Label: PENTOXIFYLLINE tablet, film coated, extended release

-
Contains inactivated NDC Code(s)
NDC Code(s): 53808-0516-1 - Packager: State of Florida DOH Central Pharmacy
- This is a repackaged label.
- Source NDC Code(s): 60505-0033
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated May 31, 2010
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
Each extended-release tablet, for oral administration, contains 400 mg of pentoxifylline and the following inactive ingredients: D&C Red #30 Aluminum Lake, FD&C Blue #2 Aluminum Lake, FD&C Yellow #6 Aluminum Lake, hydroxyethyl cellulose, hypromellose, magnesium stearate, microcrystalline cellulose, polydextrose, polyethylene glycol, povidone, titanium dioxide and triacetin in an extended-release formulation. Pentoxifylline is a tri-substituted xanthine derivative designated chemically as 1-(5-oxohexyl)-3,7-dimethylxanthine that, unlike theophylline, is a hemorrheologic agent, i.e. an agent that affects blood viscosity. Pentoxifylline is soluble in water and ethanol, and sparingly soluble in toluene.
The structural formula is:
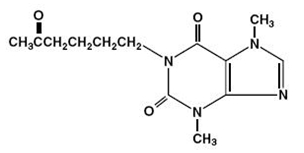
C13H18N4O3 M.W. 278.31
Pentoxifylline Extended-release Tablets, USP meet USP Drug Release Test 6.
-
CLINICAL PHARMACOLOGY
Mode of Action
Pentoxifylline and its metabolites improve the flow properties of blood by decreasing its viscosity. In patients with chronic peripheral arterial disease, this increases blood flow to the affected microcirculation and enhances tissue oxygenation. The precise mode of action of pentoxifylline and the sequence of events leading to clinical improvement are still to be defined. Pentoxifylline administration has been shown to produce dose related hemorrheologic effects, lowering blood viscosity, and improving erythrocyte flexibility. Leukocyte properties of hemorrheologic importance have been modified in animal and in vitro human studies. Pentoxifylline has been shown to increase leukocyte deformability and to inhibit neutrophil adhesion and activation. Tissue oxygen levels have been shown to be significantly increased by therapeutic doses of pentoxifylline in patients with peripheral arterial disease.
Pharmacokinetics and Metabolism
After oral administration in aqueous solution pentoxifylline is almost completely absorbed. It undergoes a first-pass effect and the various metabolites appear in plasma very soon after dosing. Peak plasma levels of the parent compound and its metabolites are reached within 1 hour. The major metabolites are Metabolite 1 (1-[5-hydroxyhexyl]-3,7-di-methylxanthine) and Metabolite V (1-[3-carboxypropyl]-3,7-dimethylxanthine), and plasma levels of these metabolites are 5 and 8 times greater, respectively, than pentoxifylline.
Following oral administration of aqueous solutions containing 100 to 400 mg of pentoxifylline, the pharmacokinetics of the parent compound and Metabolite 1 are dose-related and not proportional (non-linear), with half-life and area under the blood-level time curve (AUC) increasing with dose. The elimination kinetics of Metabolite V are not dose-dependent. The apparent plasma half-life of pentoxifylline varies from 0.4 to 0.8 hours and the apparent plasma half-lives of its metabolites vary from 1 to 1.6 hours. There is no evidence of accumulation or enzyme induction (Cytochrome P450) following multiple oral doses.
Excretion is almost totally urinary; the main biotransformation product is Metabolite V. Essentially no parent drug is found in the urine. Despite large variations in plasma levels of parent compound and its metabolites, the urinary recovery of Metabolite V is consistent and shows dose proportionality. Less than 4% of the administered dose is recovered in feces. Food intake shortly before dosing delays absorption of an immediate release dosage form but does not affect total absorption. The pharmacokinetics and metabolism of pentoxifylline have not been studied in patients with renal and/or hepatic dysfunction, but AUC was increased and elimination rate decreased in an older population (60 to 68 years) compared to younger individuals (22 to 30 years).
After administration of a 400 mg pentoxifylline extended-release tablet, plasma levels of the parent compound and its metabolites reach their maximum within 2 to 4 hours and remain constant over an extended period of time. Coadministration of pentoxifylline extended-release tablets with meals resulted in an increase in mean Cmax and AUC by about 28% and 13% for pentoxifylline, respectively. Cmax for Metabolite 1 also increased by about 20%. The controlled release of pentoxifylline from the tablet eliminates peaks and troughs in plasma levels for improved gastrointestinal tolerance.
-
INDICATIONS AND USAGE
Pentoxifylline extended-release tablets are indicated for the treatment of patients with intermittent claudication on the basis of chronic occlusive arterial disease of the limbs. Pentoxifylline can improve function and symptoms but is not intended to replace more definitive therapy, such as surgical bypass, or removal of arterial obstructions when treating peripheral vascular disease.
