Label: CLEVIPREX- clevidipine emulsion









- NDC Code(s): 18124-011-00, 18124-011-25, 18124-011-50
- Packager: Fresenius Kabi Austria GmbH
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated November 9, 2022
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use Cleviprex safely and effectively. See full prescribing information for Cleviprex.
Cleviprex (clevidipine) injectable emulsion, for intravenous use
Initial U.S. Approval: 2008INDICATIONS AND USAGE
Cleviprex is a dihydropyridine calcium channel blocker indicated for the reduction of blood pressure when oral therapy is not feasible or not desirable. (1)
DOSAGE AND ADMINISTRATION
For intravenous use: Cleviprex is intended for intravenous use. Titrate Cleviprex to achieve the desired blood pressure reduction. Individualize dosage depending on the blood pressure response of the patient and the goal blood pressure. (2.2)
Monitoring: Monitor blood pressure and heart rate during infusion, and until vital signs stabilize. (2.1)
Initial dose: Initiate intravenous infusion of Cleviprex at 1- 2 mg/hour. (2.2)
Dose titration: Double the dose at short (90 second) intervals initially. As the blood pressure approaches goal, increase the dose by less than doubling and lengthen the time between dose adjustments to every 5-10 minutes. An approximately 1-2 mg/hour increase will generally produce an additional 2-4 mmHg decrease in systolic pressure. (2.2)
Maintenance dose: Most patients will achieve the desired therapeutic response at approximately 4-6 mg/hour. Severe hypertension is likely to require higher doses. (2.2)
Maximum dose: Most patients have received maximum doses of 16 mg/hour or less. There is limited experience with short-term dosing as high as 32 mg/hour. Because of lipid load restrictions, no more than 1000 mL or an average of 21 mg/hour of Cleviprex infusion is recommended per 24 hour period. There is little experience beyond 72 hours at any dose. (2.2)DOSAGE FORMS AND STRENGTHS
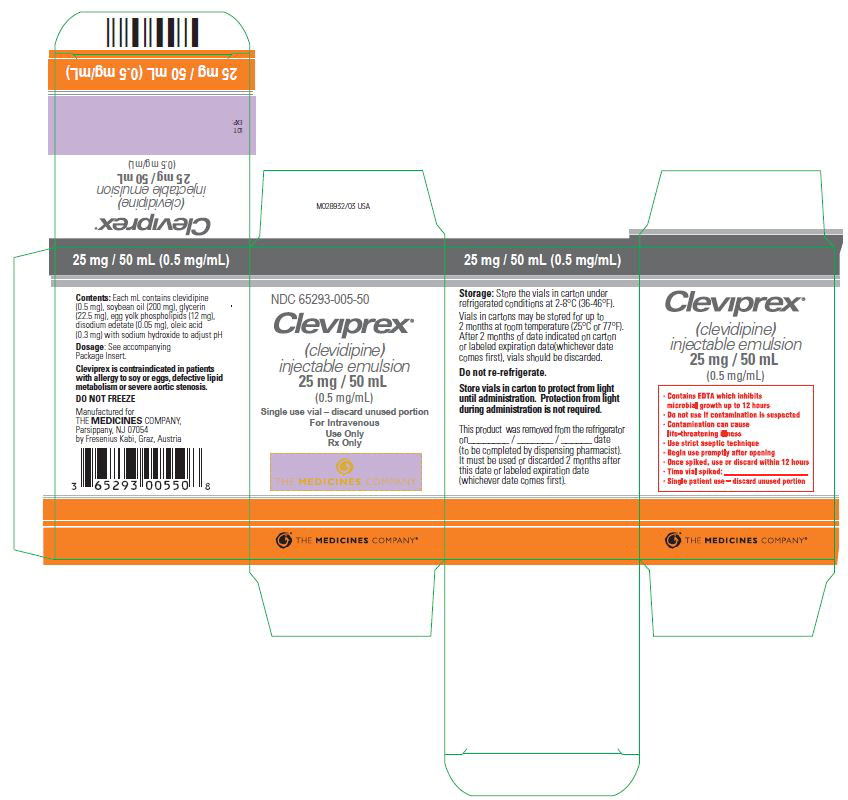
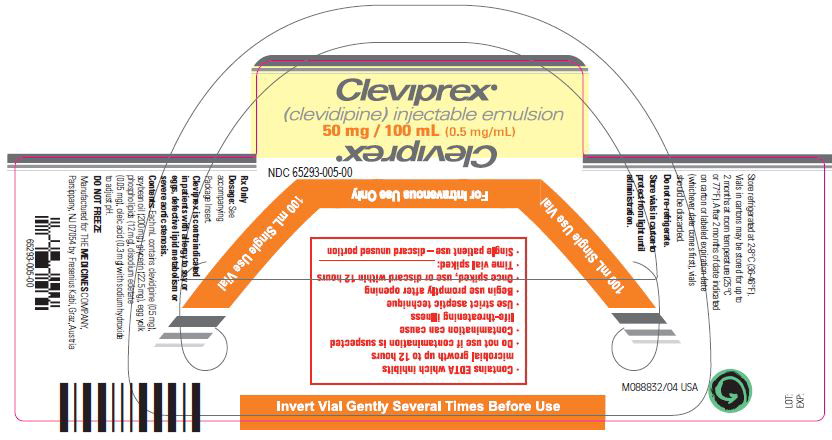
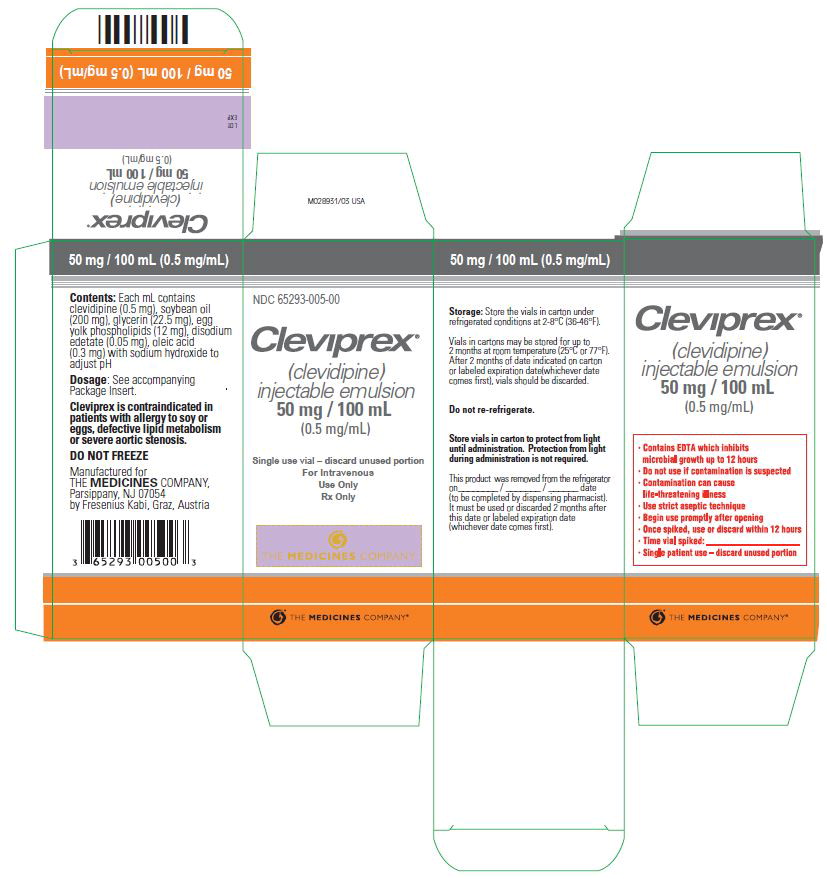
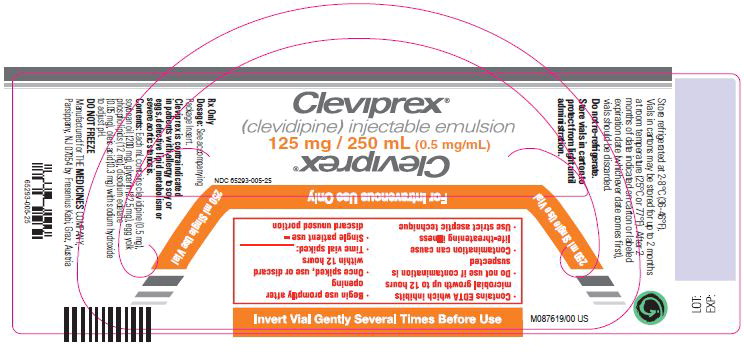
Injectible Emulsion. Single-use vials. 50 mL, or 100 mL, or 250 mL. Concentration is 0.5 mg/mL. (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Maintain aseptic technique. Discard unused portion 12 hours after stopper puncture. (5.1)
- Hypotension and reflex tachycardia are potential consequences of rapid upward titration of Cleviprex. (5.2)
- Dihydropyridine calcium channel blockers can produce negative inotropic effects and exacerbate heart failure. Monitor heart failure patients carefully. (5.4)
