gastrointestinal tract function (e.g., intestinal obstruction, paralytic ileus, constipation) when pregabalin was co-administered with medications that have the potential to produce constipation, such as opioid analgesics.
-
7. Drug Interactions
Since pregabalin is predominantly excreted unchanged in the urine, undergoes negligible metabolism in humans (less than 2% of a dose recovered in urine as metabolites), and does not bind to plasma proteins, its pharmacokinetics are unlikely to be affected by other agents through metabolic interactions or protein binding displacement. In vitro and in vivo studies showed that pregabalin is unlikely to be involved in significant pharmacokinetic drug interactions. Specifically, there are no pharmacokinetic interactions between pregabalin and the following antiepileptic drugs: carbamazepine, valproic acid, lamotrigine, phenytoin, phenobarbital, and topiramate. Important pharmacokinetic interactions would also not be expected to occur between pregabalin and commonly used antiepileptic drugs [see Clinical Pharmacology (12)].
Pharmacodynamics
Multiple oral doses of pregabalin were co-administered with oxycodone, lorazepam, or ethanol. Although no pharmacokinetic interactions were seen, additive effects on cognitive and gross motor functioning were seen when pregabalin was co-administered with these drugs. No clinically important effects on respiration were seen.
-
8. Use in Specific Populations
8.1 Pregnancy
Pregnancy Exposure RegistryThere is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to pregabalin during pregnancy. To provide information regarding the effects of in utero exposure to pregabalin, physicians are advised to recommend that pregnant patients taking pregabalin enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry. This can be done by calling the toll free number 1-888-233-2334, and must be done by patients themselves. Information on the registry can also be found at the website http://www.aedpregnancyregistry.org/.
Risk Summary
There are no adequate and well-controlled studies with pregabalin in pregnant women.
However, in animal reproduction studies, increased incidences of fetal structural abnormalities and other manifestations of developmental toxicity, including skeletal malformations, retarded ossification, and decreased fetal body weight were observed in the offspring of rats and rabbits given pregabalin orally during organogenesis, at doses that produced plasma pregabalin exposures (AUC) greater than or equal to 16 times human exposure at the maximum recommended dose (MRD) of 600 mg/day [see Data]. In an animal development study, lethality, growth retardation, and nervous and reproductive system functional impairment were observed in the offspring of rats given pregabalin during gestation and lactation. The no-effect dose for developmental toxicity was approximately twice the human exposure at MRD. The background risk of major birth defects and miscarriage for the indicated populations are unknown. However, the background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies. Advise pregnant women of the potential risk to a fetus.
Data
Animal Data
When pregnant rats were given pregabalin (500, 1,250, or 2,500 mg/kg) orally throughout the period of organogenesis, incidences of specific skull alterations attributed to abnormally advanced ossification (premature fusion of the jugal and nasal sutures) were increased at greater than or equal to 1,250 mg/kg, and incidences of skeletal variations and retarded ossification were increased at all doses. Fetal body weights were decreased at the highest dose. The low dose in this study was associated with a plasma exposure (AUC) approximately 17 times human exposure at the MRD of 600 mg/day. A no-effect dose for rat embryo-fetal developmental toxicity was not established.
When pregnant rabbits were given pregabalin (250, 500, or 1,250 mg/kg) orally throughout the period of organogenesis, decreased fetal body weight and increased incidences of skeletal malformations, visceral variations, and retarded ossification were observed at the highest dose. The no-effect dose for developmental toxicity in rabbits (500 mg/kg) was associated with a plasma exposure approximately 16 times human exposure at the MRD.
In a study in which female rats were dosed with pregabalin (50, 100, 250, 1,250, or 2,500 mg/kg) throughout gestation and lactation, offspring growth was reduced at greater than or equal to 100 mg/kg and offspring survival was decreased at greater than or equal to 250 mg/kg. The effect on offspring survival was pronounced at doses greater than or equal to 1,250 mg/kg, with 100% mortality in high-dose litters. When offspring were tested as adults, neurobehavioral abnormalities (decreased auditory startle responding) were observed at greater than or equal to 250 mg/kg and reproductive impairment (decreased fertility and litter size) was seen at 1,250 mg/kg. The no-effect dose for pre- and postnatal developmental toxicity in rats (50 mg/kg) produced a plasma exposure approximately 2 times human exposure at the MRD.
In the prenatal-postnatal study in rats, pregabalin prolonged gestation and induced dystocia at exposures greater than or equal to 50 times the mean human exposure (AUC(0 to 24) of 123 mcg•hr/mL) at the MRD.
8.2 Lactation
Risk SummarySmall amounts of pregabalin have been detected in the milk of lactating women. A pharmacokinetic study in lactating women detected pregabalin in breast milk at average steady-state concentrations approximately 76% of those in maternal plasma. The estimated average daily infant dose of pregabalin from breast milk (assuming mean milk consumption of 150 mL/kg/day) was 0.31 mg/kg/day, which on a mg/kg basis would be approximately 7% of the maternal dose [see Data]. The study did not evaluate the effects of pregabalin on milk production or the effects of pregabalin on the breastfed infant.
Based on animal studies, there is a potential risk of tumorigenicity with pregabalin exposure via breast milk to the breastfed infant [see Nonclinical Toxicology (13.1)]. Available clinical study data in patients greater than 12 years of age do not provide a clear conclusion about the potential risk of tumorigenicity with pregabalin [see Warnings and Precautions (5.9)]. Because of the potential risk of tumorigenicity, breastfeeding is not recommended during treatment with pregabalin.
Data
A pharmacokinetic study in ten lactating women, who were at least 12 weeks postpartum, evaluated the concentrations of pregabalin in plasma and breast milk. Pregabalin 150 mg oral capsule was given every 12 hours (300 mg daily dose) for a total of four doses. Pregabalin was detected in breast milk at average steady-state concentrations approximately 76% of those in maternal plasma. The estimated average daily infant dose of pregabalin from breast milk (assuming mean milk consumption of 150 mL/kg/day) was 0.31 mg/kg/day, which on a mg/kg basis would be approximately 7% of the maternal dose. The study did not evaluate the effects of pregabalin on milk production. Infants did not receive breast milk obtained during the dosing period, therefore, the effects of pregabalin on the breast fed infant were not evaluated.
8.3 Females and Males of Reproductive Potential
InfertilityMale
Effects on Spermatogenesis
In a randomized, double-blind, placebo-controlled non-inferiority study to assess the effect of pregabalin on sperm characteristics, healthy male subjects received pregabalin at a daily dose up to 600 mg (n=111) or placebo (n=109) for 13 weeks (one complete sperm cycle) followed by a 13-week washout period (off-drug). A total of 65 subjects in the pregabalin group (59%) and 62 subjects in the placebo group (57%) were included in the per protocol (PP) population. These subjects took study drug for at least 8 weeks, had appropriate timing of semen collections and did not have any significant protocol violations. Among these subjects, approximately 9% of the pregabalin group (6/65) vs. 3% in the placebo group (2/62) had greater than or equal to 50% reduction in mean sperm concentrations from baseline at Week 26 (the primary endpoint). The difference between pregabalin and placebo was within the pre-specified non-inferiority margin of 20%. There were no adverse effects of pregabalin on sperm morphology, sperm motility, serum FSH or serum testosterone levels as compared to placebo. In subjects in the PP population with greater than or equal to 50% reduction in sperm concentration from baseline, sperm concentrations were no longer reduced by greater than or equal to 50% in any affected subject after an additional 3 months off-drug. In one subject, however, subsequent semen analyses demonstrated reductions from baseline of greater than or equal to 50% at 9 and 12 months off-drug. The clinical relevance of these data is unknown.
