Label: LEVETIRACETAM injection, solution

-
Contains inactivated NDC Code(s)
NDC Code(s): 14789-400-05 - Packager: Nexus Pharmaceuticals Inc
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated August 9, 2011
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LEVETIRACETAM safely and effectively.
See full prescribing information for LEVETIRACETAM.
Levetiracetam Injection for intravenous use.
Initial U.S. Approval : 1999RECENT MAJOR CHANGES
INDICATIONS AND USAGE
LEVETIRACETAM injection is an antiepileptic drug indicated for adjunct therapy in adults (≥16 years of age) with the following seizure types when oral administration of LEVETIRACETAM is temporarily not feasible:
DOSAGE AND ADMINISTRATION
LEVETIRACETAM injection should be diluted in 100 mL of a compatible diluent and administered intravenously as a 15-minute infusion (2.1).
Initial Exposure To LEVETIRACETAM (2.2):
- Partial Onset Seizures: 1000 mg/day, given as twice-daily dosing (500 mg twice daily), increased as needed and as tolerated in increments of 1000 mg/day additional every 2 weeks to a maximum recommended daily dose of 3000 mg.
- Myoclonic Seizures in Patients with Juvenile Myoclonic Epilepsy: 1000 mg/day, given as twice-daily dosing (500 mg twice daily), increased by 1000 mg/day every 2 weeks to the recommended daily dose of 3000 mg. The effectiveness of doses lower than 3000 mg/day has not been adequately studied.
- Replacement Therapy (2.3): When switching from oral LEVETIRACETAM, the initial total daily intravenous dosage of LEVETIRACETAM should be equivalent to the total daily dosage and frequency of oral LEVETIRACETAM. At the end of the intravenous treatment period, the patient may be switched to equivalent daily dosage and frequency of the intravenous administration.
See full prescribing information for dosing instructions (2.5), adult patients with impaired renal function (2.6), and compatibility and stability (2.7).
DOSAGE FORMS AND STRENGTHS
- 500 mg/5 ml single-use vial (3)
CONTRAINDICATIONS
None (4)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
- Most common adverse reactions (difference in incidence rate is ≥5% between LEVETIRACETAM-treated patients and placebo-treated patients and occurred more frequently in LEVETIRACETAM-treated patients) include: somnolence, asthenia, infection, and dizziness (6.1) .
- Important behavioral adverse reactions (incidence of • LEVETIRACETAM-treated patients > placebo-treated patients, but <5%) include depression, nervousness, anxiety, and emotional lability (6.1) .
To Report SUSPECTED ADVERSE REACTIONS, contact Nexus Pharma at 877-913-2720 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatchUSE IN SPECIFIC POPULATIONS
- To ensure broad program access and reach, either a healthcare provider or the patient can initiate enrollment in the North American Antieplileptic Drug Pregnancy Registry by calling (888) 233-2334 (toll free). (8.1).
- A dose adjustment is recommended for patient’s estimated creatinine clearance (8.6).
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2010
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Partial Onset Seizures
1.2 Myoclonic Seizures in Patients with Juvenile Myoclonic Epilepsy
2 DOSAGE AND ADMINISTRATION
2.1 General Information
2.2 Initial Exposure to LEVETIRACETAM
2.3 Replacement Therapy
2.4 Switching to Oral Dosing
2.5 Dosing Instructions
2.6 Adult Patients with Impaired Renal Function
2.7 Compatibility and Stability
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Neuropsychiatric Adverse Reactions
5.2 Withdrawal Seizures
5.3 Hematologic Abnormalities
5.4 Hepatic Abnormalities
5.5 Laboratory Tests
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 General Information
7.2 Phenytoin
7.3 Valproate
7.4 Other Antiepileptic Drug
7.5 Oral Contraceptives
7.6 Digoxin
7.7 Warfarin
7.8 Probenecid
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Labor and Delivery
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Use in Patients with Impaired Renal Function
9 DRUG ABUSE AND DEPENDENCE
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Pharmacology and/or Toxicology
14 CLINICAL STUDIES
14.1 Partial Onset Seizures
14.2 Myoclonic Seizures in Patients with Juvenile Myoclonic Epilepsy
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Information
LEVETIRACETAM injection is for intravenous use only and must be diluted prior to administration. LEVETIRACETAM injection (500 mg/5 mL) should be diluted in 100 mL of a compatible diluent [see Dosage and Administration (2.7)] and administered intravenously as a 15-minute IV infusion.
Product with particulate matter or discoloration should not be used.
Any unused portion of the LEVETIRACETAM injection vial contents should be discarded.
2.2 Initial Exposure to LEVETIRACETAM
LEVETIRACETAM can be initiated with either intravenous or oral administration.
Partial Onset Seizures - In clinical trials of oral LEVETIRACETAM, daily doses of 1000 mg, 2000 mg, and 3000 mg, given as twice-daily dosing, were shown to be effective. Although in some studies there was a tendency toward greater response with higher dose [see Clinical Studies (14.1)], a consistent increase in response with increased dose has not been shown. Treatment should be initiated with a daily dose of 1000 mg/day, given as twice-daily dosing (500 mg twice daily). Additional dosing increments may be given (1000 mg/day additional every 2 weeks) to a maximum recommended daily dose of 3000 mg. Doses greater than 3000 mg/day have been used in open-label studies with LEVETIRACETAM tablets for periods of 6 months and longer. There is no evidence that doses greater than 3000 mg/day confer additional benefit.
Myoclonic Seizures in Patients with Juvenile Myoclonic Epilepsy - Treatment should be initiated with a dose of 1000 mg/day, given as twice-daily dosing (500 mg twice daily). Dosage should be increased by 1000 mg/day every 2 weeks to the recommended daily dose of 3000 mg. The effectiveness of doses lower than 3000 mg/day has not been studied.
2.3 Replacement Therapy
When switching from oral LEVETIRACETAM, the initial total daily intravenous dosage of LEVETIRACETAM should be equivalent to the total daily dosage and frequency of oral LEVETIRACETAM and should be administered as a 15-minute intravenous infusion following dilution in 100 mL of a compatible diluent.
2.4 Switching to Oral Dosing
At the end of the intravenous treatment period, the patient may be switched to LEVETIRACETAM oral administration at the equivalent daily dosage and frequency of the intravenous administration.
2.5 Dosing Instructions
LEVETIRACETAM injection is for intravenous use only and must be diluted prior to administration. One vial of LEVETIRACETAM injection contains 500 mg levetiracetam (500 mg/5mL). See Table 1 for the recommended preparation and administration of LEVETIRACETAM injection to achieve a dose of 500 mg, 1000 mg, or 1500 mg.