- Cleviprex gives no protection against the effects of abrupt beta-blocker withdrawal. (5.5)
- Patients who receive prolonged Cleviprex infusions and are not transitioned to other antihypertensive therapies should be monitored for the possibility of rebound hypertension for at least 8 hours after the infusion is stopped. (5.6)
ADVERSE REACTIONS
Most common adverse reactions (>2%) are headache, nausea, and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact The Medicines Company at 1-888-977-MDCO (6326) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
At clinically relevant concentrations, clevidipine and its metabolites do not inhibit or induce any CYP450 enzymes. The potential of clevidipine to interact with other drugs is low. (7)
USE IN SPECIFIC POPULATIONS
Pediatric use: Safety and effectiveness of Cleviprex in children under 18 years of age have not been established. (8.4)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2016
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Monitoring
2.2 Recommended Dosing
2.3 Instructions for Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Known Allergy
4.2 Defective Lipid Metabolism
4.3 Severe Aortic Stenosis
5 WARNINGS AND PRECAUTIONS
5.1 Need for Aseptic Technique
5.2 Hypotension and Reflex Tachycardia
5.3 Lipid Intake
5.4 Negative Inotropy
5.5 Beta-Blocker Withdrawal
5.6 Rebound Hypertension
5.7 Pheochromocytoma
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-Marketing and Other Clinical Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Labor and Delivery
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.3 Developmental Toxicology
14 CLINICAL STUDIES
14.1 Perioperative Hypertension
14.2 Severe Hypertension
14.3 Essential Hypertension
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Monitoring
Monitor blood pressure and heart rate continually during infusion, and then until vital signs are stable. Patients who receive prolonged Cleviprex infusions and are not transitioned to other antihypertensive therapies should be monitored for the possibility of rebound hypertension for at least 8 hours after the infusion is stopped. These patients may need follow-up adjustments in blood pressure control.
2.2 Recommended Dosing
Cleviprex is intended for intravenous use. Titrate drug to achieve the desired blood pressure reduction. Individualize dosage depending on the blood pressure to be obtained and the response of the patient.
Initial dose: Initiate the intravenous infusion of Cleviprex at 1-2 mg/hour.
