Label: DARZALEX- daratumumab injection, solution, concentrate
DARZALEX IV- daratumumab injection, solution, concentrate


- NDC Code(s): 57894-502-05, 57894-502-20, 57894-505-05, 57894-505-20
- Packager: Janssen Biotech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated April 15, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use DARZALEX safely and effectively. See full prescribing information for DARZALEX.
DARZALEX ®(daratumumab) injection, for intravenous use
Initial U.S. Approval: 2015INDICATIONS AND USAGE
DARZALEX is a CD38-directed cytolytic antibody indicated for the treatment of adult patients with multiple myeloma:
- in combination with lenalidomide and dexamethasone in newly diagnosed patients who are ineligible for autologous stem cell transplant and in patients with relapsed or refractory multiple myeloma who have received at least one prior therapy
- in combination with bortezomib, melphalan and prednisone in newly diagnosed patients who are ineligible for autologous stem cell transplant
- in combination with bortezomib, thalidomide, and dexamethasone in newly diagnosed patients who are eligible for autologous stem cell transplant
- in combination with bortezomib and dexamethasone in patients who have received at least one prior therapy
- in combination with carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma who have received one to three prior lines of therapy
- in combination with pomalidomide and dexamethasone in patients who have received at least two prior therapies including lenalidomide and a proteasome inhibitor
- as monotherapy, in patients who have received at least three prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory agent or who are double-refractory to a PI and an immunomodulatory agent. ( 1)
DOSAGE AND ADMINISTRATION
- Pre-medicate with corticosteroids, antipyretics and antihistamines. ( 2.3)
- Dilute and administer as an intravenous infusion. ( 2.5)
- Recommended dose is 16 mg/kg actual body weight. See full prescribing information for drugs used in combination and schedule. ( 2.2)
- Administer post-infusion medications. ( 2.3)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Patients with a history of severe hypersensitivity to daratumumab or any of the components of the formulation. ( 4)
WARNINGS AND PRECAUTIONS
- Infusion-related reactions: Interrupt DARZALEX infusion for infusion-related reactions of any severity. Permanently discontinue the infusion in case of anaphylactic reactions or life-threatening infusion-related reactions and institute appropriate emergency care. ( 2.4, 5.1)
- Interference with cross-matching and red blood cell antibody screening: Type and screen patients prior to starting treatment. Inform blood banks that a patient has received DARZALEX. ( 5.2, 7.1)
- Neutropenia: Monitor complete blood cell counts periodically during treatment. Monitor patients with neutropenia for signs of infection. Dose delay may be required to allow recovery of neutrophils. ( 5.3)
- Thrombocytopenia: Monitor complete blood cell counts periodically during treatment. Dose delay may be required to allow recovery of platelets. ( 5.4)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise pregnant women of the potential risk to a fetus and advise females of reproductive potential to use effective contraception. ( 5.6, 8.1, 8.3)
ADVERSE REACTIONS
The most frequently reported adverse reactions (incidence ≥20%) are: upper respiratory infection, neutropenia, infusion-related reactions, thrombocytopenia, diarrhea, constipation, anemia, peripheral sensory neuropathy, fatigue, peripheral edema, nausea, cough, pyrexia, dyspnea, and asthenia. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-526-7736 (1-800-JANSSEN) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 4/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
2.2 Recommended Dosage
2.3 Recommended Concomitant Medications
2.4 Dosage Modifications for Adverse Reactions
2.5 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions
5.2 Interference with Serological Testing
5.3 Neutropenia
5.4 Thrombocytopenia
5.5 Interference with Determination of Complete Response
5.6 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effects of Daratumumab on Laboratory Tests
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Newly Diagnosed Multiple Myeloma
14.2 Relapsed/Refractory Multiple Myeloma
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
DARZALEX is indicated for the treatment of adult patients with multiple myeloma:
- in combination with lenalidomide and dexamethasone in newly diagnosed patients who are ineligible for autologous stem cell transplant and in patients with relapsed or refractory multiple myeloma who have received at least one prior therapy.
- in combination with bortezomib, melphalan and prednisone in newly diagnosed patients who are ineligible for autologous stem cell transplant.
- in combination with bortezomib, thalidomide, and dexamethasone in newly diagnosed patients who are eligible for autologous stem cell transplant.
- in combination with bortezomib and dexamethasone in patients who have received at least one prior therapy.
- in combination with carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma who have received one to three prior lines of therapy.
- in combination with pomalidomide and dexamethasone in patients who have received at least two prior therapies including lenalidomide and a proteasome inhibitor.
- as monotherapy, in patients who have received at least three prior lines of therapy including a proteasome inhibitor (PI) and an immunomodulatory agent or who are double-refractory to a PI and an immunomodulatory agent.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Dosing Information
- Administer pre-infusion and post-infusion medications [see Dosage and Administration (2.3)] .
- Administer only as an intravenous infusion after dilution in 0.9% Sodium Chloride Injection [see Dosage and Administration (2.5)].
- DARZALEX should be administered by a healthcare provider, with immediate access to emergency equipment and appropriate medical support to manage infusion-related reactions if they occur [see Warnings and Precautions (5.1)].
- Type and screen patients prior to starting DARZALEX [see Warnings and Precautions (5.2)] .
2.2 Recommended Dosage
Monotherapy and In Combination with Lenalidomide (D-Rd) or Pomalidomide (D-Pd) and Dexamethasone
The DARZALEX dosing schedule in Table 1 is for combination therapy (4-week cycle regimens) and monotherapy as follows:
- combination therapy with lenalidomide and low-dose dexamethasone for newly diagnosed patients ineligible for autologous stem cell transplant (ASCT) and in patients with relapsed/refractory multiple myeloma
- combination therapy with pomalidomide and low-dose dexamethasone for patients with relapsed/refractory multiple myeloma
- monotherapy for patients with relapsed/refractory multiple myeloma.
The recommended dose of DARZALEX is 16 mg/kg actual body weight administered as an intravenous infusion according to the following dosing schedule:
Table 1: DARZALEX Dosing Schedule in Combination With Lenalidomide or Pomalidomide (4-Week Cycle) and Low-Dose Dexamethasone and for Monotherapy Weeks Schedule Weeks 1 to 8 weekly (total of 8 doses) Weeks 9 to 24 * every two weeks (total of 8 doses) Week 25 onwards until disease progression † every four weeks For dosing instructions of combination agents administered with DARZALEX, see Clinical Studies (14)and manufacturer's prescribing information.
In Combination with Bortezomib, Melphalan and Prednisone (D-VMP)
The DARZALEX dosing schedule in Table 2 is for combination therapy with bortezomib, melphalan and prednisone (6-week cycle regimen) for patients with newly diagnosed multiple myeloma ineligible for ASCT.
The recommended dose of DARZALEX is 16 mg/kg actual body weight administered as an intravenous infusion according to the following dosing schedule:
Table 2: DARZALEX Dosing Schedule in Combination With Bortezomib, Melphalan and Prednisone ([VMP], 6-Week Cycle) Weeks Schedule Weeks 1 to 6 weekly (total of 6 doses) Weeks 7 to 54 * every three weeks (total of 16 doses) Week 55 onwards until disease progression † every four weeks For dosing instructions of combination agents administered with DARZALEX see Clinical Studies (14.1).
In Combination with Bortezomib, Thalidomide and Dexamethasone (D-VTd)
The DARZALEX dosing schedule in Table 3 is for combination therapy with bortezomib, thalidomide, and dexamethasone (4-week cycle regimen) for patients with newly diagnosed multiple myeloma eligible for ASCT.
The recommended dose of DARZALEX is 16 mg/kg actual body weight administered as an intravenous infusion according to the following dosing schedule:
Table 3: DARZALEX Dosing Schedule in Combination With Bortezomib, Thalidomide and Dexamethasone ([VTd]; 4-Week Cycle) Treatment phase Weeks Schedule Induction Weeks 1 to 8 weekly (total of 8 doses) Weeks 9 to 16 * every two weeks (total of 4 doses) Stop for high dose chemotherapy and ASCT Consolidation Weeks 1 to 8 † every two weeks (total of 4 doses) For dosing instructions of combination agents administered with DARZALEX, see Clinical Studies (14.1)and the manufacturer's prescribing information.
In Combination with Bortezomib and Dexamethasone (D-Vd)
The DARZALEX dosing schedule in Table 4 is for combination therapy with bortezomib and dexamethasone (3-week cycle) for patients with relapsed/refractory multiple myeloma.
The recommended dose of DARZALEX is 16 mg/kg actual body weight administered as an intravenous infusion according to the following dosing schedule:
Table 4: DARZALEX Dosing Schedule With Bortezomib and Dexamethasone (3-Week Cycle) Weeks Schedule Weeks 1 to 9 weekly (total of 9 doses) Weeks 10 to 24 * every three weeks (total of 5 doses) Week 25 onwards until disease progression † every four weeks For dosing instructions of combination agents administered with DARZALEX see Clinical Studies (14.2)and manufacturer's prescribing information .
In Combination with Carfilzomib and Dexamethasone (DKd)
The recommended dosage for DARZALEX when administered in combination with carfilzomib and dexamethasone (4-week cycle) for patients with relapsed/refractory multiple myeloma is provided in Table 5.
Table 5: DARZALEX Dosing Schedule With Carfilzomib and Dexamethasone (4-Week Cycle) Weeks DARZALEX Dose * Schedule Week 1 8 mg/kg days 1 and 2 (total 2 doses) Weeks 2 to 8 16 mg/kg weekly (total of 7 doses) Weeks 9 to 24 † 16 mg/kg every two weeks (total of 8 doses) Week 25 onwards until disease progression ‡ 16 mg/kg every four weeks For dosing instructions of combination agents administered with DARZALEX see Clinical Studies (14.2)and manufacturer's prescribing information .
Infusion Rates
Administer DARZALEX intravenously at the infusion rate described below in Table 6. Consider incremental escalation of the infusion rate only in the absence of infusion-related reactions.
The recommended dose of 16 mg/kg to be administered on Day 1 when DARZALEX is administered as monotherapy or in combination may be split over two consecutive days, such that an 8 mg/kg dose is administered on Day 1 and Day 2, respectively.
Table 6: Infusion Rates for DARZALEX (16 mg/kg) Administration Dilution volume Initial rate (first hour) Rate increment * Maximum rate - *
- Consider incremental escalation of the infusion rate only in the absence of infusion-related reactions.
- †
- Use a dilution volume of 500 mL for the 16 mg/kg dose only if there were no infusion-related reactions the previous week. Otherwise, use a dilution volume of 1,000 mL.
- ‡
- Use a modified initial rate (100 mL/hour) for subsequent infusions (i.e. Week 3 onwards) only if there were no infusion-related reactions during the previous infusion. Otherwise, continue to use instructions indicated in the table for the Week 2 infusion rate.
Week 1 Infusion Option 1 (Single dose infusion) Week 1 Day 1 (16 mg/kg) 1,000 mL 50 mL/hour 50 mL/hour every hour 200 mL/hour Option 2 (Split dose infusion) Week 1 Day 1 (8 mg/kg) 500 mL 50 mL/hour 50 mL/hour every hour 200 mL/hour Week 1 Day 2 (8 mg/kg) 500 mL 50 mL/hour 50 mL/hour every hour 200 mL/hour Week 2 (16 mg/kg)† 500 mL 50 mL/hour 50 mL/hour every hour 200 mL/hour Week 3 onwards (16 mg/kg)‡ 500 mL 100 mL/hour 50 mL/hour every hour 200 mL/hour 2.3 Recommended Concomitant Medications
Pre-infusion Medication
Administer the following pre-infusion medications 1 hour to 3 hours before every DARZALEX infusion:
- Corticosteroid (long- or intermediate-acting)
Monotherapy:
Administer methylprednisolone 100 mg (or equivalent) intravenously. Following the second infusion, consider reducing the dose to 60 mg (or equivalent) administered either orally or intravenously.
In Combination:
Administer dexamethasone 20 mg (or equivalent) orally or intravenously.
When dexamethasone is the background regimen-specific corticosteroid, the dexamethasone dose that is part of the background regimen will serve as pre-medication on DARZALEX infusion days [see Clinical Studies (14)].
Do not administer background regimen-specific corticosteroids (e.g. prednisone) on DARZALEX infusion days when patients have received dexamethasone (or equivalent) as a pre-medication.- Acetaminophen 650 mg to 1,000 mg orally
- Diphenhydramine 25 mg to 50 mg (or equivalent) orally or intravenously.
Post-infusion Medication
Administer the following post-infusion medications:
-
Monotherapy:
Administer methylprednisolone 20 mg (or an equivalent dose of an intermediate- or long-acting corticosteroid) orally for 2 days starting the day after the administration of DARZALEX.
In Combination:
Consider administering oral methylprednisolone at a dose of less than or equal to 20 mg (or an equivalent dose of an intermediate- or long-acting corticosteroid) beginning the day after the administration of a DARZALEX infusion.
If a background regimen-specific corticosteroid (e.g. dexamethasone, prednisone) is administered the day after the DARZALEX infusion, additional corticosteroids may not be needed [see Clinical Studies (14)].
For patients with a history of chronic obstructive pulmonary disease, consider prescribing short and long-acting bronchodilators and inhaled corticosteroids. Following the first 4 DARZALEX infusions, consider discontinuing these additional post-infusion medications, if the patient does not experience a major infusion-related reaction.
Prophylaxis for Herpes Zoster Reactivation
Initiate antiviral prophylaxis to prevent herpes zoster reactivation within 1 week after starting DARZALEX and continue for 3 months following the end of treatment [see Adverse Reactions (6.1)].
2.4 Dosage Modifications for Adverse Reactions
No dose reductions of DARZALEX are recommended. Consider withholding DARZALEX to allow recovery of blood cell counts in the event of myelosuppression [see Warnings and Precautions (5.3, 5.4)] .
For information concerning drugs given in combination with DARZALEX, see manufacturer's prescribing information.
Infusion-Related Reactions
For infusion-related reactions of any grade/severity, immediately interrupt the DARZALEX infusion and manage symptoms. Management of infusion-related reactions may further require reduction in the rate of infusion, or treatment discontinuation of DARZALEX as outlined below [see Warnings and Precautions (5.1)] .
- Grade 1–2 (mild to moderate): Once reaction symptoms resolve, resume the infusion at no more than half the rate at which the reaction occurred. If the patient does not experience any further reaction symptoms, infusion rate escalation may resume at increments and intervals as clinically appropriate up to the maximum rate of 200 mL/hour (Table 6).
- Grade 3 (severe): Once reaction symptoms resolve, consider restarting the infusion at no more than half the rate at which the reaction occurred. If the patient does not experience additional symptoms, resume infusion rate escalation at increments and intervals as outlined in Table 6. Repeat the procedure above in the event of recurrence of Grade 3 symptoms. Permanently discontinue DARZALEX upon the third occurrence of a Grade 3 or greater infusion-related reaction.
- Grade 4 (life-threatening): Permanently discontinue DARZALEX.
2.5 Preparation and Administration
Preparation
DARZALEX is for single dose only.
Prepare the solution for infusion using aseptic technique as follows:
- Calculate the dose (mg), total volume (mL) of DARZALEX solution required and the number of DARZALEX vials needed based on patient actual body weight.
- DARZALEX vials of the same strength with different NDCs are available and can be admixed in the same infusion bag [see Description (11), How Supplied/Storage and Handling (16)] .
- Check that the DARZALEX solution is colorless to pale yellow. Do not use if opaque particles, discoloration or other foreign particles are present.