Table 1 : Preparation and Administration of LEVETIRACETAM Injection Dose Withdraw Volume Volume of Diluent Infusion Time 500 mg 5 mL (5 mL vial) 100 mL 15 minutes 1000 mg 10 mL (two 5 mL vials) 100 mL 15 minutes 1500 mg 15 mL (three 5 mL vials) 100 mL 15 minutes For example, to prepare a 1000 mg dose, dilute 10 mL of LEVETIRACETAM injection in 100 mL of a compatible diluent [ see Dosage and Administration (2.7)] and administer intravenously as a 15-minute infusion.
2.6 Adult Patients with Impaired Renal Function
LEVETIRACETAM dosing must be individualized according to the patient's renal function status. Recommended doses and adjustment for dose for adults are shown in Table 2. To use this dosing table, an estimate of the patient's creatinine clearance (CLcr) in mL/min is needed. CLcr in mL/min may be estimated from serum creatinine (mg/dL) determination using the following formula:
1. For female patients
[140-age (years)] x weight (kg)
CLcr= - - - - - - - - - - - - - - - - - - - - - - x 10.85
72 x serum creatinine (mg/dL)1For female patients
1Following dialysis, a 250 to 500 mg supplemental dose is recommended.
Table 2 : Dosing Adjustment Regimen for Adult Patients with Impaired Renal Function
Group
Creatinine Clearance(mL/min)
Dosage(mg)
Frequency Normal > 80 500 to 1,500 Every 12 h Mild 50 to 80 500 to 1,000 Every 12 h Moderate 30 to 50 250 to 750 Every 12 h Severe < 30 250 to 500 Every 12 h ESRD patients using dialysis --- 500 to 1,000 1Every 24 h 2.7 Compatibility and Stability
LEVETIRACETAM injection was found to be physically compatible and chemically stable when mixed with the following diluents and antiepileptic drugs for at least 24 hours and stored in polyvinyl chloride (PVC) bags at controlled room temperature 15 to 30°C (59 to 86°F).
DiluentsSodium chloride (0.9%) injection, USP
Lactated Ringer’s injection
Dextrose 5% injection, USP
Other Antiepileptic DrugsLorazepam
Valproate sodium
Diazepam
There is no data to support the physical compatibility of LEVETIRACETAM injection with antiepileptic drugs that are not listed above.Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Neuropsychiatric Adverse Reactions
Partial Onset Seizures - In some adults experiencing partial onset seizures, LEVETIRACETAM causes the occurrence of central nervous system adverse reactions that can be classified into the following categories: 1) somnolence and fatigue, 2) coordination difficulties, and 3) behavioral abnormalities.
In controlled trials of adult patients with epilepsy experiencing partial onset seizures, 14.8% of LEVETIRACETAM-treated patients reported somnolence, compared to 8.4% of placebo patients. There was no clear dose response up to 3000 mg/day. In a study where there was no titration, about 45% of patients receiving 4000 mg/day reported somnolence. The somnolence was considered serious in 0.3% of the treated patients, compared to 0% in the placebo group. About 3% of LEVETIRACETAM-treated patients discontinued treatment due to somnolence,compared to 0.7% of placebo patients. In 1.4% of treated patients and in 0.9% of placebo patients the dose was reduced, while 0.3% of the treated patients were hospitalized due to somnolence.
In controlled trials of adult patients with epilepsy experiencing partial onset seizures, 14.7% of treated patients reported asthenia, compared to 9.1% of placebo patients. Treatment was discontinued in 0.8% of treated patients as compared to 0.5% of placebo patients. In 0.5% of treated patients and in 0.2% of placebo patients the dose was reduced.
A total of 3.4% of LEVETIRACETAM-treated patients experienced coordination difficulties, (reported as either ataxia, abnormal gait, or incoordination) compared to 1.6% of placebo patients. A total of 0.4% of patients in controlled trials discontinued LEVETIRACETAM treatment due to ataxia, compared to 0% of placebo patients. In 0.7% of treated patients and in 0.2% of placebo patients the dose was reduced due to coordination difficulties, while one of the treated patients was hospitalized due to worsening of pre-existing ataxia.
Somnolence, asthenia and coordination difficulties occurred most frequently within the first 4 weeks of treatment.
In controlled trials of patients with epilepsy experiencing partial onset seizures, 5 (0.7%) of LEVETIRACETAM-treated patients experienced psychotic symptoms compared to 1 (0.2%) placebo patient. Two (0.3%) LEVETIRACETAM-treated patients were hospitalized and their treatment was discontinued. Both events, reported as psychosis, developed within the first week of treatment and resolved within 1 to 2 weeks following treatment discontinuation. Two other events, reported as hallucinations , occurred after 1-5 months and resolved within 2-7 days while the patients remained on treatment. In one patient experiencing psychotic depression occurring within a month, symptoms resolved within 45 days while the patient continued treatment. A total of 13.3% of LEVETIRACETAM patients experienced other behavioral symptoms (reported as aggression, agitation, anger, anxiety, apathy, depersonalization, depression, emotional lability, hostility, irritability, etc.) compared to 6.2% of placebo patients. Approximately half of these patients reported these events within the first 4 weeks. A total of 1.7% of treated patients discontinued treatment due to these events, compared to 0.2% of placebo patients. The treatment dose was reduced in 0.8% of treated patients and in 0.5% of placebo patients. A total of 0.8% of treated patients had a serious behavioral event (compared to 0.2% of placebo patients) and were hospitalized.
In addition, 4 (0.5%) of treated patients attempted suicide compared to 0% of placebo patients. One of these patients completed suicide. In the other 3 patients, the events did not lead to discontinuation or dose reduction. The events occurred after patients had been treated for between 4 weeks and 6 months [see Patient Counseling Information (17)].
Myoclonic Seizures - During clinical development, the number of patients with myoclonic seizures exposed to LEVETIRACETAM was considerably smaller than the number with partial seizures. Therefore, under-reporting of certain adverse reactions was more likely to occur in the myoclonic seizure population. In some patients experiencing myoclonic seizures, LEVETIRACETAM causes somnolence and behavioral abnormalities. It is expected that the events seen in partial seizure patients would occur in patients with JME.
In the double-blind, controlled trial in patients with juvenile myoclonic epilepsy experiencing myoclonic seizures, 11.7% of LEVETIRACETAM-treated patients experienced somnolence compared to 1.7% of placebo patients. No patient discontinued treatment as a result of somnolence. In 1.7% of LEVETIRACETAM-treated patients and in 0% of placebo patients the dose was reduced as a result of somnolence.