Dose titration: The dose may be doubled at short (90 second) intervals initially. As the blood pressure approaches goal, the increase in doses should be less than doubling and the time between dose adjustments should be lengthened to every 5-10 minutes. An approximately 1-2 mg/hour increase will generally produce an additional 2-4 mmHg decrease in systolic pressure.
Maintenance dose: The desired therapeutic response for most patients occurs at doses of 4-6 mg/hour. Patients with severe hypertension may require doses up to 32 mg/hour, but there is limited experience at this dose rate.
Maximum dose: Most patients were treated with maximum doses of 16 mg/hour or less.There is limited short-term experience with doses up to 32 mg/hour. Because of lipid load restrictions, no more than 1000 mL or an average of 21 mg/hour of Cleviprex infusion is recommended per 24 hour period. In clinical trials, 55 hypertensive patients were treated with >500mL of Cleviprex infusion per 24 hour period. There is little experience with infusion durations beyond 72 hours at any dose.
Transition to an oral antihypertensive agent: Discontinue Cleviprex or titrate downward while appropriate oral therapy is established. When an oral antihypertensive agent is being instituted, consider the lag time of onset of the oral agent’s effect. Continue blood pressure monitoring until desired effect is achieved.
Special populations: Special populations were not specifically studied. In clinical trials, 78 patients with abnormal hepatic function (one or more of the following: elevated serum bilirubin, AST/SGOT, ALT/SGPT) and 121 patients with moderate to severe renal impairment were treated with Cleviprex. An initial Cleviprex infusion rate of 1-2 mg/hour is appropriate in these patients.
Table 1 is a guideline for dosing conversion from mg/hour to mL/hour.
Table 1. Dose conversion Dose
(mg/hour)
Dose
(mL/hour)
1
2
2
4
4
8
6
12
8
16
10
20
12
24
14
28
16
32
18
36
20
40
22
44
24
48
26
52
28
56
30
60
32
64
2.3 Instructions for Administration
Maintain aseptic technique while handling Cleviprex. Cleviprex is a single-use parenteral product. Do not use if contamination is suspected. Once the stopper is punctured, use within 12 hours and discard any unused portion.
Cleviprex is supplied in sterile, pre-mixed, ready-to-use 50 mL, or 100 mL, or 250 mL vials. Invert vial gently several times before use to ensure uniformity of the emulsion prior to administration. Inspect parenteral drug products for particulate matter and discoloration prior to administration whenever solution and container permit. Administer Cleviprex using an infusion device allowing calibrated infusion rates. Commercially available standard plastic cannulae may be used to administer the infusion. Administer Cleviprex by a central line or a peripheral line.