In the animal fertility study with pregabalin in male rats, adverse reproductive and developmental effects were observed [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Neuropathic Pain Associated with Diabetic Peripheral Neuropathy, Postherpetic Neuralgia, and Neuropathic Pain Associated with Spinal Cord InjurySafety and effectiveness in pediatric patients have not been established.
Fibromyalgia
Safety and effectiveness in pediatric patients have not been established.
A 15-week, placebo-controlled trial was conducted with 107 pediatric patients with fibromyalgia, ages 12 through 17 years, at pregabalin total daily doses of 75 to 450 mg per day. The primary efficacy endpoint of change from baseline to Week 15 in mean pain intensity (derived from an 11-point numeric rating scale) showed numerically greater improvement for the pregabalin-treated patients compared to placebo-treated patients, but did not reach statistical significance. The most frequently observed adverse reactions in the clinical trial included dizziness, nausea, headache, weight increased, and fatigue. The overall safety profile in adolescents was similar to that observed in adults with fibromyalgia.
Adjunctive Therapy for Partial-Onset Seizures
Safety and effectiveness in pediatric patients below the age of 1 month have not been established.
Juvenile Animal Data
In studies in which pregabalin (50 to 500 mg/kg) was orally administered to young rats from early in the postnatal period (Postnatal Day 7) through sexual maturity, neurobehavioral abnormalities (deficits in learning and memory, altered locomotor activity, decreased auditory startle responding and habituation) and reproductive impairment (delayed sexual maturation and decreased fertility in males and females) were observed at doses greater than or equal to 50 mg/kg. The neurobehavioral changes of acoustic startle persisted at greater than or equal to 250 mg/kg and locomotor activity and water maze performance at greater than or equal to 500 mg/kg in animals tested after cessation of dosing and, thus, were considered to represent long-term effects. The low effect dose for developmental neurotoxicity and reproductive impairment in juvenile rats (50 mg/kg) was associated with a plasma pregabalin exposure (AUC) approximately equal to human exposure at the maximum recommended dose of 600 mg/day. A no-effect dose was not established.
Information describing a clinical study in which efficacy was not demonstrated in patients is approved for Pfizer Inc.’s Lyrica® (pregabalin) products. Additional pediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
8.5 Geriatric Use
In controlled clinical studies of pregabalin in neuropathic pain associated with diabetic peripheral neuropathy, 246 patients were 65 to 74 years of age, and 73 patients were 75 years of age or older.In controlled clinical studies of pregabalin in neuropathic pain associated with postherpetic neuralgia, 282 patients were 65 to 74 years of age, and 379 patients were 75 years of age or older.
In controlled clinical studies of pregabalin in epilepsy, there were only 10 patients 65 to 74 years of age, and 2 patients who were 75 years of age or older.
No overall differences in safety and efficacy were observed between these patients and younger patients.
In controlled clinical studies of pregabalin in fibromyalgia, 106 patients were 65 years of age or older. Although the adverse reaction profile was similar between the two age groups, the following neurological adverse reactions were more frequent in patients 65 years of age or older: dizziness, vision blurred, balance disorder, tremor, confusional state, coordination abnormal, and lethargy.
Pregabalin is known to be substantially excreted by the kidney, and the risk of toxic reactions to pregabalin may be greater in patients with impaired renal function. Because pregabalin is eliminated primarily by renal excretion, adjust the dose for elderly patients with renal impairment [see Dosage and Administration (2.7)].
8.6 Renal Impairment
Pregabalin is eliminated primarily by renal excretion and dose adjustment is recommended for adult patients with renal impairment [see Dosage and Administration (2.7) and Clinical Pharmacology (12.3)]. The use of pregabalin in pediatric patients with compromised renal function has not been studied. -
9. Drug Abuse and Dependence
9.1 Controlled Substance
Pregabalin is a Schedule V controlled substance.
Pregabalin is not known to be active at receptor sites associated with drugs of abuse. As with any CNS active drug, carefully evaluate patients for history of drug abuse and observe them for signs of pregabalin misuse or abuse (e.g., development of tolerance, dose escalation, drug-seeking behavior).
9.2 Abuse
In a study of recreational users (N=15) of sedative/hypnotic drugs, including alcohol, pregabalin (450 mg, single dose) received subjective ratings of "good drug effect," "high" and "liking" to a degree that was similar to diazepam (30 mg, single dose). In controlled clinical studies in over 5,500 patients, 4% of pregabalin-treated patients and 1 % of placebo-treated patients overall reported euphoria as an adverse reaction, though in some patient populations studied, this reporting rate was higher and ranged from 1% to 12%.
9.3 Dependence
In clinical studies, following abrupt or rapid discontinuation of pregabalin, some patients reported symptoms including insomnia, nausea, headache or diarrhea [see Warnings and Precautions (5.6)], consistent with physical dependence. In the postmarketing experience, in addition to these reported symptoms there have also been reported cases of anxiety and hyperhidrosis.
-
10. Overdosage
Signs, Symptoms and Laboratory Findings of Acute Overdosage in Humans
In the postmarketing experience, the most commonly reported adverse events observed with pregabalin when taken in overdose include reduced consciousness, depression/anxiety, confusional state, agitation, and restlessness. Seizures and heart block have also been reported. Deaths have been reported in the setting of lone pregabalin overdose and in combination with other CNS depressants.
Treatment or Management of Overdose
There is no specific antidote for overdose with pregabalin. If indicated, elimination of unabsorbed drug may be attempted by emesis or gastric lavage; observe usual precautions to maintain the airway. General supportive care of the patient is indicated including monitoring of vital signs and observation of the clinical status of the patient. Contact a Certified Poison Control Center for up-to-date information on the management of overdose with pregabalin.
Pregabalin can be removed by hemodialysis. Standard hemodialysis procedures result in significant clearance of pregabalin (approximately 50% in 4 hours).
-
11. Description
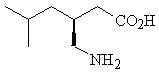
Pregabalin is described chemically as (S)-3-(aminomethyl)-5-methylhexanoic acid. The molecular formula is C8H17NO2 and the molecular weight is 159.23. The chemical structure of pregabalin is:
Pregabalin is a white to off-white, crystalline solid with a pKa1 of 4.2 and a pKa2 of 10.6. It is freely soluble in water and both basic and acidic aqueous solutions. The log of the partition coefficient (n-octanol/0.05M phosphate buffer) at pH 7.4 is – 1.35.
Pregabalin Capsules are administered orally and are supplied as imprinted hard-shell capsules containing 25 mg, 50 mg, 75 mg, 100 mg, 150 mg, 200 mg, 225 mg, and 300 mg of pregabalin, along with pregelatinized starch and talc. The capsule shells contain gelatin and titanium dioxide. In addition, the orange capsule shells contain red iron oxide and white capsule shells contain sodium lauryl sulfate.
Each capsule shell is imprinted with black pharmaceutical ink which contains: butyl alcohol, dehydrated alcohol, ferrosoferric oxide, isopropyl alcohol, propylene glycol, potassium hydroxide, purified water, strong ammonia solution and shellac.
-
12. Clinical Pharmacology
12.1 Mechanism of Action
Pregabalin binds with high affinity to the alpha2-delta site (an auxiliary subunit of voltage-gated calcium channels) in central nervous system tissues. Although the mechanism of action of pregabalin has not been fully elucidated, results with genetically modified mice and with compounds structurally related to pregabalin (such as gabapentin) suggest that binding to the alpha2-delta subunit may be involved in pregabalin's anti-nociceptive and antiseizure effects in animals. In animal models of nerve damage, pregabalin has been shown to reduce calcium-dependent release of pro-nociceptive neurotransmitters in the spinal cord, possibly by disrupting alpha2-delta containing-calcium channel trafficking and/or reducing calcium currents. Evidence from other animal models of nerve damage and persistent pain suggest the anti-nociceptive activities of pregabalin may also be mediated through interactions with descending noradrenergic and serotonergic pathways originating from the brainstem that modulate pain transmission in the spinal cord.