Non-psychotic behavioral disorders (reported as aggression and irritability) occurred in 5% of the LEVETIRACETAM-treated patients compared to 0% of placebo patients. Non-psychotic mood disorders (reported as depressed mood, depression, and mood swings) occurred in 6.7% of LEVETIRACETAM-treated patients compared to 3.3% of placebo patients. A total of 5.0% of LEVETIRACETAM-treated patients had a reduction in dose or discontinued treatment due to behavioral or psychiatric events (reported as anxiety, depressed mood, depression, irritability, and nervousness), compared to 1.7% of placebo patients.
5.2 Withdrawal Seizures
Antiepileptic drugs, including LEVETIRACETAM, should be withdrawn gradually to minimize the potential of increased seizure frequency.
5.3 Hematologic Abnormalities
Partial Onset Seizures - Minor, but statistically significant, decreases compared to placebo in total mean RBC count (0.03 x 106/mm3 ), mean hemoglobin (0.09 g/dL), and mean hematocrit (0.38%), were seen in LEVETIRACETAM-treated patients in controlled trials.
A total of 3.2% of treated and 1.8% of placebo patients had at least one possibly significant (≤2.8 x 109/L) decreased WBC, and 2.4% of treated and 1.4% of placebo patients had at least one possibly significant (≤1.0 x 109/L) decreased neutrophil count. Of the treated patients with a low neutrophil count, all but one rose towards or to baseline with continued treatment. No patient was discontinued secondary to low neutrophil counts.
Juvenile Myoclonic Epilepsy - Although there were no obvious hematologic abnormalities observed in patients with JME, the limited number of patients makes any conclusion tentative. The data from the partial seizure patients should be considered to be relevant for JME patients.
5.4 Hepatic Abnormalities
There were no meaningful changes in mean liver function tests (LFT) in controlled trials in adult patients; lesser LFT abnormalities were similar in drug and placebo treated patients in controlled trials (1.4%). No patients were discontinued from controlled trials for LFT abnormalities except for 1 (0.07%) adult epilepsy patient receiving open treatment.
-
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The adverse reactions that result from LEVETIRACETAM injection use include all of those reported for LEVETIRACETAM tablets and oral solution. Equivalent doses of intravenous (IV) levetiracetam and oral levetiracetam result in equivalent Cmax, Cmin, and total systemic exposure to levetiracetam when the IV levetiracetam is administered as a 15 minute infusion. The prescriber should be aware that the adverse reaction incidence figures in the following tables, obtained when LEVETIRACETAM was added to concurrent AED therapy, cannot be used to predict the frequency of adverse experiences in the course of usual medical practice where patient characteristics and other factors may differ from those prevailing during clinical studies.Similarly, the cited frequencies cannot be directly compared with figures obtained from other clinical investigations involving different treatments, uses, or investigators. An inspection of these frequencies, however, does provide the prescriber with one basis to estimate the relative contribution of drug and non-drug factors to the adverse reaction incidences in the population studied.
Partial Onset Seizures - In well-controlled clinical studies using LEVETIRACETAM tablets in adults with partial onset seizures, the most frequently reported adverse reactions in patients receiving LEVETIRACETAM in combination with other AEDs, not seen at an equivalent frequency among placebo-treated patients, were somnolence, asthenia, infection and dizziness. Of the most frequently reported adverse reactions in placebo-controlled studies using LEVETIRACETAM tablets in adults experienc ing partial onset seizures, asthenia, somnolence and dizziness appeared to occur predominantly during the first 4 weeks of treatment with LEVETIRACETAM.
Table 3 lists treatment-emergent adverse reactions that occurred in at least 1% of adult epilepsy patients treated with LEVETIRACETAM tablets participating in placebo-controlled studies and were numerically more common than in patients treated with placebo. In these studies, either LEVETIRACETAM or placebo was added to concurrent AED therapy. Adverse reactions were usually mild to moderate in intensity.
Table 3: Incidence (%) of Treatment-Emergent Adverse Reactions in Placebo-Controlled, Add-On Studies in Adults Experiencing Partial Onset Seizures by Body System (Adverse Reactions Occurred in at Least 1% of LEVETIRACETAM-Treated Patients and Occurred More Frequently than Placebo-Treated Patients)
Body System/
Adverse Reaction
LEVETIRACETAM
(N=769)
%
Placebo
(N=439)
%
Body as a Whole Asthenia 15 9 Headache
14 13 Infection
13 8 Pain
7 6 Digestive System Anorexia
3 2 Nervous System Somnolence
15 8 Dizziness
9 4 Depression
4 2 Nervousness
4 2 Ataxia
3 1 Vertigo
3 1 Amnesia
2 1 Anxiety
2 1 Hostility
2 1 Paresthesia
2 1 Emotional Lability
2 0 Respiratory System Pharyngitis
6 4 Rhinitis
4 3 Cough Increased
2 1 Sinusitis
2 1 Special Senses Diplopia
2 1 Myoclonic Seizures - Although the pattern of adverse reactions in this study seems somewhat different from that seen in patients with partial seizures, this is likely due to the much smaller number of patients in this study compared to partial seizure studies. The adverse reaction pattern for patients with JME is expected to be essentially the same as for patients with partial seizures.
In the well-controlled clinical study using LEVETIRACETAM tablets in patients with myoclonic seizures, the most frequently reported adverse reactions in patients using LEVETIRACETAM in combination with other AEDs, not seen at an equivalent frequency among placebo-treated patients, were somnolence, neck pain, and pharyngitis.
Table 4 lists treatment-emergent adverse reactions that occurred in at least 5% of juvenile myoclonic epilepsy patients experiencing myoclonic seizures treated with LEVETIRACETAM tablets and were numerically more common than in patients treated with placebo. In this study, either LEVETIRACETAM or placebo was added to concurrent AED therapy. Adverse reactions were usually mild to moderate in intensity.
Table 4: Incidence (%) of Treatment-Emergent Adverse Reactions in a Placebo-Controlled, Add-On Study in Patients with Myoclonic Seizures by Body System (Adverse Reactions Occurred in at Least 5% of LEVETIRACETAM-Treated Patients and Occurred More Frequently than Placebo-Treated Patients)
Body System/
Adverse Reaction
LEVETIRACETAM
(N=60)
%
Placebo
(N=60)
%
Ear and labyrinth disorders Vertigo
5 3 Infections and infestations Pharyngitis
7 0 Influenza
5 2 Musculoskeletal and connective tissue disorders Neck pain
8 2 Nervous System disorders Somnolence
12 2 Psychiatric disorders Depression
5 2 Discontinuation or Dose Reduction in Well-Controlled Clinical Studies
Partial Onset Seizures - In well-controlled adult clinical studies using LEVETIRACETAM tablets, 15.0% of patients receiving LEVETIRACETAM and 11.6% receiving placebo either discontinued or had a dose reduction as a result of an adverse event. Table 5 lists the most common (>1%) adverse reactions that resulted in discontinuation or dose reduction and that occurred more frequently in LEVETIRACETAM-treated patients than in placebo-treated patients.