- Remove a volume of 0.9% Sodium Chloride Injection from the infusion bag/container that is equal to the required volume of DARZALEX solution.
- Withdraw the necessary amount of DARZALEX solution and dilute to the appropriate volume by adding to the infusion bag/container containing 0.9% Sodium Chloride Injection as specified in Table 6 [see Dosage and Administration (2.2)] . Infusion bags/containers must be made of either polyvinylchloride (PVC), polypropylene (PP), polyethylene (PE) or polyolefin blend (PP+PE). Dilute under appropriate aseptic conditions. Discard any unused portion left in the vial.
- Gently invert the bag/container to mix the solution. Do not shake.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. The diluted solution may develop very small, translucent to white proteinaceous particles, as daratumumab is a protein. Do not use if visibly opaque particles, discoloration or foreign particles are observed.
- If not used immediately, store the diluted solution refrigerated for up to 24 hours at 2°C to 8°C (36°F to 46°F) and/or at room temperature up to 15 hours at 15°C to 25°C (59°F to 77°F). The room temperature storage includes infusion time. Protect from light during storage. Do not freeze.
Administration
- If stored in the refrigerator, allow the solution to come to room temperature. Administer the diluted solution by intravenous infusion using an infusion set fitted with a flow regulator and with an in-line, sterile, non-pyrogenic, low protein-binding polyethersulfone (PES) filter (pore size 0.22 micrometer or 0.2 micrometer). Administration sets must be made of either polyurethane (PU), polybutadiene (PBD), PVC, PP or PE.
- Do not store any unused portion of the infusion solution for reuse. Any unused product or waste material should be disposed of in accordance with local requirements.
- Do not infuse DARZALEX concomitantly in the same intravenous line with other agents.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
DARZALEX is contraindicated in patients with a history of severe hypersensitivity (e.g. anaphylactic reactions) to daratumumab or any of the components of the formulation [see Warnings and Precautions (5.1)] .
-
5 WARNINGS AND PRECAUTIONS
5.1 Infusion-Related Reactions
DARZALEX can cause severe and/or serious infusion-related reactions including anaphylactic reactions. These reactions can be life-threatening and fatal outcomes have been reported [see Adverse Reactions (6.2)] .
In clinical trials (monotherapy and combination: N=2,066), infusion-related reactions occurred in 37% of patients with the Week 1 (16 mg/kg) infusion, 2% with the Week 2 infusion, and cumulatively 6% with subsequent infusions. Less than 1% of patients had a Grade 3/4 infusion-related reaction at Week 2 or subsequent infusions. The median time to onset was 1.5 hours (range: 0 to 73 hours). The incidence of infusion modification due to reactions was 36%. Median durations of 16 mg/kg infusions for the Week 1, Week 2, and subsequent infusions were approximately 7, 4, and 3 hours respectively. Nearly all reactions occurred during infusion or within 4 hours of completing DARZALEX. Prior to the introduction of post-infusion medication in clinical trials, infusion-related reactions occurred up to 48 hours after infusion.
Severe reactions have occurred, including bronchospasm, hypoxia, dyspnea, hypertension, tachycardia, headache, laryngeal edema, pulmonary edema, and ocular adverse reactions, including choroidal effusion, acute myopia, and acute angle closure glaucoma. Signs and symptoms may include respiratory symptoms, such as nasal congestion, cough, throat irritation, as well as chills, vomiting and nausea. Less common signs and symptoms were wheezing, allergic rhinitis, pyrexia, chest discomfort, pruritus, hypotension, and blurred vision [see Adverse Reactions (6.1)] .
When DARZALEX dosing was interrupted in the setting of ASCT (CASSIOPEIA) for a median of 3.75 months (range: 2.4 to 6.9 months), upon re-initiation of DARZALEX, the incidence of infusion-related reactions was 11% for the first infusion following ASCT. Infusion rate/dilution volume used upon re-initiation was that used for the last DARZALEX infusion prior to interruption for ASCT. Infusion-related reactions occurring at re-initiation of DARZALEX following ASCT were consistent in terms of symptoms and severity (Grade 3 or 4: <1%) with those reported in previous studies at Week 2 or subsequent infusions.
In EQUULEUS, patients receiving combination treatment (n=97) were administered the first 16 mg/kg dose at Week 1 split over two days i.e. 8 mg/kg on Day 1 and Day 2, respectively. The incidence of any grade infusion-related reactions was 42%, with 36% of patients experiencing infusion-related reactions on Day 1 of Week 1, 4% on Day 2 of Week 1, and 8% with subsequent infusions. The median time to onset of a reaction was 1.8 hours (range: 0.1 to 5.4 hours). The incidence of infusion interruptions due to reactions was 30%. Median durations of infusions were 4.2 hours for Week 1-Day 1, 4.2 hours for Week 1-Day 2, and 3.4 hours for the subsequent infusions.
Pre-medicate patients with antihistamines, antipyretics and corticosteroids. Frequently monitor patients during the entire infusion [see Dosage and Administration (2.3)] . Interrupt DARZALEX infusion for reactions of any severity and institute medical management as needed. Permanently discontinue DARZALEX therapy if an anaphylactic reaction or life-threatening (Grade 4) reaction occurs and institute appropriate emergency care. For patients with Grade 1, 2, or 3 reactions, reduce the infusion rate when re-starting the infusion [see Dosage and Administration (2.4)] .
To reduce the risk of delayed infusion-related reactions, administer oral corticosteroids to all patients following DARZALEX infusions [see Dosage and Administration (2.3)] . Patients with a history of chronic obstructive pulmonary disease may require additional post-infusion medications to manage respiratory complications. Consider prescribing short- and long-acting bronchodilators and inhaled corticosteroids for patients with chronic obstructive pulmonary disease [see Dosage and Administration (2.3)] .
Ocular adverse reactions, including acute myopia and narrowing of the anterior chamber angle due to ciliochoroidal effusions with potential for increased intraocular pressure or glaucoma, have occurred with DARZALEX infusion. If ocular symptoms occur, interrupt DARZALEX infusion and seek immediate ophthalmologic evaluation prior to restarting DARZALEX.
5.2 Interference with Serological Testing
Daratumumab binds to CD38 on red blood cells (RBCs) and results in a positive Indirect Antiglobulin Test (Indirect Coombs test). Daratumumab-mediated positive indirect antiglobulin test may persist for up to 6 months after the last daratumumab infusion. Daratumumab bound to RBCs masks detection of antibodies to minor antigens in the patient's serum [see References (15)] . The determination of a patient's ABO and Rh blood type are not impacted [see Drug Interactions (7.1)] .
Notify blood transfusion centers of this interference with serological testing and inform blood banks that a patient has received DARZALEX. Type and screen patients prior to starting DARZALEX [see Dosage and Administration (2.1)] .
5.3 Neutropenia
DARZALEX may increase neutropenia induced by background therapy [see Adverse Reactions (6.1)].
Monitor complete blood cell counts periodically during treatment according to manufacturer's prescribing information for background therapies. Monitor patients with neutropenia for signs of infection. Consider withholding DARZALEX until recovery of neutrophils.
5.4 Thrombocytopenia
DARZALEX may increase thrombocytopenia induced by background therapy [see Adverse Reactions (6.1)].
Monitor complete blood cell counts periodically during treatment according to manufacturer's prescribing information for background therapies. Consider withholding DARZALEX until recovery of platelets.
5.5 Interference with Determination of Complete Response
Daratumumab is a human IgG kappa monoclonal antibody that can be detected on both, the serum protein electrophoresis (SPE) and immunofixation (IFE) assays used for the clinical monitoring of endogenous M-protein [see Drug Interactions (7.1)] . This interference can impact the determination of complete response and of disease progression in some patients with IgG kappa myeloma protein.
5.6 Embryo-Fetal Toxicity
Based on the mechanism of action, DARZALEX can cause fetal harm when administered to a pregnant woman. DARZALEX may cause depletion of fetal immune cells and decreased bone density. Advise pregnant women of the potential risk to a fetus. Advise females with reproductive potential to use effective contraception during treatment with DARZALEX and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)] .
The combination of DARZALEX with lenalidomide, pomalidomide, or thalidomide is contraindicated in pregnant women, because lenalidomide, pomalidomide, and thalidomide may cause birth defects and death of the unborn child. Refer to the lenalidomide, pomalidomide, or thalidomide prescribing information on use during pregnancy.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Infusion-related reactions [see Warnings and Precautions (5.1)] .
- Neutropenia [see Warnings and Precautions (5.3)] .
- Thrombocytopenia [see Warnings and Precautions (5.4)] .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described below reflects exposure to DARZALEX (16 mg/kg) in 2,459 patients with multiple myeloma including 2,303 patients who received DARZALEX in combination with background regimens and 156 patients who received DARZALEX as monotherapy. In this pooled safety population, the most common adverse reactions (≥20%) were upper respiratory infection, neutropenia, infusion-related reactions, thrombocytopenia, diarrhea, constipation, anemia, peripheral sensory neuropathy, fatigue, peripheral edema, nausea, cough, pyrexia, dyspnea, and asthenia.
Newly Diagnosed Multiple Myeloma Ineligible for Autologous Stem Cell Transplant
Combination Treatment with Lenalidomide and Dexamethasone (DRd)
The safety of DARZALEX in combination with lenalidomide and dexamethasone was evaluated in MAIA [see Clinical Studies (14.1)]. Adverse reactions described in Table 7 reflect exposure to DARZALEX for a median treatment duration of 25.3 months (range: 0.1 to 40.44 months) for daratumumab-lenalidomide-dexamethasone (DRd) and of 21.3 months (range: 0.03 to 40.64 months) for lenalidomide-dexamethasone (Rd).
Serious adverse reactions with a 2% greater incidence in the DRd arm compared to the Rd arm were pneumonia (DRd 15% vs Rd 8%), bronchitis (DRd 4% vs Rd 2%) and dehydration (DRd 2% vs Rd <1%).
Table 7: Adverse Reactions Reported in ≥10% of Patients and With at Least a 5% Greater Frequency in the DRd Arm in MAIA Body System
Adverse ReactionDRd (N=364) Rd (N=365) All Grades (%) Grade 3 (%) Grade 4 (%) All Grades (%) Grade 3 (%) Grade 4 (%) Key: D=daratumumab, Rd=lenalidomide-dexamethasone. - *
- Acute sinusitis, Bacterial rhinitis, Laryngitis, Metapneumovirus infection, Nasopharyngitis, Oropharyngeal candidiasis, Pharyngitis, Respiratory syncytial virus infection, Respiratory tract infection, Respiratory tract infection viral, Rhinitis, Rhinovirus infection, Sinusitis, Tonsillitis, Tracheitis, Upper respiratory tract infection, Viral pharyngitis, Viral rhinitis, Viral upper respiratory tract infection
- †
- Bronchiolitis, Bronchitis, Bronchitis viral, Respiratory syncytial virus bronchiolitis, Tracheobronchitis
- ‡
- Atypical pneumonia, Bronchopulmonary aspergillosis, Lung infection, Pneumocystis jirovecii infection, Pneumocystis jirovecii pneumonia, Pneumonia, Pneumonia aspiration, Pneumonia pneumococcal, Pneumonia viral, Pulmonary mycosis
- §
- Infusion-related reaction includes terms determined by investigators to be related to infusion
- ¶
- Generalized edema, Gravitational edema, Edema, Peripheral edema, Peripheral swelling
- #
- Dyspnea, Dyspnea exertional
- Þ
- Cough, Productive cough
- ß
- Blood pressure increased, Hypertension
Gastrointestinal disorders Diarrhea 57 7 0 46 4 0 Constipation 41 1 <1 36 <1 0 Nausea 32 1 0 23 1 0 Vomiting 17 1 0 12 <1 0 Infections Upper respiratory tract infection * 52 2 <1 36 2 <1 Bronchitis † 29 3 0 21 1 0 Pneumonia ‡ 26 14 1 14 7 1 Urinary tract infection 18 2 0 10 2 0 General disorders and administration site conditions Infusion-related reactions § 41 2 <1 0 0 0 Peripheral edema ¶ 41 2 0 33 1 0 Fatigue 40 8 0 28 4 0 Asthenia 32 4 0 25 3 <1 Pyrexia 23 2 0 18 2 0 Chills 13 0 0 2 0 0 Musculoskeletal and connective tissue disorders Back pain 34 3 <1 26 3 <1 Muscle spasms 29 1 0 22 1 0 Respiratory, thoracic and mediastinal disorders Dyspnea # 32 3 <1 20 1 0 Cough Þ 30 <1 0 18 0 0 Nervous system disorders Peripheral sensory neuropathy 24 1 0 15 0 0 Headache 19 1 0 11 0 0 Paresthesia 16 0 0 8 0 0 Metabolism and nutrition disorders Decreased appetite 22 1 0 15 <1 <1 Hyperglycemia 14 6 1 8 3 1 Hypocalcemia 14 1 <1 9 1 1 Vascular disorders Hypertension ß 13 6 <1 7 4 0 Laboratory abnormalities worsening during treatment from baseline listed in Table 8.
Table 8: Treatment-Emergent Hematology Laboratory Abnormalities in MAIA DRd (N=364) Rd (N=365) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, Rd=lenalidomide-dexamethasone. Leukopenia 90 30 5 82 20 4 Neutropenia 91 39 17 77 28 11 Lymphopenia 84 41 11 75 36 6 Thrombocytopenia 67 6 3 58 7 4 Anemia 47 13 0 57 24 0 Combination Treatment with Bortezomib, Melphalan and Prednisone
The safety of DARZALEX in combination with bortezomib, melphalan and prednisone was evaluated in ALCYONE [see Clinical Studies (14.1)]. Adverse reactions described in Table 9 reflect exposure to DARZALEX for a median treatment duration of 14.7 months (range: 0 to 25.8 months) for daratumumab, bortezomib, melphalan and prednisone (D-VMP) and of 12 months (range: 0.1 to 14.9 months) for VMP.
Serious adverse reactions with at least a 2% greater incidence in the D-VMP arm compared to the VMP arm were pneumonia (D-VMP 11% vs VMP 4%), upper respiratory tract infection (D-VMP 5% vs VMP 1%), and pulmonary edema (D-VMP 2% vs VMP 0%).
Table 9: Adverse Reactions Reported in ≥10% of Patients and With at Least a 5% Greater Frequency in the D-VMP Arm in ALCYONE Body System
Adverse ReactionD-VMP (N=346) VMP (N=354) All Grades (%) Grade 3 (%) Grade 4 (%) All Grades (%) Grade 3 (%) Grade 4 (%) Key: D=daratumumab, VMP=bortezomib-melphalan-prednisone. - *
- upper respiratory tract infection, bronchitis, bronchitis bacterial, epiglottitis, laryngitis, laryngitis bacterial, metapneumovirus infection, nasopharyngitis, oropharyngeal candidiasis, pharyngitis, pharyngitis streptococcal, respiratory syncytial virus infection, respiratory tract infection, respiratory tract infection viral, rhinitis, sinusitis, tonsillitis, tracheitis, tracheobronchitis, viral pharyngitis, viral rhinitis, viral upper respiratory tract infection
- †
- pneumonia, lung infection, pneumonia aspiration, pneumonia bacterial, pneumonia pneumococcal, pneumonia streptococcal, pneumonia viral, and pulmonary sepsis
- ‡
- Infusion-related reaction includes terms determined by investigators to be related to infusion
- §
- edema peripheral, generalized edema, peripheral swelling
- ¶
- cough, productive cough
- #
- dyspnea, dyspnea exertional
- Þ
- hypertension, blood pressure increased
Infections Upper respiratory tract infection * 48 5 0 28 3 0 Pneumonia † 16 12 < 1 6 5 < 1 General disorders and administration site conditions Infusion-related reactions ‡ 28 4 1 0 0 0 Peripheral edema § 21 1 < 1 14 1 0 Respiratory, thoracic and mediastinal disorders Cough ¶ 16 < 1 0 8 < 1 0 Dyspnea # 13 2 1 5 1 0 Vascular disorders Hypertension Þ 10 4 < 1 3 2 0 Laboratory abnormalities worsening during treatment from baseline listed in Table 10.