While pregabalin is a structural derivative of the inhibitory neurotransmitter gamma-aminobutyric acid (GABA), it does not bind directly to GABAA, GABAB, or benzodiazepine receptors, does not augment GABAA responses in cultured neurons, does not alter rat brain GABA concentration or have acute effects on GABA uptake or degradation. However, in cultured neurons prolonged application of pregabalin increases the density of GABA transporter protein and increases the rate of functional GABA transport. Pregabalin does not block sodium channels, is not active at opiate receptors, and does not alter cyclooxygenase enzyme activity. It is inactive at serotonin and dopamine receptors and does not inhibit dopamine, serotonin, or noradrenaline reuptake.
12.3 Pharmacokinetics
Pregabalin is well absorbed after oral administration, is eliminated largely by renal excretion, and has an elimination half-life of about 6 hours.
Absorption and Distribution
Following oral administration of pregabalin capsules under fasting conditions, peak plasma concentrations occur within 1.5 hours. Pregabalin oral bioavailability is greater than or equal to 90% and is independent of dose. Following single- (25 mg to 300 mg) and multiple-dose (75 to 900 mg/day) administration, maximum plasma concentrations (Cmax) and area under the plasma concentration-time curve (AUC) values increase linearly. Following repeated administration, steady-state is achieved within 24 to 48 hours. Multiple-dose pharmacokinetics can be predicted from single-dose data.
The rate of pregabalin absorption is decreased when given with food, resulting in a decrease in Cmax of approximately 25% to 30% and an increase in Tmax to approximately 3 hours. However, administration of pregabalin with food has no clinically relevant effect on the total absorption of pregabalin. Therefore, pregabalin can be taken with or without food.
Pregabalin does not bind to plasma proteins. The apparent volume of distribution of pregabalin following oral administration is approximately 0.5 L/kg. Pregabalin is a substrate for system L transporter which is responsible for the transport of large amino acids across the blood brain barrier. Although there are no data in humans, pregabalin has been shown to cross the blood brain barrier in mice, rats, and monkeys. In addition, pregabalin has been shown to cross the placenta in rats and is present in the milk of lactating rats.
Metabolism and Elimination
Pregabalin undergoes negligible metabolism in humans. Following a dose of radiolabeled pregabalin, approximately 90% of the administered dose was recovered in the urine as unchanged pregabalin. The N-methylated derivative of pregabalin, the major metabolite of pregabalin found in urine, accounted for 0.9% of the dose. In preclinical studies, pregabalin (S-enantiomer) did not undergo racemization to the R-enantiomer in mice, rats, rabbits, or monkeys.
Pregabalin is eliminated from the systemic circulation primarily by renal excretion as unchanged drug with a mean elimination half-life of 6.3 hours in subjects with normal renal function. Mean renal clearance was estimated to be 67.0 to 80.9 mL/min in young healthy subjects. Because pregabalin is not bound to plasma proteins this clearance rate indicates that renal tubular reabsorption is involved. Pregabalin elimination is nearly proportional to creatinine clearance (CLcr) [see Dosage and Administration (2.7)].
Pharmacokinetics in Specific Populations
Race
In population pharmacokinetic analyses of the clinical studies in various populations, the pharmacokinetics of pregabalin were not significantly affected by race (Caucasians, Blacks, and Hispanics).
Gender
Population pharmacokinetic analyses of the clinical studies showed that the relationship between daily dose and pregabalin drug exposure is similar between genders.
Renal Impairment and Hemodialysis
Pregabalin clearance is nearly proportional to creatinine clearance (CLcr). Dosage reduction in patients with renal dysfunction is necessary. Pregabalin is effectively removed from plasma by hemodialysis. Following a 4-hour hemodialysis treatment, plasma pregabalin concentrations are reduced by approximately 50%. For patients on hemodialysis, dosing must be modified [see Dosage and Administration (2.7)].
Elderly
Pregabalin oral clearance tended to decrease with increasing age. This decrease in pregabalin oral clearance is consistent with age-related decreases in CLcr. Reduction of pregabalin dose may be required in patients who have age-related compromised renal function [see Dosage and Administration (2.7)].
Pediatric Pharmacokinetics
Pediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
Drug Interactions
In Vitro Studies
Pregabalin, at concentrations that were, in general, 10-times those attained in clinical trials, does not inhibit human CYP1A2, CYP2A6, CYP2C9, CYP2C19, CYP2D6, CYP2E1, and CYP3A4 enzyme systems. In vitro drug interaction studies demonstrate that pregabalin does not induce CYP1A2 or CYP3A4 activity. Therefore, an increase in the metabolism of co-administered CYP1A2 substrates (e.g., theophylline, caffeine) or CYP 3A4 substrates (e.g., midazolam, testosterone) is not anticipated.
In Vivo Studies
The drug interaction studies described in this section were conducted in healthy adults, and across various patient populations.
Gabapentin
The pharmacokinetic interactions of pregabalin and gabapentin were investigated in 12 healthy subjects following concomitant single-dose administration of 100-mg pregabalin and 300-mg gabapentin and in 18 healthy subjects following concomitant multiple-dose administration of 200-mg pregabalin every 8 hours and 400-mg gabapentin every 8 hours. Gabapentin pharmacokinetics following single- and multiple-dose administration were unaltered by pregabalin co-administration. The extent of pregabalin absorption was unaffected by gabapentin co-administration, although there was a small reduction in rate of absorption.
Oral Contraceptive
Pregabalin co-administration (200 mg three times a day) had no effect on the steady-state pharmacokinetics of norethindrone and ethinyl estradiol (1 mg/35 mcg, respectively) in healthy subjects.
Lorazepam
Multiple-dose administration of pregabalin (300 mg twice a day) in healthy subjects had no effect on the rate and extent of lorazepam single-dose pharmacokinetics and single-dose administration of lorazepam (1 mg) had no effect on the steady-state pharmacokinetics of pregabalin.
Oxycodone
Multiple-dose administration of pregabalin (300 mg twice a day) in healthy subjects had no effect on the rate and extent of oxycodone single-dose pharmacokinetics. Single-dose administration of oxycodone (10 mg) had no effect on the steady-state pharmacokinetics of pregabalin.
Ethanol
Multiple-dose administration of pregabalin (300 mg twice a day) in healthy subjects had no effect on the rate and extent of ethanol single-dose pharmacokinetics and single-dose administration of ethanol (0.7 g/kg) had no effect on the steady-state pharmacokinetics of pregabalin.
Phenytoin, carbamazepine, valproic acid, and lamotrigine
Steady-state trough plasma concentrations of phenytoin, carbamazepine and carbamazepine 10,11 epoxide, valproic acid, and lamotrigine were not affected by concomitant pregabalin (200 mg three times a day) administration.
Population pharmacokinetic analyses in patients treated with pregabalin and various concomitant medications suggest the following:
Therapeutic class
Specific concomitant drug studied
Concomitant drug has no effect on the pharmacokinetics of pregabalin
Hypoglycemics
Glyburide, insulin, metformin
Diuretics
Furosemide
Antiepileptic Drugs
Tiagabine
Concomitant drug has no effect on the pharmacokinetics of pregabalin and pregabalin has no effect on the pharmacokinetics of concomitant drug
Antiepileptic Drugs
Carbamazepine, lamotrigine, phenobarbital, phenytoin, topiramate, valproic acid
-
13. Non Clinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
A dose-dependent increase in the incidence of malignant vascular tumors (hemangiosarcomas) was observed in two strains of mice (B6C3F1 and CD-1) given pregabalin (200, 1,000, or 5,000 mg/kg) in the diet for two years. Plasma pregabalin exposure (AUC) in mice receiving the lowest dose that increased hemangiosarcomas was approximately equal to the human exposure at the maximum recommended dose (MRD) of 600 mg/day. A no-effect dose for induction of hemangiosarcomas in mice was not established. No evidence of carcinogenicity was seen in two studies in Wistar rats following dietary administration of pregabalin for two years at doses (50, 150, or 450 mg/kg in males and 100, 300, or 900 mg/kg in females) that were associated with plasma exposures in males and females up to approximately 14 and 24 times, respectively, human exposure at the MRD.