Table 5: Adverse Reactions that Most Commonly Resulted in Discontinuation or Dose Reduction that Occurred More Frequently in LEVETIRACETAM-Treated Patients in Placebo-Controlled Studies in Adult Patients Experiencing Partial Onset Seizures Adverse Reaction LEVETIRACETAM
(N=769)
n (%)
Placebo
(N=439)
n (%)
Asthenia
10 (1.3%)
3 (0.7%)
Dizziness
11 (1.4%)
0 Somnolence
34 (4.4%)
7 (1.6%)
Myoclonic Seizures - In the placebo-controlled study using LEVETIRACETAM tablets, 8.3% of patients receiving LEVETIRACETAM and 1.7% receiving placebo either discontinued or had a dose reduction as a result of an adverse event. The adverse reactions that led to discontinuation or dose reduction in the well-controlled study and that occurred more frequently in LEVETIRACETAM-treated patients than in placebo-treated patients are presented in Table 6.
Table 6: Adverse Reactions that Resulted in Discontinuation or Dose Reduction that Occurred More Frequently in LEVETIRACETAM-Treated Patients in the Placebo-Controlled Study in Patients with Juvenile Myoclonic Epilepsy Adverse Reaction LEVETIRACETAM
(N=60)
n (%)
Placebo
(N=60)
n (%)
Anxiety
2 (3.3%)
1 (1.7%)
Depressed mood
1 (1.7%)
0 Depression
1 (1.7%)
0
Diplopia
1 (1.7%) 0 Hypersomnia
1 (1.7%) 0 Insomnia
1 (1.7%) 0 Irritability
1 (1.7%) 0 Nervousness
1 (1.7%) 0 Somnolence
1 (1.7%) 0 This study was too small to adequately characterize the adverse reactions leading to discontinuation. It is expected that the adverse reactions that would lead to discontinuation in this population would be similar to those resulting in discontinuation in other epilepsy trials (see tables 5 - 6).
Comparison of Gender, Age and Race - The overall adverse experience profile of LEVETIRACETAM was similar between females and males. There are insufficient data to support a statement regarding the distribution of adverse experience reports by age and race.
6.2 Postmarketing Experience
The following adverse events have been identified during postapproval use of LEVETIRACETAM. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a casual relationship to drug exposure.
In addition to the adverse reactions listed above [see Adverse Reactions (6.1)], the following adverse events have been reported in patients receiving marketed LEVETIRACETAM worldwide.
The listing is alphabetized: abnormal liver function test, hepatic failure, hepatitis, leukopenia, neutropenia, pancreatitis, pancytopenia (with bone marrow suppression identified in some of these cases), thrombocytopenia and weight loss. Alopecia has been reported with LEVETIRACETAM use; recovery was observed in majority of cases where LEVETIRACETAM was discontinued. There have been reports of suicidal behavior (including completed suicide, suicide attempt and suicidal ideation) with marketed LEVETIRACETAM [see Patient Counseling Information (17)].
-
7 DRUG INTERACTIONS
7.1 General Information
In vitro data on metabolic interactions indicate that LEVETIRACETAM is unlikely to produce, or be subject to, pharmacokinetic interactions. Levetiracetam and its major metabolite, at concentrations well above Cmax levels achieved within the therapeutic dose range, are neither inhibitors of nor high affinity substrates for human liver cytochrome P450 isoforms, epoxide hydrolase or UDP-glucuronidation enzymes. In addition, levetiracetam does not affect the in vitro glucuronidation of valproic acid.
Levetiracetam circulates largely unbound (<10% bound) to plasma proteins; clinically significant interactions with other drugs through competition for protein binding sites are therefore unlikely.
Potential pharmacokinetic interactions were assessed in clinical pharmacokinetic studies (phenytoin, valproate, oral contraceptive, digoxin, warfarin, probenecid) and through pharmacokinetic screening in the placebo-controlled clinical studies in epilepsy patients.
7.2 Phenytoin
LEVETIRACETAM (3000 mg daily) had no effect on the pharmacokinetic disposition of phenytoin in patients with refractory epilepsy. Pharmacokinetics of levetiracetam were also not affected by phenytoin.
7.3 Valproate
LEVETIRACETAM (1500 mg twice daily) did not alter the pharmacokinetics of valproate in healthy volunteers. Valproate 500 mg twice daily did not modify the rate or extent of levetiracetam absorption or its plasma clearance or urinary excretion. There also was no effect on exposure to and the excretion of the primary metabolite, ucb L057.
7.4 Other Antiepileptic Drug
Potential drug interactions between LEVETIRACETAM and other AEDs (carbamazepine, gabapentin, lamotrigine, phenobarbital, phenytoin, primidone and valproate) were also assessed by evaluating the serum concentrations of levetiracetam and these AEDs during placebo-controlled clinical studies. These data indicate that levetiracetam does not influence the plasma concentration of other AEDs and that these AEDs do not influence the pharmacokinetics of levetiracetam.
7.5 Oral Contraceptives
LEVETIRACETAM (500 mg twice daily) did not influence the pharmacokinetics of an oral contraceptive containing 0.03 mg ethinyl estradiol and 0.15 mg levonorgestrel, or of the luteinizing hormone and progesterone levels, indicating that impairment of contraceptive efficacy is unlikely. Coadministration of this oral contraceptive did not influence the pharmacokinetics of levetiracetam.
7.6 Digoxin
LEVETIRACETAM (1000 mg twice daily) did not influence the pharmacokinetics and pharmacodynamics (ECG) of digoxin given as a 0.25 mg dose every day. Coadministration of digoxin did not influence the pharmacokinetics of levetiracetam.
7.7 Warfarin
LEVETIRACETAM (1000 mg twice daily) did not influence the pharmacokinetics of R and S warfarin. Prothrombin time was not affected by levetiracetam. Coadministration of warfarin did not affect the pharmacokinetics of levetiracetam.
7.8 Probenecid
Probenecid, a renal tubular secretion blocking agent, administered at a dose of 500 mg four times a day, did not change the pharmacokinetics of levetiracetam 1000 mg twice daily. Cssmax of the metabolite, ucb L057, was approximately doubled in the presence of probenecid while the fraction of drug excreted unchanged in the urine remained the same. Renal clearance of ucb L057 in the presence of probenecid decreased 60%, probably related to competitive inhibition of tubular secretion of ucb L057. The effect of LEVETIRACETAM on probenecid was not studied.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C - There are no adequate and well-controlled studies in pregnant women. In animal studies, levetiracetam produced evidence of developmental toxicity, including teratogenic effects, at doses similar to or greater than human therapeutic doses. LEVETIRACETAM should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus. As with other antiepileptic drugs, physiological changes during pregnancy may affect levetiracetam concentration. There have been reports of decreased levetiracetam concentration during pregnancy. Discontinuation of antiepileptic treatments may result in disease worsening, which can be harmful to the mother and the fetus.