Cleviprex should not be administered in the same line as other medications.
Cleviprex should not be diluted, but it can be administered with the following:
- Water for Injection, USP
- Sodium Chloride (0.9%) Injection, USP
- Dextrose (5%) Injection, USP
- Dextrose (5%) in Sodium Chloride (0.9%) Injection, USP
- Dextrose (5%) in Ringers Lactate Injection, USP
- Lactated Ringers Injection, USP
- 10% amino acid
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 Known Allergy
Cleviprex is contraindicated in patients with allergies to soybeans, soy products, eggs, or egg products.
-
5 WARNINGS AND PRECAUTIONS
5.1 Need for Aseptic Technique
Use aseptic technique and discard any unused product within 12 hours of stopper puncture [see Dosage and Administration (2.3)].
5.2 Hypotension and Reflex Tachycardia
Cleviprex may produce systemic hypotension and reflex tachycardia. If either occurs, decrease the dose of Cleviprex. There is limited experience with short-duration therapy with beta-blockers as a treatment for Cleviprex-induced tachycardia. Beta-blocker use for this purpose is not recommended.
5.3 Lipid Intake
Cleviprex contains approximately 0.2 g of lipid per mL (2.0 kcal). Lipid intake restrictions may be necessary for patients with significant disorders of lipid metabolism. For these patients, a reduction in the quantity of concurrently administered lipids may be necessary to compensate for the amount of lipid infused as part of the Cleviprex formulation.
5.4 Negative Inotropy
Dihydropyridine calcium channel blockers can produce negative inotropic effects and exacerbate heart failure. Monitor heart failure patients carefully.
5.5 Beta-Blocker Withdrawal
Cleviprex is not a beta-blocker, does not reduce heart rate, and gives no protection against the effects of abrupt beta-blocker withdrawal. Beta-blockers should be withdrawn only after a gradual reduction in dose.
-
6 ADVERSE REACTIONS
The following risk is discussed elsewhere in the labeling:
- Hypotension and Reflex Tachycardia [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Cleviprex clinical development included 19 studies, with 99 healthy subjects and 1307 hypertensive patients who received at least one dose of clevidipine (1406 total exposures). Clevidipine was evaluated in 15 studies in hypertensive patients: 1099 patients with perioperative hypertension, 126 with severe hypertension and 82 patients with essential hypertension.
The desired therapeutic response was achieved at doses of 4-6 mg/hour. Cleviprex was infused for <24 hours in the majority of patients (n=1199); it was infused as a continuous infusion in an additional 93 patients for durations between 24 and 72 hours.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Perioperative Hypertension
The placebo-controlled experience with Cleviprex in the perioperative setting was both small and brief (about 30 minutes). Table 2 shows treatment-emergent adverse reactions and the category of “any common adverse event” in ESCAPE-1 and ESCAPE-2 where the rate on Cleviprex exceeded the rate on placebo by at least 5% (common adverse reactions).
Table 2. Common adverse reactions in placebo-controlled perioperative studies. ESCAPE-1
ESCAPE-2
CLV
N=53(%)
PBO
N=51(%)
CLV
N=61(%)
PBO
N=49(%)
Any common adverse event 27 (51%)
21 (41%)
32 (53%)
24 (49%)
Acute renal failure 5 (9%)
1 (2%)
--
--
Atrial fibrillation --
--
13 (21%)
6 (12%)
Nausea --
--
13 (21%)
6 (12%)
Three randomized, parallel, open-label studies called ECLIPSE, with longer exposure in cardiac surgery patients define the adverse reactions for patients with perioperative hypertension. Each ECLIPSE study compared Cleviprex (n=752) to an active comparator: nitroglycerin (NTG, n=278), sodium nitroprusside (SNP, n=283), or nicardipine (NIC, n=193). The pooled mean maximum dose in these studies was 10 mg/hour and the mean duration of treatment was 8 hours.
There were many adverse events associated with the operative procedure in the clinical studies of Cleviprex and relatively few plausibly related to the drugs used to lower blood pressure. Thus, the ability to differentiate the adverse event profile between treatments is limited. The adverse events observed within one hour of the end of the infusion were similar in patients who received Cleviprex and in those who received comparator agents. There was no adverse reaction that was more than 2% more common on Cleviprex than on the average of all comparators.
Serious Adverse Events and Discontinuation – Perioperative Hypertension Studies
The incidence of adverse events leading to study drug discontinuation in patients with perioperative hypertension receiving Cleviprex was 5.9% versus 3.2% for all active comparators. For patients receiving Cleviprex and all active comparators the incidence of serious adverse events within one hour of drug infusion discontinuation was similar.Severe Hypertension
The adverse events for patients with severe hypertension are based on an uncontrolled study in patients with severe hypertension (VELOCITY, n=126).
