Label: NEXAVAR- sorafenib tablet, film coated


- NDC Code(s): 50419-488-58, 50419-489-01
- Packager: Bayer HealthCare Pharmaceuticals Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated August 28, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NEXAVAR safely and effectively. See full prescribing information for NEXAVAR.
NEXAVAR (sorafenib) tablets, for oral use
Initial U.S. Approval: 2005INDICATIONS AND USAGE
DOSAGE FORMS AND STRENGTHS
Tablets: 200 mg (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- •
- Cardiovascular Events: Consider temporary or permanent discontinuation of NEXAVAR. (2.2, 5.1)
- •
- Hemorrhage: Discontinue NEXAVAR if needed. (5.2)
- •
- Hypertension: Monitor blood pressure weekly during the first 6 weeks and periodically thereafter. Consider temporary or permanent discontinuation for severe or persistent hypertension despite antihypertensive therapy. (5.3)
- •
- Dermatologic Toxicities: Interrupt and/or decrease dose. Discontinue for severe or persistent reactions, or if Stevens-Johnson syndrome and toxic epidermal necrolysis is suspected. (5.4)
- •
- Gastrointestinal Perforation: Discontinue NEXAVAR. (5.5)
- •
- Risk of Impaired Wound Healing: Withhold NEXAVAR for at least 10 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of NEXAVAR after resolution of wound healing complications has not been established. (5.7)
- •
- QT Prolongation: Monitor electrocardiograms and electrolytes in patients at increased risk for ventricular arrhythmias. Correct electrolytes. Interrupt if QTc greater than 500 msec or increases greater than 60 msec from baseline. (2.2, 5.9, 12.2)
- •
- Drug-Induced Liver Injury: Monitor liver function tests regularly; discontinue for unexplained transaminase elevations. (5.10)
- •
- Embryo-Fetal Toxicity: NEXAVAR may cause fetal harm. Advise patients of the potential risk to a fetus and to use effective contraception. (5.11, 8.1, 8.3)
- •
- Impairment of Thyroid Stimulating Hormone Suppression (TSH) in DTC: Monitor TSH monthly and adjust thyroid replacement therapy in patients with thyroid cancer. (5.12)
ADVERSE REACTIONS
The most common adverse reactions (≥20%) are diarrhea, fatigue, infection, alopecia, hand-foot skin reaction, rash, weight loss, decreased appetite, nausea, gastrointestinal and abdominal pains, hypertension, and hemorrhage. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Bayer HealthCare Pharmaceuticals Inc. at 1-888-842-2937 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 8/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Hepatocellular Carcinoma
1.2 Renal Cell Carcinoma
1.3 Differentiated Thyroid Carcinoma
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Dosage Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Events
5.2 Hemorrhage
5.3 Hypertension
5.4 Dermatologic Toxicities
5.5 Gastrointestinal Perforation
5.6 Increased Risk of Bleeding with Concomitant Use of Warfarin
5.7 Risk of Impaired Wound Healing
5.8 Increased Mortality Observed with NEXAVAR Administered in Combination with Carboplatin/Paclitaxel and Gemcitabine/Cisplatin in Squamous Cell Lung Cancer
5.9 QT Interval Prolongation
5.10 Drug-Induced Liver Injury
5.11 Embryo-Fetal Toxicity
5.12 Impairment of Thyroid Stimulating Hormone Suppression in Differentiated Thyroid Carcinoma
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on NEXAVAR
7.2 Concomitant Use of Warfarin
7.3 Drugs That Prolong the QT Interval
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Hepatocellular Carcinoma
14.2 Renal Cell Carcinoma
14.3 Differentiated Thyroid Carcinoma
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of NEXAVAR is 400 mg orally twice daily without food (at least 1 hour before or 2 hours after a meal) until the patient is no longer clinically benefiting from therapy or until unacceptable toxicity.
2.2 Dosage Modifications for Adverse Reactions
Recommended Dosage Modifications
The recommended dosage modifications for adverse reactions are provided in Tables 1, 2, and 3.
Table 1: Recommended Dose Reductions for Adverse Reactions Dose Reduction
Hepatocellular Carcinoma and
Renal Cell Carcinoma
Differentiated Thyroid Carcinoma
First Dose Reduction
400 mg orally once daily
400 mg orally in the morning and 200 mg orally in the evening about 12 hours apart
OR
200 mg orally in the morning and 400 mg orally in the evening about 12 hours apart
Second Dose Reduction
200 mg orally once daily
OR
400 every other day
200 mg orally twice daily
Third Dose Reduction
None
200 mg orally once daily
Table 2: Recommended Dosage Modifications of NEXAVAR for Adverse Reactions Adverse Reaction
Severity1
NEXAVAR Dosage Modification
Cardiovascular Events [see Warnings and Precautions (5.1)]
Cardiac Ischemia and/or Infarction
Grade 2 and above
Permanently discontinue.
Congestive Heart Failure
Grade 3
Interrupt2 until Grade 1 or less, resume at reduced dose by 1 dose level.3
Grade 4
Permanently discontinue.
Hemorrhage [see Warnings and Precautions (5.2)]
Grade 2 and above requiring medical intervention
Permanently discontinue.
Hypertension [see Warnings and Precautions (5.3)]
Grade 2 (symptomatic/persistent)
OR
Grade 2 symptomatic increase by greater than 20 mm Hg (diastolic) or greater than 140/90 mm Hg if previously within normal limits
OR
Grade 3
Interrupt until symptoms resolve and diastolic blood pressure less than 90 mm Hg, then resume at reduced dose by 1 dose level.3
If needed, reduce another dose level.3
Grade 4
Permanently discontinue.
Gastrointestinal Perforation
[see Warnings and Precautions (5.5)]Any grade
Permanently discontinue.
QT Interval Prolongation
[see Warnings and Precautions (5.9)]Greater than 500 milliseconds
OR
Increase from baseline of 60 milliseconds or greater
Interrupt and correct electrolyte abnormalities (magnesium, potassium, calcium).
Use medical judgement before restarting.
Drug-Induced Liver Injury [see Warnings and Precautions (5.10)]
Grade 3 ALT or higher in the absence of another cause4
OR
AST/ALT greater than 3 × upper limit normal (ULN) with bilirubin greater than 2 × ULN in the absence of another cause4
Permanently discontinue.
Non-hematological toxicities [see Adverse Reactions (6.1)]
Grade 2
Continue treatment at reduced dose by 1 dose level.
Grade 3
1st occurrence
Interrupt until Grade 2 or less, then resume at reduced dose by 1 dose level.
No improvement within 7 days
OR
2nd or 3rd occurrence
Interrupt until Grade 2 or less, then resume at reduced dose by 2 dose levels.
4th occurrence
Interrupt until Grade 2 or less, then resume at reduced dose by 2 dose levels for HCC and RCC or 3 dose levels for DTC.
Grade 4
Permanently discontinue.
- 1
- Adverse reactions graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0).
- 2
- If no recovery after 30 day interruption, discontinue treatment unless the patient is deriving clinical benefit.
- 3
- If more than 2 dose reductions are required, permanently discontinue treatment.
- 4
- In addition, any grade increased alkaline phosphatase in the absence of known bone pathology and Grade 2 or worse increased bilirubin; any 1 of the following: INR of 1.5 or greater, ascites and/or encephalopathy in the absence of underlying cirrhosis or other organ failure considered to be due to drug-induced liver injury.
Table 3: Recommended Dosage Modifications for Dermatologic Toxicities Dermatologic Toxicity Grade
Occurrence
NEXAVAR Dosage Modification
Hepatocellular and
Renal Cell CarcinomaDifferentiated Thyroid Carcinoma
Grade 2: Painful erythema and swelling of the hands or feet and/or discomfort affecting the patient’s normal activities
1st occurrence
Continue NEXAVAR and consider topical therapy for symptomatic relief.
If no improvement within 7 days, see below.
Decrease NEXAVAR to 600 mg daily. If no improvement within 7 days, see below.
No improvement within 7 days at reduced dose
OR
2nd and 3rd occurrence
Interrupt NEXAVAR until resolved or improved to Grade 0 to 1.
Interrupt NEXAVAR until completely resolved or improved to Grade1.
When resuming treatment, decrease dose by 1 dose level.
When resuming treatment, decrease dose by 1 dose level for 2nd occurrence and 2 doses levels for 3rd occurrence.
4th occurrence
Discontinue NEXAVAR treatment.
Grade 3: Moist desquamation, ulceration, blistering, or severe pain of the hands or feet, resulting in inability to work or perform activities of daily living
1st occurrence
Interrupt NEXAVAR until resolved or improved to Grade 0 to 1
Interrupt NEXAVAR until completely resolved or improved to Grade 1.
When resuming treatment, decrease dose by 1 dose level.
When resuming treatment, decrease dose by 1 dose level.
2nd occurrence
Interrupt NEXAVAR until resolved or improved to
Grade 0 to 1Interrupt NEXAVAR until completely resolved or improved to Grade 1.
When resuming treatment, decrease dose by 1 dose level.
When resuming treatment, decrease dose by 2 dose levels.
3rd occurrence
Discontinue NEXAVAR treatment.
Following improvement of Grade 2 or 3 dermatologic toxicity to Grade 0 or 1 for at least 28 days on a reduced dose of NEXAVAR, the dose of NEXAVAR may be increased 1 dose level from the reduced dose. Approximately 50% of patients requiring a dose reduction for dermatologic toxicity are expected to meet these criteria for resumption of the higher dose and roughly 50% of patients resuming the previous dose are expected to tolerate the higher dose (that is, maintain the higher dose level without recurrent Grade 2 or higher dermatologic toxicity).
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
- •
- NEXAVAR is contraindicated in patients with known severe hypersensitivity to sorafenib or any other component of NEXAVAR.
