Label: permax- Pergolide tablet
-
Contains inactivated NDC Code(s)
NDC Code(s): 0187-0839-01, 0187-0840-02, 0187-0841-02 - Packager: Valeant Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
Drug Label Information
Updated March 30, 2007
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- N/A - Section Title Not Found In Database
-
BOXED WARNING
(What is this?)
WARNING
Cardiac Valvulopathy and Fibrotic Complications
Cardiac Valvulopathy
The use of pergolide has been shown to increase the risk of cardiac valvular disease involving one or more valves. Some patients have required valve replacement, and deaths have been reported. Cases have been reported after exposures to pergolide ranging from several months to several years. The histopathology of explanted valves is similar to that of other drug-induced valvulopathies. Precise risk estimates of pergolide-induced cardiac valvular disease are not available.
Specific risk factors predisposing patients to developing cardiac valvular disease with pergolide have not been identified. Cardiac valvulopathy has been reported with all doses of pergolide; however, available data suggest that the risk may be greater with higher doses. Doses of pergolide above 5 mg/day are not recommended (seeDOSAGE & ADMINISTRATION).
Pergolide is not recommended for use in patients with a history of cardiac valvulopathy.
Before initiating treatment with pergolide, all patients should undergo a cardiovascular evaluation, including an echocardiogram, to determine whether valvular disease is present and to provide a baseline for subsequent monitoring. Although the risk of disease progression in patients with asymptomatic valvular disease is unknown, pergolide ordinarily should not be initiated if valvulopathy is detected at screening.
All patients taking pergolide should undergo periodic echocardiograms to screen for the development of valvulopathy. Patients should also be monitored for signs and symptoms of valvulopathy, including dyspnea, edema, congestive heart failure and new cardiac murmurs. If a patient develops these signs or symptoms, consideration should be given to suspending treatment with pergolide until a full diagnostic evaluation, including echocardiogram, has been performed. Pergolide should ordinarily be discontinued if a patient is diagnosed with cardiac valvular disease. In some cases, signs and/or symptoms of cardiac valvulopathy improved after discontinuation of pergolide.
Fibrotic Complications
Case reports have demonstrated that pergolide increases the risk of fibrotic complications including pulmonary, pleural, and/or retroperitoneal fibrosis, pericarditis, pleuritis, and pericardial and/or pleural effusions. Cases have been reported after exposures to pergolide ranging from about one to several years. Precise risk estimates of pergolide-induced fibrotic complications are not available.
Specific risk factors predisposing patients to developing fibrotic complications with pergolide have not been identified. Fibrotic complications have been reported with all therapeutic doses of pergolide.
Pergolide is not recommended for use in patients with a history of fibrotic conditions.
Patients should also be monitored for signs and symptoms of fibrotic complications, including dyspnea, persistent edema, cough, congestive heart failure, new cardiac rub, and/or signs of urinary tract obstruction. If a patient develops these signs or symptoms, consideration should be given to suspending treatment with pergolide until a full diagnostic evaluation has been performed. Pergolide should ordinarily be discontinued if a patient is diagnosed with a specific fibrotic complication. In some cases, signs and/or symptoms of fibrotic complications improved after discontinuation of pergolide.
-
DESCRIPTION
Permax® (Pergolide Tablets, USP) is an ergot derivative dopamine receptor agonist at both D1 and D2 receptor sites. Pergolide mesylate is chemically designated as 8β-[(Methylthio)methyl]-6-propylergoline monomethanesulfonate; the structural formula is as follows:
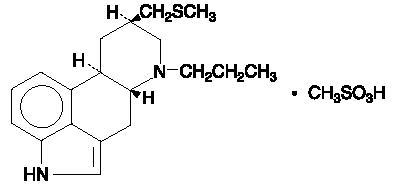
The empirical formula is C19H26N2S∙CH4O3S, representing a molecular weight of 410.60.
Permax is provided for oral administration in tablets containing 0.05 mg (0.159 µmol), 0.25 mg (0.795 µmol), or 1 mg (3.18 µmol) pergolide as the base. The tablets also contain croscarmellose sodium, iron oxide, lactose, magnesium stearate, and povidone. The 0.05 mg tablet also contains L-methionine, and the 0.25 mg tablet also contains FD&C Blue No. 2.
-
CLINICAL PHARMACOLOGY
Pharmacodynamic Information
Pergolide is a potent dopamine receptor agonist. Pergolide is 10 to 1000 times more potent than bromocriptine on a milligram per milligram basis in various in vitro and in vivo test systems. Pergolide inhibits the secretion of prolactin in humans; it causes a transient rise in serum concentrations of growth hormone and a decrease in serum concentrations of luteinizing hormone. In Parkinson's disease, pergolide is believed to exert its therapeutic effect by directly stimulating postsynaptic dopamine receptors in the nigrostriatal system.