Mutagenesis
Pregabalin was not mutagenic in bacteria or in mammalian cells in vitro, was not clastogenic in mammalian systems in vitro and in vivo, and did not induce unscheduled DNA synthesis in mouse or rat hepatocytes.
Impairment of Fertility
In fertility studies in which male rats were orally administered pregabalin (50 to 2,500 mg/kg) prior to and during mating with untreated females, a number of adverse reproductive and developmental effects were observed. These included decreased sperm counts and sperm motility, increased sperm abnormalities, reduced fertility, increased preimplantation embryo loss, decreased litter size, decreased fetal body weights, and an increased incidence of fetal abnormalities. Effects on sperm and fertility parameters were reversible in studies of this duration (3 to 4 months). The no-effect dose for male reproductive toxicity in these studies (100 mg/kg) was associated with a plasma pregabalin exposure (AUC) approximately 3 times human exposure at the maximum recommended dose (MRD) of 600 mg/day.
In addition, adverse reactions on reproductive organ (testes, epididymides) histopathology were observed in male rats exposed to pregabalin (500 to 1,250 mg/kg) in general toxicology studies of four weeks or greater duration. The no-effect dose for male reproductive organ histopathology in rats (250 mg/kg) was associated with a plasma exposure approximately 8 times human exposure at the MRD.
In a fertility study in which female rats were given pregabalin (500, 1,250, or 2,500 mg/kg) orally prior to and during mating and early gestation, disrupted estrous cyclicity and an increased number of days to mating were seen at all doses, and embryolethality occurred at the highest dose. The low dose in this study produced a plasma exposure approximately 9 times that in humans receiving the MRD. A no-effect dose for female reproductive toxicity in rats was not established.
13.2 Animal Toxicology and/or Pharmacology
Dermatopathy
Skin lesions ranging from erythema to necrosis were seen in repeated-dose toxicology studies in both rats and monkeys. The etiology of these skin lesions is unknown. At the maximum recommended human dose (MRD) of 600 mg/day, there is a 2-fold safety margin for the dermatological lesions. The more severe dermatopathies involving necrosis were associated with pregabalin exposures (as expressed by plasma AUCs) of approximately 3 to 8 times those achieved in humans given the MRD. No increase in incidence of skin lesions was observed in clinical studies.
Ocular Lesions
Ocular lesions (characterized by retinal atrophy [including loss of photoreceptor cells] and/or corneal inflammation/mineralization) were observed in two lifetime carcinogenicity studies in Wistar rats. These findings were observed at plasma pregabalin exposures (AUC) greater than or equal to 2 times those achieved in humans given the maximum recommended dose of 600 mg/day. A no-effect dose for ocular lesions was not established. Similar lesions were not observed in lifetime carcinogenicity studies in two strains of mice or in monkeys treated for 1 year.
-
14. Clinical Studies
14.1 Neuropathic Pain Associated with Diabetic Peripheral Neuropathy
The efficacy of the maximum recommended dose of pregabalin for the management of neuropathic pain associated with diabetic peripheral neuropathy was established in three double-blind, placebo-controlled, multicenter studies with three times a day dosing, two of which studied the maximum recommended dose. Patients were enrolled with either Type 1 or Type 2 diabetes mellitus and a diagnosis of painful distal symmetrical sensorimotor polyneuropathy for 1 to 5 years. A total of 89% of patients completed Studies DPN 1 and DPN 2. The patients had a minimum mean baseline pain score of greater than or equal to 4 on an 11-point numerical pain rating scale ranging from 0 (no pain) to 10 (worst possible pain). The baseline mean pain scores across the two studies ranged from 6.1 to 6.7. Patients were permitted up to 4 grams of acetaminophen per day as needed for pain, in addition to pregabalin. Patients recorded their pain daily in a diary.
Study DPN 1: This 5-week study compared pregabalin 25 mg, 100 mg, or 200 mg three times a day with placebo. Treatment with pregabalin 100 mg and 200 mg three times a day statistically significantly improved the endpoint mean pain score and increased the proportion of patients with at least a 50% reduction in pain score from baseline. There was no evidence of a greater effect on pain scores of the 200 mg three times a day dose than the 100 mg three times a day dose, but there was evidence of dose dependent adverse reactions [see Adverse Reactions (6.1)]. For a range of levels of improvement in pain intensity from baseline to study endpoint, Figure 1 shows the fraction of patients achieving that level of improvement. The figure is cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
Figure 1: Patients Achieving Various Levels of Improvement in Pain Intensity – Study DPN 1
Study DPN 2: This 8-week study compared pregabalin 100 mg three times a day with placebo. Treatment with pregabalin 100 mg three times a day statistically significantly improved the endpoint mean pain score and increased the proportion of patients with at least a 50% reduction in pain score from baseline. For various levels of improvement in pain intensity from baseline to study endpoint, Figure 2 shows the fraction of patients achieving that level of improvement. The figure is cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
Figure 2: Patients Achieving Various Levels of Improvement in Pain Intensity– Study DPN 2
14.2 Postherpetic Neuralgia
The efficacy of pregabalin for the management of postherpetic neuralgia was established in three double-blind, placebo-controlled, multicenter studies. These studies enrolled patients with neuralgia persisting for at least 3 months following healing of herpes zoster rash and a minimum baseline score of greater than or equal to 4 on an 11-point numerical pain rating scale ranging from 0 (no pain) to 10 (worst possible pain). Seventy-three percent of patients completed the studies. The baseline mean pain scores across the 3 studies ranged from 6 to 7. Patients were permitted up to 4 grams of acetaminophen per day as needed for pain, in addition to pregabalin. Patients recorded their pain daily in a diary.