- •
- NEXAVAR in combination with carboplatin and paclitaxel is contraindicated in patients with squamous cell lung cancer [see Warnings and Precautions (5.8)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Events
In the SHARP (HCC) study, the incidence of cardiac ischemia/infarction was 2.7% in NEXAVAR-treated patients compared with 1.3% in those receiving placebo; in the TARGET (RCC) study, the incidence of cardiac ischemia/infarction was higher in the NEXAVAR-treated group (2.9%) compared with patients receiving placebo (0.4%), and in the DECISION (DTC) study, the incidence of cardiac ischemia/infarction was 1.9% in the NEXAVAR-treated group compared with 0% in patients receiving placebo. Patients with unstable coronary artery disease or recent myocardial infarction were excluded from this study. In multiple clinical trials, congestive heart failure has been reported in 1.9% of NEXAVAR-treated patients (N=2276) [see Adverse Reactions (6.2)].
Consider temporary or permanent discontinuation of NEXAVAR in patients who develop cardiovascular events [see Dosage and Administration (2.2)].
5.2 Hemorrhage
An increased risk of bleeding may occur following NEXAVAR administration. In the SHARP (HCC) study, the rates of bleeding from esophageal varices (2.4% and 4%) and of bleeding with a fatal outcome from any site (2.4% and 4%) were similar in NEXAVAR-treated patients and those receiving placebo, respectively . In the TARGET (RCC) study, bleeding was reported in 15.3% of patients in the NEXAVAR-treated group and 8.2% of patients receiving placebo. The incidence of Grade 3 and 4 bleeding was 2% and 0%, respectively, in NEXAVAR-treated patients, and 1.3% and 0.2%, respectively, in those receiving placebo. There was one fatal hemorrhage in each treatment group in the TARGET (RCC) study. In the DECISION (DTC) study, bleeding was reported in 17.4% of NEXAVAR-treated patients and 9.6% of those receiving placebo; however, the incidence of Grade 3 bleeding was similar (1% and 1.4%) in NEXAVAR-treated patients and in those receiving placebo.
If any bleeding necessitates medical intervention, consider permanent discontinuation of NEXAVAR [see Dosage and Administration (2.2)]. Due to the potential risk of bleeding, treat tracheal, bronchial, and esophageal infiltration with local therapy prior to administering NEXAVAR in patients with DTC.
5.3 Hypertension
In the SHARP (HCC) study, hypertension was reported in 9.4% of NEXAVAR-treated patients and 4.3% of patients receiving placebo. In the TARGET (RCC) study, hypertension was reported in 16.9% of NEXAVAR-treated patients and 1.8% of patients receiving placebo. In the DECISION (DTC) study, hypertension was reported in 40.6% of NEXAVAR-treated patients and 12.4% of patients receiving placebo. Hypertension was usually mild to moderate, occurred early in the course of treatment, and was managed with standard antihypertensive therapy. Permanent discontinuation due to hypertension occurred in 1 of 297 NEXAVAR-treated patients in the SHARP (HCC) study, 1 of 451 NEXAVAR-treated patients in the TARGET (RCC) study, and 1 of 207 NEXAVAR-treated patients in the DECISION (DTC) study.
Monitor blood pressure weekly during the first 6 weeks of NEXAVAR. Thereafter, monitor blood pressure and treat hypertension, if required, in accordance with standard medical practice. In cases of severe or persistent hypertension despite institution of antihypertensive therapy, consider temporary or permanent discontinuation of NEXAVAR [see Dosage and Administration (2.2)].
5.4 Dermatologic Toxicities
Hand-foot skin reaction and rash represent the most common adverse reactions attributed to NEXAVAR. Rash and hand-foot skin reaction are usually Grade 1 and 2 and generally appear during the first six weeks of treatment with NEXAVAR. Permanent discontinuation of therapy due to hand-foot skin reaction occurred in 4 (1.3%) of 297 NEXAVAR-treated patients with HCC, 3 (0.7%) of 451 NEXAVAR-treated patients with RCC, and 11 (5.3%) of 207 NEXAVAR-treated patients with DTC.
Management of dermatologic toxicities may include topical therapies for symptomatic relief, temporary treatment interruption and/or dose reduction of NEXAVAR, or in severe or persistent cases, permanent discontinuation of NEXAVAR [see Dosage and Administration (2.2)].
There have been reports of severe dermatologic toxicities, including Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN). These cases may be life-threatening. Discontinue NEXAVAR if SJS or TEN are suspected.
5.5 Gastrointestinal Perforation
Gastrointestinal perforation has been reported in less than 1% of patients taking NEXAVAR. In some cases this was not associated with apparent intra-abdominal tumor. In the event of a gastrointestinal perforation, permanently discontinue NEXAVAR.
5.6 Increased Risk of Bleeding with Concomitant Use of Warfarin
Infrequent bleeding or elevations in the International Normalized Ratio (INR) have been reported in some patients taking warfarin while on NEXAVAR. Monitor patients taking concomitant warfarin regularly for changes in prothrombin time (PT), INR or clinical bleeding episodes.
5.7 Risk of Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the VEGF signaling pathway. Therefore, NEXAVAR has the potential to adversely affect wound healing.
Withhold NEXAVAR for at least 10 days prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of NEXAVAR after resolution of wound healing complications has not been established.
5.8 Increased Mortality Observed with NEXAVAR Administered in Combination with Carboplatin/Paclitaxel and Gemcitabine/Cisplatin in Squamous Cell Lung Cancer
In a subset analysis of two randomized controlled trials in chemo-naive patients with Stage IIIB-IV non-small cell lung cancer, patients with squamous cell carcinoma experienced higher mortality with the addition of NEXAVAR compared to those treated with carboplatin/paclitaxel alone (HR 1.81; 95% CI 1.19, 2.74) and gemcitabine/cisplatin alone (HR 1.22; 95% CI 0.82, 1.80). The use of NEXAVAR in combination with carboplatin/paclitaxel is contraindicated in patients with squamous cell lung cancer. NEXAVAR in combination with gemcitabine/cisplatin is not recommended in patients with squamous cell lung cancer. The safety and effectiveness of NEXAVAR has not been established in patients with non-small cell lung cancer.
5.9 QT Interval Prolongation
NEXAVAR can prolong the QT/QTc interval. QT/QTc interval prolongation increases the risk for ventricular arrhythmias.
Avoid NEXAVAR in patients with congenital long QT syndrome. Monitor electrolytes and electrocardiograms in patients with congestive heart failure, bradyarrhythmias, drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics. Correct electrolyte abnormalities (magnesium, potassium, calcium). Interrupt NEXAVAR if QTc interval is greater than 500 milliseconds or for an increase from baseline of 60 milliseconds or greater [see Clinical Pharmacology (12.2)].
5.10 Drug-Induced Liver Injury
Sorafenib-induced hepatitis is characterized by a hepatocellular pattern of liver damage with significant increases of transaminases which may result in hepatic failure and death. Increases in bilirubin and INR may also occur. The incidence of severe drug-induced liver injury, defined as elevated transaminase levels above 20 times the upper limit of normal or transaminase elevations with significant clinical sequelae (for example, elevated INR, ascites, fatal, or transplantation), was two of 3,357 patients (0.06%) in a global monotherapy database.
Monitor liver function tests regularly. In case of significantly increased transaminases without alternative explanation, such as viral hepatitis or progressing underlying malignancy, discontinue NEXAVAR [see Dosage and Administration (2.2)].
5.11 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animals, NEXAVAR may cause fetal harm when administered to a pregnant woman. Sorafenib caused embryo-fetal toxicities in animals at maternal exposures that were significantly lower than the human exposures at the recommended dose of 400 mg twice daily. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the last dose of NEXAVAR. Advise male patients with female partners of reproductive potential and pregnant partners to use effective contraception during treatment and for 3 months following the last dose of NEXAVAR [see Use in Specific Populations (8.1, 8.3)].
5.12 Impairment of Thyroid Stimulating Hormone Suppression in Differentiated Thyroid Carcinoma
NEXAVAR impairs exogenous thyroid suppression. In the DECISION (DTC) study, 99% of patients had a baseline thyroid stimulating hormone (TSH) level less than 0.5 mU/L. Elevation of TSH level above 0.5 mU/L was observed in 41% of NEXAVAR-treated patients as compared with 16% of those receiving placebo patients. For patients with impaired TSH suppression while receiving NEXAVAR, the median maximal TSH was 1.6 mU/L and 25% had TSH levels greater than 4.4 mU/L.
Monitor TSH levels monthly and adjust thyroid replacement medication as needed in patients with DTC.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are discussed elsewhere in the labeling:
- •
- Cardiovascular events [see Warnings and Precautions (5.1)]
- •
- Hemorrhage [see Warnings and Precautions (5.2)]
- •
- Hypertension [see Warnings and Precautions (5.3)]
- •
- Dermatologic toxicities [see Warnings and Precautions (5.4)]
- •
- Gastrointestinal perforation [see Warnings and Precautions (5.5)]
- •
- QT interval prolongation [see Warnings and Precautions (5.9) and Clinical Pharmacology (12.2)]
- •
- Drug-induced liver injury [see Warnings and Precautions (5.10)]
- •
- Impairment of TSH suppression in DTC [see Warnings and Precautions (5.12)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described reflect exposure to NEXAVAR in 955 patients who participated in placebo-controlled studies in hepatocellular carcinoma (N=297), advanced renal cell carcinoma (N=451), or differentiated thyroid carcinoma (N = 207). The most common adverse reactions (≥20%), which were considered to be related to NEXAVAR, in patients with HCC, RCC or DTC are diarrhea, fatigue, infection, alopecia, hand-foot skin reaction, rash, weight loss, decreased appetite, nausea, gastrointestinal and abdominal pains, hypertension, and hemorrhage.
Hepatocellular Carcinoma
Table 4 shows the percentage of patients in the SHARP (HCC) study experiencing adverse reactions that were reported in at least 10% of patients and at a higher rate in the NEXAVAR-treated group than in those receiving placebo.