Pharmacokinetic Information (Absorption, Distribution, Metabolism, and Elimination)
Information on oral systemic bioavailability of pergolide is unavailable because of the lack of a sufficiently sensitive assay to detect the drug after the administration of a single dose. However, following oral administration of 14C radiolabeled pergolide, approximately 55% of the administered radioactivity can be recovered from the urine and 5% from expired CO2, suggesting that a significant fraction is absorbed. Nothing can be concluded about the extent of presystemic clearance, if any.
Data on postabsorption distribution of pergolide are unavailable.
At least 10 metabolites have been detected, including N-despropylpergolide, pergolide sulfoxide, and pergolide sulfone. Pergolide sulfoxide and pergolide sulfone are dopamine agonists in animals. The other detected metabolites have not been identified and it is not known whether any other metabolites are active pharmacologically.
The major route of excretion is the kidney.
Pergolide is approximately 90% bound to plasma proteins. This extent of protein binding may be important to consider when pergolide is coadministered with other drugs known to affect protein binding.
-
INDICATIONS AND USAGE
Permax is indicated as adjunctive treatment to levodopa/carbidopa in the management of the signs and symptoms of Parkinson's disease.
Evidence to support the efficacy of pergolide as an antiparkinsonian adjunct was obtained in a multicenter study enrolling 376 patients with mild to moderate Parkinson's disease who were intolerant to l-dopa/carbidopa as manifested by moderate to severe dyskinesia and/or on-off phenomena. On average, the patients evaluated had been on l-dopa/carbidopa for 3.9 years (range, 2 days to 16.8 years). The administration of pergolide permitted a 5% to 30% reduction in the daily dose of l-dopa. On average, these patients treated with pergolide maintained an equivalent or better clinical status than they exhibited at baseline.
- CONTRAINDICATIONS
-
WARNINGS
Cardiac Valvulopathy and Fibrotic Complications (SEEBOXED WARNING).
Falling Asleep During Activities of Daily Living—Patients treated with Permax have reported falling asleep while engaged in activities of daily living, including the operation of motor vehicles which sometimes resulted in accidents. Although many of these patients reported somnolence while on Permax, some perceived that they had no warning signs such as excessive drowsiness, and believed that they were alert immediately prior to the event. Some of these events had been reported as late as 1 year after the initiation of treatment.
Somnolence is a common occurrence in patients receiving Permax. Many clinical experts believe that falling asleep while engaged in activities of daily living always occurs in a setting of preexisting somnolence, although patients may not give such a history. For this reason, prescribers should continually reassess patients for drowsiness or sleepiness, especially since some of the events occur well after the start of treatment. Prescribers should also be aware that patients may not acknowledge drowsiness or sleepiness until directly questioned about drowsiness or sleepiness during specific activities.
Before initiating treatment with Permax, patients should be advised of the potential to develop drowsiness and specifically asked about factors that may increase the risk with Permax such as concomitant sedating medications or the presence of sleep disorders. If a patient develops significant daytime sleepiness or episodes of falling asleep during activities that require participation (e.g., conversations, eating, etc.), Permax should ordinarily be discontinued. If a decision is made to continue Permax, patients should be advised to not drive and to avoid other potentially dangerous activities.
While dose reduction may reduce the degree of somnolence, there is insufficient information to establish that dose reduction will eliminate episodes of falling asleep while engaged in activities of daily living.
Symptomatic Hypotension — In clinical trials, approximately 10% of patients taking pergolide with l-dopa versus 7% taking placebo with l-dopa experienced symptomatic orthostatic and/or sustained hypotension, especially during initial treatment. With gradual dosage titration, tolerance to the hypotension usually develops. It is therefore important to warn patients of the risk, to begin therapy with low doses, and to increase the dosage in carefully adjusted increments over a period of 3 to 4 weeks (seeDOSAGE AND ADMINISTRATION).
Hallucinosis— In controlled trials, pergolide with l-dopa caused hallucinosis in about 14% of patients as opposed to 3% taking placebo with l-dopa. This was of sufficient severity to cause discontinuation of treatment in about 3% of those enrolled; tolerance to this untoward effect was not observed.
Fatalities— In the placebo-controlled trial, 2 of 187 patients treated with placebo died as compared with 1 of 189 patients treated with pergolide. Of the 2299 patients treated with pergolide in premarketing studies evaluated as of October 1988, 143 died while on the drug or shortly after discontinuing it. Because the patient population under evaluation was elderly, ill, and at high risk for death, it seems unlikely that pergolide played any role in these deaths, but the possibility that pergolide shortens survival of patients cannot be excluded with absolute certainty.