The common adverse reactions for Cleviprex in severe hypertension included headache (6.3%), nausea (4.8%), and vomiting (3.2%). The incidence of adverse events leading to study drug discontinuation for Cleviprex in severe hypertension was 4.8%.
Less Common Adverse Reactions in Patients with Severe or Essential Hypertension
Adverse reactions that were reported in <1% of patients with severe or essential hypertension included:
Cardiac: myocardial infarction, cardiac arrest
Nervous system: syncope
Respiratory: dyspnea6.2 Post-Marketing and Other Clinical Experience
Because adverse reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate reliably their frequency or to establish a causal relationship to drug exposure. The following adverse reactions have been identified during post-approval use of Cleviprex: increased blood triglycerides, ileus, hypersensitivity, hypotension, nausea, decreased oxygen saturation (possible pulmonary shunting) and reflex tachycardia.
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Category C
There are no adequate and well-controlled studies of Cleviprex use in pregnant women. In animal studies, clevidipine caused increases in maternal and fetal mortality and length of gestation. Cleviprex should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.There was decreased fetal survival when pregnant rats and rabbits were treated with clevidipine during organogenesis at doses 0.7 times (on a body surface area basis) the maximum recommended human dose (MRHD) in rats and 2 times the MRHD in rabbits.
In pregnant rats dosed with clevidipine during late gestation and lactation, there were dose-related increases in maternal mortality, length of gestation and prolonged parturition at doses greater than or equal to 1/6 of the MRHD based on body surface area. When offspring of these dams were mated, they had a conception rate lower than that of controls. Clevidipine has been shown to cross the placenta in rats [see Nonclinical Toxicology (13.3)].
8.2 Labor and Delivery
Cleviprex in the labor and delivery setting has not been established as safe and effective. Other calcium channel blockers suppress uterine contractions in humans. Pregnant rats treated with clevidipine during late gestation had an increased rate of prolonged parturition.
8.3 Nursing Mothers
It is not known whether clevidipine is excreted in human milk. Because many drugs are excreted in human milk, consider possible infant exposure when Cleviprex is administered to a nursing woman.
8.4 Pediatric Use
The safety and effectiveness of Cleviprex in children under 18 years of age have not been established.
8.5 Geriatric Use
Of the 1406 subjects (1307 with hypertension) treated with Cleviprex in clinical studies, 620 were ≥65 years of age and 232 were ≥75 years of age. No overall differences in safety or effectiveness were observed between these and younger patients. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, for an elderly patient doses should be titrated cautiously, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal or cardiac function, and of concomitant disease or other drug therapy.
-
10 OVERDOSAGE
There has been no experience of overdosage in human clinical trials. In clinical trials, doses of Cleviprex up to 106 mg/hour or 1153 mg maximum total dose were administered. The expected major effects of overdose would be hypotension and reflex tachycardia.
Discontinuation of Cleviprex leads to a reduction in antihypertensive effects within 5 to 15 minutes [see Clinical Pharmacology (12.2)]. In case of suspected overdosage, Cleviprex should be discontinued immediately and the patient’s blood pressure should be supported.
-
11 DESCRIPTION
Cleviprex is a sterile, milky-white emulsion containing 0.5 mg/mL of clevidipine suitable for intravenous administration. Clevidipine is a dihydropyridine calcium channel blocker. Chemically, the active substance, clevidipine, is butyroxymethyl methyl 4-(2´,3´-dichlorophenyl)-1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate. It is a racemic mixture with a molecular weight of 456.3 g/mol. Each enantiomer has equipotent antihypertensive activity. The structure and formula are:
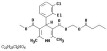
Clevidipine is practically insoluble in water and is formulated in an oil-in-water emulsion. In addition to the active ingredient, clevidipine, Cleviprex contains soybean oil (200 mg/mL), glycerin (22.5 mg/mL), purified egg yolk phospholipids (12 mg/mL),oleic acid (0.3 mg/mL), disodium edetate (0.05 mg/mL), and sodium hydroxide to adjust pH. Cleviprex has a pH of 6.0 – 8.0 and is a ready-to-use emulsion.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Clevidipine is a dihydropyridine L-type calcium channel blocker. L-type calcium channels mediate the influx of calcium during depolarization in arterial smooth muscle. Experiments in anesthetized rats and dogs show that clevidipine reduces mean arterial blood pressure by decreasing systemic vascular resistance. Clevidipine does not reduce cardiac filling pressure (pre-load), confirming lack of effects on the venous capacitance vessels.