Table 10: Treatment-Emergent Hematology Laboratory Abnormalities in ALCYONE D-VMP (N=346) VMP (N=354) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, VMP=bortezomib-melphalan-prednisone. Thrombocytopenia 88 27 11 88 26 16 Neutropenia 86 34 10 87 32 11 Lymphopenia 85 46 12 83 44 9 Anemia 47 18 0 50 21 0 Newly Diagnosed Multiple Myeloma Eligible for Autologous Stem Cell Transplant
Combination Treatment with Bortezomib, Thalidomide and Dexamethasone (DVTd)
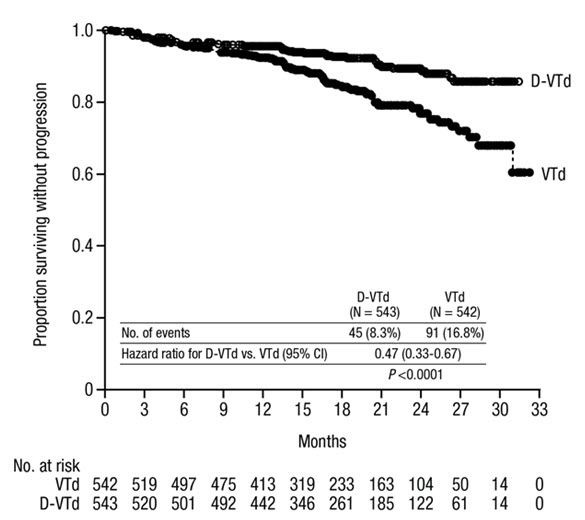
The safety of DARZALEX in combination with bortezomib, thalidomide and dexamethasone was evaluated in CASSIOPEIA [see Clinical Studies (14.1)]. Adverse reactions described in Table 11 reflect exposure to DARZALEX up to day 100 post-transplant. The median duration of induction/ASCT/consolidation treatment was 8.9 months (range: 7.0 to 12.0 months) for DVTd and 8.7 months (range: 6.4 to 11.5 months) for VTd.
Serious adverse reactions with a 2% greater incidence in the DVTd arm compared to the VTd arm were bronchitis (DVTd 2% vs VTd <1%) and pneumonia (DVTd 6% vs VTd 4%).
Table 11: Adverse Reactions Reported in ≥ 10% of Patients and With at Least a 5% Greater Frequency in the DVTd Arm in CASSIOPEIA Body System
Adverse ReactionDVTd (N=536) VTd (N=538) All Grades (%) Grade 3 (%) Grade 4 (%) All Grades (%) Grade 3 (%) Grade 4 (%) Key: D=daratumumab, VTd=bortezomib-thalidomide -dexamethasone.
Note: Hematology laboratory related toxicities were excluded and reported separately in the table below.- *
- Infusion-related reaction includes terms determined by investigators to be related to infusion
- †
- Laryngitis, Laryngitis viral, Metapneumovirus infection, Nasopharyngitis, Oropharyngeal candidiasis, Pharyngitis, Respiratory syncytial virus infection, Respiratory tract infection, Respiratory tract infection viral, Rhinitis, Rhinovirus infection, Sinusitis, Tonsillitis, Tracheitis, Upper respiratory tract infection, Viral pharyngitis, Viral rhinitis, Viral upper respiratory tract infection
- ‡
- Bronchiolitis, Bronchitis, Bronchitis chronic, Respiratory syncytial virus bronchitis, Tracheobronchitis
- §
- Cough, Productive cough
General disorders and administration site conditions Infusion-related reactions * 35 3 <1 0 0 0 Pyrexia 26 2 <1 21 2 0 Gastrointestinal disorders Nausea 30 4 0 24 2 <1 Vomiting 16 2 0 10 2 0 Infections Upper respiratory tract infection † 27 1 0 17 1 0 Bronchitis ‡ 20 1 0 13 1 0 Respiratory, thoracic and mediastinal disorders Cough § 17 0 0 9 0 0 Vascular disorders Hypertension 10 4 0 5 2 0 Table 12: Treatment-Emergent Hematology Laboratory Abnormalities in CASSIOPEIA DVTd (N=536) VTd (N=538) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, VTd=bortezomib-thalidomide -dexamethasone. Lymphopenia 95 44 15 91 37 10 Leukopenia 82 14 10 57 6 9 Thrombocytopenia 81 9 5 58 8 3 Neutropenia 63 19 14 41 10 9 Anemia 36 4 0 35 5 0 Relapsed/Refractory Multiple Myeloma
Combination Treatment with Lenalidomide and Dexamethasone
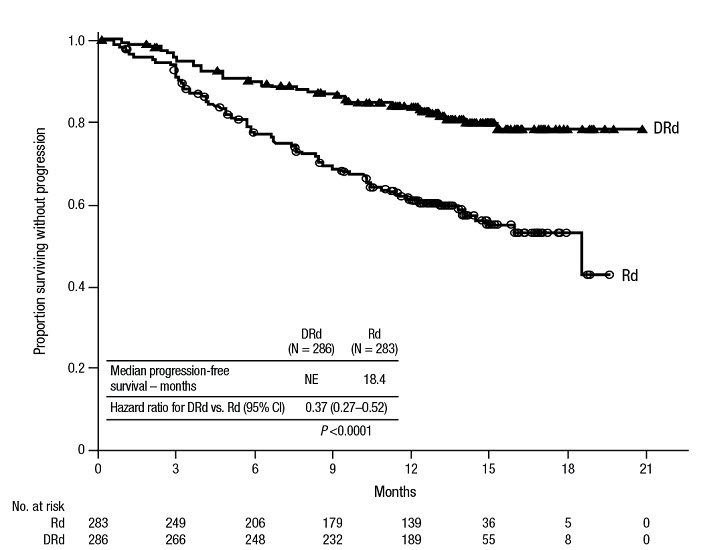
The safety of DARZALEX in combination with lenalidomide and dexamethasone was evaluated in POLLUX [see Clinical Studies (14.2)]. Adverse reactions described in Table 13 reflect exposure to DARZALEX for a median treatment duration of 13.1 months (range: 0 to 20.7 months) for daratumumab-lenalidomide-dexamethasone (DRd) and of 12.3 months (range: 0.2 to 20.1 months) for lenalidomide-dexamethasone (Rd).
Serious adverse reactions occurred in 49% of patients in the DRd arm compared with 42% in the Rd arm. Serious adverse reactions with at least a 2% greater incidence in the DRd arm compared to the Rd arm were pneumonia (DRd 12% vs Rd 10%), upper respiratory tract infection (DRd 7% vs Rd 4%), influenza and pyrexia (DRd 3% vs Rd 1% for each).
Adverse reactions resulted in discontinuations for 7% (n=19) of patients in the DRd arm versus 8% (n=22) in the Rd arm.
Table 13: Adverse Reactions Reported in ≥ 10% of Patients and With at Least a 5% Greater Frequency in the DRd Arm in POLLUX Adverse Reaction DRd (N=283) Rd (N=281) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, Rd=lenalidomide-dexamethasone. - *
- upper respiratory tract infection, bronchitis, sinusitis, respiratory tract infection viral, rhinitis, pharyngitis, respiratory tract infection, metapneumovirus infection, tracheobronchitis, viral upper respiratory tract infection, laryngitis, respiratory syncytial virus infection, staphylococcal pharyngitis, tonsillitis, viral pharyngitis, acute sinusitis, nasopharyngitis, bronchiolitis, bronchitis viral, pharyngitis streptococcal, tracheitis, upper respiratory tract infection bacterial, bronchitis bacterial, epiglottitis, laryngitis viral, oropharyngeal candidiasis, respiratory moniliasis, viral rhinitis, acute tonsillitis, rhinovirus infection
- †
- Infusion-related reaction includes terms determined by investigators to be related to infusion
- ‡
- cough, productive cough, allergic cough
- §
- dyspnea, dyspnea exertional
Infections Upper respiratory tract infection * 65 6 < 1 51 4 0 General disorders and administration site conditions Infusion-related reactions † 48 5 0 0 0 0 Fatigue 35 6 < 1 28 2 0 Pyrexia 20 2 0 11 1 0 Gastrointestinal disorders Diarrhea 43 5 0 25 3 0 Nausea 24 1 0 14 0 0 Vomiting 17 1 0 5 1 0 Respiratory, thoracic and mediastinal disorders Cough ‡ 30 0 0 15 0 0 Dyspnea § 21 3 < 1 12 1 0 Musculoskeletal and connective tissue disorders Muscle spasms 26 1 0 19 2 0 Nervous system disorders Headache 13 0 0 7 0 0 Laboratory abnormalities worsening during treatment from baseline listed in Table 14.
Table 14: Treatment-Emergent Hematology Laboratory Abnormalities in POLLUX DRd (N=283) Rd (N=281) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, Rd=lenalidomide-dexamethasone. Lymphopenia 95 42 10 87 32 6 Neutropenia 92 36 17 87 32 8 Thrombocytopenia 73 7 6 67 10 5 Anemia 52 13 0 57 19 0 Combination Treatment with Bortezomib and Dexamethasone
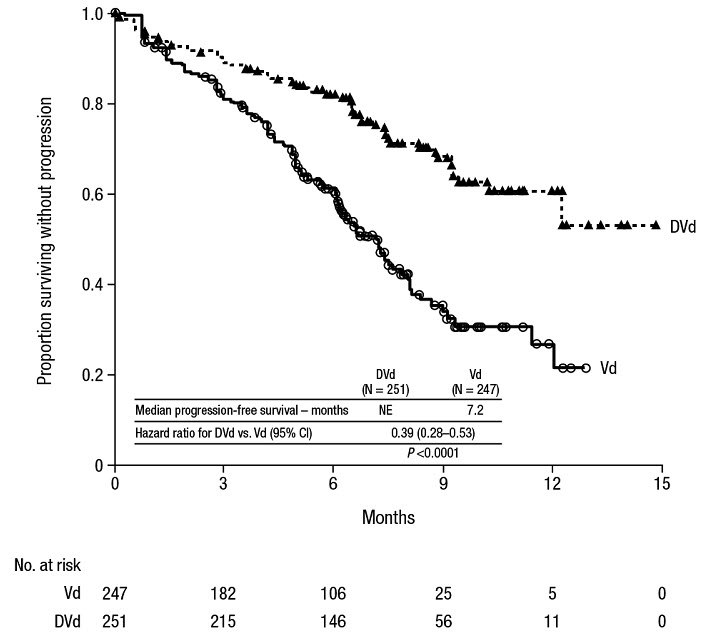
The safety of DARZALEX in combination with bortezomib and dexamethasone was evaluated in CASTOR [see Clinical Studies (14.2)]. Adverse reactions described in Table 15 reflect exposure to DARZALEX for a median treatment duration of 6.5 months (range: 0 to 14.8 months) for daratumumab-bortezomib-dexamethasone (DVd) and of 5.2 months (range: 0.2 to 8.0 months) for bortezomib-dexamethasone (Vd) arm.
Serious adverse reactions occurred in 42% of patients in the DVd arm compared with 34% in the Vd arm. Serious adverse reactions with at least a 2% greater incidence in the DVd arm compared to the Vd arm were upper respiratory tract infection (DVd 5% vs Vd 2%), diarrhea and atrial fibrillation (DVd 2% vs Vd 0% for each).
Adverse reactions resulted in discontinuations for 7% (n=18) of patients in the DVd arm versus 9% (n=22) in the Vd arm.
Table 15: Adverse Reactions Reported in ≥10% of Patients and With at Least a 5% Greater Frequency in the DVd Arm CASTOR Adverse Reaction DVd (N=243) Vd (N=237) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, Vd=bortezomib-dexamethasone. - *
- Infusion-related reaction includes terms determined by investigators to be related to infusion
- †
- edema peripheral, edema, generalized edema, peripheral swelling
- ‡
- upper respiratory tract infection, bronchitis, sinusitis, respiratory tract infection viral, rhinitis, pharyngitis, respiratory tract infection, metapneumovirus infection, tracheobronchitis, viral upper respiratory tract infection, laryngitis, respiratory syncytial virus infection, staphylococcal pharyngitis, tonsillitis, viral pharyngitis, acute sinusitis, nasopharyngitis, bronchiolitis, bronchitis viral, pharyngitis streptococcal, tracheitis, upper respiratory tract infection bacterial, bronchitis bacterial, epiglottitis, laryngitis viral, oropharyngeal candidiasis, respiratory moniliasis, viral rhinitis, acute tonsillitis, rhinovirus infection
- §
- cough, productive cough, allergic cough
- ¶
- dyspnea, dyspnea exertional
Nervous system disorders Peripheral sensory neuropathy 47 5 0 38 6 < 1 General disorders and administration site conditions Infusion-related reactions * 45 9 0 0 0 0 Peripheral edema † 22 1 0 13 0 0 Pyrexia 16 1 0 11 1 0 Infections Upper respiratory tract infection ‡ 44 6 0 30 3 < 1 Gastrointestinal disorders Diarrhea 32 3 < 1 22 1 0 Vomiting 11 0 0 4 0 0 Respiratory, thoracic and mediastinal disorders Cough § 27 0 0 14 0 0 Dyspnea ¶ 21 4 0 11 1 0 Laboratory abnormalities worsening during treatment are listed in Table 16.
Table 16: Treatment-Emergent Hematology Laboratory Abnormalities in CASTOR DVd (N=243) Vd (N=237) All Grades
(%)Grade 3
(%)Grade 4
(%)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, Vd=bortezomib-dexamethasone. Thrombocytopenia 90 28 19 85 22 13 Lymphopenia 89 41 7 81 24 3 Neutropenia 58 12 3 40 5 < 1 Anemia 48 13 0 56 14 0 Combination Treatment with Twice-Weekly (20/56 mg/m 2) Carfilzomib and Dexamethasone
The safety of DARZALEX in combination with twice weekly carfilzomib and dexamethasone was evaluated in CANDOR [see Clinical Studies (14.2)] . Adverse reactions described in Table 17 reflect exposure to DARZALEX for a median treatment duration of 16.1 months (range: 0.1 to 23.7 months) for the daratumumab-carfilzomib-dexamethasone (DKd) group and median treatment duration of 9.3 months (range: 0.1 to 22.4 months) for the carfilzomib-dexamethasone group (Kd).
Serious adverse reactions occurred in 56% of patients who received DARZALEX in combination with Kd and 46% of patients who received Kd. The most frequent serious adverse reactions reported in the DKd arm as compared with the Kd arm were pneumonia (DKd 14% vs Kd 9%), pyrexia (DKd 4.2% vs Kd 2.0%), influenza (DKd 3.9% vs Kd 1.3%), sepsis (DKd 3.9% vs Kd 1.3%), anemia (DKd 2.3% vs Kd 0.7%), bronchitis (DKd 1.9% vs Kd 0%), and diarrhea (DKd 1.6% vs Kd 0%). Fatal adverse reactions within 30 days of the last dose of any study treatment occurred in 10% of 308 patients who received DARZALEX in combination with Kd versus 5% of 153 patients who received Kd. The most frequent fatal adverse reaction was infection (4.5% vs 2.6%).