- CONTRAINDICATIONS
-
PRECAUTIONS
General
Patients with chronic occlusive arterial disease of the limbs frequently show other manifestations of arteriosclerotic disease. Pentoxifylline has been used safely for treatment of peripheral arterial disease in patients with concurrent coronary artery and cerebrovascular diseases, but there have been occasional reports of angina, hypotension, and arrhythmia. Controlled trials do not show that pentoxifylline causes such adverse effects more often than placebo, but, as it is a methylxanthine derivative, it is possible some individuals will experience such responses. Patients on warfarin should have more frequent monitoring of prothrombin times, while patients with other risk factors complicated by hemorrhage (e.g., recent surgery, peptic ulceration, cerebral and/or retinal bleeding) should have periodic examinations for bleeding including hematocrit and/or hemoglobin.
Drug Interactions
Although a causal relationship has not been established, there have been reports of bleeding and/or prolonged prothrombin time in patients treated with pentoxifylline with and without anticoagulants or platelet aggregation inhibitors. Patients on warfarin should have more frequent monitoring of prothrombin times, while patients with other risk factors complicated by hemorrhage (e.g., recent surgery, peptic ulceration) should have periodic examinations for bleeding including hematocrit and/or hemoglobin. Concomitant administration of pentoxifylline and theophylline-containing drugs leads to increased theophylline levels and theophylline toxicity in some individuals. Such patients should be closely monitored for signs of toxicity and have their theophylline dosage adjusted as necessary. Pentoxifylline has been used concurrently with antihypertensive drugs, beta blockers, digitalis, diuretics, antidiabetic agents, and antiarrhythmics, without observed problems. Small decreases in blood pressure have been observed in some patients treated with pentoxifylline; periodic systemic blood pressure monitoring is recommended for patients receiving concomitant antihypertensive therapy. If indicated, dosage of the antihypertensive agents should be reduced.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term studies of the carcinogenic potential of pentoxifylline were conducted in mice and rats by dietary administration of the drug at doses up to 450 mg/kg (approximately 19 times the maximum recommended human daily dose [MRHD] in both species when based on body-weight; 1.5 times the MRHD in the mouse and 3.3 times the MRHD in the rat when based on body-surface area). In mice, the drug was administered for 18 months, whereas in rats, the drug was administered for 18 months followed by an additional 6 months without drug exposure. In the rat study, there was a statistically significant increase in benign mammary fibroadenomas in females of the 450 mg/kg group. The relevance of this finding to human use is uncertain. Pentoxifylline was devoid of mutagenic activity in various strains of Salmonella (Ames test) and in cultured mammalian cells (unscheduled DNA synthesis test) when tested in the presence and absence of metabolic activation. It was also negative in the in vivo mouse micronucleus test.
Teratogenic Effects. Category C
Teratogenicity studies have been performed in rats and rabbits, using oral doses up to 576 and 264 mg/kg, respectively. On a weight basis, these doses are 24 and 11 times the maximum recommended human dose (MRHD); on a body-surface-area basis, they are 4.2 and 3.5 times the MRHD. No evidence of fetal malformation was observed. Increased resorption was seen in rats of the 576 mg/kg group. There are no adequate and well controlled studies in pregnant women. Pentoxifylline should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
Pentoxifylline and its metabolites are excreted in human milk. Because of the potential for tumorigenicity shown for pentoxifylline in rats, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Clinical studies of pentoxifylline did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
The active metabolite is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
-
ADVERSE REACTIONS
Clinical trials were conducted using either extended-release pentoxifylline tablets for up to 60 weeks or immediate-release pentoxifylline capsules for up to 24 weeks. Dosage ranges in the tablet studies were 400 mg bid to tid and in the capsule studies, 200 to 400 mg tid.
The table summarizes the incidence (in percent) of adverse reactions considered drug related, as well as the numbers of patients who received extended-release pentoxifylline tablets, immediate-release pentoxifylline capsules, or the corresponding placebos. The incidence of adverse reactions was higher in the capsule studies (where dose related increases were seen in digestive and nervous system side effects) than in the tablet studies. Studies with the capsule include domestic experience, whereas studies with the extended-release tablets were conducted outside the U.S.
The table indicates that in the tablet studies few patients discontinued because of adverse effects.
INCIDENCE (%) OF SIDE EFFECTS Extended-Release Tablets Immediate-Release Capsules Commercially Available Used only for Controlled Clinical Trials Pentoxifylline Placebo Pentoxifylline Placebo (Numbers of Patients at Risk) (321) (128) (177) (138) Discontinued for Side Effect 3.1 0 9.6 7.2 CARDIOVASCULAR SYSTEM Angina/Chest Pain 0.3 - 1.1 2.2 Arrhythmia/Palpitation - - 1.7 0.7 Flushing - - 2.3 0.7 DIGESTIVE SYSTEM Abdominal Discomfort - - 4.0 1.4 Belching/Flatus/Bloating 0.6 - 9.0 3.6 Diarrhea - - 3.4 2.9 Dyspepsia 2.8 4.7 9.6 2.9 Nausea 2.2 0.8 28.8 8.7 Vomiting 1.2 - 4.5 0.7 NERVOUS SYSTEM Agitation/Nervousness - - 1.7 0.7 Dizziness 1.9 3.1 11.9 4.3 Drowsiness - - 1.1 5.8 Headache 1.2 1.6 6.2 5.8 Insomnia - - 2.3 2.2 Tremor 0.3 0.8 - - Blurred Vision - - 2.3 1.4 Pentoxifylline has been marketed in Europe and elsewhere since 1972. In addition to the above symptoms, the following have been reported spontaneously since marketing or occurred in other clinical trials with an incidence of less than 1%; the causal relationship was uncertain:
Cardiovascular: dyspnea, edema, hypotension.