Table 4: Adverse Reactions Reported in at Least 10% of Patients and at a Higher Rate in NEXAVAR Arm than the Placebo Arm – SHARP (HCC) Adverse Reaction1
NEXAVAR
N=297
Placebo
N=302
All
Grades
%Grade
3
%Grade
4
%All
Grades
%Grade
3
%Grade
4
%Any Adverse Reaction
98
39
6
96
24
8
Gastrointestinal
Diarrhea
55
10
<1
25
2
0
Anorexia
29
3
0
18
3
<1
Nausea
24
1
0
20
3
0
Vomiting
15
2
0
11
2
0
Constipation
14
0
0
10
0
0
Constitutional symptoms
Fatigue
46
9
1
45
12
2
Weight loss
30
2
0
10
1
0
Pain
Pain, abdomen
31
9
0
26
5
1
Dermatology/skin
Hand-foot skin reaction
21
8
0
3
<1
0
Rash/desquamation
19
1
0
14
0
0
Alopecia
14
0
0
2
0
0
Pruritus
14
<1
0
11
<1
0
Dry skin
10
0
0
6
0
0
Hepatobiliary/pancreas
Liver dysfunction
11
2
1
8
2
1
- 1
- Adverse reactions graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0).
Hypertension was reported in 9% of patients treated with NEXAVAR and 4% of those receiving placebo. Grade 3 hypertension was reported in 4% of NEXAVAR-treated patients and 1% of those receiving placebo.
Hemorrhage/bleeding was reported in 18% of those receiving NEXAVAR and 20% of patients receiving placebo. The rates of Grade 3 and 4 bleeding were also higher in patients receiving placebo (Grade 3 – 3% NEXAVAR and 5% placebo and Grade 4 – 2% NEXAVAR and 4% placebo). Bleeding from esophageal varices was reported in 2.4% in NEXAVAR-treated patients and 4% of patients receiving placebo.
Renal failure was reported in <1% of patients treated with NEXAVAR and 3% of patients receiving placebo. Clinical pancreatitis was reported in 1 of 297 NEXAVAR-treated patients (Grade 2).
The rate of adverse reactions (including those associated with progressive disease) resulting in permanent discontinuation was similar in both the NEXAVAR-treated patients and those receiving placebo (32% of NEXAVAR-treated patients and 35% of patients receiving placebo).
Laboratory test abnormalities reported in SHARP are presented in Table 5.
Table 5: Laboratory Test Abnormalities Reported in SHARP (HCC) Laboratory Parameter1
NEXAVAR
N=297
Placebo
N=302
All Grades
(%)
Grade 3 or 4 (%)
All Grades
(%)
Grade 3 or 4 (%)
Hypoalbuminemia
59
0
47
0
Elevated Lipase
40
9
37
9
Lymphopenia
47
NR
42
NR
- Thrombocytopenia
46
4
41
<1
Elevated INR
42
4
34
2
Hypophosphatemia
35
11
11
2
Elevated Amylase
34
2
29
2
Hypocalcemia
27
2.4
15
1
Hypokalemia
10
<1
6
<1
- 1-
- Laboratory parameters graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0).
NR = not reported
Renal Cell Carcinoma
Table 6 shows the percentage of patients in the TARGET (RCC) study experiencing adverse reactions that were reported in at least 10% of patients and at a higher rate in NEXAVAR-treated patients arm than in those receiving placebo.
The rate of adverse reactions (including those associated with progressive disease) resulting in permanent discontinuation was similar in both the NEXAVAR-treated patients and patients receiving placebo (10% and 8%, respectively).
Clinical pancreatitis was reported in 3 of 451 NEXAVAR-treated patients (one Grade 2 and two Grade 4).
Table 6: Adverse Reactions Reported in at Least 10% of Patients and at a Higher Rate in NEXAVAR Arm than the Placebo Arm – TARGET (RCC) Adverse Reaction1
NEXAVAR
N=451
Placebo
N=451
All Grades
%
Grade 3
%
Grade 4
%
All Grades
%
Grade 3
%
Grade 4
%
Any Adverse Reactions
95
31
7
86
22
6
Gastrointestinal symptoms
Diarrhea
43
2
0
13
<1
0
Nausea
23
<1
0
19
<1
0
Anorexia
16
<1
0
13
1
0
Vomiting
16
<1
0
12
1
0
Constipation
15
<1
0
11
<1
0
Dermatology/skin
Rash/desquamation
40
<1
0
16
<1
0
Hand-foot skin reaction
30
6
0
7
0
0
Alopecia
27
<1
0
3
0
0
Pruritus
19
<1
0
6
0
0
Dry skin
11
0
0
4
0
0
Constitutional symptoms
Fatigue
37
5
<1
28
3
<1
Weight loss
10
<1
0
6
0
0
Cardiovascular, General
Hypertension
17
3
<1
2
<1
0
Hemorrhage/bleeding
Hemorrhage – all sites
15
2
0
8
1
<1
Pulmonary
Dyspnea
14
3
<1
12
2
<1
Neurology
Neuropathy-sensory
13
<1
0
6
<1
0
Pain
Pain, abdomen
11
2
0
9
2
0
Pain, headache
10
<1
0
6
<1
0
Pain, joint
10
2
0
6
<1
0
- 1
- Adverse reactions graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0).
Laboratory test abnormalities reported in TARGET are presented in Table 7.
Table 7: Laboratory Test Abnormalities Reported in TARGET (RCC) Laboratory Parameter1
NEXAVAR
N=451
Placebo
N=451
All Grades
(%)
Grade 3 or 4 (%)
All Grades
(%)
Grade 3 or 4 (%)
Hypophosphatemia
45
13
11
3
Anemia
44
2
49
4
Elevated Lipase
41
12
30
7
Elevated Amylase
30
1
23
3
Lymphopenia
23
13
13
7
Neutropenia
18
5
10
2
- Thrombocytopenia
12
1
5
0
Hypocalcemia
12
2
8
<1
Hypokalemia
5
1
<1
<1
- 1
- Laboratory parameters graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0).
Differentiated Thyroid Carcinoma
The safety of NEXAVAR was evaluated in DECISION in 416 patients with locally recurrent or metastatic, progressive differentiated thyroid carcinoma (DTC) refractory to radioactive iodine (RAI) treatment randomized to receive 400 mg twice daily NEXAVAR (n=207) or matching placebo (n=209) until disease progression or intolerable toxicity in a double-blind trial [see Clinical Studies (14.3)]. The data described below reflect a median exposure to NEXAVAR for 46 weeks (range 0.3 to 135). The population exposed to NEXAVAR was 50% male, and had a median age of 63 years.
Dose interruptions for adverse reactions were required in 66% of patients receiving NEXAVAR and dose reductions were required in 64% of patients. Adverse reactions that resulted in treatment discontinuation were reported in 14% of NEXAVAR-treated patients compared to 1.4% of patients receiving placebo.
Table 8 shows the percentage of DTC patients experiencing adverse reactions at a higher rate in NEXAVAR-treated patients than in patients receiving placebo in the double-blind phase of the DECISION study. Grade 3 adverse reactions occurred in 53% of NEXAVAR-treated patients compared to 23% of patients receiving placebo. Grade 4 adverse reactions occurred in 12% of NEXAVAR-treated patients compared to 7% of patients receiving placebo.
Table 8: Selected Adverse Reactions Occurring at a Higher Incidence in NEXAVAR-Treated Patients [Between Arm Difference of ≥ 5% (All Grades)1 or ≥ 2% (Grades 3 and 4)] Adverse Reaction NEXAVAR
N = 207Placebo
N = 209All Grades
(%)Grades 3 and 4
(%)All Grades
(%)Grades 3 and 4
(%)Skin and subcutaneous tissue disorders
PPES5
69
19
8
0
Alopecia
67
0
8
0
Rash
35
5
7
0
Pruritus
20
0.5
11
0
Dry skin
13
0.5
5
0
Erythema
10
0
0.5
0
Hyperkeratosis
7
0
0
0
Gastrointestinal disorders
Diarrhea
68
6
15
1
Stomatitis3
24
2
3
0
Nausea
21
0
12
0
Abdominal pain2
20
1
7
1
Constipation
16
0
8
0.5
Oral pain4
14
0.5
6
0
Vomiting
11
0
3
0
Investigations
Weight loss
49
6
14
1
General disorders and administration site conditions
Fatigue
41
5
20
1
Asthenia
12
0
7
0
Pyrexia
11
1
5
0
Vascular disorders
Hypertension6
41
10
12
2
Metabolism and nutrition disorders
Decreased appetite
30
2
5
0
Nervous system disorders
Headache
17
0
6
0
Dysgeusia
6
0
0
0
Musculoskeletal and connective tissue disorders
Pain in extremity
15
1
7
0
Muscle spasms
10
0
3
0
Respiratory, thoracic and mediastinal disorders
Dysphonia
13
0.5
3
0
Epistaxis
7
0
1
0
Neoplasms benign, malignant and unspecified
Squamous cell carcinoma of skin
3
3
0
0
- 1
- National Cancer Institute Common Terminology Criteria for Adverse Events Version 3.0
- 2
- Includes the following terms: abdominal pain, abdominal discomfort, hepatic pain, esophageal pain, esophageal discomfort, abdominal pain lower, abdominal pain upper, abdominal tenderness, abdominal rigidity
- 3
- Includes the following terms: stomatitis, aphthous stomatitis, mouth ulceration, mucosal inflammation
- 4
- Includes the following terms: oral pain, oropharyngeal discomfort, glossitis, burning mouth syndrome, glossodynia
- 5
- Palmar-plantar erythrodysesthesia syndrome (Hand-foot skin reaction)
- 6
- Includes the following terms: hypertension, blood pressure increased, blood pressure systolic increased
The relative increase for the following laboratory abnormalities observed in NEXAVAR-treated patients as compared to patients receiving placebo in the DECISION study is similar to that observed in the RCC and HCC studies: lipase, amylase, hypokalemia, hypophosphatemia, neutropenia, lymphopenia, anemia, and thrombocytopenia. Hypocalcemia was more frequent and more severe in patients with DTC, especially those with a history of hypoparathyroidism, compared to patients with RCC or HCC. Other laboratory test abnormalities reported in DECISION are presented in Table 9
Table 9: Laboratory Test Abnormalities Reported in DECISION (DTC Laboratory Parameter1
NEXAVAR
N=207
Placebo
N=209
All Grades
(%)
Grade 3 or 4 (%)
All Grades
(%)
Grade 3 or 4 (%)
Elevated ALT
59
4
24
0
Elevated AST
54
2
15
0
Hypocalcemia
36
10
11
3
- 1
- Laboratory parameters graded according to National Cancer Institute Common Terminology Criteria for Adverse Events version 3.0 (NCI CTCAE v3.0).