Administration to female rats throughout pregnancy and lactation led to increased incidences of minor fetal skeletal abnormalities and retarded offspring growth pre- and/or postnatally at doses ≥350 mg/kg/day (approximately equivalent to the maximum recommended human dose of 3000 mg [MRHD] on a mg/m2 basis) and with increased pup mortality and offspring behavioral alterations at a dose of 1800 mg/kg/day (6 times the MRHD on a mg/m2 basis). The developmental no effect dose was 70 mg/kg/day (0.2 times the MRHD on a mg/m2 basis). There was no overt maternal toxicity at the doses used in this study.
There was no overt maternal toxicity at the doses used in this study.
Treatment of pregnant rabbits during the period of organogenesis resulted in increased embryofetal mortality and increased incidences of minor fetal skeletal abnormalities at doses ≥600 mg/kg/day (approximately 4 times MRHD on a mg/m2 basis) and in decreased fetal weights and increased incidences of fetal malformations at a dose of 1800 mg/kg/day (12 times the MRHD on a mg/m2 basis). The developmental no effect dose was 200 mg/kg/day (1.3 times the MRHD on a mg/m2 basis). Maternal toxicity was also observed at 1800 mg/kg/day.
When pregnant rats were treated during the period of organogenesis, fetal weights were decreased and the incidence of fetal skeletal variations was increased at a dose of 3600 mg/kg/day (12 times the MRHD). 1200 mg/kg/day (4 times the MRHD) was a developmental no effect dose. There was no evidence of maternal toxici ty in this study.
Treatment of rats during the last third of gestation and throughout lactation produced no adverse developmental or maternal effects at doses of up to 1800 mg/kg/day (6 times the MRHD on a mg/m2 basis).
Patients may enroll in the North American Antiepileptic Drug Pregnancy Registry by calling (888) 233-2334 (toll free).
8.3 Nursing Mothers
Levetiracetam is excreted in breast milk. Because of the potential for serious adverse reactions in nursing infants from LEVETIRACETAM, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
Safety and effectiveness of LEVETIRACETAM injection in patients below the age of 16 years have not been established.8.5 Geriatric Use
Of the total number of subjects in clinical studies of levetiracetam, 347 were 65 and over. No overall differences in safety were observed between these subjects and younger subjects. There were insufficient numbers of elderly subjects in controlled trials of epilepsy to adequately assess the effectiveness of LEVETIRACETAM in these patients. A study in 16 elderly subjects (age 61-88 years) with oral administration of single dose and multiple twice-daily doses for 10 days showed no pharmacokinetic differences related to age alone. Levetiracetam is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
8.6 Use in Patients with Impaired Renal Function
Clearance of levetiracetam is decreased in patients with renal impairment and is correlated with creatinine clearance. Caution should be taken in dosing patients with moderate and severe renal impairment and in patients undergoing hemodialysis. The dosage should be reduced in patients with impaired renal function receiving LEVETIRACETAM and supplemental doses should be given to patients after dialysis [ see Clinical Pharmacology (12.3) and Dosage and Administration (2.6)].
- 9 DRUG ABUSE AND DEPENDENCE
-
10 OVERDOSAGE
Signs, Symptoms and Laboratory Findings of Acute Overdosage in Humans
The highest known dose of oral LEVETIRACETAM received in the clinical development program was 6000 mg/day. Other than drowsiness, there were no adverse reactions in the few known cases of overdose in clinical trials. Cases of somnolence, agitation, aggression, depressed level of consciousness, respiratory depression and coma were observed with LEVETIRACETAM overdoses in postmarketing use.
Treatment or Management of Overdose - There is no specific antidote for overdose with LEVETIRACETAM. If indicated, elimination of unabsorbed drug should be attempted by emesis or gastric lavage; usual precautions should be observed to maintain airway. General supportive care of the patient is indicated including monitoring of vital signs and observation of the patient’s clinical status. A Certified Poison Control Center should be contacted for up to date information on the management of overdose with LEVETIRACETAM.
Hemodialysis - Standard hemodialysis procedures result in significant clearance of levetiracetam (approximately 50% in 4 hours) and should be considered in cases of overdose. Although hemodialysis has not been performed in the few known cases of overdose, it may be indicated by the patient's clinical state or in patients with significant renal impairment.
-
11 DESCRIPTION
LEVETIRACETAM injection is an antiepileptic drug available as a clear, colorless, sterile solution (100 mg/mL) for intravenous administration.
The chemical name of levetiracetam, a single enantiomer, is (-)-(S)-α-ethyl-2-oxo-1-pyrrolidine acetamide, its molecular formula is C8H14N2O2 and its molecular weight is 170.21. Levetiracetam is chemically unrelated to existing antiepileptic drugs (AEDs). It has the following structural formula:
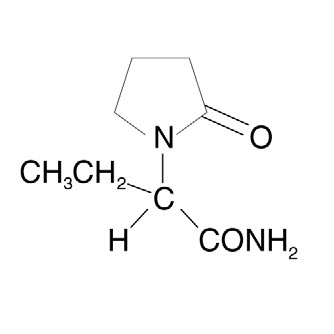
Levetiracetam is a white to off-white crystalline powder with a faint odor and a bitter taste. It is very soluble in water (104.0 g/100 mL). It is freely soluble in chloroform (65.3 g/100 mL) and in methanol (53.6 g/100 mL), soluble in ethanol (16.5 g/100 mL), sparingly soluble in acetonitrile (5.7 g/100 mL) and practically insoluble in n-hexane. (Solubility limits are expressed as g/100 mL solvent.)
LEVETIRACETAM injection contains 100 mg of levetiracetam per mL. It is supplied in single-use 5 mL vials containing 500 mg levetiracetam, water for injection, 45 mg sodium chloride, and buffered at approximately pH 5.5 with glacial acetic acid and 8.2 mg sodium acetate trihydrate. LEVETIRACETAM injection must be diluted prior to intravenous infusion [see Dosage and Administration (2.1)].