Permanent discontinuation of DARZALEX due to an adverse reaction occurred in 9% of patients. Adverse reactions (>1%) which resulted in permanent discontinuation of DARZALEX included pneumonia.
Infusion-related reactions that occurred on the day of administration of any DARZALEX dose or on the next day occurred in 18% of patients and that occurred on the day of administration of the first DARZALEX dose or the next day occurred in 12%.
Table 17: Adverse Reactions (≥15%) in Patients Who Received DARZALEX in Combination with Carfilzomib and Dexamethasone (DKd) in CANDOR Adverse Reaction DKd (N=308) Kd (N=153) All Grades Grades 3 or 4 All Grades Grades 3 or 4 (%) (%) (%) (%) Key: D=daratumumab; Kd=carfilzomib-dexamethasone. - *
- The incidence of infusion-related reactions is based on a group of symptoms (including hypertension, pyrexia, rash, myalgia, hypotension, blood pressure increased, urticaria, acute kidney injury, bronchospasm, face edema, hypersensitivity, rash, syncope, wheezing, eye pruritus, eyelid edema, renal failure, swelling face) related to infusion reactions which occurred within 1 day after DKd or Kd administration.
- †
- Fatigue includes fatigue and asthenia.
- ‡
- Respiratory tract infection includes respiratory tract infection, lower respiratory tract infection, upper respiratory tract infection and viral upper respiratory tract infection.
- §
- Includes fatal adverse reactions.
- ¶
- Thrombocytopenia includes platelet count decreased and thrombocytopenia.
- #
- Anemia includes anemia, hematocrit decreased and hemoglobin decreased.
- Þ
- Cough includes productive cough and cough.
General Disorders and Administration Site Conditions Infusion-related reactions * 41 12 28 5 Fatigue † 32 11 28 8 Pyrexia 20 1.9 15 0.7 Infections Respiratory tract infection ‡ 40 § 7 29 3.3 Pneumonia 18 § 13 12 9 Bronchitis 17 2.6 12 1.3 Blood and lymphatic system disorders Thrombocytopenia ¶ 37 25 30 16 Anemia # 33 17 31 14 Gastrointestinal disorders Diarrhea 32 3.9 14 0.7 Nausea 18 0 13 0.7 Vascular Disorders Hypertension 31 18 28 13 Respiratory, Thoracic and Mediastinal Disorders Cough Þ 21 0 21 0 Dyspnea 20 3.9 22 2.6 Psychiatric disorders Insomnia 18 3.9 11 2 Musculoskeletal and connective tissue disorders Back pain 16 1.9 10 1.3 Adverse Reactions Occurring at a Frequency of < 15%
- Blood and lymphatic system disorders:neutropenia, lymphopenia, leukopenia, febrile neutropenia
- Cardiac disorders:atrial fibrillation
- Gastrointestinal disorders:vomiting, constipation
- General disorders and administration site conditions:peripheral edema, asthenia, chills
- Infections:influenza, urinary tract infection, sepsis, septic shock
- Metabolism and nutrition disorders:decreased appetite, hyperglycemia, hypocalcemia, dehydration
- Musculoskeletal and connective tissue disorders:muscle spasms, arthralgia, musculoskeletal chest pain
- Nervous system disorders:headache, dizziness, peripheral sensory neuropathy, paresthesia, posterior reversible encephalopathy syndrome
- Respiratory, thoracic and mediastinal disorders:pulmonary edema
- Skin and subcutaneous tissue disorders:rash, pruritus
Combination Treatment with Once-Weekly (20/70 mg/m 2) Carfilzomib and Dexamethasone
The safety of DARZALEX in combination with once-weekly carfilzomib and dexamethasone was evaluated in EQUULEUS [see Clinical Studies (14.2)] . Adverse reactions described in Table 18 reflect exposure to DARZALEX for a median treatment duration of 19.8 months (range: 0.3 to 34.5 months).
Serious adverse reactions were reported in 48% of patients. The most frequent serious adverse reactions reported were pneumonia (4.7%), upper respiratory tract infection (4.7%), basal cell carcinoma (4.7%), influenza (3.5%), general physical health deterioration (3.5%), and hypercalcemia (3.5%). Fatal adverse reactions within 30 days of the last dose of any study treatment occurred in 3.5% of patients who died of general physical health deterioration, multi-organ failure secondary to pulmonary aspergillosis, and disease progression.
Permanent discontinuation of DARZALEX due to an adverse reaction occurred in 8% of patients. No adverse reactions which resulted in permanent discontinuation of DARZALEX occurred in more than one patient.
Infusion-related reactions that occurred on the day of administration of any DARZALEX dose or on the next day occurred in 44% of patients. For patients who received the split first dose of DARZALEX, infusion-related reactions that occurred in 36% and 4% on the first and second day of administration of DARZALEX, respectively.
Table 18: Adverse Reactions (≥15%) of Patients Who Received DARZALEX in Combination with Carfilzomib and Dexamethasone in EQUULEUS Adverse Reaction DKd (N=85) All Grades (%) Grades 3 or 4 (%) Key: D=daratumumab; Kd=carfilzomib-dexamethasone. - *
- Thrombocytopenia includes platelet count decreased and thrombocytopenia.
- †
- Anemia includes anemia, hematocrit decreased and hemoglobin decreased.
- ‡
- Neutropenia includes neutrophil count decreased and neutropenia.
- §
- Lymphopenia includes lymphocyte count decreased and lymphopenia
- ¶
- Fatigue includes fatigue and asthenia.
- #
- The incidence of infusion-related reactions is based on a group of symptoms (including hypertension, pyrexia, rash, myalgia, hypotension, blood pressure increased, urticaria, acute kidney injury, bronchospasm, face edema, hypersensitivity, rash, syncope, wheezing, eye pruritus, eyelid edema, renal failure, swelling face) related to infusion reactions which occurred within 1 day after DKd administration.
- Þ
- Respiratory tract infection includes respiratory tract infection, lower respiratory tract infection, upper respiratory tract infection and viral upper respiratory tract infection.
- ß
- Cough includes productive cough and cough.
Blood and lymphatic system disorders Thrombocytopenia * 68 32 Anemia † 52 21 Neutropenia ‡ 31 21 Lymphopenia § 29 25 General disorder and administration site conditions Fatigue ¶ 54 18 Infusion-related reactions # 53 12 Pyrexia 37 1.2 Infections Respiratory tract infection Þ 53 3.5 Bronchitis 19 0 Nasopharyngitis 18 0 Influenza 17 3.5 Gastrointestinal disorders Nausea 42 1.2 Vomiting 40 1.2 Diarrhea 38 2.4 Constipation 17 0 Respiratory, thoracic and mediastinal disorders Dyspnea 35 3.5 Cough ß 33 0 Vascular disorders Hypertension 33 20 Psychiatric disorders Insomnia 33 4.7 Nervous system disorders Headache 27 1.2 Musculoskeletal and connective tissue disorders Back pain 25 0 Pain in extremity 15 0 Adverse Reactions Occurring at a Frequency of < 15%
- Blood and lymphatic system disorders:leukopenia, febrile neutropenia
- Cardiac disorders:atrial fibrillation
- Gastrointestinal disorders:pancreatitis
- General disorders and administration site conditions:peripheral edema, chills
- Infections:pneumonia, urinary tract infection, sepsis, septic shock
- Metabolism and nutrition disorders:decreased appetite, hyperglycemia, dehydration, hypocalcemia
- Musculoskeletal and connective tissue disorders:muscle spasms, musculoskeletal chest pain, arthralgia
- Nervous system disorders:dizziness, paresthesia, peripheral sensory neuropathy
- Skin and subcutaneous tissue disorders:pruritus, rash
Combination Treatment with Pomalidomide and Dexamethasone
The safety of DARZALEX in combination with pomalidomide and dexamethasone was evaluated in EQUULEUS [see Clinical Studies (14.2)] . Adverse reactions described in Table 19 reflect exposure to DARZALEX, pomalidomide and dexamethasone (DPd) for a median treatment duration of 6 months (range: 0.03 to 16.9 months).
The overall incidence of serious adverse reactions was 49%. Serious adverse reactions reported in ≥5% patients included pneumonia (7%). Adverse reactions resulted in discontinuations for 13% of patients.
Table 19: Adverse Reactions With Incidence ≥10% Reported in EQUULEUS Adverse Reaction DPd (N=103) All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, Pd=pomalidomide-dexamethasone. - *
- Infusion-related reaction includes terms determined by investigators to be related to infusion
- †
- edema, edema peripheral, peripheral swelling
- ‡
- acute tonsillitis, bronchitis, laryngitis, nasopharyngitis, pharyngitis, respiratory syncytial virus infection, rhinitis, sinusitis, tonsillitis, upper respiratory tract infection
- §
- lung infection, pneumonia, pneumonia aspiration
- ¶
- cough, productive cough, allergic cough
- #
- dyspnea, dyspnea exertional
General disorders and administration site conditions Fatigue 50 10 0 Infusion-related reactions * 50 4 0 Pyrexia 25 1 0 Chills 20 0 0 Edema peripheral † 17 4 0 Asthenia 15 0 0 Non-cardiac chest pain 15 0 0 Pain 11 0 0 Infections Upper respiratory tract infection ‡ 50 4 1 Pneumonia § 15 8 2 Respiratory, thoracic and mediastinal disorders Cough ¶ 43 1 0 Dyspnea # 33 6 1 Nasal congestion 16 0 0 Gastrointestinal disorders Diarrhea 38 3 0 Constipation 33 0 0 Nausea 30 0 0 Vomiting 21 2 0 Musculoskeletal and connective tissue disorders Muscle spasms 26 1 0 Back pain 25 6 0 Arthralgia 22 2 0 Pain in extremity 15 0 0 Bone pain 13 4 0 Musculoskeletal chest pain 13 2 0 Psychiatric disorders Insomnia 23 2 0 Anxiety 13 0 0 Nervous system disorders Dizziness 21 2 0 Tremor 19 3 0 Headache 17 0 0 Metabolism and nutrition disorders Hypokalemia 16 3 0 Hyperglycemia 13 5 1 Decreased appetite 11 0 0 Laboratory abnormalities worsening during treatment are listed in Table 20.
Table 20: Treatment-Emergent Hematology Laboratory Abnormalities in EQUULEUS DPd
(N=103)All Grades
(%)Grade 3
(%)Grade 4
(%)Key: D=daratumumab, Pd=pomalidomide-dexamethasone. Neutropenia 95 36 46 Lymphopenia 94 45 26 Thrombocytopenia 75 10 10 Anemia 57 30 0 Monotherapy
The safety of DARZALEX was evaluated in 156 adult patients with relapsed and refractory multiple myeloma in three open-label, clinical trials. Patients received DARZALEX 16 mg/kg. The median duration of exposure was 3.3 months (range: 0.03 to 20.04 months).
Serious adverse reactions were reported in 51 (33%) patients. The most frequent serious adverse reactions were pneumonia (6%), general physical health deterioration (3%), and pyrexia (3%).
Adverse reactions resulted in treatment delay for 24 (15%) patients, most frequently for infections. Adverse reactions resulted in discontinuations for 6 (4%) patients.
Adverse reactions occurring in at least 10% of patients are presented in Table 21. Table 22 describes Grade 3–4 laboratory abnormalities reported at a rate of ≥10%.
Table 21: Adverse Reactions With Incidence ≥10% in Patients With Multiple Myeloma Treated With DARZALEX 16 mg/kg Adverse Reaction DARZALEX
(N=156)All Grades
(%)Grade 3
(%)Grade 4
(%)General disorders and administration site conditions Infusion-related reaction * 48 3 0 Fatigue 39 2 0 Pyrexia 21 1 0 Chills 10 0 0 Gastrointestinal disorders Nausea 27 0 0 Diarrhea 16 1 0 Constipation 15 0 0 Vomiting 14 0 0 Musculoskeletal and connective tissue disorders Back pain 23 2 0 Arthralgia 17 0 0 Pain in extremity 15 1 0 Musculoskeletal chest pain 12 1 0 Respiratory, thoracic and mediastinal disorders Cough 21 0 0 Nasal congestion 17 0 0 Dyspnea 15 1 0 Infections Upper respiratory tract infection 20 1 0 Nasopharyngitis 15 0 0 Pneumonia † 11 6 0 Metabolism and nutrition disorders Decreased appetite 15 1 0 Nervous system disorders Headache 12 1 0 Vascular disorders Hypertension 10 5 0 Table 22: Treatment-Emergent Grade 3–4 Laboratory Abnormalities (≥10%) Daratumumab 16 mg/kg (N=156) All Grades
(%)Grade 3
(%)Grade 4
(%)Lymphopenia 72 30 10 Neutropenia 60 17 3 Thrombocytopenia 48 10 8 Anemia 45 19 0 Herpes Zoster Virus Reactivation
Prophylaxis for Herpes Zoster Virus reactivation was recommended for patients in some clinical trials of DARZALEX. In monotherapy studies, herpes zoster was reported in 3% of patients. In the combination therapy studies, herpes zoster was reported in 2–5% of patients receiving DARZALEX.
Infections
Grade 3 or 4 infections were reported as follows:
- Relapsed/refractory patient studies: DVd: 21% vs. Vd: 19%; DRd: 28% vs. Rd: 23%; DPd: 28%; DKd 1: 37%, Kd 1: 29%; DKd 2: 21%
- Newly diagnosed patient studies: D-VMP: 23%, VMP: 15%; DRd: 32%, Rd: 23%; DVTd: 22%; VTd: 20%.
Pneumonia was the most commonly reported severe (Grade 3 or 4) infection across studies. In active controlled studies, discontinuations from treatment due to infections occurred in 1–4% of patients.
Fatal infections (Grade 5) were reported as follows:
- Relapsed/refractory patient studies: DVd: 1%, Vd: 2%; DRd: 2%, Rd: 1%; DPd: 2%; DKd 1: 5%, Kd 1: 3%; DKd 2: 0%
- Newly diagnosed patient studies: D-VMP: 1%, VMP: 1%; DRd: 2%, Rd: 2%; DVTd: 0%, VTd: 0%.
Fatal infections were generally infrequent and balanced between the DARZALEX containing regimens and active control arms. Fatal infections were primarily due to pneumonia and sepsis.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of daratumumab. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System disorders:Anaphylactic reaction, IRR (including deaths)
Gastrointestinal disorders:Pancreatitis
Infections:Cytomegalovirus, Listeriosis
-
7 DRUG INTERACTIONS
7.1 Effects of Daratumumab on Laboratory Tests
Interference with Indirect Antiglobulin Tests (Indirect Coombs Test)
Daratumumab binds to CD38 on RBCs and interferes with compatibility testing, including antibody screening and cross matching. Daratumumab interference mitigation methods include treating reagent RBCs with dithiothreitol (DTT) to disrupt daratumumab binding [see References (15)] or genotyping. Since the Kell blood group system is also sensitive to DTT treatment, supply K-negative units after ruling out or identifying alloantibodies using DTT-treated RBCs.
If an emergency transfusion is required, administer non-cross-matched ABO/RhD-compatible RBCs per local blood bank practices.