Additional Data from Multiple Clinical Trials
The following additional drug-related adverse reactions and laboratory abnormalities were reported from clinical trials of NEXAVAR (very common 10% or greater, common 1 to less than 10%, uncommon 0.1% to less than 1%, rare less than 0.1 %):
Cardiovascular: Common: congestive heart failure*†, myocardial ischemia and/or infarction Uncommon: hypertensive crisis* Rare: QT prolongation*
Dermatologic: Very common: erythema Common: exfoliative dermatitis, acne, flushing, folliculitis, hyperkeratosis Uncommon: eczema, erythema multiforme
Digestive: Very common: increased lipase, increased amylase Common: mucositis, stomatitis (including dry mouth and glossodynia), dyspepsia, dysphagia, gastrointestinal reflux Uncommon: pancreatitis, gastritis, gastrointestinal perforations*, cholecystitis, cholangitis
Note that elevations in lipase are very common (41%, see below); a diagnosis of pancreatitis should not be made solely on the basis of abnormal laboratory values
General Disorders: Very common: infection, hemorrhage (including gastrointestinal* and respiratory tract* and uncommon cases of cerebral hemorrhage*), asthenia, pain (including mouth, bone, and tumor pain), pyrexia, decreased appetite Common: influenza-like illness
Hematologic: Very common: leukopenia, lymphopenia Common: anemia, neutropenia, thrombocytopenia Uncommon: INR abnormal
Hepatobiliary disorders: Rare: drug-induced liver injury (including hepatic failure and death)
Hypersensitivity: Uncommon: hypersensitivity reactions (including skin reactions and urticaria), anaphylactic reaction
Metabolic and Nutritional: Very common: hypophosphatemia Common: transient increases in transaminases, hypocalcemia, hypokalemia, hyponatremia, hypothyroidism Uncommon: dehydration, transient increases in alkaline phosphatase, increased bilirubin (including jaundice), hyperthyroidism
Musculoskeletal: Very common: arthralgia Common: myalgia, muscle spasms
Nervous System and Psychiatric: Common: depression, dysgeusia Uncommon: tinnitus, reversible posterior leukoencephalopathy*
Renal and Genitourinary: Common: renal failure, proteinuria Rare: nephrotic syndrome
Reproductive: Common: erectile dysfunction Uncommon: gynecomastia
Respiratory: Common: rhinorrhea Uncommon: interstitial lung disease-like events (includes reports of pneumonitis, radiation pneumonitis, acute respiratory distress, interstitial pneumonia, pulmonitis and lung inflammation)
In addition, the following medically significant adverse reactions were uncommon during clinical trials of NEXAVAR: transient ischemic attack, arrhythmia, and thromboembolism. For these adverse reactions, the causal relationship to NEXAVAR has not been established.
*adverse reactions may have a life-threatening or fatal outcome.
†reported in 1.9% of patients treated with NEXAVAR (N= 2276).
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of NEXAVAR. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic disorders: Thrombotic microangiopathy (TMA)
Dermatologic: Stevens-Johnson syndrome and toxic epidermal necrolysis (TEN)
Hypersensitivity: Angioedema
Musculoskeletal: Rhabdomyolysis, osteonecrosis of the jaw
Respiratory: Interstitial lung disease-like events (which may have a life-threatening or fatal outcome)
Vascular: Arterial (including aortic) aneurysms, dissections, and rupture
-
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on NEXAVAR
Strong CYP3A4 Inducers
The concomitant use of NEXAVAR with rifampin, a strong CYP3A4 inducer decreased the mean AUC of sorafenib, which may decrease the antitumor activity [seeClinical Pharmacology (12.3)]. Avoid concomitant use of NEXAVAR with strong CYP3A4 inducers, when possible, because these drugs can decrease the systemic exposure to sorafenib.
Neomycin
The concomitant use of NEXAVAR with neomycin decreased the mean AUC of sorafenib, which may decrease the antitumor activity. Avoid concomitant use of NEXAVAR with neomycin. The effects of other antibiotics on the pharmacokinetics of sorafenib have not been studied [see Clinical Pharmacology (12.3)].
7.2 Concomitant Use of Warfarin
The concomitant use of NEXAVAR and warfarin may increase the risk of bleeding or increased the INR. Monitor INR and for clinical bleeding episodes in patients taking warfarin while receiving NEXAVAR [see Warnings and Precautions (5.6)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action[see Clinical Pharmacology (12.1)], NEXAVAR may cause fetal harm when administered to a pregnant woman. There are no available data in pregnant women to inform a drug-associated risk. In animal reproduction studies, oral administration of sorafenib to pregnant rats and rabbits during the period of organogenesis resulted in embryo-fetal toxicities at maternal exposures that were significantly lower than human exposures at the recommended dose of 400 mg twice daily (see Data). Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In animal reproduction studies, sorafenib was teratogenic and induced embryo-fetal toxicity (including increased post-implantation loss, resorptions, skeletal retardations, and retarded fetal weight) when administered orally to pregnant rats and rabbits during the period of organogenesis. The effects occurred at doses considerably below the recommended human dose of 400 mg twice daily (approximately 500 mg/m2/day on a body surface area basis). Adverse intrauterine development effects were seen at doses >0.2 .mg/kg/day (1.2 mg/m2/day) in rats and ≥0.3 mg/kg/day (≥3.6 mg/m2/day) in rabbits. These doses result in exposures (AUC) that are approximately 0.008 times the AUC in patients at the recommended dose.
8.2 Lactation
Risk Summary
There are no data on the presence of sorafenib or its metabolites in human milk, or its effects on the breast-fed child or on milk production. Sorafenib was present in milk of lactating rats (see Data). Because of the potential for serious adverse reactions in a breastfed child from NEXAVAR, advise women not to breastfeed during treatment with NEXAVAR and for 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
NEXAVAR may cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to the initiation of NEXAVAR.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the last dose of NEXAVAR.
Males
Based on genotoxicity and findings in animal reproduction studies, advise males with female partners of reproductive potential and pregnant partners to use effective contraception during treatment with NEXAVAR and for 3 months following the last dose of NEXAVAR [see Use in Specific Populations (8.1), Nonclinical Toxicology (13.1)].
Infertility
Males
Based on findings in animal studies, NEXAVAR may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of NEXAVAR have not been established in pediatric patients.
Juvenile Animal Toxicity Data
Repeat dosing of sorafenib to young and growing dogs resulted in irregular thickening of the femoral growth plate at daily sorafenib doses ≥600 mg/m2 (approximately 0.3 times the AUC at the recommended human dose), hypocellularity of the bone marrow adjoining the growth plate at 200 mg/m2/day (approximately 0.1 times the AUC at the recommended human dose), and alterations of the dentin composition at 600 mg/m2/day. Similar effects were not observed in adult dogs when dosed for 4 weeks or less.
8.5 Geriatric Use
In total, 59% of HCC patients treated with NEXAVAR were age 65 years or older and 19% were 75 and older. In total, 32% of RCC patients treated with NEXAVAR were age 65 years or older and 4% were 75 and older. No differences in safety or efficacy were observed between older and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dose adjustment is necessary for patients with mild, moderate or severe renal impairment who are not on dialysis.The pharmacokinetics of sorafenib have not been studied in patients who are on dialysis [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment is necessary for patients with mild or moderate hepatic impairment. The pharmacokinetics of sorafenib have not been studied in patients with severe (Child-Pugh C) hepatic impairment [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
The adverse reactions observed at a dose of 800 mg twice daily (2 times the recommended dose) were primarily diarrhea and dermatologic. No information is available on symptoms of acute overdose in animals because of the saturation of absorption in oral acute toxicity studies conducted in animals.
In cases of suspected overdose, withhold NEXAVAR and institute supportive care.
-
11 DESCRIPTION
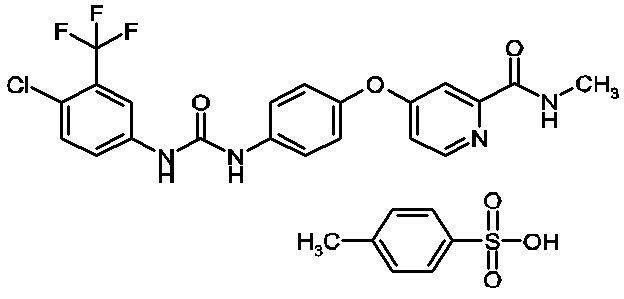
Sorafenib, a kinase inhibitor, is the tosylate salt of sorafenib. Sorafenib tosylate has the chemical name 4-(4-{3-[4-Chloro-3-(trifluoromethyl)phenyl]ureido}phenoxy)N2-methylpyridine-2-carboxamide 4-methylbenzenesulfonate. The molecular formula of sorafenib tosylate is C21H16ClF3N4O3 x C7H8O3S and the molecular weight of sorafenib tosylate is 637.0 g/mole. Its structural formula is:
Sorafenib tosylate is a white to yellowish or brownish solid. Sorafenib tosylate is practically insoluble in aqueous media, slightly soluble in ethanol and soluble in PEG 400.
NEXAVAR (sorafenib), for oral use is supplied as film-coated tablets containing 200 mg sorafenib equivalent to 274 mg sorafenib tosylate and the following inactive ingredients: croscarmellose sodium, ferric oxide red, hypromellose, magnesium stearate, microcrystalline cellulose, polyethylene glycol sodium lauryl sulphate, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Sorafenib is a kinase inhibitor that decreases tumor cell proliferation in vitro. Sorafenib was shown to inhibit multiple intracellular (c-CRAF, BRAF and mutant BRAF) and cell surface kinases (KIT, FLT- 3, RET, RET/PTC, VEGFR-1, VEGFR- 2, VEGFR- 3, and PDGFR-ß). Several of these kinases are thought to be involved in tumor cell signaling, angiogenesis and apoptosis. Sorafenib inhibited tumor growth of HCC, RCC, and DTC human tumor xenografts in immunocompromised mice. Reductions in tumor angiogenesis were seen in models of HCC and RCC upon sorafenib treatment, and increases in tumor apoptosis were observed in models of HCC, RCC, and DTC.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of NEXAVAR 400 mg twice daily on the QTc interval was evaluated in a multi-center, open-label, non-randomized trial in 53 patients with advanced cancer. No large changes in the mean QTc intervals (that is, >20 ms) from baseline were detected in the trial. After one 28-day treatment cycle, the largest mean QTc interval change of 8.5 ms (upper bound of two-sided 90% confidence interval, 13.3 ms) was observed at 6 hours post-dose on day 1 of cycle 2 [see Warnings and Precautions (5.9), Drug Interactions (7.3)].