Study PHN 1: This 13-week study compared pregabalin 75 mg, 150 mg, and 300 mg twice daily with placebo. Patients with creatinine clearance (CLcr) between 30 to 60 mL/min were randomized to 75 mg, 150 mg, or placebo twice daily. Patients with creatinine clearance greater than 60 mL/min were randomized to 75 mg, 150 mg, 300 mg or placebo twice daily. In patients with creatinine clearance greater than 60 mL/min treatment with all doses of pregabalin statistically significantly improved the endpoint mean pain score and increased the proportion of patients with at least a 50% reduction in pain score from baseline. Despite differences in dosing based on renal function, patients with creatinine clearance between 30 to 60 mL/min tolerated pregabalin less well than patients with creatinine clearance greater than 60 mL/min as evidenced by higher rates of discontinuation due to adverse reactions. For various levels of improvement in pain intensity from baseline to study endpoint, Figure 3 shows the fraction of patients achieving that level of improvement. The figure is cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
Figure 3: Patients Achieving Various Levels of Improvement in Pain Intensity– Study PHN 1
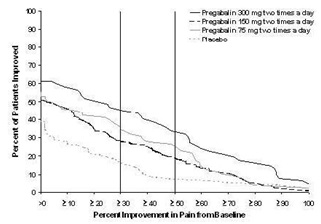
Study PHN 2: This 8-week study compared pregabalin 100 mg or 200 mg three times a day with placebo, with doses assigned based on creatinine clearance. Patients with creatinine clearance between 30 to 60 mL/min were treated with 100 mg three times a day, and patients with creatinine clearance greater than 60 mL/min were treated with 200 mg three times daily. Treatment with pregabalin statistically significantly improved the endpoint mean pain score and increased the proportion of patients with at least a 50% reduction in pain score from baseline. For various levels of improvement in pain intensity from baseline to study endpoint, Figure 4 shows the fraction of patients achieving those levels of improvement. The figure is cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
Figure 4: Patients Achieving Various Levels of Improvement in Pain Intensity – Study PHN 2
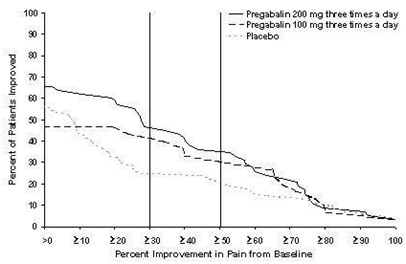
Study PHN 3: This 8-week study compared pregabalin 50 mg or 100 mg three times a day with placebo with doses assigned regardless of creatinine clearance. Treatment with pregabalin 50 mg and 100 mg three times a day statistically significantly improved the endpoint mean pain score and increased the proportion of patients with at least a 50% reduction in pain score from baseline. Patients with creatinine clearance between 30 to 60 mL/min tolerated pregabalin less well than patients with creatinine clearance greater than 60 mL/min as evidenced by markedly higher rates of discontinuation due to adverse reactions. For various levels of improvement in pain intensity from baseline to study endpoint, Figure 5 shows the fraction of patients achieving that level of improvement. The figure is cumulative, so that patients whose change from baseline is, for example, 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
Figure 5: Patients Achieving Various Levels of Improvement in Pain Intensity– Study PHN 3
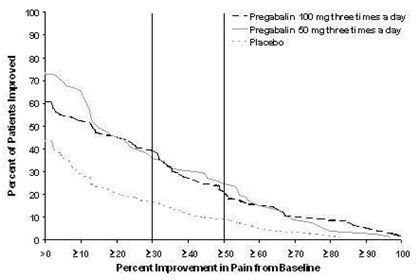
14.3 Adjunctive Therapy for Partial-Onset Seizures in Patients 17 Years of Age and Older
Adjunctive Therapy for Partial-Onset Seizures in Adult Patients
The efficacy of pregabalin as adjunctive therapy for partial-onset seizures in adult patients was established in three 12-week, randomized, double-blind, placebo-controlled, multicenter studies. Patients were enrolled who had partial-onset seizures with or without secondary generalization and were not adequately controlled with 1 to 3 concomitant antiepileptic drugs (AEDs). Patients taking gabapentin were required to discontinue gabapentin treatment 1 week prior to entering baseline. During an 8-week baseline period, patients had to experience at least 6 partial-onset seizures with no seizure-free period exceeding 4 weeks. The mean duration of epilepsy was 25 years in these 3 studies and the mean and median baseline seizure frequencies were 22.5 and 10 seizures per month, respectively. Approximately half of the patients were taking 2 concurrent AEDs at baseline. Among the pregabalin-treated patients, 80% completed the double-blind phase of the studies.
Table 11 shows median baseline seizure rates and median percent reduction in seizure frequency by dose.
Table 11. Seizure Response in Controlled, Adjunctive Epilepsy Studies in Adults
Daily Dose of Pregabalin Dosing Regimen N Baseline Seizure Frequency/mo Median % Change from Baseline p-value, vs. placebo Study E1 Placebo BID 100 9.5 0 50 mg/day BID 88 10.3 -9 0.4230 150 mg/day BID 86 8.8 -35 0.0001 300 mg/day BID 90 9.8 -37 0.0001 600 mg/day BID 89 9.0 -51 0.0001 Study E2 Placebo TID 96 9.3 1 150 mg/day TID 99 11.5 -17 0.0007 600 mg/day TID 92 12.3 -43 0.0001 Study E3
Placebo BID/TID 98 11 -1 600 mg/day BID 103 9.5 -36 0.0001 600 mg/day TID 111 10 -48 0.0001 A secondary outcome measure included the responder rate (proportion of patients with greater than or equal to 50% reduction from baseline in partial seizure frequency). The following figure displays responder rate by dose for two of the studies.In the first study (E1), there was evidence of a dose-response relationship for total daily doses of pregabalin between 150 and 600 mg/day; a dose of 50 mg/day was not effective. In the first study (E1), each daily dose was divided into two equal doses (twice a day dosing). In the second study (E2), each daily dose was divided into three equal doses (three times a day dosing). In the third study (E3), the same total daily dose was divided into two equal doses for one group (twice a day dosing) and three equal doses for another group (three times a day dosing). While the three times a day dosing group in Study E3 performed numerically better than the twice a day dosing group, this difference was small and not statistically significant.
Figure 6: Responder Rate by Adjunctive Epilepsy Study
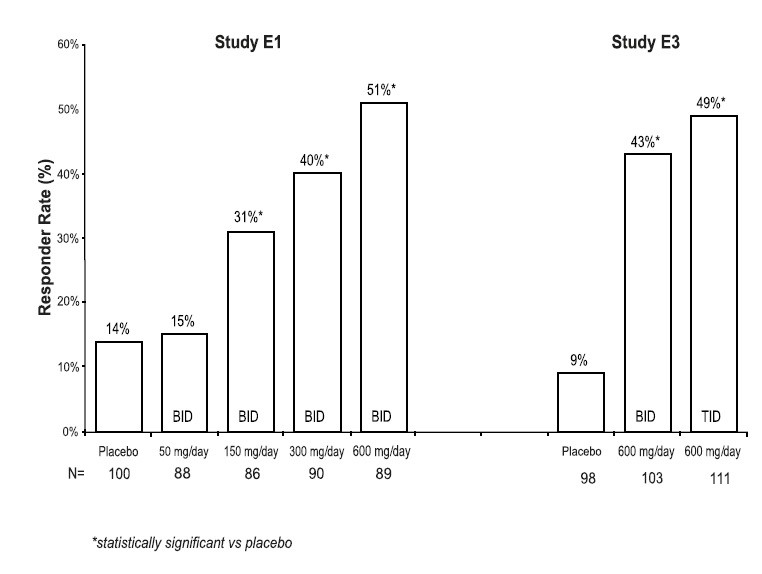
Figure 7: Seizure Reduction by Dose (All Partial-Onset Seizures) for Studies E1, E2, and E3
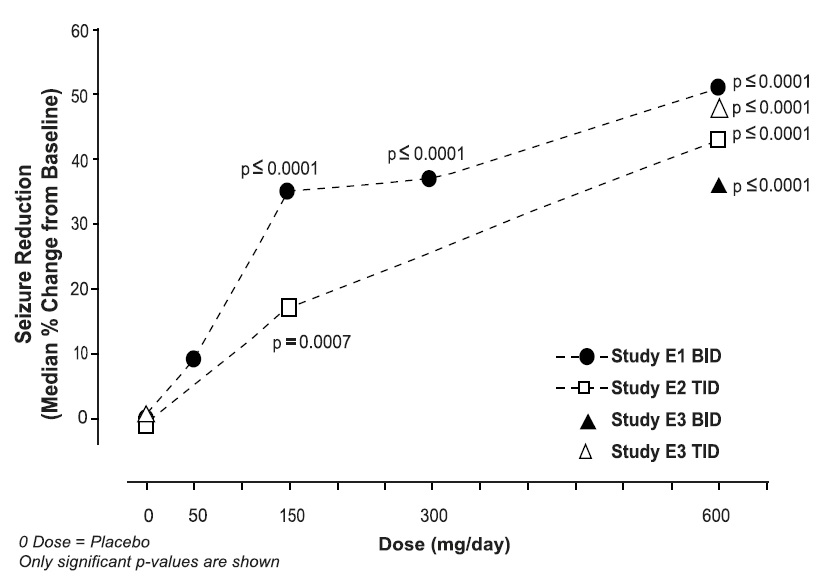
Subset evaluations of the antiseizure efficacy of pregabalin showed no clinically important differences as a function of age, gender, or race.
Pediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
14.4 Management of Fibromyalgia
The efficacy of pregabalin for management of fibromyalgia was established in one 14-week, double-blind, placebo-controlled, multicenter study (F1) and one six-month, randomized withdrawal study (F2). Studies F1 and F2 enrolled patients with a diagnosis of fibromyalgia using the American College of Rheumatology (ACR) criteria (history of widespread pain for 3 months, and pain present at 11 or more of the 18 specific tender point sites). The studies showed a reduction in pain by visual analog scale. In addition, improvement was demonstrated based on a patient global assessment (PGIC), and on the Fibromyalgia Impact Questionnaire (FIQ).
Study F1: This 14-week study compared pregabalin total daily doses of 300 mg, 450 mg and 600 mg with placebo. Patients were enrolled with a minimum mean baseline pain score of greater than or equal to 4 on an 11-point numeric pain rating scale and a score of greater than or equal to 40 mm on the 100 mm pain visual analog scale (VAS). The baseline mean pain score in this trial was 6.7. Responders to placebo in an initial one-week run-in phase were not randomized into subsequent phases of the study. A total of 64% of patients randomized to pregabalin completed the study. There was no evidence of a greater effect on pain scores of the 600 mg daily dose than the 450 mg daily dose, but there was evidence of dose-dependent adverse reactions [see Adverse Reactions (6.1)]. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study. The results are summarized in Figure 9 and Table 14.
For various levels of improvement in pain intensity from baseline to study endpoint, Figure 9 shows the fraction of patients achieving that level of improvement. The figure is cumulative. Patients who did not complete the study were assigned 0% improvement. Some patients experienced a decrease in pain as early as Week 1, which persisted throughout the study.
Figure 9: Patients Achieving Various Levels of Improvement in Pain Intensity – Fibromyalgia Study F1
Table 14. Patient Global Response in Fibromyalgia Study F1
Patient Global Impression of Change Treatment Group (mg/day) % Any Improvement 95% CI Placebo 47.6 (40.0, 55.2) PGB 300 68.1 (60.9, 75.3) PGB 450 77.8 (71.5, 84.0) PGB 600 66.1 (59.1, 73.1) Study F2: This randomized withdrawal study compared pregabalin with placebo. Patients were titrated during a 6-week open-label dose optimization phase to a total daily dose of 300 mg, 450 mg, or 600 mg. Patients were considered to be responders if they had both: 1) at least a 50% reduction in pain (VAS) and, 2) rated their overall improvement on the PGIC as "much improved" or "very much improved.” Those who responded to treatment were then randomized in the double-blind treatment phase to either the dose achieved in the open-label phase or to placebo. Patients were treated for up to 6 months following randomization. Efficacy was assessed by time to loss of therapeutic response, defined as 1) less than 30% reduction in pain (VAS) from open-label baseline during two consecutive visits of the double-blind phase, or 2) worsening of FM symptoms necessitating an alternative treatment. Fifty-four percent of patients were able to titrate to an effective and tolerable dose of pregabalin during the 6-week open-label phase. Of the patients entering the randomized treatment phase assigned to remain on pregabalin, 38% of patients completed 26 weeks of treatment versus 19% of placebo-treated patients.PGB = Pregabalin
When considering return of pain or withdrawal due to adverse events as loss of response (LTR), treatment with pregabalin resulted in a longer time to loss of therapeutic response than treatment with placebo. Fifty-three percent of the pregabalin-treated subjects compared to 33% of placebo patients remained on study drug and maintained a therapeutic response to Week 26 of the study. Treatment with pregabalin also resulted in a longer time to loss of response based on the FIQ1, and longer time to loss of overall assessment of patient status, as measured by the PGIC2.
1 Time to worsening of the FIQ was defined as the time to a 1-point increase from double-blind baseline in each of the subscales, and a 5-point increase from double-blind baseline evaluation for the FIQ total score.
2 Time to PGIC lack of improvement was defined as time to PGIC assessments indicating less improvement than “much improvement.”
Figure 10: Time to Loss of Therapeutic Response, Fibromyalgia Study F2 (Kaplan-Meier Analysis)
14.5 Management of Neuropathic Pain Associated with Spinal Cord Injury
The efficacy of pregabalin for the management of neuropathic pain associated with spinal cord injury was established in two double-blind, placebo-controlled, multicenter studies. Patients were enrolled with neuropathic pain associated with spinal cord injury that persisted continuously for at least three months or with relapses and remissions for at least six months. A total of 63% of patients completed study 1 and 84% completed study 2. The patients had a minimum mean baseline pain score of greater than or equal to 4 on an 11-point numerical pain rating scale ranging from 0 (no pain) to 10 (worst possible pain). The baseline mean pain scores across the two studies ranged from 6.5 to 6.7.
Patients were allowed to take opioids, non-opioid analgesics, antiepileptic drugs, muscle relaxants, and antidepressant drugs if the dose was stable for 30 days prior to screening. Patients were allowed to take acetaminophen and nonsteroidal anti-inflammatory drugs during the studies.
Study SCI 1: This 12-week, randomized, double-blind, parallel-group, multicenter, flexible dose (150 to 600 mg/day) study compared pregabalin with placebo. The 12-week study consisted of a 3-week dose adjustment phase and a 9-week dose maintenance phase. Treatment with pregabalin 150 to 600 mg/day statistically significantly improved the endpoint weekly mean pain score, and increased the proportion of patients with at least a 30% and 50% reduction in pain score from baseline. The fraction of patients achieving various levels of improvement in pain intensity from baseline to Week 12 is presented in Figure 11. Some patients experienced a decrease in pain as early as week 1, which persisted throughout the study.
Figure 11: Patients Achieving Various Levels of Improvement in Pain Intensity – Study SCI 1
Study SCI 2: This 16-week, randomized, double-blind, placebo-controlled, parallel-group, multicenter, flexible dose (150 to 600 mg/day, in increments of 150 mg) study compared the efficacy, safety and tolerability of pregabalin with placebo. The 16-week study consisted of a 4-week dose adjustment phase and a 12-week dose maintenance phase. Treatment with pregabalin statistically significantly improved the endpoint weekly mean pain score, and increased the proportion of patients with at least a 30% and 50% reduction in pain score from baseline. The fraction of patients achieving various levels of improvement in pain intensity from baseline to Week 16 is presented in Figure 12. Some patients experienced a decrease in pain as early as week 1, which persisted throughout the study.
Figure 12: Patients Achieving Various Levels of Improvement in Pain Intensity – Study SCI 2
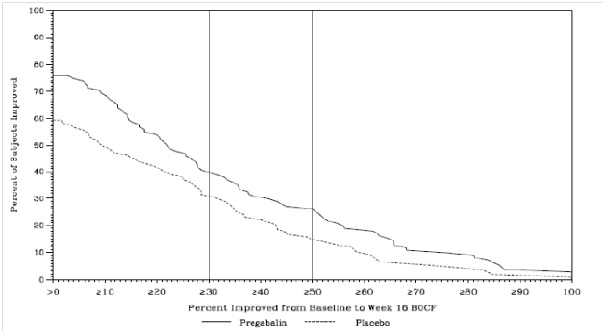
-
16. How Supplied/Storage and Handling
Pregabalin capsules, 100 mg are supplied as orange, hard gelatin capsule printed with black ink “AN” on cap & “1313” on body. They are available as follows:
Bottles of 90 Capsules NDC 80425-0123-03
Storage and Handling
Store at 20° to 25°C (68° to 77°F); excursions permitted between 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Dispense in tight (USP), child-resistant containers.