Interference with Serum Protein Electrophoresis and Immunofixation Tests
Daratumumab may be detected on serum protein electrophoresis (SPE) and immunofixation (IFE) assays used for monitoring disease monoclonal immunoglobulins (M protein). False positive SPE and IFE assay results may occur for patients with IgG kappa myeloma protein impacting initial assessment of complete responses by International Myeloma Working Group (IMWG) criteria. In patients with persistent very good partial response, where daratumumab interference is suspected, consider using a FDA-approved daratumumab-specific IFE assay to distinguish daratumumab from any remaining endogenous M protein in the patient's serum, to facilitate determination of a complete response.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
DARZALEX can cause fetal harm when administered to a pregnant woman. The assessment of associated risks with daratumumab products is based on the mechanism of action and data from target antigen CD38 knockout animal models (see Data) . There are no available data on the use of DARZALEX in pregnant women to evaluate drug-associated risk of major birth defects, miscarriage or adverse maternal or fetal outcomes. Animal reproduction studies have not been conducted.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
The combination of DARZALEX and lenalidomide, pomalidomide, or thalidomide is contraindicated in pregnant women, because lenalidomide, pomalidomide, and thalidomide may cause birth defects and death of the unborn child. Lenalidomide, pomalidomide, and thalidomide are only available through a REMS program. Refer to the lenalidomide, pomalidomide, or thalidomide prescribing information on use during pregnancy.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Immunoglobulin G1 (IgG1) monoclonal antibodies are transferred across the placenta. Based on its mechanism of action, DARZALEX may cause depletion of fetal CD38 positive immune cells and decreased bone density. Defer administering live vaccines to neonates and infants exposed to DARZALEX in uterountil a hematology evaluation is completed.
Data
Animal Data
Mice that were genetically modified to eliminate all CD38 expression (CD38 knockout mice) had reduced bone density at birth that recovered by 5 months of age. Data from studies using CD38 knockout animal models also suggest the involvement of CD38 in regulating humoral immune responses (mice), feto-maternal immune tolerance (mice), and early embryonic development (frogs).
8.2 Lactation
Risk Summary
There is no data on the presence of daratumumab in human milk, the effects on the breastfed child, or the effects on milk production. Maternal immunoglobulin G is known to be present in human milk. Published data suggest that antibodies in breast milk do not enter the neonatal and infant circulations in substantial amounts. Because of the potential for serious adverse reactions in the breastfed child when DARZALEX is administered with lenalidomide, pomalidomide, or thalidomide, advise women not to breastfeed during treatment with DARZALEX. Refer to lenalidomide, pomalidomide, or thalidomide prescribing information for additional information.
8.3 Females and Males of Reproductive Potential
DARZALEX can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)] .
8.4 Pediatric Use
Safety and effectiveness of DARZALEX in pediatric patients have not been established.
The safety and effectiveness of DARZALEX in combination with chemotherapy were assessed but not established in a single open-label trial (DELPHINUS; NCT03384654) in 34 pediatric patients (2 to <17 years of age) with relapsed/refractory acute lymphoblastic leukemia or lymphoblastic lymphoma. No new safety signals were observed in these pediatric patients. The pharmacokinetic parameters in these pediatric patients were within range of values previously observed in adults with multiple myeloma given the same dose based on body weight.
8.5 Geriatric Use
Of the 2,459 patients who received DARZALEX at the recommended dose, 38% were 65 to 74 years of age, and 15% were 75 years of age and older. No overall differences in effectiveness were observed between these patients and younger patients. The incidence of serious adverse reactions was higher in older than in younger patients [see Adverse Reactions (6.1)] . Among patients with relapsed and refractory multiple myeloma (n=1,213), the serious adverse reactions that occurred more frequently in patients 65 years and older were pneumonia and sepsis. Within the DKd group in CANDOR, fatal adverse reactions occurred in 14% of patients 65 years and older compared to 6% of patients less than 65 years. Among patients with newly diagnosed multiple myeloma who are ineligible for autologous stem cell transplant (n=710), the serious adverse reaction that occurred more frequently in patients 75 years and older was pneumonia.
-
11 DESCRIPTION
Daratumumab is an immunoglobulin G1 kappa (IgG1κ) human monoclonal antibody that binds to CD38 antigen. It is produced in Chinese Hamster Ovary (CHO) cells using recombinant DNA technology. The molecular weight of daratumumab is approximately 148 kDa.
DARZALEX ®(daratumumab) injection is supplied as a colorless to pale yellow preservative-free solution for intravenous use in a single-dose vial. The pH is 5.5.
Each DARZALEX 20 mL single-dose vial contains (NDC 57894-502-20) 400 mg daratumumab, glacial acetic acid (3.7 mg), mannitol (510 mg), polysorbate 20 (8 mg), sodium acetate trihydrate (59.3 mg), sodium chloride (70.1 mg), and Water for Injection, USP.
Each DARZALEX 5 mL single-dose vial contains (NDC 57894-502-05) 100 mg daratumumab, glacial acetic acid (0.9 mg), mannitol (127.5 mg), polysorbate 20 (2 mg), sodium acetate trihydrate (14.8 mg), sodium chloride (17.5 mg), and Water for Injection, USP.
Each DARZALEX 20 mL single-dose vial contains (NDC 57894-505-20) 400 mg daratumumab, L-histidine (7 mg), L-histidine hydrochloride monohydrate (32.6 mg), L-methionine (20 mg), polysorbate 20 (8 mg), sorbitol (1093 mg), and Water for Injection, USP.
Each DARZALEX 5 mL single-dose vial contains (NDC 57894-505-05) 100 mg daratumumab, L-histidine (1.8 mg), L-histidine hydrochloride monohydrate (8.2 mg), L-methionine (5 mg), polysorbate 20 (2 mg), sorbitol (273.3 mg), and Water for Injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CD38 is a transmembrane glycoprotein (48 kDa) expressed on the surface of hematopoietic cells, including multiple myeloma and other cell types and tissues and has multiple functions, such as receptor mediated adhesion, signaling, and modulation of cyclase and hydrolase activity. Daratumumab is an IgG1κ human monoclonal antibody (mAb) that binds to CD38 and inhibits the growth of CD38 expressing tumor cells by inducing apoptosis directly through Fc mediated cross linking as well as by immune-mediated tumor cell lysis through complement dependent cytotoxicity (CDC), antibody dependent cell mediated cytotoxicity (ADCC) and antibody dependent cellular phagocytosis (ADCP). A subset of myeloid derived suppressor cells (CD38+MDSCs), regulatory T cells (CD38+T regs) and B cells (CD38+B regs) are decreased by daratumumab.
12.2 Pharmacodynamics
NK cells express CD38 and are susceptible to daratumumab mediated cell lysis. Decreases in absolute counts and percentages of total NK cells (CD16+CD56+) and activated (CD16+CD56 dim) NK cells in peripheral whole blood and bone marrow were observed with DARZALEX treatment.
12.3 Pharmacokinetics
Daratumumab area under the concentration-time curve (AUC) increases more than proportionally over a dosage range from 1 to 24 mg/kg (0.06 to 1.5 times the approved recommended dosage) as monotherapy or 1 to 16 mg/kg (0.06 to 1 time the approved recommended dosage) as combination therapy.
Following administration of the approved recommended dosage of DARZALEX as monotherapy or in combination therapy, the mean serum maximal concentration (C max) was approximately 2.7 to 3-fold higher at the end of weekly dosing compared to the first dose. The mean ± standard deviation (SD) trough serum concentration (C min) at the end of weekly dosing was 573 ± 332 µg/mL when DARZALEX was administered as monotherapy and 502 ± 196 to 607 ± 231 µg/mL when DARZALEX was administered as combination therapy. Split dosing of the first dose resulted in a different PK profile in the first day compared to single dosing; however, similar C max and C min concentrations were both predicted and observed following the administration of the second split dose on Week 1 Day 2.
When DARZALEX was administered as monotherapy, daratumumab steady state was achieved approximately 5 months into the every 4-week dosing period (by the 21 stinfusion). At steady state, daratumumab mean ± SD accumulation ratio for C max was 1.6 ± 0.5.
Distribution
Daratumumab volume of distribution was 4.7 ± 1.3 L as monotherapy and 4.4 ± 1.5 L as combination therapy following administration of the approved dosage.
Elimination
Daratumumab clearance decreased with increasing dose and with multiple dosing. The mean ± SD linear clearance was estimated to be 171.4 ± 95.3 mL/day and the mean ± SD estimated terminal half-life associated with linear clearance was 18 ± 9 days following administration of the approved recommended dosage of DARZALEX as monotherapy. Terminal half-life was similar when DARZALEX was administered as combination therapy.
Specific Populations
No clinically significant differences in the pharmacokinetics of daratumumab as monotherapy or as combination therapy were observed based on sex, age (31 to 93 years), mild [total bilirubin 1 to 1.5 times upper limit of normal (ULN) or aspartate aminotransaminase (AST)>ULN] and moderate (total bilirubin 1.5 to 3 times ULN and any AST) hepatic impairment, or renal impairment [Creatinine clearance (CLcr) 15–89 mL/min]. The effect of severe (total bilirubin >3 times ULN and any AST) hepatic impairment on daratumumab pharmacokinetics is unknown.
12.6 Immunogenicity
The observed incidence of anti-drug antibody (ADA, including neutralizing antibody) is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of ADA in the studies described below with the incidence of ADA in other studies, including those of daratumumab or of other daratumumab products.
With the median DARZALEX treatment ranging from 3.3 to 48 months across 10 clinical trials of patients with multiple myeloma treated with DARZALEX as monotherapy or as combination therapies, the incidence of anti-daratumumab antibody development was 0.6% (14/2,179) and 12 patients tested positive for neutralizing antibodies. Because of the low occurrence of anti-drug antibodies, the effect of these antibodies on the pharmacokinetics, pharmacodynamics, safety, and/or effectiveness of daratumumab products is unknown.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or genotoxicity studies have been conducted with daratumumab. No animal studies have been performed to evaluate the potential effects of daratumumab on reproduction or development, or to determine potential effects on fertility in males or females.
-
14 CLINICAL STUDIES
14.1 Newly Diagnosed Multiple Myeloma
Combination Treatment with Lenalidomide and Dexamethasone in Patients Ineligible for Autologous Stem Cell Transplant
MAIA (NCT02252172), an open-label, randomized, active-controlled trial, compared treatment with DARZALEX 16 mg/kg in combination with lenalidomide and low-dose dexamethasone (DRd) to treatment with lenalidomide and low-dose dexamethasone (Rd) in patients with newly diagnosed multiple myeloma ineligible for autologous stem cell transplant. Lenalidomide (25 mg once daily orally on Days 1–21 of repeated 28-day [4-week] cycles) was given with low dose oral or intravenous dexamethasone 40 mg/week (or a reduced dose of 20 mg/week for patients >75 years or body mass index [BMI] <18.5). On DARZALEX infusion days, the dexamethasone dose was given as a pre-infusion medication. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 737 patients were randomized: 368 to the DRd arm and 369 to the Rd arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The median age was 73 (range: 45–90) years, with 44% of the patients ≥75 years of age. Fifty-two percent (52%) of patients were male, 92% White, 4% Black or African American, and 1% Asian. Three percent (3%) of patients reported an ethnicity of Hispanic or Latino. Thirty-four (34%) had an Eastern Cooperative Oncology Group (ECOG) performance score of 0, 50% had an ECOG performance score of 1 and 17% had an ECOG performance score of ≥2. Twenty-seven percent had International Staging System (ISS) Stage I, 43% had ISS Stage II and 29% had ISS Stage III disease. Efficacy was evaluated by progression free survival (PFS) based on International Myeloma Working Group (IMWG) criteria.
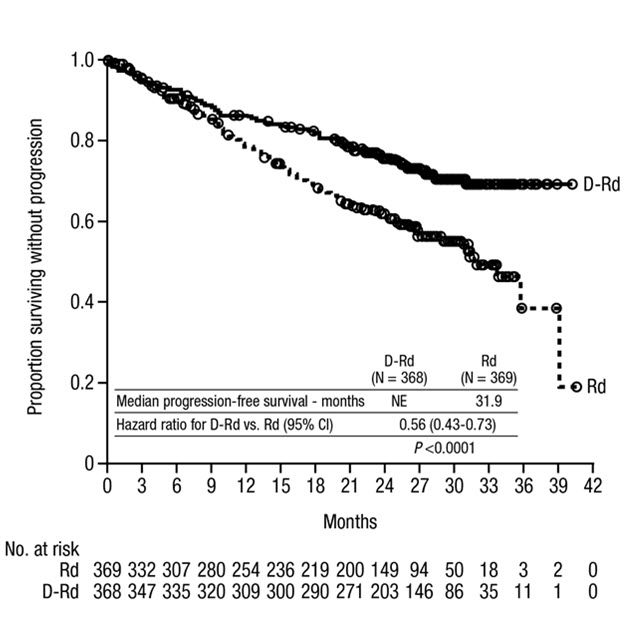
MAIA demonstrated an improvement in Progression Free Survival (PFS) in the DRd arm as compared to the Rd arm; the median PFS had not been reached in the DRd arm and was 31.9 months in the Rd arm (hazard ratio [HR]=0.56; 95% CI: 0.43, 0.73; p<0.0001), representing 44% reduction in the risk of disease progression or death in patients treated with DRd. After a median follow-up of 64 months, the median PFS was 61.9 months (95% CI: 54.8, NE) in the DRd arm and 34.4 months (95% CI: 29.6, 39.2) in the Rd arm.
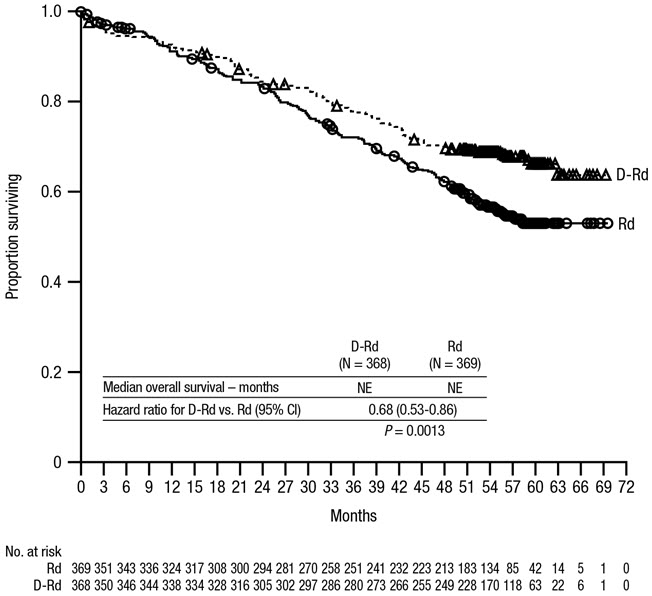
After a median follow-up of 56 months, MAIA demonstrated an improvement in overall survival (OS) in the DRd arm as compared to the Rd arm (HR=0.68; 95% CI: 0.53, 0.86; p=0.0013), representing a 32% reduction in the risk of death in patients treated in the DRd arm. Median OS had not been reached for either arm.
After a median follow-up of 89 months, the median OS was 90.3 months (95% CI: 80.8, NE) in the DRd arm and 64.1 months (95% CI: 56.0, 70.8) in the Rd arm.
Additional efficacy results from MAIA are presented in Table 23.