12.3 Pharmacokinetics
Multiple doses of NEXAVAR for 7 days resulted in a 2.5- to 7-fold accumulation compared to a single dose. Steady-state plasma sorafenib concentrations were achieved within 7 days, with a peak-to-trough ratio of mean concentrations of less than 2.
The steady-state concentrations of sorafenib following administration of NEXAVAR 400 mg twice daily were evaluated in DTC, RCC and HCC patients. Patients with DTC have mean steady-state concentrations that are 1.8-fold higher than patients with HCC and 2.3-fold higher than those with RCC. The reason for increased sorafenib concentrations in DTC patients is unknown.
Mean Cmax and AUC increased less than proportionally beyond oral doses of 400 mg administered twice daily.
Absorption
After administration of NEXAVAR tablets, the mean relative bioavailability was 38–49% when compared to an oral solution. Following oral administration, sorafenib reached peak plasma levels in approximately 3 hours.
Elimination
The mean elimination half-life of sorafenib was approximately 25 to 48 hours.
Metabolism
Sorafenib undergoes oxidative metabolism by hepatic CYP3A4, as well as glucuronidation by UGT1A9.
Excretion
Sorafenib accounted for approximately 70–85% of the circulating analytes in plasma at steady-state. Eight metabolites of sorafenib have been identified, of which 5 have been detected in plasma. The main circulating metabolite of sorafenib, the pyridine N-oxide that comprises approximately 9–16% of circulating analytes at steady-state, showed in vitro potency similar to that of sorafenib.
Following oral administration of a 100 mg dose of a solution formulation of sorafenib, 96% of the dose was recovered within 14 days, with 77% of the dose excreted in feces and 19% of the dose excreted in urine as glucuronidated metabolites. Unchanged sorafenib, accounting for 51% of the dose, was found in feces but not in urine.
Specific Populations
A study of the pharmacokinetics of sorafenib indicated that the mean AUC of sorafenib in Asians (N=78) was 30% lower than in Whites (N=40). Sex and age do not have a clinically meaningful effect on the pharmacokinetics of sorafenib.
Patients with Renal Impairment
Mild (CLcr 50-80 mL/min), moderate (CLcr 30 - <50 mL/min), and severe (CLcr <30 mL/min) renal impairment do not affect the pharmacokinetics of sorafenib [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
Mild (Child-Pugh A) and moderate (Child-Pugh B) hepatic impairment do not affect the pharmacokinetics of sorafenib [see Use in Specific Populations (8.7)].
Drug Interactions Studies
Effect of Strong CYP3A4 Inhibitors on Sorafenib: Ketoconazole, a strong inhibitor of CYP3A4 and P-glycoprotein, administered at a dose of 400 not alter the mean AUC of a single oral dose of NEXAVAR 50 mg in healthy subjects.
Effect of Strong CYP3A4 Inducers on Sorafenib: Concomitant use of NEXAVAR with rifampin administered at a dose of 600 mg once daily for 5 days with a single oral dose of NEXAVAR 400 mg in healthy volunteers resulted in a 37% decrease in the mean AUC of sorafenib.
Effect of Neomycin on Sorafenib: Neomycin administered as an oral dose of 1 g three times daily for 5 days decreased the mean AUC of sorafenib by 54% in healthy subjects administered a single oral dose of NEXAVAR 400 mg.
Effect of Sorafenib on Other Drugs: NEXAVAR 400 mg twice daily for 28 days did not increase the systemic exposure of concomitantly administered midazolam (CYP3A4 substrate), dextromethorphan (CYP2D6 substrate), and omeprazole (CYP2C19 substrate) [see Clinical Pharmacology (12.3)].
Drugs that Increase Gastric pH: The aqueous solubility of sorafenib is pH dependent, with higher pH resulting in lower solubility. However, omeprazole, a proton pump inhibitor, administered at a dose of 40 mg once daily for 5 days, did not result in a clinically meaningful change in sorafenib single dose exposure.
In Vitro Studies
Sorafenib competitively inhibited CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4 in vitro. However, NEXAVAR 400 mg twice daily for 28 days with substrates of CYP3A4, CYP2D6 and CYP2C19 did not increase the systemic exposure of these substrates [see Drug Interactions (7.3)].
Sorafenib did not increase CYP1A2 and CYP3A4 activities, suggesting that sorafenib is unlikely to induce CYP1A2 or CYP3A4 in humans.
Sorafenib inhibits glucuronidation by UGT1A1 and UGT1A9 in vitro. NEXAVAR could increase the systemic exposure of concomitantly administered drugs that are UGT1A1 or UGT1A9 substrates.
Sorafenib inhibited P-glycoprotein in vitro. NEXAVAR could increase the concentrations of concomitantly administered drugs that are P-glycoprotein substrates.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with sorafenib. Sorafenib was clastogenic when tested in an in vitro mammalian cell assay (Chinese hamster ovary) in the presence of metabolic activation. Sorafenib was not mutagenic in the in vitro Ames bacterial cell assay or clastogenic in an in vivo mouse micronucleus assay. One intermediate in the manufacturing process, which is also present in the final drug substance (<0.15%), was positive for mutagenesis in an in vitro bacterial cell assay (Ames test) when tested independently.
No specific studies with sorafenib have been conducted in animals to evaluate the effect on fertility. However, results from the repeat-dose toxicity studies suggest there is a potential for sorafenib to impair reproductive function and fertility. Multiple adverse effects were observed in male and female reproductive organs, with the rat being more susceptible than mice or dogs. Typical changes in rats consisted of testicular atrophy or degeneration, degeneration of epididymis, prostate, and seminal vesicles, central necrosis of the corpora lutea and arrested follicular development. Sorafenib-related effects on the reproductive organs of rats were manifested at daily oral doses ≥ 5 mg/kg (30 mg/m2). This dose results in an exposure (AUC) that is approximately 0.5 times the AUC in patients at the recommended human dose. Dogs showed tubular degeneration in the testes at 30 mg/kg/day (600 mg/m2/day). This dose results in an exposure that is approximately 0.3 times the AUC at the recommended human dose. Oligospermia was observed in dogs at 60 mg/kg/day (1200 mg/m2/day) of sorafenib.
-
14 CLINICAL STUDIES
14.1 Hepatocellular Carcinoma
The SHARP (HCC) study (NCT00105443) was an international, multicenter, randomized, double blind, placebo-controlled trial in patients with unresectable hepatocellular carcinoma. Overall survival was the primary endpoint. A total of 602 patients were randomized; 299 to NEXAVAR 400 mg twice daily and 303 to matching placebo. All 602 randomized subjects were included in the ITT population for the efficacy analyses.
Demographics and baseline disease characteristics were similar between the NEXAVAR and placebo arms with regard to age, gender, race, performance status, etiology (including hepatitis B, hepatitis C and alcoholic liver disease), TNM stage (stage I: <1% vs. <1%; stage II: 10.4% vs. 8.3%; stage III: 37.8% vs. 43.6%; stage IV: 50.8% vs. 46.9%), absence of both macroscopic vascular invasion and extrahepatic tumor spread (30.1% vs. 30.0%), and Barcelona Clinic Liver Cancer stage (stage B: 18.1% vs. 16.8%; stage C: 81.6% vs. 83.2%; stage D: <1% vs. 0%). Liver impairment by Child-Pugh score was comparable between the NEXAVAR and placebo arms (Class A: 95% vs. 98%; B: 5% vs. 2%). Only one patient with Child-Pugh class C was entered. Prior treatments included surgical resection procedures (19.1% vs. 20.5%), locoregional therapies (including radiofrequency ablation, percutaneous ethanol injection and transarterial chemoembolization; 38.8% vs. 40.6%), radiotherapy (4.3% vs. 5.0%) and systemic therapy (3.0% vs. 5.0%).
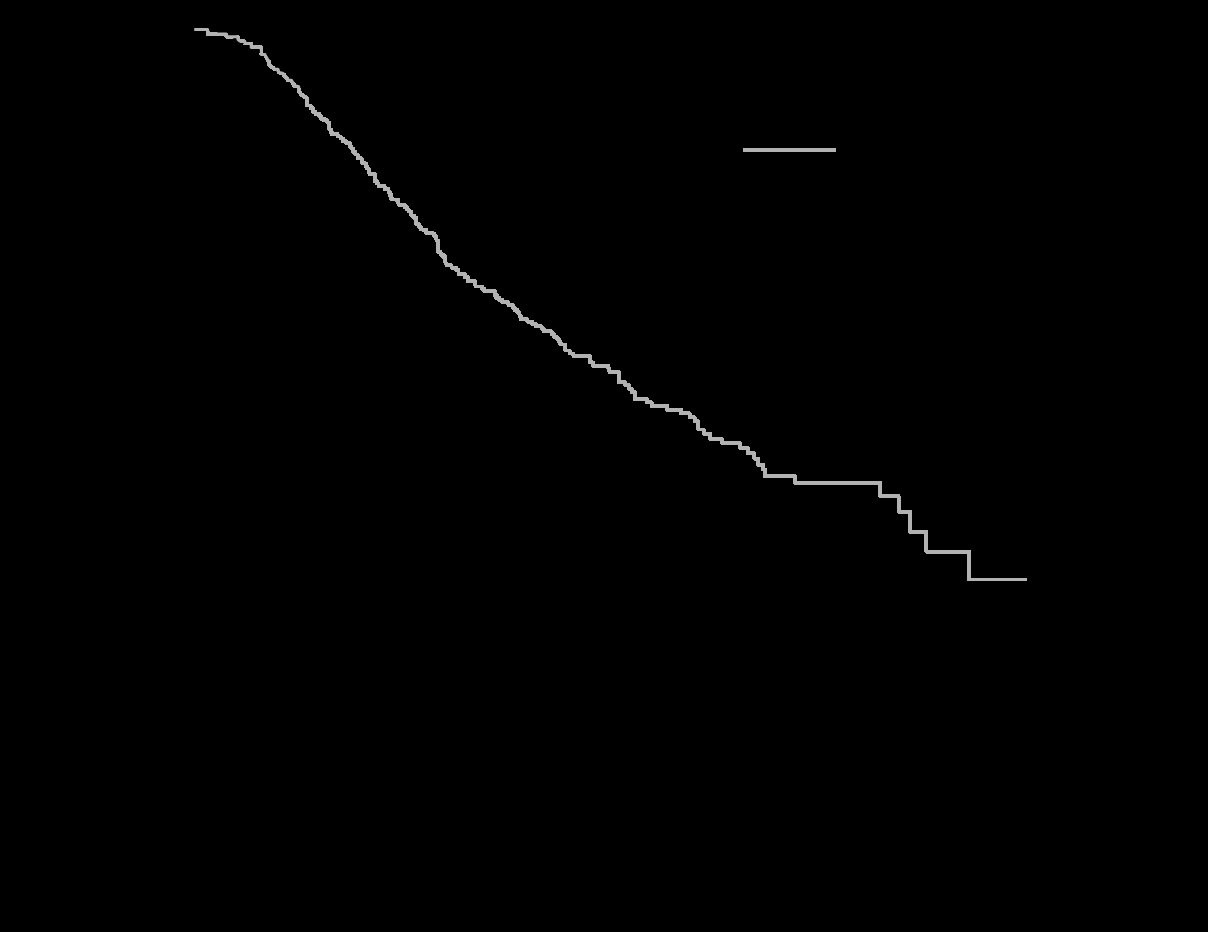
The trial was stopped for efficacy following a pre-specified second interim analysis for survival showing a statistically significant advantage for NEXAVAR over placebo for overall survival (HR: 0.69, p= 0.00058) (see Table 10 and Figure 1). This advantage was consistent across all subsets analyzed.