-
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Angioedema
Advise patients that pregabalin may cause angioedema, with swelling of the face, mouth (lip, gum, tongue) and neck (larynx and pharynx) that can lead to life-threatening respiratory compromise. Instruct patients to discontinue pregabalin and immediately seek medical care if they experience these symptoms [see Warnings and Precautions (5.1)].
Hypersensitivity
Advise patients that pregabalin has been associated with hypersensitivity reactions such as wheezing, dyspnea, rash, hives, and blisters. Instruct patients to discontinue pregabalin and immediately seek medical care if they experience these symptoms [see Warnings and Precautions (5.2)].
Suicidal Thinking and Behavior
Patients, their caregivers, and families should be counseled that AEDs, including pregabalin, may increase the risk of suicidal thoughts and behavior and should be advised of the need to be alert for the emergence or worsening of symptoms of depression, any unusual changes in mood or behavior, or the emergence of suicidal thoughts, behavior, or thoughts about self-harm. Report behaviors of concern immediately to healthcare providers [see Warnings and Precautions (5.3)].
Respiratory Depression
Inform patients about the risk of respiratory depression. Include information that the risk is greatest for those using concomitant central nervous system (CNS) depressants (such as opioid analgesics) or in those with underlying respiratory impairment. Teach patients how to recognize respiratory depression and advise them to seek medical attention immediately if it occurs [see Warnings and Precautions (5.4)].
Dizziness and Somnolence
Counsel patients that pregabalin may cause dizziness, somnolence, blurred vision and other CNS signs and symptoms. Accordingly, advise patients not to drive, operate complex machinery, or engage in other hazardous activities until they have gained sufficient experience on pregabalin to gauge whether or not it affects their mental, visual, and/or motor performance adversely [see Warnings and Precautions (5.5)].
CNS Depressants
Inform patients who require concomitant treatment with central nervous system depressants such as opiates or benzodiazepines that they may experience additive CNS side effects, such as respiratory depression, somnolence, and dizziness [see Warnings and Precautions (5.4, 5.5) and Drug Interactions (7)]. Advise patients to avoid consuming alcohol while taking pregabalin, as pregabalin may potentiate the impairment of motor skills and sedating effects of alcohol.
Adverse Reactions with Abrupt or Rapid Discontinuation
Advise patients to take pregabalin as prescribed. Abrupt or rapid discontinuation may result in increased seizure frequency in patients with seizure disorders, and insomnia, nausea, headache, anxiety, hyperhidrosis, or diarrhea [see Warnings and Precautions (5.6)].
Missed Dose
Counsel patients if they miss a dose, they should take it as soon as they remember. If it is almost time for the next dose, they should skip the missed dose and take the next dose at their regularly scheduled time. Instruct patients not to take two doses at the same time.
Weight Gain and Edema
Counsel patients that pregabalin may cause edema and weight gain. Advise patients that concomitant treatment with pregabalin and a thiazolidinedione antidiabetic agent may lead to an additive effect on edema and weight gain. For patients with preexisting cardiac conditions, this may increase the risk of heart failure [see Warnings and Precautions (5.7, 5.8)].
Ophthalmological Effects
Counsel patients that pregabalin may cause visual disturbances. Inform patients that if changes in vision occur, they should notify their physician [see Warnings and Precautions (5.10)].
Creatine Kinase Elevations
Instruct patients to promptly report unexplained muscle pain, tenderness, or weakness, particularly if accompanied by malaise or fever [see Warnings and Precautions (5.11)].
Pregnancy
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to pregabalin during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Advise nursing mothers that breastfeeding is not recommended during treatment with pregabalin [see Use in Specific Populations (8.2)].
Male Fertility
Inform men being treated with pregabalin who plan to father a child of the potential risk of male-mediated teratogenicity. In preclinical studies in rats, pregabalin was associated with an increased risk of male-mediated teratogenicity. The clinical significance of this finding is uncertain [see Nonclinical Toxicology (13.1) and Use in Specific populations (8.3)].
Dermatopathy
Instruct diabetic patients to pay particular attention to skin integrity while being treated with pregabalin and to inform their healthcare provider about any sores or skin problems. Some animals treated with pregabalin developed skin ulcerations, although no increased incidence of skin lesions associated with pregabalin was observed in clinical trials [see Nonclinical Toxicology (13.2)].
This product’s label may have been updated. For current full prescribing information, please visit www.amneal.com.
Manufactured by:
Amneal Pharmaceuticals Pvt. Ltd.
Oral Solid Dosage Unit
Ahmedabad 382213, INDIADistributed by:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807Distributed by:
Advanced Rx Pharmacy of Tennessee LLC
Nashville, TN 37211
Rev. 06-2020-01
-
Medication Guide Section
MEDICATION GUIDE
Pregabalin (pree gabʹ a lin) Capsules, CV
Read this Medication Guide before you start taking pregabalin capsules and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. If you have any questions about pregabalin capsules, ask your healthcare provider or pharmacist.
What is the most important information I should know about pregabalin capsules?
Pregabalin capsules may cause serious side effects including:
serious, even life-threatening, allergic reactions
swelling of your hands, legs and feet
suicidal thoughts or actions
dizziness and sleepiness
serious breathing problems
These serious side effects are described below:
Serious, even life-threatening, allergic reactions.
Stop taking pregabalin capsules and call your healthcare provider right away if you have any of these signs of a serious allergic reaction:
swelling of your face, mouth, lips, gums, tongue, throat or neck
trouble breathing
rash, hives (raised bumps) or blistersLike other antiepileptic drugs, pregabalin capsules may cause suicidal thoughts or actions in a very small number of people, about 1 in 500. Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
thoughts about suicide or dying
trouble sleeping (insomnia)
attempts to commit suicide
new or worse irritability
new or worse depression
acting aggressive, being angry, or violent
new or worse anxiety
acting on dangerous impulses
feeling agitated or restless
an extreme increase in activity and talking (mania)
panic attacks
other unusual changes in behavior or mood
If you have suicidal thoughts or actions, do not stop pregabalin capsules without first talking to a healthcare provider.
Stopping pregabalin capsules suddenly can cause serious problems.
Suicidal thoughts or actions can be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.How can I watch for early symptoms of suicidal thoughts and actions?
Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
Keep all follow-up visits with your healthcare provider as scheduled.
Call your healthcare provider between visits as needed, especially if you are worried about symptoms.Serious breathing problems can occur when pregabalin capsules is taken with other medicines that can cause severe sleepiness or decreased awareness, or when it is taken by someone who already has breathing problems. Watch for increased sleepiness or decreased breathing when starting pregabalin capsules or when the dose is increased. Get help right away if breathing problems occur.
Swelling of your hands, legs and feet. This swelling can be a serious problem for people with heart problems.
Dizziness and sleepiness. Do not drive a car, work with machines, or do other dangerous activities until you know how pregabalin capsule affects you. Ask your healthcare provider about when it will be okay to do these activities.What are pregabalin capsules?
Pregabalin capsules are a prescription medicine used in adults, 18 years of age and older to treat:
pain from damaged nerves (neuropathic pain) that happens with diabetes
pain from damaged nerves (neuropathic pain) that follows healing of shingles
fibromyalgia (pain all over your body)
pain from damaged nerves (neuropathic pain) that follows spinal cord injuryIt is not known if pregabalin capsules are safe and effective in people under 18 years of age for the treatment of fibromyalgia and neuropathic pain with diabetes, shingles, or spinal cord injury.