Table 23: Additional Efficacy Results From MAIA * DRd (N=368) Rd (N=369) DRd=daratumumab-lenalidomide-dexamethasone; Rd=lenalidomide-dexamethasone; MRD=minimal residual disease; CI=confidence interval Overall response (sCR+CR+VGPR+PR) n(%) * 342 (92.9%) 300 (81.3%) p-value † <0.0001 Stringent complete response (sCR) 112 (30.4%) 46 (12.5%) Complete response (CR) 63 (17.1%) 46 (12.5%) Very good partial response (VGPR) 117 (31.8%) 104 (28.2%) Partial response (PR) 50 (13.6%) 104 (28.2%) CR or better (sCR + CR) 175 (47.6%) 92 (24.9%) p-value † <0.0001 VGPR or better (sCR + CR + VGPR) 292 (79.3%) 196 (53.1%) p-value † <0.0001 MRD negativity rate *,‡n(%) 89 (24.2%) 27 (7.3%) 95% CI (%) (19.9%, 28.9%) (4.9%, 10.5%) p-value § <0.0001 MRD negativity rate in patients with CR or better ‡ Number of patients with CR or better N=175 N=92 MRD negativity rate n(%) 89 (50.9%) 27 (29.3%) 95% CI (%) (43.2%, 58.5%) (20.3%, 39.8%) In responders, the median time to response was 1.05 months (range: 0.2 to 12.1 months) in the DRd group and 1.05 months (range: 0.3 to 15.3 months) in the Rd group. The median duration of response had not been reached in the DRd group and was 34.7 months (95% CI: 30.8, not estimable) in the Rd group.
Combination Treatment with Bortezomib, Melphalan and Prednisone (VMP) in Patients Ineligible for Autologous Stem Cell Transplant
ALCYONE (NCT02195479), an open-label, randomized, active-controlled trial, compared treatment with DARZALEX 16 mg/kg in combination with bortezomib, melphalan and prednisone (D-VMP), to treatment with VMP in patients with newly diagnosed multiple myeloma ineligible for autologous stem cell transplant. Bortezomib was administered by subcutaneous (SC) injection at a dose of 1.3 mg/m 2 body surface area twice weekly at Weeks 1, 2, 4 and 5 for the first 6-week cycle (Cycle 1; 8 doses), followed by once weekly administrations at Weeks 1, 2, 4 and 5 for eight more 6-week cycles (Cycles 2–9; 4 doses per cycle). Melphalan at 9 mg/m 2, and prednisone at 60 mg/m 2 were orally administered on Days 1 to 4 of the nine 6-week cycles (Cycles 1–9). DARZALEX was continued until disease progression or unacceptable toxicity.
A total of 706 patients were randomized: 350 to the D-VMP arm and 356 to the VMP arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The median age was 71 (range: 40–93) years, with 30% of the patients ≥75 years of age. The majority were white (85%), female (54%), 25% had an ECOG performance score of 0, 50% had an ECOG performance score of 1 and 25% had an ECOG performance score of 2. Nineteen percent of patients had ISS Stage I, 42% had ISS Stage II and 38% had ISS Stage III disease. Efficacy was evaluated by PFS based on IMWG criteria and overall survival (OS).
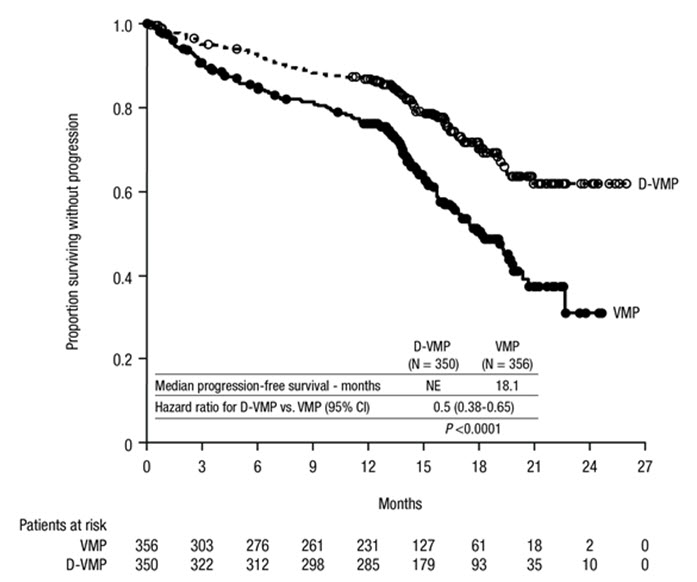
ALCYONE demonstrated an improvement in PFS in the D-VMP arm as compared to the VMP arm (HR=0.50; 95% CI: 0.38, 0.65; p<0.0001), representing a 50% reduction in the risk of disease progression or death in patients treated with D-VMP. After a median follow-up of 40 months, the median PFS was 36.4 months (95% CI: 32.1, 45.9) in the D-VMP arm and 19.3 months (95% CI: 18.0, 20.4) in the VMP arm.
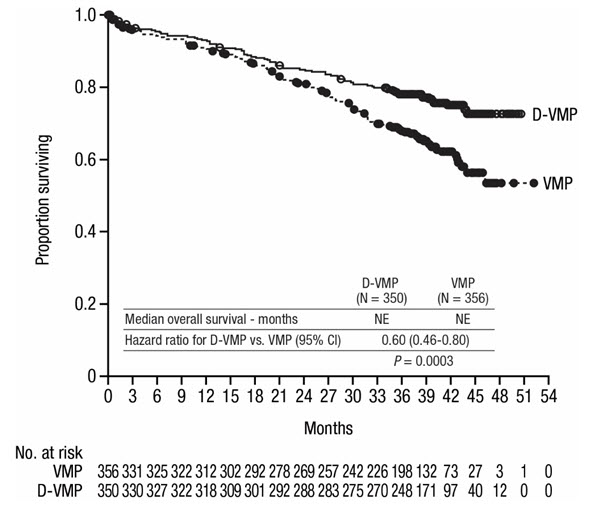
After a median follow-up of 40 months, ALCYONE demonstrated an improvement in overall survival (OS) in the D-VMP arm as compared to the VMP arm (HR=0.60; 95% CI: 0.46, 0.80; p=0.0003), representing a 40% reduction in the risk of death in patients treated in the D-VMP arm. Median OS had not been reached for either arm.
After a median follow-up of 87 months, the median OS was 83 months (95% CI: 72.5, NE) in the D-VMP arm and 53.6 months (95% CI: 46.3, 60.9) in the VMP arm.
Additional efficacy results from ALCYONE are presented in Table 24.
Table 24: Additional Efficacy Results From ALCYONE D-VMP (N=350) VMP (N=356) D-VMP = daratumumab-bortezomib-melphalan-prednisone; VMP = bortezomib-melphalan-prednisone; MRD = minimal residual disease; CI = confidence interval Overall response (sCR+CR+VGPR+PR) n(%) * 318 (90.9%) 263 (73.9%) p-value † <0.0001 Stringent complete response (sCR) 63 (18.0%) 25 (7.0%) Complete response (CR) 86 (24.6%) 62 (17.4%) Very good partial response (VGPR) 100 (28.6%) 90 (25.3%) Partial response (PR) 69 (19.7%) 86 (24.2%) MRD negativity rate *,‡n(%) 78 (22.3%) 22 (6.2%) 95% CI (%) (18.0, 27.0) (3.9, 9.2) p-value § <0.0001 MRD negativity rate in patients with CR or better ‡ Number of patients with CR or better N=149 N=87 MRD negativity rate n(%) 74 (49.7%) 22 (25.3%) 95% CI (%) (41.4, 58.0) (16.6, 35.7) In responders, the median time to response was 0.79 months (range: 0.4 to 15.5 months) in the D-VMP group and 0.82 months (range: 0.7 to 12.6 months) in the VMP group. The median duration of response had not been reached in the D-VMP group and was 21.3 months (range: 0.5+, 23.7+) in the VMP group.
Combination Treatment with Bortezomib, Thalidomide and Dexamethasone in Patients Eligible for Autologous Stem Cell Transplant (ASCT)
CASSIOPEIA (NCT02541383), an open-label, randomized, active-controlled trial compared induction and consolidation treatment with DARZALEX 16 mg/kg in combination with bortezomib, thalidomide and dexamethasone (DVTd) to treatment with bortezomib, thalidomide and dexamethasone (VTd) in patients with newly diagnosed multiple myeloma eligible for ASCT. The consolidation phase of treatment began a minimum of 30 days post-ASCT, when the patient had recovered sufficiently, and engraftment was complete. The trial was limited to patients 65 years of age and younger. Bortezomib was administered by subcutaneous (SC) injection or intravenous (IV) injection at a dose of 1.3 mg/m 2 body surface area twice weekly for two weeks (Days 1, 4, 8, and 11) of repeated 28-day (4-week) induction treatment cycles (Cycles 1–4) and two consolidation cycles (Cycles 5 and 6) following ASCT after Cycle 4. Thalidomide was administered orally at 100 mg daily during the six bortezomib cycles. Dexamethasone (oral or intravenous) was administered at 40 mg on Days 1, 2, 8, 9, 15, 16, 22 and 23 of Cycles 1 and 2, and at 40 mg on Days 1–2 and 20 mg on subsequent dosing days (Days 8, 9, 15, 16) of Cycles 3–4. Dexamethasone 20 mg was administered on Days 1, 2, 8, 9, 15, 16 in Cycles 5 and 6. On the days of DARZALEX infusion, the dexamethasone dose was administered intravenously as a pre-infusion medication.
A total of 1,085 patients were randomized: 543 to the DVTd arm and 542 to the VTd arm. The baseline demographic and disease characteristics were similar between the two treatment groups. The median age was 58 years (range: 22 to 65 years). The majority were male (59%), 48% had an ECOG performance score of 0, 42% had an ECOG performance score of 1 and 10% had an ECOG performance score of 2. Forty percent had ISS Stage I, 45% had ISS Stage II and 15% had ISS Stage III disease.
Efficacy was evaluated by stringent Complete Response (sCR) rate at Day 100 post-transplant, Complete Response Rate (CR) at Day 100 post-transplant, and Progression-Free Survival (PFS).
Table 25: Efficacy Results From CASSIOPEIA at Day 100 Post-Transplant DVTd (N=543) VTd (N=542) D-VTd = daratumumab-bortezomib-thalidomide-dexamethasone; VTd = bortezomib-thalidomide-dexamethasone Overall response (sCR+CR+VGPR+PR) n(%) * 503 (92.6%) 487 (89.9%) Stringent complete response (sCR) 157 (28.9%) 110 (20.3%) p-value † 0.0010 Complete response (CR) 54 (9.9%) 31 (5.7%) Very good partial response (VGPR) 242 (44.6%) 282 (52.0%) Partial response (PR) 50 (9.2%) 64 (11.8%) CASSIOPEIA demonstrated an improvement in PFS in the DVTd arm as compared to the VTd arm; with a median follow up of 18.8 months, the median PFS had not been reached in either arm. Treatment with DVTd resulted in a reduction in the risk of progression or death by 53% compared to VTd alone (HR=0.47; 95% CI: 0.33, 0.67; p<0.0001).
14.2 Relapsed/Refractory Multiple Myeloma
Combination Treatment with Lenalidomide and Dexamethasone
POLLUX (NCT02076009), an open-label, randomized, active-controlled trial, compared treatment with DARZALEX 16 mg/kg in combination with lenalidomide and low-dose dexamethasone (DRd) to treatment with lenalidomide and low-dose dexamethasone (Rd) in patients with multiple myeloma who had received at least one prior therapy. Lenalidomide (25 mg once daily orally on Days 1–21 of repeated 28-day [4-week] cycles) was given with low dose oral or intravenous dexamethasone 40 mg/week (or a reduced dose of 20 mg/week for patients >75 years or BMI <18.5). On DARZALEX infusion days, 20 mg of the dexamethasone dose was given as a pre-infusion medication and the remainder given the day after the infusion. For patients on a reduced dexamethasone dose, the entire 20 mg dose was given as a DARZALEX pre-infusion medication. Dose adjustments for lenalidomide and dexamethasone were applied according to manufacturer's prescribing information. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 569 patients were randomized; 286 to the DRd arm and 283 to the Rd arm. The baseline demographic and disease characteristics were similar between the DARZALEX and the control arm. The median patient age was 65 years (range 34 to 89 years), 11% were ≥75 years, 59% were male; 69% White, 18% Asian, and 3% African American. Patients had received a median of 1 prior line of therapy. Sixty-three percent (63%) of patients had received prior autologous stem cell transplantation (ASCT). The majority of patients (86%) received a prior PI, 55% of patients had received a prior immunomodulatory agent, including 18% of patients who had received prior lenalidomide; and 44% of patients had received both a prior PI and immunomodulatory agent. At baseline, 27% of patients were refractory to the last line of treatment. Eighteen percent (18%) of patients were refractory to a PI only, and 21% were refractory to bortezomib. Efficacy was evaluated by PFS based on IMWG criteria.
POLLUX demonstrated an improvement in PFS in the DRd arm as compared to the Rd arm (HR=0.37; 95% CI: 0.27, 0.52; p<0.0001), representing a 63% reduction in the risk of disease progression or death in patients treated with DRd. After a median follow-up of 55 months, the median PFS was 45.0 months (95% CI: 34.1, 53.9) in the DRd arm and was 17.5 months (95% CI: 13.9, 20.8) in the Rd arm.
After a median follow-up of 80 months, POLLUX demonstrated an improvement in overall survival (OS) in the DRd arm as compared to the Rd arm (HR=0.73; 95% CI: 0.58, 0.91; p=0.0044), representing a 27% reduction in the risk of death in patients treated in the DRd arm. The median OS was 67.6 months in the DRd arm and 51.8 months in the Rd arm.
Figure 7: Kaplan-Meier Curve of OS in POLLUX
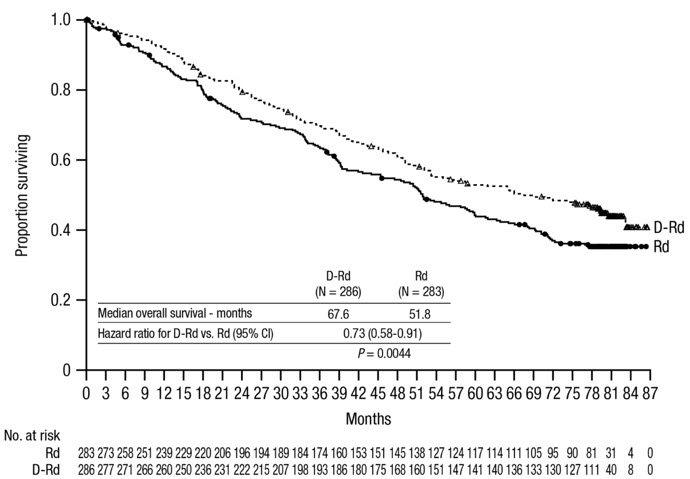
Additional efficacy results from POLLUX are presented in Table 26.
Table 26: Additional Efficacy Results From POLLUX * DRd (N=286) Rd (N=283) DRd = daratumumab- lenalidomide-dexamethasone; Rd = lenalidomide-dexamethasone Overall response (sCR+CR+VGPR+PR) 261 (91.3%) 211 (74.6%) p-value † <0.0001 Stringent complete response (sCR) 51 (17.8%) 20 (7.1%) Complete response (CR) 70 (24.5%) 33 (11.7%) Very good partial response (VGPR) 92 (32.2%) 69 (24.4%) Partial response (PR) 48 (16.8%) 89 (31.4%) In responders, the median time to response was 1 month (range: 0.9 to 13 months) in the DRd group and 1.1 months (range: 0.9 to 10 months) in the Rd group. The median duration of response had not been reached in the DRd group (range: 1+ to 19.8+ months) and was 17.4 months (range: 1.4 to 18.5+ months) in the Rd group.