Final analysis of time to tumor progression (TTP) based on data from an earlier time point (by independent radiologic review) also was significantly longer in the NEXAVAR arm (HR: 0.58, p=0.000007) (see Table 10).
Table 10: Efficacy Results from SHARP (HCC) Efficacy Parameter
NEXAVAR
(N=299)
Placebo
(N=303)
Overall Survival
Number of Events
143
178
Median, months
10.7
7.9
(95% CI)
(9.4, 13.3)
(6.8, 9.1)
Hazard Ratio1 (95% CI)
0.69 (0.55, 0.87)
P-value (log-rank test2)
0.00058
Time to Progression 3
Number of Events
107
156
Median, months
5.5
2.8
(95% CI)
(4.1, 6.9)
(2.7, 3.9)
Hazard Ratio1 (95% CI)
0.58
(0.45, 0.74)
P-value (log-rank test2)
0.000007
CI=Confidence interval
- 1
- Hazard ratio, sorafenib/placebo, stratified Cox model
- 2
- Stratified log rank (for the interim analysis of survival, the stopping boundary one-sided alpha = 0.0077)
- 3
- The time-to-progression (TTP) analysis, based on independent radiologic review, was based on data from an earlier time point than the survival analysis
14.2 Renal Cell Carcinoma
The safety and efficacy of NEXAVAR in the treatment of advanced renal cell carcinoma (RCC) were studied in the following two randomized controlled clinical trials.
TARGET
TARGET (NCT00073307) was an international, multicenter, randomized, double blind, placebo-controlled trial in patients with advanced renal cell carcinoma who had received one prior systemic therapy. Primary study endpoints included overall survival and progression-free survival (PFS). Tumor response rate was a secondary endpoint. The PFS analysis included 769 patients, per protocol, stratified by MSKCC (Memorial Sloan Kettering Cancer Center) prognostic risk category (low or intermediate) and country and randomized to NEXAVAR 400 mg twice daily (N=384) or to placebo (N=385).
Table 11 summarizes the demographic and disease characteristics of the study population analyzed. Baseline demographics and disease characteristics were well balanced for both treatment groups. The median time from initial diagnosis of RCC to randomization was 1.6 and 1.9 years for the NEXAVAR and placebo arms, respectively.
Table 11: Demographic and Disease Characteristics – TARGET (RCC) Characteristics
NEXAVAR
N=384
Placebo
N=385
N
(%)
N
(%)
Gender
- Male
267
(70)
287
(75)
- Female
116
(30)
98
(25)
Race
- White
276
(72)
278
(73)
- Black/Asian/
11
(3)
10
(2)
- Not reported 1
97
(25)
97
(25)
Age group
- < 65 years
255
(67)
280
(73)
- ≥ 65 years
127
(33)
103
(27)
ECOG performance status at baseline
- 0
184
(48)
180
(47)
- 1
191
(50)
201
(52)
- 2
6
(2)
1
(<1)
- Not reported
3
(<1)
3
(<1)
MSKCC prognostic risk category
- Low
200
(52)
194
(50)
- Intermediate
184
(48)
191
(50)
Prior IL-2 and/or interferon
- Yes
319
(83)
313
(81)
- No
65
(17)
72
(19)
- 1
- Race was not collected from the 186 patients enrolled in France due to local regulations. In 8 other patients, race was not available at the time of analysis.
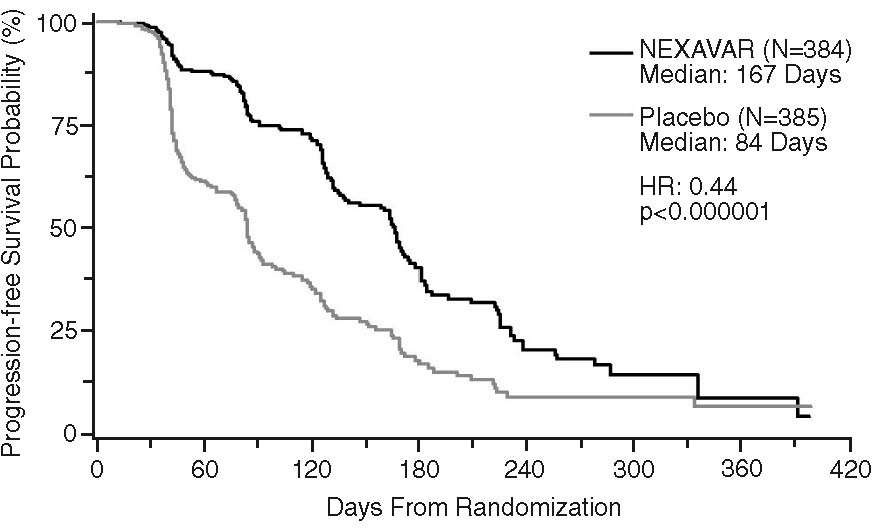
Progression-free survival, defined as the time from randomization to progression or death from any cause, whichever occurred earlier, was evaluated by blinded independent radiological review using RECIST criteria. Figure 2 depicts Kaplan-Meier curves for PFS. The PFS analysis was based on a two-sided Log-Rank test stratified by MSKCC prognostic risk category and country.
- NOTE: HR is from Cox regression model with the following covariates: MSKCC prognostic risk category and country. P-value is from two-sided Log-Rank test stratified by MSKCC prognostic risk category and country.
The median PFS for patients randomized to NEXAVAR was 167 days compared to 84 days for patients randomized to placebo. The estimated hazard ratio (immediate risk of progression or death with NEXAVAR compared to placebo) was 0.44 (95% CI: 0.35, 0.55).
A series of patient subsets were examined in exploratory univariate analyses of PFS. The subsets included age above or below 65 years, ECOG PS 0 or 1, MSKCC prognostic risk category, whether the prior therapy was for progressive metastatic disease or for an earlier disease setting and time from diagnosis of less than or greater than 1.5 years. The effect of NEXAVAR on PFS was consistent across these subsets, including patients with no prior IL-2 or interferon therapy (N=137; 65 patients receiving NEXAVAR and 72 placebo), for whom the median PFS was 172 days in the NEXAVAR arm compared to 85 days in the placebo arm.
Tumor response was determined by independent radiologic review according to RECIST criteria. Overall, of 672 patients who were evaluable for response, 7 (2%) patients in the NEXAVAR and no (0%) patients in the placebo arms had a confirmed partial response. Thus the gain in PFS primarily reflects the stable disease population.
At the time of a planned interim survival analysis, based on 220 deaths, overall survival was longer for those randomized to NEXAVAR compared with placebo with a hazard ratio of 0.72. This analysis did not meet the prespecified criteria for statistical significance. Additional analyses are planned as the survival data mature.
BAY43-9006
BAY43-9006 (NCT00101413) was a randomized discontinuation trial in patients with metastatic malignancies, including RCC. The primary endpoint was the percentage of randomized patients remaining progression-free at 24 weeks. All patients received NEXAVAR for the first 12 weeks. Radiologic assessment was repeated at week 12. Patients with <25% change in bi-dimensional tumor measurements from baseline were randomized to NEXAVAR or placebo for a further 12 weeks. Patients who were randomized to placebo were permitted to cross over to open-label NEXAVAR upon progression. Patients with tumor shrinkage ≥25% continued NEXAVAR, whereas patients with tumor growth ≥25% discontinued treatment.
A total of 202 patients with advanced RCC were enrolled into BAY43-9006, including patients who had received no prior therapy and patients with tumor histology other than clear cell carcinoma. After the initial 12 weeks of NEXAVAR, 79 patients with RCC continued on open-label NEXAVAR, and 65 patients were randomized to NEXAVAR or placebo. After an additional 12 weeks, at week 24, for the 65 randomized patients, the progression-free rate was significantly higher in patients randomized to NEXAVAR (16/32, 50%) than in patients randomized to placebo (6/33, 18%) (p=0.0077). Progression-free survival was significantly longer in the NEXAVAR arm (163 days) than in the those randomized to placebo (41 days) (p=0.0001, HR=0.29).