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The precise mechanism(s) by which levetiracetam exerts its antiepileptic effect is unknown. The antiepileptic activity of levetiracetam was assessed in a number of animal models of epileptic seizures. Levetiracetam did not inhibit single seizures induced by maximal stimulation with electrical current or different chemoconvulsants and showed only minimal activity in submaximal stimulation and in threshold tests. Protection was observed, however, against secondarily generalized activity from focal seizures induced by pilocarpine and kainic acid, two chemoconvulsants that induce seizures that mimic some features of human complex partial seizures with secondary generalization. Levetiracetam also displayed inhibitory properties in the kindling model in rats, another model of human complex partial seizures, both during kindling development and in the fully kindled state. The predictive value of these animal models for specific types of human epilepsy is uncertain.
In vitro and in vivo recordings of epileptiform activity from the hippocampus have shown that levetiracetam inhibits burst firing without affecting normal neuronal excitability, suggesting that levetiracetam may selectively prevent hypersynchronization of epileptiform burst firing and propagation of seizure activity.
Levetiracetam at concentrations of up to 10μM did not demonstrate binding affinity for a variety of known receptors, such as those associated with benzodiazepines, GABA (gamma-aminobutyric acid), glycine, NMDA (N-methyl-D-aspartate), re-uptake sites, and second messenger systems. Furthermore, in vitro studies have failed to find an effect of levetiracetam on neuronal voltage-gated sodium or T-type calcium current s and levetiracetam does not appear to directly facilitate GABAergic neurotransmission. However, in vitro studies have demonstrated that levetiracetam opposes the activity of negative modulators of GABA- and glycine-gated currents and partially inhibits N-type calcium currents in neuronal cells.
A saturable and stereoselective neuronal binding site in rat brain tissue has been described for levetiracetam. Experimental data indicate that this binding site is the synaptic vesicle protein SV2A, thought to be involved in the regulation of vesicle exocytosis. Although the molecular significance of levetiracetam binding to synaptic vesicle protein SV2A is not understood, levetiracetam and related analogs showed a rank order of affinity for SV2A which correlated with the potency of their antiseizure activity in audiogenic seizure-prone mice. These findings suggest that the interaction of levetiracetam with the SV2A protein may contribute to the antiepileptic mechanism of action of the drug.
12.3 Pharmacokinetics
Equivalent doses of intravenous (IV) levetiracetam and oral levetiracetam result in equivalent Cmax, Cmin, and total systemic exposure to levetiracetam when the IV levetiracetam is administered as a 15 minute infusion.
The pharmacokinetics of levetiracetam have been studied in healthy adult subjects, adults and pediatric patients with epilepsy, elderly subjects and subjects with renal and hepatic impairment.
Overview - Levetiracetam is rapidly and almost completely absorbed after oral administration. Levetiracetam injection and tablets are bioequivalent. The pharmacokinetics of levetiracetam are linear and time-invariant, with low intra- and inter-subject variability. Levetiracetam is not significantly protein-bound (<10% bound) and its volume of distribution is close to the volume of intracellular and extracellular water. Sixty-six percent (66%) of the dose is renally excreted unchanged. The major metabolic pathway of levetiracetam (24% of dose) is an enzymatic hydrolysis of the acetamide group. It is not liver cytochrome P450 dependent. The metabolites have no known pharmacological activity and are renally excreted. Plasma half-life of levetiracetam across studies is approximately 6-8 hours. It is increased in the elderly (primarily due to impaired renal clearance) and in subjects with renal impairment.
Distribution - The equivalence of levetiracetam injection and the oral formulation was demonstrated in a bioavailability study of 17 healthy volunteers. In this study, levetiracetam 1500 mg was diluted in 100 mL 0.9% sterile saline solution and was infused over 15 minutes. The selected infusion rate provided plasma concentrations of levetiracetam at the end of the infusion period similar to those achieved at Tmax after an equivalent oral dose. It is demonstrated that levetiracetam 1500 mg intravenous infusion is equivalent to levetiracetam 3 x 500 mg oral tablets. The time independent pharmacokinetic profile of levetiracetam was demonstrated following 1500 mg intravenous infusion for 4 days with BID dosing. The AUC(0-12) at steady-state was equivalent to AUCinf following an equivalent single dose. Levetiracetam and its major metabolite are less than 10% bound to plasma proteins; clinically significant interactions with other drugs through competition for protein binding sites are therefore unlikely.
Metabolism - Levetiracetam is not extensively metabolized in humans. The major metabolic pathway is the enzymatic hydrolysis of the acetamide group, which produces the carboxylic acid metabolite, ucb L057 (24% of dose) and is not dependent on any liver cytochrome P450 isoenzymes. The major metabolite is inactive in animal seizure models. Two minor metabolites were identified as the product of hydroxylation of the 2-oxo-pyrrolidine ring (2% of dose) and opening of the 2-oxo-pyrrolidine ring in position 5 (1% of dose). There is no enantiomeric interconversion of levetiracetam or its major metabolite.
Elimination - Levetiracetam plasma half-life in adults is 7 ± 1 hour and is unaffected by either dose, route of administration or repeated administration. Levetiracetam is eliminated from the systemic circulation by renal excretion as unchanged drug which represents 66% of administered dose. The total body clearance is 0.96 mL/min/kg and the renal clearance is 0.6 mL/min/kg. The mechanism of excretion is glomerular filtration with subsequent partial tubular reabsorption. The metabolite ucb L057 is excreted by glomerular filtration and active tubular secretion with a renal clearance of 4 mL/min/kg. Levetiracetam elimination is correlated to creatinine clearance. Levetiracetam clearance is reduced in patients with impaired renal function [see Use in Specific Populations (8.6) and Dosage and Administration (2.6)].
Pharmacokinetic Interactions - In vitro data on metabolic interactions indicate that levetiracetam is unlikely to produce, or be subject to, pharmacokinetic interactions. Levetiracetam and its major metabolite, at concentrations well above Cmax levels achieved within the therapeutic dose range, are neither inhibitors of, nor high affinity substrates for, human liver cytochrome P450 isoforms, epoxide hydrolase or UDP-glucuronidation enzymes. In addition, levetiracetam does not affect the in vitro glucuronidation of valproic acid.
Potential pharmacokinetic interactions of or with levetiracetam were assessed in clinical pharmacokinetic studies (phenytoin, valproate, warfarin, digoxin, oral contraceptive, probenecid) and through pharmacokinetic screening in the placebo-controlled clinical studies in epilepsy patients [see Drug Interactions (7)].
Special Populations
Elderly - Pharmacokinetics of levetiracetam were evaluated in 16 elderly subjects (age 61 to 88 years) with creatinine clearance ranging from 30 to 74 mL/min. Following oral administration of twice-daily dosing for 10 days, total body clearance decreased by 38% and the half-life was 2.5 hours longer in the elderly compared to healthy adults. This is most likely due to the decrease in renal function in these subjects.