Combination Treatment with Bortezomib and Dexamethasone
CASTOR (NCT02136134), an open-label, randomized, active-controlled Phase 3 trial, compared treatment with DARZALEX 16 mg/kg in combination with bortezomib and dexamethasone (DVd), to treatment with bortezomib and dexamethasone (Vd) in patients with multiple myeloma who had received at least one prior therapy. Bortezomib was administered by SC injection or IV injection at a dose of 1.3 mg/m 2 body surface area twice weekly for two weeks (Days 1, 4, 8, and 11) of repeated 21 day (3-week) treatment cycles, for a total of 8 cycles. Dexamethasone was administered orally at a dose of 20 mg on Days 1, 2, 4, 5, 8, 9, 11, and 12 of each of the 8 bortezomib cycles (80 mg/week for two out of three weeks of the bortezomib cycle) or a reduced dose of 20 mg/week for patients >75 years, BMI <18.5, poorly controlled diabetes mellitus or prior intolerance to steroid therapy. On the days of DARZALEX infusion, 20 mg of the dexamethasone dose was administered as a pre-infusion medication. For patients on a reduced dexamethasone dose, the entire 20 mg dose was given as a DARZALEX pre-infusion medication. Bortezomib and dexamethasone were given for 8 three-week cycles in both treatment arms; whereas DARZALEX was given until disease progression. However, dexamethasone 20 mg was continued as a DARZALEX pre-infusion medication in the DVd arm. Dose adjustments for bortezomib and dexamethasone were applied according to manufacturer's prescribing information.
A total of 498 patients were randomized; 251 to the DVd arm and 247 to the Vd arm. The baseline demographic and disease characteristics were similar between the DARZALEX and the control arm. The median patient age was 64 years (range 30 to 88 years); 12% were ≥75 years, 57% were male; 87% White, 5% Asian and 4% African American. Patients had received a median of 2 prior lines of therapy and 61% of patients had received prior autologous stem cell transplantation (ASCT). Sixty-nine percent (69%) of patients had received a prior PI (66% received bortezomib) and 76% of patients received an immunomodulatory agent (42% received lenalidomide). At baseline, 32% of patients were refractory to the last line of treatment and the proportions of patients refractory to any specific prior therapy were in general well balanced between the treatment groups. Thirty-three percent (33%) of patients were refractory to an immunomodulatory agent only, with 24% patients in the DVd arm and 33% of patients in the Vd arm respectively refractory to lenalidomide. Efficacy was evaluated by PFS based on IMWG criteria.
CASTOR demonstrated an improvement in PFS in the DVd arm as compared to the Vd arm (HR=0.39; 95% CI: 0.28, 0.53; p<0.0001), representing a 61% reduction in the risk of disease progression or death for patients treated with DVd versus Vd. After a median follow-up of 50 months, the median PFS was 16.7 months (95% CI: 13.1, 19.4) in the DVd arm and was 7.1 months (95% CI: 6.2, 7.7) in the Vd arm.
After a median follow-up of 73 months, CASTOR demonstrated an improvement in overall survival (OS) in the DVd arm as compared to the Vd arm (HR=0.74; 95% CI: 0.59, 0.92; p=0.0075), representing a 26% reduction in the risk of death in patients treated in the DVd arm. The median OS was 49.6 months in the DVd arm and 38.5 months in the Vd arm.
Figure 9: Kaplan-Meier Curve of OS in CASTOR
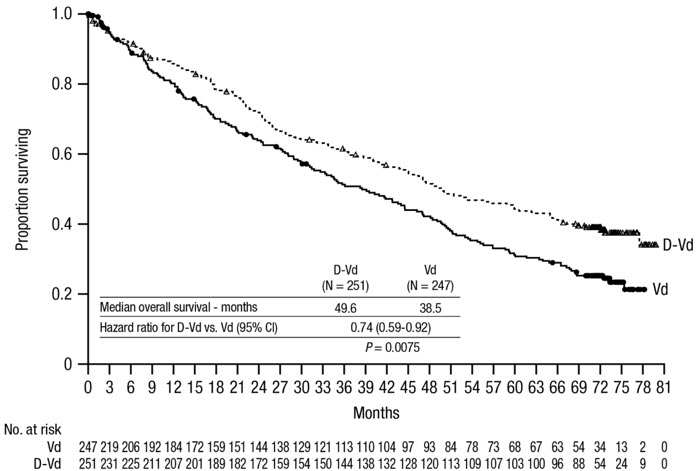
Additional efficacy results from CASTOR are presented in Table 27.
Table 27: Additional Efficacy Results From CASTOR * DVd (N=251) Vd (N=247) DVd = daratumumab- bortezomib-dexamethasone; Vd = bortezomib-dexamethasone Overall response (sCR+CR+VGPR+PR) 199 (79.3%) 148 (59.9%) P-value † <0.0001 Stringent complete response (sCR) 11 (4.4%) 5 (2.0%) Complete response (CR) 35 (13.9%) 16 (6.5%) Very good partial response (VGPR) 96 (38.2%) 47 (19.0%) Partial response (PR) 57 (22.7%) 80 (32.4%) In responders, the median time to response was 0.8 months (range: 0.7 to 4 months) in the DVd group and 1.5 months (range: 0.7 to 5 months) in the Vd group. The median duration of response had not been reached in the DVd group (range: 1.4+ to 14.1+ months) and was 7.9 months (1.4+ to 12+ months) in the Vd group.
Combination Treatment with Twice-Weekly (20/56 mg/m 2) Carfilzomib and Dexamethasone
CANDOR (NCT03158688) was a randomized, open-label, multicenter trial which evaluated the combination of DARZALEX with twice-weekly carfilzomib and dexamethasone (DKd) versus twice-weekly carfilzomib and dexamethasone (Kd) in patients with relapsed or refractory multiple myeloma who had received at least 1 to 3 prior lines of therapy. Patients who had the following were excluded from the trial: known moderate or severe persistent asthma within the past 2 years, known chronic obstructive pulmonary disease (COPD) with a FEV1 <50% of predicted normal, and active congestive heart failure. Randomization was stratified by the ISS (stage 1 or 2 vs stage 3) at screening, prior proteasome inhibitor exposure (yes vs no), number of prior lines of therapy (1 vs ≥2), or prior cluster differentiation antigen 38 (CD38) antibody therapy (yes vs no).
DARZALEX was administered intravenously at a dose of 8 mg/kg in Cycle 1 on Days 1 and 2. Thereafter, DARZALEX was administered intravenously at a dose of 16 mg/kg on Days 8, 15 and 22 of Cycle 1; Days 1, 8 and 15 and 22 of Cycle 2; Days 1 and 15 of Cycles 3 to 6; and Day 1 of each 28-day cycle until disease progression. Carfilzomib was administered intravenously at a dose of 20 mg/m 2 in Cycle 1 on Days 1 and 2; at a dose of 56 mg/m 2 in Cycle 1 on Days 8, 9, 15, and 16 ; and at a dose 56 mg/m 2 on Days 1, 2, 8, 9, 15, and 16 of each 28-day cycle thereafter. Dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 8, 9, 15 and 16 and then 40 mg orally or intravenously on Day 22 of each 28-day cycle. For patients >75 years on a reduced dexamethasone dose of 20 mg, the entire 20 mg dose was given as a DARZALEX pre-infusion medication on days when DARZALEX was administered. Dosing of dexamethasone was otherwise split across days when carfilzomib was administered in both study arms. Treatment was continued in both arms until disease progression or unacceptable toxicity.
A total of 466 patients were randomized; 312 to the DKd arm and 154 to the Kd arm. The baseline demographic and disease characteristics were similar between arms. The median age was 64 years (range 29 to 84 years), 9% were ≥75 years, 58% were male; 79% White, 14% Asian, and 2% Black. Patients had received a median of 2 prior lines of therapy and 58% of patients had received prior autologous stem cell transplantation (ASCT). The majority of patients (92%) received a prior PI and of those 34% were refractory to PI including regimen. Fourty-two percent (42%) of patients had received prior lenalidomide and of those, 33% were refractory to a lenalidomide containing regimen.
Efficacy was evaluated by IRC evaluation of PFS based on the IMWG response criteria. Efficacy results are provided in Figure 10. CANDOR demonstrated an improvement in PFS in the DKd arm as compared to the Kd arm; the median PFS had not been reached in the DKd arm and was 15.8 months in the Kd arm (hazard ratio [HR]=0.63; 95% CI: 0.46, 0.85; p=0.0014), representing 37% reduction in the risk of disease progression or death for patients treated with DKd versus Kd.
Figure 10: Kaplan-Meier Curve of PFS in CANDOR
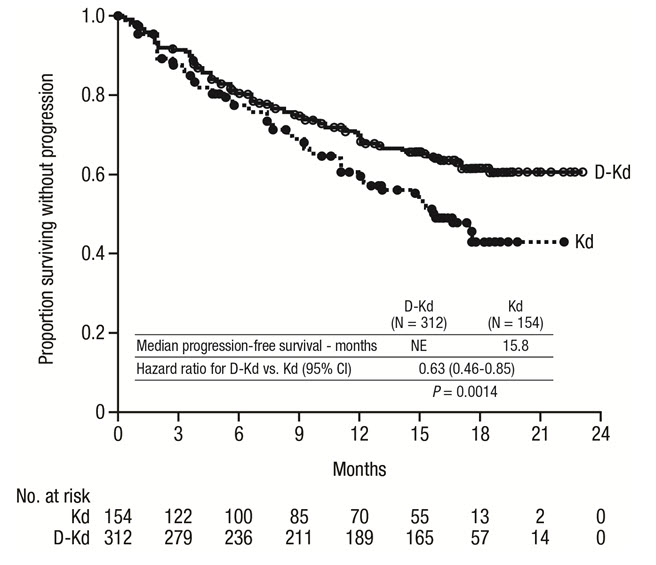
Additional efficacy results from CANDOR are presented in Table 28.
Table 28: Additional Efficacy Results From CANDOR (Intent-to-Treat Population) DKd (N=312) Kd (N=154) DKd = daratumumab-carfilzomib-dexamethasone; Kd =carfilzomib-dexamethasone; MRD [-] CR=minimal residual disease; CI=confidence interval Overall response (sCR+CR+VGPR+PR) n(%) 263 (84%) 115 (75%) 95% CI (%) (80, 88) (67, 81) p-value *(1-sided) 0.0040 Complete response (CR) 89 (28%) 16 (10%) Very good partial response (VGPR) 127 (41%) 59 (38%) Partial response (PR) 47 (15%) 40 (26%) MRD [-] CR rate at 12 months n(%) † 39 (12%) 2 (1.3%) 95% CI (%) (9, 17) (0.2, 4.6) p-value *(1-sided) <0.0001 MRD [-] CR † 43 (14%) 5 (3.2%) The median time to response was 1 month (range: 1 to 14 months) in the DKd group and 1 month (range: 1 to 10 months) in the Kd group. The median duration of response had not been reached in the DKd group and was 16.6 months (95% CI: 13.9, not estimable) in the Kd group.
Combination Treatment with Once-Weekly (20/70 mg/m 2) Carfilzomib and Dexamethasone
EQUULEUS (NCT01998971) was an open-label, multi-cohort trial which evaluated the combination of DARZALEX with one-weekly carfilzomib and dexamethasone in patients with relapsed or refractory multiple myeloma who had received at least 1 to 3 prior lines of therapy. Patients who had the following were excluded from the trial: known moderate or severe persistent asthma within the past 2 years, known chronic obstructive pulmonary disease (COPD) with a FEV1 <50% of predicted normal, or active congestive heart failure (defined as New York Heart Association Class III–IV).
Ten patients were administered DARZALEX at a dose of 16 mg/kg intravenously on Cycle 1, Day 1 and the remaining patients were administered DARZALEX at a dose of 8 mg/kg intravenously on Cycle 1, Days 1 and 2. Thereafter, DARZALEX was administered intravenously at a dose of 16 mg/kg on Days 8, 15 and 22 of Cycle 1; Days 1, 8, 15 and 22 of Cycle 2; Days 1 and 15 of Cycles 3 to 6; and then Day 1 for the remaining cycles of each 28 day cycle. Carfilzomib was administered intravenously once weekly at a dose of 20 mg/m 2 on Cycle 1 Day 1 and escalated to dose of 70 mg/m 2 on Cycle 1 Days 8 and 15, and Days 1, 8, and 15 of each subsequent 28-day cycle. In Cycles 1 and 2, dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 8, 9, 15, 16, 22 and 23; in cycles 3 to 6, dexamethasone 20 mg was administered orally or intravenously on Days 1, 2, 15 and 16 and at a dose of 40 mg on Day 8 and 22; and in cycles 7 and thereafter, dexamethasone 20 mg was administered orally or intravenously on Days 1 and 2 and at a dose of 40 mg on Days 8, 15, and 22. For patients >75 years of age, dexamethasone 20 mg was administered orally or intravenously weekly after the first week. Treatment continued until disease progression or unacceptable toxicity.
The EQUULEUS trial enrolled 85 patients. The median patient age was 66 years (range: 38 to 85 years) with 9% of patients ≥75 years of age; 54% were male; 80% were White, 3.5% were Black and 3.5% were Asian. Patients in the study had received a median of 2 prior lines of therapy. Seventy-three percent (73%) of patients had received prior ASCT. All patients received prior bortezomib, and 95% of patients received prior lenalidomide. Fifty-nine percent (59%) of patients were refractory to lenalidomide and 29% of patients were refractory to both a PI an IMiD.
Efficacy results were based on overall response rate using IMWG criteria. Efficacy results are provided in Table 29. The median time to response was 0.95 months (range: 0.9, 14.3). The median duration of response was 28 months (95% CI: 20.5, not estimable).
Table 29: Efficacy results for EQUULEUS N=85 ORR = sCR+CR+VGPR+PR
CI = confidence intervalOverall response rate (ORR) 69 (81%) 95% CI (%) (71, 89) Stringent complete response (sCR) 18 (21%) Complete response (CR) 12 (14%) Very good partial response (VGPR) 28 (33%) Partial response (PR) 11 (13%) Combination Treatment with Pomalidomide and Dexamethasone
EQUULEUS (NCT01998971) was an open-label trial in which 103 patients with multiple myeloma who had received a prior PI and an immunomodulatory agent, received 16 mg/kg DARZALEX in combination with pomalidomide and low-dose dexamethasone until disease progression. Pomalidomide (4 mg once daily orally on Days 1–21 of repeated 28-day [4-week] cycles) was given with low dose oral or intravenous dexamethasone 40 mg/week (reduced dose of 20 mg/week for patients >75 years or BMI <18.5). On DARZALEX infusion days, 20 mg of the dexamethasone dose was given as a pre-infusion medication and the remainder given the day after the infusion. For patients on a reduced dexamethasone dose, the entire 20 mg dose was given as a DARZALEX pre-infusion medication.
The median patient age was 64 years (range: 35 to 86 years) with 8% of patients ≥75 years of age. Patients in the study had received a median of 4 prior lines of therapy. Seventy-four percent (74%) of patients had received prior ASCT. Ninety-eight percent (98%) of patients received prior bortezomib treatment, and 33% of patients received prior carfilzomib. All patients received prior lenalidomide treatment, with 98% of patients previously treated with the combination of bortezomib and lenalidomide. Eighty nine percent (89%) of patients were refractory to lenalidomide and 71% refractory to bortezomib; 64% of patients were refractory to bortezomib and lenalidomide.
Efficacy results were based on overall response rate as determined by Independent Review Committee using IMWG criteria (see Table 30).
Table 30: Efficacy Results for EQUULEUS N=103 ORR = sCR+CR+VGPR+PR
CI = Confidence IntervalOverall response rate (ORR) 61 (59.2%) 95% CI (%) (49.1, 68.8) Stringent complete response (sCR) 8 (7.8%) Complete response (CR) 6 (5.8%) Very good partial response (VGPR) 29 (28.2%) Partial response (PR) 18 (17.5%) The median time to response was 1 month (range: 0.9 to 2.8 months). The median duration of response was 13.6 months (range: 0.9+ to 14.6+ months).