14.3 Differentiated Thyroid Carcinoma
The safety and effectiveness of NEXAVAR was evaluated in a multicenter, randomized (1:1), double-blind, placebo-controlled trial (DECISION; NCT00984282) conducted in 417 patients with locally recurrent or metastatic, progressive differentiated thyroid carcinoma (DTC) refractory to radioactive iodine (RAI) treatment. Randomization was stratified by age (< 60 years versus ≥ 60 years) and geographical region (North America, Europe, and Asia). All 417 subjects were included in the ITT population for the efficacy analyses
All patients were required to have actively progressing disease defined as progression within 14 months of enrollment. RAI-refractory disease was defined based on four criteria that were not mutually exclusive. All RAI treatments and diagnostic scans were to be performed under conditions of a low iodine diet and adequate TSH stimulation. Following are the RAI-refractory criteria and the proportion of patients in the study that met each one: a target lesion with no iodine uptake on RAI scan (68%); tumors with iodine uptake and progression after RAI treatment within 16 months of enrollment (12%); tumors with iodine uptake and multiple RAI treatments with the last treatment greater than 16 months prior to enrollment, and disease progression after each of two RAI treatments administered within 16 months of each other (7%); cumulative RAI dose ≥ 600 mCi administered (34%). The major efficacy outcome measure was progression-free survival (PFS) as determined by a blinded, independent radiological review using a modified Response Evaluation Criteria in Solid Tumors v. 1.0 (RECIST). RECIST was modified by inclusion of clinical progression of bone lesions based on the need for external beam radiation (4.4% of progression events). Additional efficacy outcomes measures included overall survival (OS), tumor response rate, and duration of response.
Patients were randomized to receive NEXAVAR 400 mg twice daily (n=207) or placebo (n=210). Of the 417 patients randomized, 48% were male, the median age was 63 years, 61% were 60 years or older, 60% were white, 62% had an ECOG performance status of 0, and 99% had undergone thyroidectomy. The histological diagnoses were papillary carcinoma in 57%, follicular carcinoma (including Hürthle cell) in 25%, and poorly differentiated carcinoma in 10%, and other in 8% of the study population. Metastases were present in 96% of the patients: lungs in 86%, lymph nodes in 51%, and bone in 27%. The median cumulative RAI activity administered prior to study entry was 400 mCi.
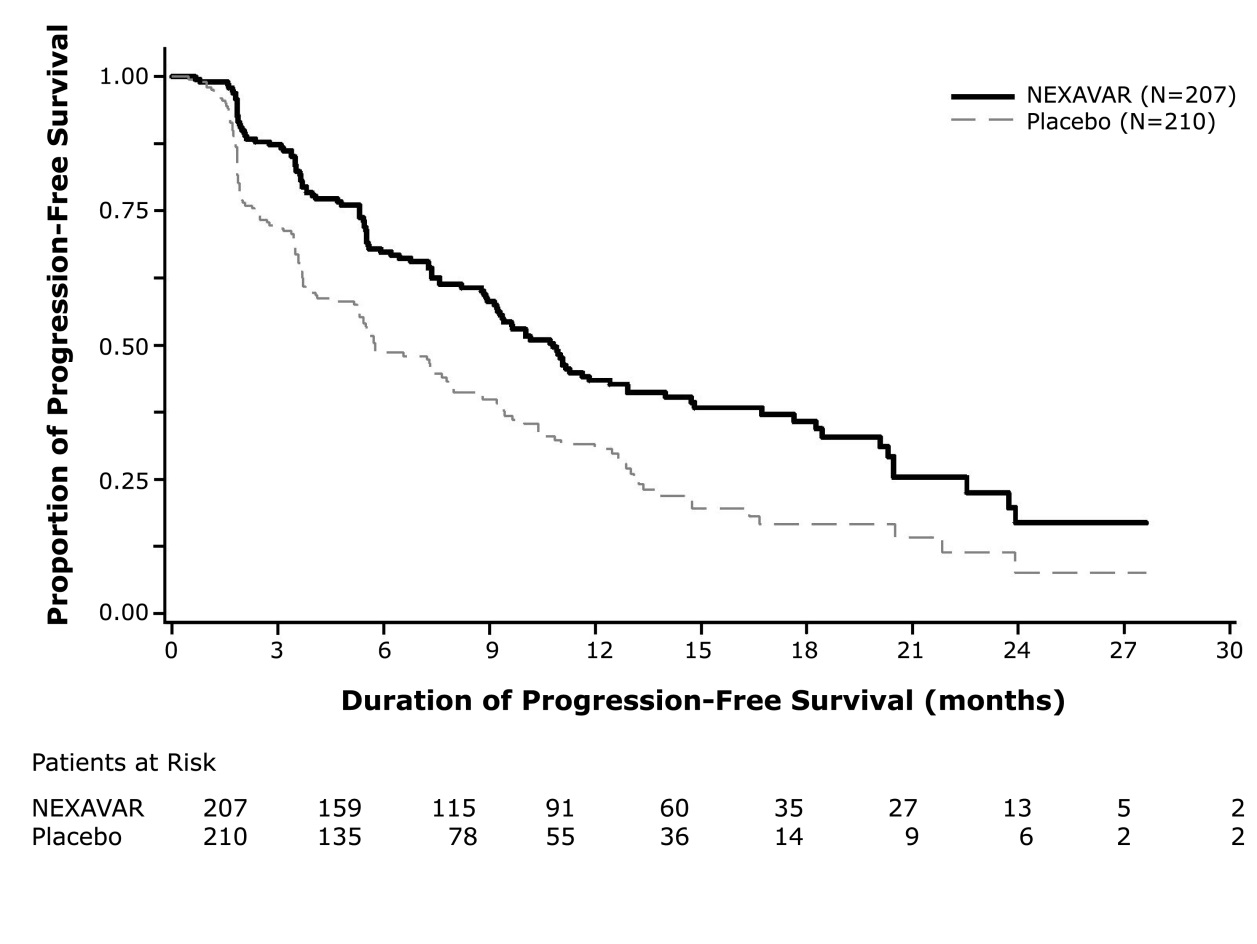
A statistically significant prolongation of PFS was demonstrated for NEXAVAR-treated patients compared to those receiving placebo (Figure 3); no statistically significant difference was seen in the final overall survival (OS) analysis (Table 12). Crossover to open label NEXAVAR occurred in 161 (77%) patients randomized to placebo after investigator-determined disease progression
Table 12: Efficacy Results from DECISION in Differentiated Thyroid Carcinoma NEXAVAR
N=207Placebo
N=210Progression-free Survival1
Number of Deaths or Progression
113 (55%)
136 (65%)
Median PFS in Months (95% CI)
10.8 (9.1, 12.9)
5.8 ( 5.3, 7.8)
Hazard Ratio (95% CI)
0.59 (0.46, 0.76)
P-value 2
<0.001
Overall Survival3
Number of Deaths
103 (49.8%)
109 (51.9%)
Median OS in Months (95% CI)
42.8 (34.6, 52.6)
39.4 (32.7, 51.4)
Hazard Ratio (95% CI)
0.92 (0.71, 1.21)
P-value2
0.570
Objective Response
Number of Objective Responders 4
24 (12%)
1 (0.5%)
(95% CI)
(7.6%, 16.8%)
(0.01%, 2.7%)
Median Duration of Response in Months (95% CI)
10.2 (7.4, 16.6)
NE
- 1
- Independent radiological review
- 2
- Two-sided log-rank test stratified by age (< 60 years, ≥ 60 years) and geographic region (North America, Europe, Asia)
- 3
- Conducted after 212 events, which occurred 36 months after the primary PFS analysis.
- 4
- All objective responses were partial responses
- NR = Not Reached, CI = Confidence interval, NE = Not Estimable
Figure 3: Kaplan-Meier Curve of Progression-Free Survival in DECISION (DTC)
Figure 3: Kaplan-Meier Curve of Progression-Free Survival in DECISION (DTC)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
NEXAVAR is supplied in bottles of 120:
- •
- 200 mg, round, biconvex, red film-coated tablets, debossed with the “Bayer cross” on one side and “200” on the other side. NDC 50419-488-58
- •
- 200 mg, round, faceted, biconvex, red film-coated tablets, debossed with the “Bayer cross” on one side and “200” on the other side. NDC 50419-489-01
Store at 20°C to 25° C (68°F to 77° F); excursions permitted to 15°C to 30° C (59°F to 86° F) [see USP controlled room temperature]. Store in a dry place.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read FDA-approved patient labeling (Patient Information).
Cardiovascular Events
Discuss with patients that cardiac ischemia and/or infarction and congestive heart failure, have been reported during NEXAVAR treatment, and that they should immediately report any episodes of chest pain or other symptoms of cardiac ischemia or congestive heart failure [see Warnings and Precautions (5.1)].
Bleeding
Inform patients that NEXAVAR can increase the risk of bleeding and that they should promptly report any episodes of bleeding [see Warnings and Precautions (5.2)].
Inform patients that bleeding or elevations in the International Normalized Ratio (INR) have been reported in some patients taking warfarin while on NEXAVAR and that their INR should be monitored regularly [see Warnings and Precautions (5.6)].
Hypertension
Inform patients that hypertension can develop during NEXAVAR treatment, especially during the first six weeks of therapy, and that blood pressure should be monitored regularly during treatment [see Warnings and Precautions (5.3)].
Skin Reactions
Advise patients of the possible occurrence of hand-foot skin reaction and rash during NEXAVAR treatment and appropriate countermeasures [see Warnings and Precautions (5.4)].
Gastrointestinal Perforation
Advise patients that cases of gastrointestinal perforation have been reported in patients taking NEXAVAR [see Warnings and Precautions (5.5)].
Risk of Impaired Wound Healing
Advise patients that NEXAVAR may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions (5.7)].
QT Interval Prolongation
Inform patients with a history of prolonged QT interval that NEXAVAR can worsen the condition [see Warnings and Precautions (5.9) and Clinical Pharmacology (12.2)].
Drug-Induced Liver Injury
Inform patients that NEXAVAR can cause hepatitis which may result in hepatic failure and death. Advise patients that liver function tests should be monitored regularly during treatment and to report signs and symptoms of hepatitis [see Warnings and Precautions (5.10)].
Embryo-Fetal Toxicity
Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of pregnancy [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with NEXAVAR and for 6 months after the last dose. Advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment with NEXAVAR and for 3 months after receiving the last dose of NEXAVAR [see Warnings and Precautions (5.11), Use in Specific Populations (8.1, 8.3)].