Pediatric Patients - Safety and effectiveness of LEVETIRACETAM injection in patients below the age of 16 years have not been established
Gender - Levetiracetam Cmax and AUC were 20% higher in women (N=11) compared to men (N=12). However, clearances adjusted for body weight were comparable.
Race - Formal pharmacokinetic studies of the effects of race have not been conducted. Cross study comparisons involving Caucasians (N=12) and Asians (N=12), however, show that pharmacokinetics of levetiracetam were comparable between the two races. Because levetiracetam is primarily renally excreted and there are no important racial differences in creatinine clearance, pharmacokinetic differences due to race are not expected.
Renal Impairment - The disposition of levetiracetam was studied in adult subjects with varying degrees of renal function. Total body clearance of levetiracetam is reduced in patients with impaired renal function by 40% in the mild group (CLcr = 50 to 80 mL/min), 50% in the moderate group (CLcr = 30 to 50 mL/min) and 60% in the severe renal impairment group (CLcr <30 mL/min). Clearance of levetiracetam is correlated with creatinine clearance.
In anuric (end stage renal disease) patients, the total body clearance decreased 70% compared to normal subjects (CLcr >80mL/min). Approximately 50% of the pool of levetiracetamin the body is removed during a standard 4 hour hemodialysis procedure.
Dosage should be reduced in patients with impaired renal function receiving levetiracetam, and supplemental doses should be given to patients after dialysis [see Dosage and Administration (2.6)].
Hepatic Impairment - In subjects with mild (Child-Pugh A) to moderate (Child-Pugh B) hepatic impairment, the pharmacokinetics of levetiracetam were unchanged. In patients with severe hepatic impairment (Child-Pugh C), total body clearance was 50% that of normal subjects, but decreased renal clearance accounted for most of the decrease. No dose adjustment is needed for patients with hepatic impairment.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis - Rats were dosed with levetiracetam in the diet for 104 weeks at doses of 50, 300 and 1800 mg/kg/day. The highest dose corresponds to 6 times the maximum recommended daily human dose (MRHD) of 3000 mg on a mg/m2 basis and it also provided systemic exposure (AUC) approximately 6 times that achieved in humans receiving the MRHD. There was no evidence of carcinogenicity. A study was conducted in which mice received levetiracetam in the diet for 80 weeks at doses of 60, 240 and 960 mg/kg/day (high dose is equivalent to 2 times the MRHD on a mg/m2 or exposure basis). Although no evidence for carcinogenicity was seen, the potential for a carcinogenic response has not been fully evaluated in that species because adequate doses have not been studied.
Mutagenesis - Levetiracetam was not mutagenic in the Ames test or in mammalian cells in vitro in the Chinese hamster ovary/HGPRT locus assay. It was not clastogenic in an in vitro analysis of metaphase chromosomes obtained from Chinese hamster ovary cells or in an in vivo mouse micronucleus assay. The hydrolysis product and major human metabolite of levetiracetam (ucb L057) was not mutagenic in the Ames test or the in vitro mouse lymphoma assay.
Impairment of Fertility - No adverse effects on male or female fertility or reproductive performance were observed in rats at doses up to 1800 mg/kg/day (approximately 6 times the maximum recommended human dose on a mg/m2 or exposure basis).
-
14 CLINICAL STUDIES
All efficacy trials utilized oral formulations. The recommendation for the parenteral formulation is based upon these studies as well as the demonstration of comparable bioavailability of the oral and the parenteral formulation [see Pharmacokinetics (12.3)].
In the following studies, statistical significance versus placebo indicates a p value < 0.05.
14.1 Partial Onset Seizures
Effectiveness in Partial Onset Seizures in Adults with Epilepsy -The effectiveness of LEVETIRACETAM as adjunctive therapy (added to other antiepileptic drugs) in adults was established in three multicenter, randomized, double-blind, placebo-controlled clinical studies in patients who had refractory partial onset seizures with or without secondary generalization. The tablet formulation was used in all these studies. In these studies, 904 patients were randomized to placebo, 1000 mg, 2000 mg, or 3000 mg/day. Patients enrolled in Study 1 or Study 2 had refractory partial onset seizures for at least two years and had taken two or more classical AEDs. Patients enrolled in Study 3 had refractory partial onset seizures for at least 1 year and had taken one classical AED. At the time of the study, patients were taking a stable dose regimen of at least one and could take a maximum of two AEDs. During the baseline period, patients had to have experienced at least two partial onset seizures during each 4-week period.
The criteria for statistical significance in all studies was a p value < 0.05.
Study 1Study 1 was a double-blind, placebo-controlled, parallel-group study conducted at 41 sites in the United States comparing LEVETIRACETAM 1000 mg/day (N=97), LEVETIRACETAM 3000 mg/day (N=101), and placebo (N=95) given in equally divided doses twice daily. After a prospective baseline period of 12 weeks, patients were randomized to one of the three treatment groups described above. The 18-week treatment period consisted of a 6-week titration period, followed by a 12-week fixed dose evaluation period, during which concomitant AED regimens were held constant. The primary measure of effectiveness was a between group comparison of the percent reduction in weekly partial seizure frequency relative to placebo over the entire randomized treatment period (titration + evaluation period). Secondary outcome variables included the responder rate (incidence of patients with ≥50% reduction from baseline in partial onset seizure frequency). The results of the analysis of Study 1 are displayed in Table 7.
Table 7: Reduction in Mean Over Placebo in Weekly Frequency of Partial Onset Seizures in Study 1
Placebo
LEVETIRACETAM
LEVETIRACETAM (N=95)
1000 mg/day
3000 mg/day
(N=97)
(N=101)
Percent reduction in partial seizure frequency over placebo
-
26.1%*
30.1%*
* Statistically significant versus placebo
The percentage of patients (y-axis) who achieved ≥50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomized treatment period (titration + evaluation period) within the three treatment groups (x-axis) is presented in Figure 1.
Figure 1: Responder Rate (≥50% Reduction from Baseline) in Study 1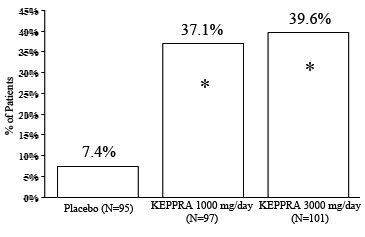
* Statistically significant versus placebo
Study 2 - Study 2 was a double-blind, placebo-controlled, crossover study conducted at 62 centers in Europe comparing LEVETIRACETAM 1000 mg/day (N=106), LEVETIRACETAM 2000 mg/day (N=105), and placebo (N=111) given in equally divided doses twice daily.