Monotherapy
SIRIUS (NCT01985126), was an open-label trial evaluating DARZALEX monotherapy in patients with relapsed or refractory multiple myeloma who had received at least 3 prior lines of therapy including a proteasome inhibitor and an immunomodulatory agent or who were double-refractory to a proteasome inhibitor and an immunomodulatory agent. In 106 patients, DARZALEX 16 mg/kg was administered with pre- and post-infusion medication. Treatment continued until unacceptable toxicity or disease progression.
The median patient age was 63.5 years (range: 31 to 84 years), 49% were male and 79% were White. Patients had received a median of 5 prior lines of therapy. Eighty percent of patients had received prior autologous stem cell transplantation (ASCT). Prior therapies included bortezomib (99%), lenalidomide (99%), pomalidomide (63%) and carfilzomib (50%). At baseline, 97% of patients were refractory to the last line of treatment, 95% were refractory to both, a proteasome inhibitor (PI) and immunomodulatory agent, and 77% were refractory to alkylating agents.
Efficacy results were based on overall response rate as determined by the Independent Review Committee assessment using IMWG criteria (see Table 31).
Table 31: Efficacy Results for SIRIUS N=106 ORR = sCR+CR+VGPR+PR
CI = confidence intervalOverall response rate (ORR) 31 (29.2%) 95% CI (%) (20.8, 38.9) Stringent complete response (sCR) 3 (2.8%) Complete response (CR) 0 Very good partial response (VGPR) 10 (9.4%) Partial response (PR) 18 (17.0%) The median time to response was 1 month (range: 0.9 to 5.6 months). The median duration of response was 7.4 months (range: 1.2 to 13.1+ months).
Study GEN501 (NCT00574288) was an open-label dose escalation trial evaluating DARZALEX monotherapy in patients with relapsed or refractory multiple myeloma who had received at least 2 different cytoreductive therapies. In 42 patients, DARZALEX 16 mg/kg was administered with pre- and post-infusion medication. Treatment continued until unacceptable toxicity or disease progression.
The median patient age was 64 years (range: 44 to 76 years), 64% were male and 76% were White. Patients in the study had received a median of 4 prior lines of therapy. Seventy-four percent of patients had received prior ASCT. Prior therapies included bortezomib (100%), lenalidomide (95%), pomalidomide (36%) and carfilzomib (19%). At baseline, 76% of patients were refractory to the last line of treatment, 64% of patients were refractory to both, a PI and an immunomodulatory agent, and 60% of patients were refractory to alkylating agents.
Overall response rate was 36% (95% CI: 21.6, 52.0%) with 1 CR and 3 VGPR. The median time to response was 1 month (range: 0.5 to 3.2 months). The median duration of response was not estimable (range: 2.2 to 13.1+ months).
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
DARZALEX ®(daratumumab) injection is a colorless to pale yellow, preservative-free solution for intravenous infusion.
NDC 57894-502-05 and NDC 57894-505-05 each contain one 100 mg/5 mL (20 mg/mL) single-dose vial
NDC 57894-502-20 and NDC 57894-505-20 each contain one 400 mg/20 mL (20 mg/mL) single-dose vial
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Infusion-Related Reactions
Advise patients to seek immediate medical attention for any of the following signs and symptoms of infusion-related reactions: itchy, runny or blocked nose; fever, chills, nausea, vomiting, throat irritation, cough, headache, dizziness or lightheadedness, tachycardia, chest discomfort, wheezing, shortness of breath or difficulty breathing, itching, and blurred vision [see Warnings and Precautions (5.1)] .
Neutropenia
Advise patients to contact their healthcare provider if they have a fever [see Warnings and Precautions (5.3)] .
Thrombocytopenia
Advise patients to contact their healthcare provider if they notice signs of bruising or bleeding [see Warnings and Precautions (5.4)] .
Interference with Laboratory Tests
Advise patients to inform their healthcare providers, including personnel at blood transfusion centers that they are taking DARZALEX, in the event of a planned transfusion [see Warnings and Precautions (5.2)] .
Advise patients that DARZALEX can affect the results of some tests used to determine complete response in some patients and additional tests may be needed to evaluate response [see Warnings and Precautions (5.5)] .
Hepatitis B Virus (HBV) Reactivation
Advise patients to inform healthcare providers if they have ever had or might have a hepatitis B infection and that DARZALEX could cause hepatitis B virus to become active again [see Adverse Reactions (6.1)] .
Embryo-Fetal Toxicity
Advise pregnant women of the potential hazard to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.6), Use in Specific Populations (8.1, 8.3)] .
Advise females of reproductive potential to avoid becoming pregnant during treatment with DARZALEX and for 3 months after the last dose [see Use in Specific Populations (8.1, 8.3)] .
Advise patients that lenalidomide, pomalidomide, or thalidomide has the potential to cause fetal harm and has specific requirements regarding contraception, pregnancy testing, blood and sperm donation, and transmission in sperm. Lenalidomide, pomalidomide, and thalidomide are only available through a REMS program [see Use in Specific Populations (8.1, 8.3)] .
Hereditary Fructose Intolerance (HFI)
DARZALEX contains sorbitol. Advise patients with HFI of the risks related to sorbitol [see Description (11)] .
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 11/2022 PATIENT INFORMATION
DARZALEX ®(Dar'-zah-lex)
(daratumumab)
injection, for intravenous useDARZALEX may be used with other medicines called lenalidomide, thalidomide or pomalidomide. You should also read the Medication Guide that comes with lenalidomide, thalidomide or pomalidomide if you use DARZALEX with these medicines. What is DARZALEX?
DARZALEX is a prescription medicine used to treat adults with multiple myeloma:- in combination with the medicines lenalidomide and dexamethasone in people with newly diagnosed multiple myeloma who cannot receive a type of stem cell transplant that uses their own stem cells (autologous stem cell transplant) and in people whose multiple myeloma has come back or did not respond to treatment who have received at least one prior medicine to treat multiple myeloma.
- in combination with the medicines bortezomib, melphalan and prednisone, in people with newly diagnosed multiple myeloma who cannot receive a type of stem cell transplant that uses their own stem cells (autologous stem cell transplant).
- in combination with the medicines bortezomib, thalidomide, and dexamethasone in newly diagnosed people who are eligible to receive a type of stem cell transplant that uses their own stem cells (autologous stem cell transplant).
- in combination with the medicines bortezomib and dexamethasone, in people who have received at least one prior medicine to treat multiple myeloma.
- in combination with the medicines carfilzomib and dexamethasone, in people whose multiple myeloma has come back or did not respond to treatment who have received one to three prior medicines to treat multiple myeloma.
- in combination with the medicines pomalidomide and dexamethasone in people who have received at least two prior medicines to treat multiple myeloma, including lenalidomide and a proteasome inhibitor.
- alone in people who have received at least three prior medicines, including a proteasome inhibitor and an immunomodulatory agent, ordid not respond to a proteasome inhibitor and an immunomodulatory agent.
Do not receive DARZALEX: - if you have a history of a severe allergic reaction to daratumumab or any of the ingredients in DARZALEX. See the end of this leaflet for a complete list of ingredients in DARZALEX.
Before you receive DARZALEX, tell your healthcare provider about all of your medical conditions, including if you: - have a history of breathing problems.
- have had shingles (herpes zoster).
- have ever had or might now have a hepatitis B infection as DARZALEX could cause hepatitis B virus to become active again. Your healthcare provider will check you for signs of this infection before, during and for some time after treatment with DARZALEX. Tell your healthcare provider right away if you get worsening tiredness or yellowing of your skin or white part of your eyes.
- have hereditary fructose intolerance (HFI). DARZALEX contains sorbitol. Sorbitol is a source of fructose. People with HFI cannot break down fructose, which may cause serious side effects.
- are pregnant or plan to become pregnant. DARZALEX may harm your unborn baby. Tell your healthcare provider right away if you become pregnant or think that you may be pregnant during treatment with DARZALEX.
- Females who are able to become pregnant should use an effective method of birth control (contraception) during treatment and for 3 months after your last dose of DARZALEX. Talk to your healthcare provider about birth control methods that you can use during this time.
- Before starting DARZALEX in combination with lenalidomide, pomalidomide, or thalidomide, females and males must agree to the instructions in the lenalidomide, pomalidomide, or thalidomide REMS program.
- The lenalidomide, pomalidomide, and thalidomide REMS has more information about effective methods of birth control, pregnancy testing, and blood donation for females who can become pregnant.
- For males who have female partners who can become pregnant, there is information in the lenalidomide, pomalidomide, and thalidomide REMS about sperm donation and how lenalidomide, pomalidomide, and thalidomide can pass into human semen.
- are breastfeeding or plan to breastfeed. It is not known if DARZALEX passes into your breast milk. You should not breastfeed during treatment with DARZALEX. Talk to your healthcare provider about the best way to feed your baby during treatment with DARZALEX.
How will I receive DARZALEX? - DARZALEX may be given alone or together with other medicines used to treat multiple myeloma.
- DARZALEX will be given to you by your healthcare provider by intravenous (IV) infusion into your vein.
- Your healthcare provider will decide the time between doses as well as how many treatments you will receive.
- Your healthcare provider will give you medicines before each dose of DARZALEX and after each dose of DARZALEX to help reduce the risk of infusion-related reactions.
If you miss any appointments, call your healthcare provider as soon as possible to reschedule your appointment.
What are the possible side effects of DARZALEX?
DARZALEX may cause serious reactions, including:- Infusion-related reactions.Infusion-related reactions are common with DARZALEX. Serious allergic reactions and reactions due to release of certain substances by your body (systemic) that can lead to death, can happen with DARZALEX .Your healthcare provider may temporarily stop your infusion or completely stop treatment with DARZALEX if you have infusion-related reactions. Get medical help right away if you get any of the following symptoms:
- shortness of breath or trouble breathing
- dizziness or lightheadedness (hypotension)
- cough
- wheezing
- heart beating faster than usual
- low oxygen in the blood (hypoxia)
- throat tightness or irritation
- runny or stuffy nose
- headache
- itching
- high blood pressure
- eye pain
- nausea
- vomiting
- chills
- fever
- chest discomfort
- blurred vision
- Changes in blood tests.DARZALEX can affect the results of blood tests to match your blood type. These changes can last for up to 6 months after your final dose of DARZALEX. Your healthcare provider will do blood tests to match your blood type before you start treatment with DARZALEX. Tell all of your healthcare providers that you are being treated with DARZALEX before receiving blood transfusions.
- Decreases in blood cell counts.DARZALEX can decrease white blood cell counts which help fight infections and blood cells called platelets which help to clot blood. Decreases in blood cell counts are common with DARZALEX, but can be severe. Your healthcare provider will check your blood cell counts during treatment with DARZALEX. Tell your healthcare provider if you develop fever or have signs of bruising or bleeding.
- cold-like symptoms (upper respiratory infection)
- diarrhea
- constipation
- decreased red blood cells
- nerve damage causing tingling, numbness or pain
- tiredness
- swollen hands, ankles, or feet
- nausea
- cough
- fever
- shortness of breath
- feeling weak
These are not all the possible side effects of DARZALEX.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of DARZALEX.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. You can ask your pharmacist or healthcare provider for information about DARZALEX that is written for health professionals.What are the ingredients in DARZALEX?
Active ingredient:daratumumab
Inactive ingredients:may include glacial acetic acid, L-histidine, L-histidine hydrochloride monohydrate, L-methionine, mannitol, polysorbate 20, sodium acetate trihydrate, sodium chloride, sorbitol, and water for injection.
Manufactured by: Janssen Biotech, Inc., Horsham, PA 19044, USA; U.S. License Number 1864
For patent information: www.janssenpatents.com
© 2015–2021 Janssen Pharmaceutical Companies
For more information, call 1-800-526-7736 or go to www.DARZALEX.com. - PRINCIPAL DISPLAY PANEL - 100 mg/5 mL Vial Carton
- PRINCIPAL DISPLAY PANEL - 100 mg/5 mL Vial Carton - 57894-505
-
INGREDIENTS AND APPEARANCE
DARZALEX
daratumumab injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:57894-502 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DARATUMUMAB (UNII: 4Z63YK6E0E) (DARATUMUMAB - UNII:4Z63YK6E0E) DARATUMUMAB 100 mg in 5 mL Inactive Ingredients Ingredient Name Strength ACETIC ACID (UNII: Q40Q9N063P) SODIUM ACETATE (UNII: 4550K0SC9B) SODIUM CHLORIDE (UNII: 451W47IQ8X) MANNITOL (UNII: 3OWL53L36A) POLYSORBATE 20 (UNII: 7T1F30V5YH) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-502-05 1 in 1 CARTON 11/16/2015 10/31/2026 1 5 mL in 1 VIAL; Type 0: Not a Combination Product 2 NDC:57894-502-20 1 in 1 CARTON 11/16/2015 11/30/2026 2 20 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761036 11/16/2015 DARZALEX IV
daratumumab injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:57894-505 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DARATUMUMAB (UNII: 4Z63YK6E0E) (DARATUMUMAB - UNII:4Z63YK6E0E) DARATUMUMAB 100 mg in 5 mL Inactive Ingredients Ingredient Name Strength HISTIDINE (UNII: 4QD397987E) HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) METHIONINE (UNII: AE28F7PNPL) POLYSORBATE 20 (UNII: 7T1F30V5YH) SORBITOL (UNII: 506T60A25R) WATER (UNII: 059QF0KO0R) Product Characteristics Color yellow (colorless to yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-505-05 1 in 1 BOX 10/15/2021 1 5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:57894-505-20 1 in 1 BOX 10/15/2021 2 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761036 10/15/2021 Labeler - Janssen Biotech, Inc. (099091753) Establishment Name Address ID/FEI Business Operations Janssen Biotech, Inc. 038978363 analysis(57894-502, 57894-505) Establishment Name Address ID/FEI Business Operations AndersonBrecon, Inc. 053217022 pack(57894-502) Establishment Name Address ID/FEI Business Operations FUJIFILM Diosynth Biotechnologies Denmark ApS 307258082 api manufacture(57894-505) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG 312670654 manufacture(57894-502) , analysis(57894-502) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG 316126754 analysis(57894-502) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG 341629292 analysis(57894-502) Establishment Name Address ID/FEI Business Operations Vetter Pharma Fertigung GmbH & Co. KG 344217323 analysis(57894-502) Establishment Name Address ID/FEI Business Operations Janssen Biologics B.V. 409612918 analysis(57894-502, 57894-505) Establishment Name Address ID/FEI Business Operations Cilag AG 483237103 manufacture(57894-502, 57894-505) , pack(57894-502, 57894-505) , label(57894-502, 57894-505) , analysis(57894-502, 57894-505) Establishment Name Address ID/FEI Business Operations Samsung Biologics Co, LTD 557810567 api manufacture(57894-502) , analysis(57894-502) Establishment Name Address ID/FEI Business Operations Simtra US LLC 604719430 manufacture(57894-502) , label(57894-502) , pack(57894-502) Establishment Name Address ID/FEI Business Operations Biogen, MA Inc. 841087823 api manufacture(57894-502) Establishment Name Address ID/FEI Business Operations PPD Development Ireland Ltd. 985036175 analysis(57894-502) Establishment Name Address ID/FEI Business Operations Janssen Sciences Ireland Unlimited Company 986030167 analysis(57894-502, 57894-505)