12.2 Pharmacodynamics
Cleviprex is titrated to the desired reduction in blood pressure. The effect of Cleviprex appears to plateau at approximately 25% of baseline systolic pressure. The infusion rate for which half the maximal effect is observed is approximately 10 mg/hour.
Onset of Effect: In the perioperative patient population, Cleviprex produces a 4-5% reduction in systolic blood pressure within 2-4 minutes after starting a 0.4 mcg/kg/min infusion (approximately 1-2 mg/hr).
Maintenance of Effect: In studies up to 72 hours of continuous infusion, there was no evidence of tolerance or hysteresis.
Offset of Effect: In most patients, full recovery of blood pressure is achieved in 5-15 minutes after the infusion is stopped.
In studies up to 72 hours of continuous infusion, in patients that were not transitioned to other antihypertensive therapies, there was some evidence of rebound hypertension following Cleviprex discontinuation.
Hemodynamics: Cleviprex causes a dose-dependent decrease in systemic vascular resistance.
Heart Rate:An increase in heart rate is a normal response to vasodilation and decrease in blood pressure; in some patients these increases in heart rate may be pronounced [see Warnings and Precautions (5.2)].
Electrophysiologic Effects:In healthy volunteers, clevidipine or its major carboxylic acid metabolite, at therapeutic and supratherapeutic concentrations (approximately 2.8 times steady-state), did not prolong cardiac repolarization.
12.3 Pharmacokinetics
Clevidipine is rapidly distributed and metabolized resulting in a very short half life. The arterial blood concentration of clevidipine declines in a multi-phasic pattern following termination of the infusion. The initial phase half-life is approximately 1 minute, and accounts for 85-90% of clevidipine elimination. The terminal half-life is approximately 15 minutes.
Distribution: Clevidipine is >99.5% bound to proteins in plasma at 37°C. The steady-state volume of distribution was determined to be 0.17 L/kg in arterial blood.
Metabolism and Elimination: Clevidipine is rapidly metabolized by hydrolysis of the ester linkage, primarily by esterases in the blood and extravascular tissues, making its elimination unlikely to be affected by hepatic or renal dysfunction. The primary metabolites are the carboxylic acid metabolite and formaldehyde formed by hydrolysis of the ester group. The carboxylic acid metabolite is inactive as an antihypertensive. This metabolite is further metabolized by glucuronidation or oxidation to the corresponding pyridine derivative. The clearance of the primary dihydropyridine metabolite is 0.03 L/h/kg and the terminal half-life is approximately 9 hours.
In vitro studies show that clevidipine and its metabolite at the concentrations achieved in clinical practice will not inhibit or induce any CYP enzyme.
In a clinical study with radiolabeled clevidipine, 83% of the drug was excreted in urine and feces. The major fraction, 63-74% is excreted in the urine, 7-22% in the feces. More than 90% of the recovered radioactivity is excreted within the first 72 hours of collection.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Clevidipine displayed positive genotoxic potential in vitro in the Ames test, mouse lymphoma thymidine kinase locus assay, and chromosomal aberration assay, but not in vivo in the mouse micronucleus test. Formaldehyde, a metabolite of clevidipine, a known genotoxicant in vitro and a probable human carcinogen, appears to be at least partially responsible for the positive in vitro results. Long-term studies for evaluation of carcinogenic potential have not been performed with clevidipine due to the intended short-term duration of human use. There were no adverse effects on fertility or mating behavior of male rats at clevidipine doses of up to 55 mg/kg/day, approximately equivalent to the maximum recommended human dose (MRHD) of 504 mg/day (21 mg/hour x 24 hours) on a body surface area basis. Female rats demonstrated pseudopregnancy and changes in estrus cycle at doses as low as 13 mg/kg/day (about 1/4 th the MRHD); however, doses of up to 55 mg/kg/day did not affect mating performance or fertility.
13.3 Developmental Toxicology
When pregnant rats were dosed with clevidipine during late gestation and lactation, there were dose-related increases in mortality, length of gestation and prolonged parturition at dose levels as low as 13 mg/kg/day (about 1/4 th the maximum recommended human dose of 504 mg/day (21 mg/hour x 24 hours) on a body surface area basis). When offspring of these dams were mated, they had a conception rate lower than that of controls. Clevidipine crosses the placental membrane in this species and doses of 35 or more mg/kg/day (about 0.7 times the MRHD) administered during organogenesis adversely affected fetal survival. Fetal survival was also adversely affected when pregnant rabbits were treated during organogenesis with 55 mg/kg/day (about twice the MRHD on a body surface area basis).