Lactation
Advise patients not to breastfeed while taking NEXAVAR and for 2 weeks after receiving the last dose of NEXAVAR [see Use in Specific Populations (8.2)].
Missed Doses
Instruct patients that if a dose of NEXAVAR is missed, the next dose should be taken at the regularly scheduled time, and not double the dose. Instruct patients to contact their healthcare provider immediately if they take too much NEXAVAR.
-
Patient Package Insert
Patient Information
NEXAVAR® (NEX-A-VAR)
(sorafenib)
tablets, oral
What is NEXAVAR?
NEXAVAR is a prescription medicine used to treat:
- •
- a type of liver cancer called hepatocellular carcinoma (HCC) that cannot be removed by surgery
- •
- a type of kidney cancer called renal cell carcinoma (RCC)
- •
- a type of thyroid cancer called differentiated thyroid carcinoma (DTC) that can no longer be treated with radioactive iodine and is progressing
It is not known if NEXAVAR is safe and effective in children.
Do not take NEXAVAR if you:
- •
- are allergic to sorafenib or any of the other ingredients in NEXAVAR. See the end of this leaflet for a complete list of ingredients in NEXAVAR.
- •
- have squamous cell lung cancer and receive carboplatin and paclitaxel.
Before taking NEXAVAR, tell your healthcare provider about all of your medical conditions including if you:
- •
- have heart problems including a condition called “congenital long QT syndrome”
- •
- have chest pain
- •
- have abnormal magnesium, potassium, or calcium blood levels
- •
- have bleeding problems
- •
- have high blood pressure
- •
- plan to have surgery or have had a recent surgery. You should stop taking NEXAVAR at least 2 weeks before planned surgery. See “What are the possible side effects of NEXAVAR?”
- •
- are pregnant or plan to become pregnant. NEXAVAR may harm your unborn baby. Tell your healthcare provider right away if you become pregnant during treatment with NEXAVAR.
- •
- For females who are able to become pregnant:
- o
- Your healthcare should do a pregnancy test before you start treatment with NEXAVAR.
- o
- Use effective birth control (contraception) during your treatment with NEXAVAR and for 6 months after the last dose of NEXAVAR.
- •
- For males with female partners who are able to become pregnant:
- o
- Use effective birth control (contraception) during your treatment with NEXAVAR and for 3 months after the last dose of NEXAVAR.
- •
- are breast feeding or plan to breastfeed. It is not known if NEXAVAR passes into your breast milk. Do not breastfeed during treatment with NEXAVAR and for 2 weeks after receiving the last dose of NEXAVAR.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Especially tell your healthcare provider if you take the medicine warfarin.
How should I take NEXAVAR?
- •
- Take NEXAVAR exactly as your healthcare provider tells you to take it.
- •
- Take NEXAVAR 2 times a day. Your healthcare provider may change your dose, temporarily stop treatment or completely stop treatment with NEXAVAR if you have side effects.
- •
- Take NEXAVAR without food (at least 1 hour before or 2 hours after a meal).
- •
- If you miss a dose of NEXAVAR, skip the missed dose, and take your next dose at your regular time. Do not double your dose of NEXAVAR.
- •
- If you take too much NEXAVAR call your doctor or go to the nearest hospital emergency room right away.
What are the possible side effects of NEXAVAR?
NEXAVAR may cause serious side effects, including:
- •
- decreased blood flow to the heart, heart attack and heart failure. Get emergency help right away if you get symptoms such as chest pain, shortness of breath, racing heartbeat, swelling in lower legs, feet and abdomen, feel lightheaded or faint, tiredness, nausea, vomiting, or sweat a lot.
- •
- increased risk of bleeding. Bleeding is a common side effect of NEXAVAR that can be serious and can lead to death. Tell your healthcare provider right away if you have any signs of bleeding during treatment with NEXAVAR:
- o
- vomiting blood or if your vomit looks like coffee-grounds
- o
- pink or brown urine
- o
- red or black (looks like tar) stools
- o
- coughing up blood or blood clots
- o
- heavier than normal menstrual cycle
- o
- unusual vaginal bleeding
- o
- frequent nose bleeds
- o
- bruising
- •
- high blood pressure. High blood pressure is a common side effect of NEXAVAR and can be serious. Your blood pressure should be checked every week during the first 6 weeks of starting NEXAVAR. Your blood pressure should be checked regularly and any high blood pressure should be treated during treatment with NEXAVAR.
- •
- skin problems. A condition called hand-foot skin reactions and skin rash are common with NEXAVAR treatment and can be severe. NEXAVAR may also cause severe skin and mouth reactions that can be life-threatening. Tell your healthcare provider if you have any of the following symptoms:
- o
- skin rash
- o
- skin redness
- o
- pain or swelling
- o
- blistering and peeling of your skin
- o
- blistering and peeling on the inside of your mouth
- o
- blisters on the palms of your hand or soles of your feet
- •
- an opening in the wall of your stomach or intestines (gastrointestinal perforation). Tell your healthcare provider right away if you get fever, nausea, vomiting or severe stomach (abdominal) pain.
- •
- risk of wound healing problems. Wounds may not heal properly during NEXAVAR treatment. Tell your healthcare provider if you plan to have any surgery before starting or during treatment with NEXAVAR.
- o
- You should stop taking NEXAVAR at least 10 days before planned surgery.
- o
- Your healthcare provider should tell you when you may start taking NEXAVAR again after surgery.
- •
- changes in the electrical activity of your heart called QT prolongation. QT prolongation can cause irregular heartbeats that can be life-threatening. Your healthcare provider may do tests during your treatment with NEXAVAR to check the levels of potassium, magnesium, and calcium in your blood, and check the electrical activity of your heart with an electrocardiogram (ECG). Tell your healthcare provider right away if you feel faint, lightheaded, dizzy or feel your heart beating irregularly or fast during your treatment with NEXAVAR.
- •
- liver problems (drug-induced hepatitis). NEXAVAR may cause liver problems that may lead to liver failure and death. Your healthcare provider will do blood tests to check your liver function regularly during your treatment with NEXAVAR. Tell your healthcare provider right away if you develop any of the following symptoms:
- o
- yellowing of your skin or the whites of your eyes
- o
- dark “tea-colored” urine
- o
- light-colored bowel movements (stools)
- o
- worsening nausea or vomiting
- o
- pain on the right side of your stomach area
- o
- bleeding or bruising more easily than normal
- o
- loss of appetite
- •
- change in thyroid hormone levels. If you have differentiated thyroid cancer, you can have changes in your thyroid hormone levels during treatment with NEXAVAR. Your healthcare provider may need to change your dose of thyroid medicine during treatment with NEXAVAR. Your healthcare provider should check your thyroid hormone levels every month during treatment with NEXAVAR.
The most common side effects of NEXAVAR include:
- o
- diarrhea (frequent or loose bowel movements)
- o
- tiredness
- o
- infection
- o
- hair thinning or patchy hair loss
- o
- rash
- o
- weight loss
- o
- loss of appetite
- o
- nausea
- o
- stomach-area (abdomen) pain
- o
- low blood calcium levels in people with differentiated thyroid cancer
NEXAVAR may cause fertility problems in males. This may affect your ability to father a child. Talk to your healthcare provider if this is a concern for you.
These are not all of the possible side effects of NEXAVAR. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store NEXAVAR?
- •
- Store NEXAVAR tablets at room temperature between 68° F to 77° F (20° C to 25° C).
- •
- Store NEXAVAR tablets in a dry place.
Keep NEXAVAR and all medicines out of the reach of children.
General information about the safe and effective use of NEXAVAR
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use NEXAVAR for a condition for which it is not prescribed. Do not give NEXAVAR to other people even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about NEXAVAR that is written for health professionals.
What are the ingredients in NEXAVAR?
Active Ingredient: sorafenib tosylate
Inactive Ingredients: croscarmellose sodium, ferric oxide red, hypromellose, magnesium stearate, microcrystalline cellulose, sodium lauryl sulphate, polyethylene glycol, and titanium dioxide.
- Manufactured for:
Bayer HealthCare Pharmaceuticals Inc.
Whippany, NJ 07981
© 2015 Bayer HealthCare Pharmaceuticals Inc.
For more information, go to www.NEXAVAR.com, or call 1-866-639-2827.
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised 6/2020
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
NEXAVAR
sorafenib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50419-488 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SORAFENIB (UNII: 9ZOQ3TZI87) (SORAFENIB - UNII:9ZOQ3TZI87) SORAFENIB 200 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) SODIUM LAURYL SULFATE (UNII: 368GB5141J) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color RED Score no score Shape ROUND (ROUND) Size 10mm Flavor Imprint Code 200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50419-488-58 120 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 12/20/2005 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021923 12/20/2005 NEXAVAR
sorafenib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50419-489 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength SORAFENIB (UNII: 9ZOQ3TZI87) (SORAFENIB - UNII:9ZOQ3TZI87) SORAFENIB 200 mg Inactive Ingredients Ingredient Name Strength CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) SODIUM LAURYL SULFATE (UNII: 368GB5141J) MAGNESIUM STEARATE (UNII: 70097M6I30) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color RED Score no score Shape ROUND (faceted) Size 10mm Flavor Imprint Code 200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50419-489-01 120 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 10/02/2023 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021923 10/02/2023 Labeler - Bayer HealthCare Pharmaceuticals Inc. (005436809) Establishment Name Address ID/FEI Business Operations Sharp Corporation 143696495 PACK(50419-488, 50419-489) , LABEL(50419-488, 50419-489) Establishment Name Address ID/FEI Business Operations Bayer AG 314947622 ANALYSIS(50419-488, 50419-489) , MANUFACTURE(50419-488, 50419-489) , PACK(50419-488, 50419-489) , PARTICLE SIZE REDUCTION(50419-488, 50419-489) Establishment Name Address ID/FEI Business Operations Bayer AG 323208116 ANALYSIS(50419-488, 50419-489) , API MANUFACTURE(50419-488, 50419-489) Establishment Name Address ID/FEI Business Operations Patheon France S.A.S. 543127229 MANUFACTURE(50419-488, 50419-489)