The first period of the study (Period A) was designed to be analyzed as a parallel-group study. After a prospective baseline period of up to 12 weeks, patients were randomized to one of the three treatment groups described above. The 16-week treatment period consisted of the 4-week titration period followed by a 12-week fixed dose evaluation period, during which concomitant AED regimens were held constant. The primary measure of effectiveness was a between group comparison of the percent reduction in weekly partial seizure frequency relative to placebo over the entire randomized treatment period (titration + evaluation period). Secondary outcome variables included the responder rate (incidence of patients with ≥50% reduction from baseline in partial onset seizure frequency). The results of the analysis of Period A are displayed in Table 8.
Table 8: Reduction in Mean Over Placebo in Weekly Frequency of Partial Onset Seizures in Study 2: Period A
Placebo
LEVETIRACETAM
LEVETIRACETAM 1000 mg/day
2000 mg/day
(N=111) (N=106)
(N=105)
Percent reduction in partial seizure frequency over placebo
-
17.1%*
21.4%*
* Statistically significant versus placebo
The percentage of patients (y-axis) who achieved ≥50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomized treatment period (titration + evaluation period) within the three treatment groups (x-axis) is presented in Figure 2.
Figure 2: Responder Rate (≥50% Reduction from Baseline) in Study 2: Period A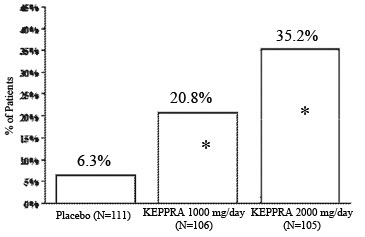
* Statistically significant versus placeboThe comparison of LEVETIRACETAM 2000 mg/day to LEVETIRACETAM 1000 mg/day for responder rate was statistically significant (P=0.02). Analysis of the trial as a cross-over yielded similar results.
Study 3Study 3 was a double-blind, placebo-controlled, parallel-group study conducted at 47 centers in Europe comparing LEVETIRACETAM 3000 mg/day (N=180) and placebo (N=104) in patients with refractory partial onset seizures, with or without secondary generalization, receiving only one concomitant AED. Study drug was given in two divided doses. After a prospective baseline period of 12 weeks, patients were randomized to one of two treatment groups described above. The 16-week treatment period consisted of a 4-week titration period, followed by a 12-week fixed dose evaluation period, during which concomitant AED doses were held constant. The primary measure of effectiveness was a between group comparison of the percent reduction in weekly seizure frequency relative to placebo over the entire randomized treatment period (titration + evaluation period). Secondary outcome variables included the responder rate (incidence of patients with ≥50% reduction from baseline in partial onset seizure frequency). Secondary outcome variables included the responder rate (incidence of patients with ≥50% reduction from baseline in partial onset seizure frequency). Table 9 displays the results of the analysis of Study 3.
Table 9: Reduction in Mean Over Placebo in Weekly Frequency of Partial Onset Seizures in Study 3
Placebo
LEVETIRACETAM
3000 mg/day
(N=104) (N=180)
Percent reduction in partial seizure frequency over placebo
-
23.0%*
* Statistically significant versus placebo
The percentage of patients (y-axis) who achieved ≥50% reduction in weekly seizure rates from baseline in partial onset seizure frequency over the entire randomized treatment period (titration + evaluation period) within the two treatment groups (x-axis) is presented in Figure 3.
Figure 3: Responder Rate (≥50% Reduction from Baseline) in Study 3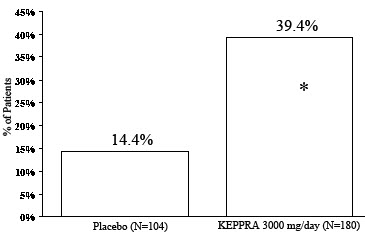
* Statistically significant versus placebo
14.2 Myoclonic Seizures in Patients with Juvenile Myoclonic Epilepsy
Effectiveness in Myoclonic Seizures in Patients with Juvenile Myoclonic Epilepsy (JME)The effectiveness of LEVETIRACETAM as adjunctive therapy (added to other antiepileptic drugs) in patients with juvenile myoclonic epilepsy (JME) experiencing myoclonic seizures was established in one multicenter, randomized, double-blind, placebo-controlled study, conducted at 37 sites in 14 countries. Of the 120 patients enrolled, 113 had a diagnosis of confirmed or suspected JME. Eligible patients on a stable dose of 1 antiepileptic drug (AED) experiencing one or more myoclonic seizures per day for at least 8 days during the prospective 8-week baseline period were randomized to either LEVETIRACETAM or placebo (LEVETIRACETAM N=60, placebo N=60). Patients were titrated over 4 weeks to a target dose of 3000 mg/day and treated at a stable dose of 3000 mg/day over 12 weeks (evaluation period). Study drug was given in 2 divided doses.
The primary measure of effectiveness was the proportion of patients with at least 50% reduction in the number of days per week with one or more myoclonic seizures during the treatment period (titration + evaluation periods) as compared to baseline. Table 10 displays the results for the 113 patients with JME in this study.
Table 10: Responder Rate (≥50% Reduction from Baseline) in Myoclonic Seizure Days per Week for Patients with JME Placebo
LEVETIRACETAM
(N=59)
(N=54)
Percentage of responders
23.7%
60.4%*
* Statistically significant versus placebo
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Patients should be advised to notify their physician if they are pregnant prior to therapy.
Patients should be advised that LEVETIRACETAM may cause dizziness and somnolence. Accordingly, patients should be advised not to drive or operate heavy machinery or engage in other hazardous activities until they have gained sufficient experience on LEVETIRACETAM to gauge whether it adversely affects their performance of these activities.
Patients should be advised that LEVETIRACETAM may cause changes in behavior (e.g. aggression, agitation, anger, anxiety, apathy , depression, hostility, and irritability) and in rare cases patients may experience psychotic symptoms.
Patients should be advised to immediately report any symptoms of depression and/or suicidal ideation to their prescribing physician as suicide, suicide attempt and suicidal ideation have been reported in patients treated with levetiracetam.
LEVETIRACETAM Injection is manufactured in the USA for Nexus Pharmaceuticals Inc., Vernon Hills, IL 60061
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LEVETIRACETAM
levetiracetam injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:14789-400 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LEVETIRACETAM (UNII: 44YRR34555) (LEVETIRACETAM - UNII:44YRR34555) LEVETIRACETAM 100 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 9 mg in 1 mL SODIUM ACETATE (UNII: 4550K0SC9B) 1.6 mg in 1 mL ACETIC ACID (UNII: Q40Q9N063P) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:14789-400-05 10 in 1 CARTON 1 5 mL in 1 VIAL Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA090813 06/03/2010 Labeler - Nexus Pharmaceuticals Inc (620714787)