-
14 CLINICAL STUDIES
14.1 Perioperative Hypertension
Cleviprex was evaluated in two double-blind, randomized, parallel, placebo-controlled, multicenter trials of cardiac surgery patients—pre-operative use in ESCAPE-1 (n=105) and post-operative use in ESCAPE-2 (n=110). Patients were undergoing coronary artery bypass grafting, with or without valve replacement. Inclusion in ESCAPE-1 required a systolic pressure ≥160 mmHg. In ESCAPE-2, the entry criterion was systolic pressure of ≥140 mmHg within 4 hours of the completed surgery. The mean baseline blood pressure was 178/77 mmHg in ESCAPE -1 and 150/71 mmHg in ESCAPE-2. The population of both studies included 27% females and 47% patients older than age 65.
Cleviprex was infused in ESCAPE-1 preoperatively for 30 minutes, until treatment failure, or until induction of anesthesia, whichever came first. Cleviprex was infused in ESCAPE-2 postoperatively for a minimum of 30 minutes unless alternative therapy was required. The maximum infusion time allowed in the ESCAPE studies was 60 minutes.
In both studies infusion of Cleviprex was started at a dose of 1- 2 mg/hour and was titrated upwards, as tolerated, in doubling increments every 90 seconds up to an infusion rate of 16 mg/hour in order to achieve the desired blood pressure-lowering effect. At doses above 16 mg/hour, increments were 7 mg/hour. The average Cleviprex infusion rate in ESCAPE-1 was 15.3 mg/hour and in ESCAPE-2 it was 5.1 mg/hour. The mean duration of exposure in the same ESCAPE studies was 30 minutes for the Cleviprex-treated patients.
Approximately 4% of Cleviprex-treated subjects in ESCAPE-1 and 41% in ESCAPE-2 were on concomitant vasodilators during the first 30 minutes of Cleviprex administration.
Cleviprex lowered blood pressure within 2-4 minutes. The change in systolic blood pressure over 30 minutes for ESCAPE-1 (preoperative) and ESCAPE-2 (postoperative) are shown in Figure 1 and 2.
Figure 1. Mean change in systolic blood pressure (mmHg) during 30-minute infusion, ESCAPE-1 (preoperative)
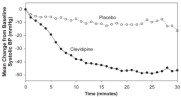
Figure 2. Mean change in systolic blood pressure (mmHg) during 30-minute infusion, ESCAPE-2 (postoperative)

The change in heart rate over 30 minutes for ESCAPE-1 (preoperative) and ESCAPE-2 (postoperative) are shown in Figure 3 and 4.
Figure 3. Mean change in heart rate (bpm) during 30-minute infusion, ESCAPE-1 (preoperative)
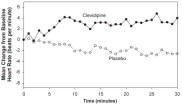
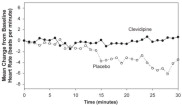
Figure 4. Mean change in heart rate (bpm) during 30-minute infusion, ESCAPE-2 (postoperative)
In three Phase 3 open-label clinical trials (ECLIPSE), 1512 patients were randomized to receive Cleviprex, nitroglycerin (perioperative hypertension), sodium nitroprusside (perioperative hypertension), or nicardipine (postoperative hypertension), for the treatment of hypertension in cardiac surgery. The mean exposure in the ECLIPSE studies was 8 hours at 4.5 mg/hour for the 752 patients who were treated with Cleviprex. Blood pressure control was assessed by measuring the magnitude and duration of SBP excursions outside the predefined pre- and post-operative SBP target range of 75-145 mmHg and the predefined intra-operative SBP range of 65-135 mmHg. In general, blood pressure control was similar with the four treatments.
14.2 Severe Hypertension
Cleviprex was evaluated in an open-label, uncontrolled clinical trial (VELOCITY) in 126 patients with severe hypertension (SBP >180 mmHg or diastolic blood pressure [DBP] >115 mmHg). Cleviprex infusion was initiated at 2 mg/hour and up-titrated every 3 minutes, doubling up to a maximum dose of 32 mg/hour as required to achieve a prespecified target blood pressure range within 30 minutes (primary endpoint). The transition to oral antihypertensive therapy was assessed for up to 6 hours following cessation of Cleviprex infusion.
The blood pressure effect in this study is shown in Figure 5. The average infusion rate was 9.5 mg/hour. The mean duration of Cleviprex exposure was 21 hours.
Figure 5. Mean percent change in SBP (%) during the first 30 minutes of infusion, VELOCITY (severe hypertension)
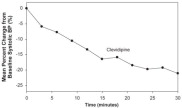
Oral antihypertensive therapy was instituted 1 hour prior to the anticipated cessation of Cleviprex infusion. Transition to oral antihypertensive therapy within 6 hours after discontinuing Cleviprex infusion was successful in 91% (115/126) of patients. No patient had IV antihypertensive therapy reinstituted following transition to oral therapy.