Digestive: anorexia, cholecystitis, constipation, dry mouth/thirst.
Nervous: anxiety, confusion, depression, seizures, aseptic meningitis.
Respiratory: epistaxis, flu-like symptoms, laryngitis, nasal congestion.
Skin and Appendages: brittle fingernails, pruritus, rash, urticaria, angioedema.
Special Senses: blurred vision, conjunctivitis, earache, scotoma.
Miscellaneous: bad taste, excessive salivation, leukopenia, malaise, sore throat/swollen neck glands, weight change.
A few rare events have been reported spontaneously worldwide since marketing in 1972. Although they occurred under circumstances in which a causal relationship with pentoxifylline could not be established, they are listed to serve as information for physicians: Cardiovascular–angina, arrhythmia, tachycardia, anaphylactoid reaction; Digestive–hepatitis, jaundice, increased liver enzymes; and Hemic and Lymphatic–decreased serum fibrinogen, pancytopenia, aplastic anemia, leukemia, purpura, thrombocytopenia.
-
OVERDOSAGE
Overdosage with pentoxifylline has been reported in pediatric patients and adults. Symptoms appear to be dose related. A report from a poison control center on 44 patients taking overdoses of enteric-coated pentoxifylline tablets noted that symptoms usually occurred 4 to 5 hours after ingestion and lasted about 12 hours. The highest amount ingested was 80 mg/kg; flushing, hypotension, convulsions, somnolence, loss of consciousness, fever, and agitation occurred. All patients recovered. In addition to symptomatic treatment and gastric lavage, special attention must be given to supporting respiration, maintaining systemic blood pressure, and controlling convulsions. Activated charcoal has been used to absorb pentoxifylline in patients who have overdosed.
-
DOSAGE AND ADMINISTRATION
The usual dosage of pentoxifylline in extended-release tablet form is one tablet (400 mg) three times a day with meals.
While the effect of pentoxifylline may be seen within 2 to 4 weeks, it is recommended that treatment be continued for at least 8 weeks. Efficacy has been demonstrated in double-blind clinical studies of 6 months duration.
Digestive and central nervous system side effects are dose related. If patients develop these effects it is recommended that the dosage be lowered to one tablet twice a day (800 mg/day). If side effects persist at this lower dosage, the administration of pentoxifylline should be discontinued.
-
HOW SUPPLIED
Pentoxifylline Extended-release Tablets, USP are available as film coated, white, unscored, elliptical-shaped tablets, each containing 400 mg of pentoxifylline. The tablet is debossed with APO on one side and 033 on the other side.
They are supplied by State of Florida DOH Central Pharmacy as follows:
NDC Strength Quantity/Form Color Source Prod. Code 53808-0516-1 400 mg 30 Tablets in a Blister Pack WHITE 60505-0033 Store at 20° to 25°C (68° to 77°F). [See USP for Controlled Room Temperature.]
Protect from light.
Dispense in a tight, light-resistant container as defined in the USP using a child-resistant closure.
This product is manufactured by:
Mylan Pharmaceuticals Inc.
Morgantown, WV 26505This Product was Repackaged By:
State of Florida DOH Central Pharmacy
104-2 Hamilton Park Drive
Tallahassee, FL 32304
United States
- 400mg Label
-
INGREDIENTS AND APPEARANCE
PENTOXIFYLLINE
pentoxifylline tablet, film coated, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:53808-0516(NDC:60505-0033) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength PENTOXIFYLLINE (UNII: SD6QCT3TSU) (PENTOXIFYLLINE - UNII:SD6QCT3TSU) PENTOXIFYLLINE 400 mg Inactive Ingredients Ingredient Name Strength D&C RED NO. 30 (UNII: 2S42T2808B) FD&C BLUE NO. 2 (UNII: L06K8R7DQK) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) HYDROXYETHYL CELLULOSE (140 CPS AT 5%) (UNII: 8136Y38GY5) HYPROMELLOSE (UNII: 3NXW29V3WO) MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYDEXTROSE (UNII: VH2XOU12IE) POLYETHYLENE GLYCOL (UNII: 3WJQ0SDW1A) POVIDONE (UNII: FZ989GH94E) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TRIACETIN (UNII: XHX3C3X673) Product Characteristics Color WHITE Score no score Shape OVAL (Elliptical) Size 16mm Flavor Imprint Code APO;033 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:53808-0516-1 30 in 1 BLISTER PACK Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA075191 07/01/2009 Labeler - State of Florida DOH Central Pharmacy (829348114) Establishment Name Address ID/FEI Business Operations State of Florida DOH Central Pharmacy 829348114 repack