Pregabalin capsules are a prescription medicine used in people 17 years of age and older to treat:
partial-onset seizures when taken together with other seizure medicines.
For the treatment of partial-onset seizures when taken together with other seizure medicines, it is not known if pregabalin capsules are safe and effective in children under 1 month of age.
Who should not take pregabalin capsules?
Do not take pregabalin capsules if you are allergic to pregabalin or any of the ingredients in pregabalin capsules.
See “What is the most important information I should know about pregabalin capsules?” for the signs of an allergic reaction. See the end of this Medication Guide for a complete list of ingredients in pregabalin capsules.
What should I tell my healthcare provider before taking pregabalin capsules?
Before taking pregabalin capsules, tell your healthcare provider about all your medical conditions, including if you:
have or have had depression, mood problems or suicidal thoughts or behavior.
have breathing problems.
have kidney problems or get kidney dialysis.
have heart problems including heart failure.
have a bleeding problem or a low blood platelet count.
have abused prescription medicines, street drugs, or alcohol in the past.
have ever had swelling of your face, mouth, tongue, lips, gums, neck, or throat (angioedema).
plan to father a child. Animal studies have shown that pregabalin, the active ingredient in pregabalin capsules, made male animals less fertile and caused sperm to change. Also, in animal studies, birth defects were seen in the offspring (babies) of male animals treated with pregabalin. It is not known if these problems can happen in people who take pregabalin capsules.
are pregnant or plan to become pregnant. Pregabalin capsules may harm your unborn baby. You and your healthcare provider will decide if you should take pregabalin capsules while you are pregnant.If you become pregnant while taking pregabalin capsules, talk to your healthcare provider about registering with the North American Antiepileptic Drug Pregnancy Registry. You can enroll in this registry by calling 1-888-233-2334. The purpose of this registry is to collect information about the safety of antiepileptic drugs during pregnancy. Information about the registry can also be found at the website, http://www.aedpregnancyregistry.org/.
are breastfeeding or plan to breastfeed. Pregabalin passes into your breast milk. It is not known if pregabalin capsules can harm your baby. Talk to your healthcare provider about the best way to feed your baby if you take pregabalin capsules. Breastfeeding is not recommended while taking pregabalin capsules.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins or herbal supplements. Pregabalin capsules and other medicines may affect each other causing side effects. Especially tell your healthcare provider if you take:
angiotensin converting enzyme (ACE) inhibitors, which are used to treat many conditions, including high blood pressure. You may have a higher chance for swelling and hives if these medicines are taken with pregabalin capsules.
Avandia (rosiglitazone) or Actos (pioglitazone) for diabetes. You may have a higher chance of weight gain or swelling of your hands or feet if these medicines are taken with pregabalin capsules.
any opioid pain medicine (such as oxycodone), or medicines for anxiety (such as lorazepam) or insomnia (such as zolpidem). You may have a higher chance for dizziness, sleepiness or serious breathing problems if these medicines are taken with pregabalin capsules.
any medicines that make you sleepy.Know the medicines you take. Keep a list of them with you to show your healthcare provider and pharmacist each time you get a new medicine. Do not start a new medicine without talking with your healthcare provider.
How should I take pregabalin capsules?
Take pregabalin capsules exactly as prescribed. Your healthcare provider will tell you how much pregabalin capsules to take and when to take it.
Pregabalin capsules may be taken with or without food.
Your healthcare provider may change your dose. Do not change your dose without talking to your healthcare provider.
Do not stop taking pregabalin capsules without talking to your healthcare provider. If you stop taking pregabalin capsules suddenly you may have headaches, nausea, diarrhea, trouble sleeping, increased sweating, or you may feel anxious. If you have epilepsy and you stop taking pregabalin capsules suddenly, you may have seizures more often. Talk with your healthcare provider about how to stop pregabalin capsules slowly.
If you miss a dose, take it as soon as you remember. If it is almost time for your next dose, just skip the missed dose. Take the next dose at your regular time. Do not take 2 doses at the same time.
If you take too much pregabalin capsules, call your healthcare provider or poison control center, or go to the nearest emergency room right away.What should I avoid while taking pregabalin capsules?
Do not drive a car, work with machines, or do other dangerous activities until you know how pregabalin capsules affects you.
Do not drink alcohol while taking pregabalin capsules. Pregabalin capsules and alcohol can affect each other and increase side effects such as sleepiness and dizziness.What are the possible side effects of pregabalin capsules?
Pregabalin capsules may cause serious side effects, including:
See "What is the most important information I should know about pregabalin capsules?"
Muscle problems, muscle pain, soreness, or weakness. If you have these symptoms, especially if you feel sick and have a fever, tell your healthcare provider right away.
Problems with your eyesight, including blurry vision. Call your healthcare provider if you have any changes in your eyesight.
Weight gain. If you have diabetes, weight gain may affect the management of your diabetes. Weight gain can also be a serious problem for people with heart problems.
Feeling "high".The most common side effects of pregabalin capsules in adults are:
dizziness
weight gain
trouble concentrating
blurry vision
sleepiness
swelling of hands and feet
dry mouth
Pregabalin capsules caused skin sores in animal studies. Skin sores did not happen in studies in people. If you have diabetes, you should pay attention to your skin while taking pregabalin capsules and tell your healthcare provider about any sores or skin problems.
Tell your healthcare provider about any side effect that bothers you or that does not go away.
These are not all the possible side effects of pregabalin capsules. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store pregabalin capsules?
Store pregabalin capsules at room temperature, 20° to 25ºC (68° to 77ºF); excursions permitted between 15º to 30ºC (59° to 86ºF) in its original package.
Safely throw away any pregabalin capsules that is out of date or no longer needed.Keep pregabalin capsules and all medicines out of the reach of children.
General information about the safe and effective use of pregabalin capsules
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use pregabalin capsules for a condition for which it was not prescribed. Do not give pregabalin capsules to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about pregabalin capsules that is written for health professionals.
What are the ingredients in pregabalin capsules?
Active ingredient: pregabalin
Inactive ingredients: pregelatinized starch and talc.
The capsule shells contain gelatin and titanium dioxide. In addition, the orange capsule shells contain red iron oxide and white capsule shells contain sodium lauryl sulfate.
Each capsule shell is imprinted with black pharmaceutical ink which contains: butyl alcohol, dehydrated alcohol, ferrosoferric oxide, isopropyl alcohol, propylene glycol, potassium hydroxide, purified water, strong ammonia solution and shellac.
Pediatric use information is approved for Pfizer’s LYRICA (pregabalin) Capsules. However, due to Pfizer’s marketing exclusivity rights, this drug product is not labeled with that pediatric information.
For more information, go to www.amneal.com or call 1-877-835-5472.
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Manufactured by:
Amneal Pharmaceuticals Pvt. Ltd.
Oral Solid Dosage Unit
Ahmedabad 382213, INDIADistributed by:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 06-2020-01 - Principal Display Panel
-
INGREDIENTS AND APPEARANCE
PREGABALIN
pregabalin capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:80425-0123(NDC:69238-1313) Route of Administration ORAL DEA Schedule CV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PREGABALIN (UNII: 55JG375S6M) (PREGABALIN - UNII:55JG375S6M) PREGABALIN 100 mg Product Characteristics Color orange Score no score Shape CAPSULE Size 16mm Flavor Imprint Code AN;1313 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:80425-0123-3 90 in 1 BOTTLE; Type 0: Not a Combination Product 07/19/2019 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA209743 07/19/2019 Labeler - Advanced Rx Pharmacy of Tennessee, LLC (117023142) Establishment Name Address ID/FEI Business Operations Advanced Rx Pharmacy of Tennessee, LLC 117023142 repack(80425-0123)