14.3 Essential Hypertension
Cleviprex was evaluated in a randomized, placebo-controlled, single-blind, parallel 72-hour continuous infusion study in 61 mild to moderate essential hypertensives. The mean baseline blood pressure was 151/86 mmHg.
Subjects were randomized to placebo or to 2, 4, 8, or 16 mg/hour. Doses above 2 mg/hour were started at 2 mg/hour and force-titrated in 2-fold increments at 3-minute intervals. Blood pressure, heart rate, and blood levels of clevidipine were measured during the infusion period. Blood levels were monitored 1 hour after the infusion was discontinued. Blood pressure and heart rate were monitored for 8 hours and also at 96 hours after the termination of infusion. Systolic blood pressure effect was related to the concentration of clevidipine and plateaued at higher measured concentrations, with the maximal effect estimated at 25% of baseline systolic blood pressure. The estimated infusion rate necessary to achieve half of this maximal effect was approximately 10 mg/hour.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Cleviprex (clevidipine) injectable emulsion is supplied as a sterile, milky white liquid emulsion product in single-use glass vials at a concentration of 0.5 mg/mL of clevidipine.
NDC 65293-005-50: 50 mL vial
NDC 65293-005-00: 100 mL vial
NDC 65293-005-25: 250 mL vialStorage
Leave vials in cartons until use. Clevidipine is photosensitive and storage in cartons protects against photodegradation. Protection from light during administration is not required.Store vials refrigerated at 2-8°C (36-46°F). Do not freeze. Vials in cartons may be transferred to 25°C (77°F, USP controlled room temperature) for a period not to exceed 2 months. Upon transfer to room temperature, mark vials in cartons “This product was removed from the refrigerator on _/_/_ date. It must be used or discarded 2 months after this date or the labeled expiration date (whichever date comes first).” Do not return to refrigerated storage after beginning room temperature storage.
Handling
Maintain aseptic technique while handling Cleviprex. Cleviprex is a single-use parenteral product that contains 0.005% disodium edetate to inhibit the rate of growth of microorganisms, for up to 12 hours, in the event of accidental contamination. However, Cleviprex can still support the growth of microorganisms, as it is not an antimicrobially preserved product under USP standards. Do not use if contamination is suspected. Once the stopper is punctured, use within 12 hours and discard any unused portion.Cleviprex inhibits microbial growth for up to 12 hours, as demonstrated by test data for representative USP microorganisms, staphylococcus epidermidis and serratiamarcescens.
-
17 PATIENT COUNSELING INFORMATION
- Advise patients with underlying hypertension that they require continued follow up for their medical condition, and, if applicable, encourage patients to continue taking their oral antihypertensive medication(s) as directed.
- Advise patients to contact a healthcare professional immediately for any of the following signs of a new hypertensive emergency: neurological symptoms, visual changes, or evidence of congestive heart failure.
Manufactured by:
Fresenius Kabi Austria GmbH, Graz, AustriaMarketed by:
The Medicines Company
Parsippany, New Jersey 07054For information call: 888-977-MDCO (6326)
US Patent 5,856,346
US Patent 5,739,152TMC [PN 1220-3 (November 08, 2013)]
M088834/04 USA
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
CLEVIPREX
clevidipine emulsionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:18124-011 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CLEVIDIPINE (UNII: 19O2GP3B7Q) (CLEVIDIPINE - UNII:19O2GP3B7Q) CLEVIDIPINE 0.5 mg in 1 mL Inactive Ingredients Ingredient Name Strength SOYBEAN OIL (UNII: 241ATL177A) GLYCERIN (UNII: PDC6A3C0OX) EGG PHOSPHOLIPIDS (UNII: 1Z74184RGV) OLEIC ACID (UNII: 2UMI9U37CP) EDETATE DISODIUM (UNII: 7FLD91C86K) SODIUM HYDROXIDE (UNII: 55X04QC32I) Product Characteristics Color white (Milky) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:18124-011-50 10 in 1 CARTON 09/15/2008 1 1 in 1 CARTON 1 50 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:18124-011-00 10 in 1 CARTON 09/15/2008 2 1 in 1 CARTON 2 100 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 3 NDC:18124-011-25 4 in 1 CARTON 09/15/2008 3 1 in 1 CARTON 3 250 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022156 09/15/2008 Labeler - Fresenius Kabi Austria GmbH (303448575) Establishment Name Address ID/FEI Business Operations Fresenius Kabi Austria GmbH 303448575 API Manufacture(18124-011) , manufacture(18124-011)