In particular, a case-by-case review of the clinical course of the patients who died failed to disclose any unique set of signs, symptoms, or laboratory results that would suggest that treatment with pergolide caused their deaths. Sixty-eight percent (68%) of the patients who died were 65 years of age or older. No death (other than a suicide) occurred within the first month of treatment; most of the patients who died had been on pergolide for years. A relative frequency of the causes of death by organ system are: Pulmonary failure/Pneumonia, 35%; Cardiovascular, 30%; Cancer, 11%; Unknown, 8.4%; Infection, 3.5%; Extrapyramidal syndrome, 3.5%; Stroke, 2.1%; Dysphagia, 2.1%; Injury, 1.4%; Suicide, 1.4%; Dehydration, 0.7%; Glomerulonephritis, 0.7%.
-
PRECAUTIONS
General
Caution should be exercised when administering pergolide to patients prone to cardiac dysrhythmias.
In a study comparing pergolide and placebo, patients taking pergolide were found to have significantly more episodes of atrial premature contractions (APCs) and sinus tachycardia.
The use of pergolide in patients on l-dopa may cause and/or exacerbate preexisting states of confusion and hallucinations (seeWARNINGS) and preexisting dyskinesia. Also, the abrupt discontinuation of pergolide in patients receiving it chronically as an adjunct to l-dopa may precipitate the onset of hallucinations and confusion; these may occur within a span of several days. Discontinuation of pergolide should be undertaken gradually whenever possible, even if the patient is to remain on l-dopa.
A symptom complex resembling the neuroleptic malignant syndrome (NMS) (characterized by elevated temperature, muscular rigidity, altered consciousness, and autonomic instability), with no other obvious etiology, has been reported in association with rapid dose reduction, withdrawal of, or changes in antiparkinsonian therapy, including pergolide.
Raynaud’s Phenomenon
Pergolide can rarely cause Raynaud’s phenomenon.
Information for Patients
Because pergolide may cause somnolence and the possibility of falling asleep during activities of daily living, patients should be cautioned about operating hazardous machinery, including automobiles, until they are reasonably certain that pergolide therapy does not affect them adversely. Patients should be advised that if increased somnolence or new episodes of falling asleep during activities of daily living (e.g., watching television, passenger in a car, etc.) are experienced at any time during treatment, they should not drive or participate in potentially dangerous activities until they have contacted their physician. Due to the possible additive sedative effects, caution should also be used when patients are taking other CNS depressants in combination with pergolide.
Patients and their families should be informed of the common adverse consequences of the use of pergolide (seeADVERSE REACTIONS) and the risk of hypotension (seeWARNINGS).
Patients should be advised to notify their physician if they become pregnant or intend to become pregnant during therapy.
Patients should be advised to notify their physician if they are breast feeding an infant.
As with other dopamine agonists, compulsive self-rewarding behavior (e.g., pathologic gambling) and libido increase have been reported in patients receiving pergolide therapy.
Laboratory Tests
No specific laboratory tests are deemed essential for the management of patients on Permax. Periodic routine evaluation of all patients, however, is appropriate.
Drug Interactions
Dopamine antagonists, such as the neuroleptics (phenothiazines, butyrophenones, thioxanthines) or metoclopramide, ordinarily should not be administered concurrently with Permax (a dopamine agonist); these agents may diminish the effectiveness of Permax.
Because pergolide is approximately 90% bound to plasma proteins, caution should be exercised if pergolide is coadministered with other drugs known to affect protein binding.
Carcinogenesis, Mutagenesis, Impairment of Fertility
A 2-year carcinogenicity study was conducted in mice using dietary levels of pergolide equivalent to oral doses of 0.6, 3.7, and 36.4 mg/kg/day in males and 0.6, 4.4, and 40.8 mg/kg/day in females. A 2-year study in rats was conducted using dietary levels equivalent to oral doses of 0.04, 0.18, and 0.88 mg/kg/day in males and 0.05, 0.28, and 1.42 mg/kg/day in females. The highest doses tested in the mice and rats were approximately 340 and 12 times the maximum human oral dose administered in controlled clinical trials (6 mg/day equivalent to 0.12 mg/kg/day).
A low incidence of uterine neoplasms occurred in both rats and mice. Endometrial adenomas and carcinomas were observed in rats. Endometrial sarcomas were observed in mice. The occurrence of these neoplasms is probably attributable to the high estrogen/progesterone ratio that would occur in rodents as a result of the prolactin-inhibiting action of pergolide. The endocrine mechanisms believed to be involved in the rodents are not present in humans. However, even though there is no known correlation between uterine malignancies occurring in pergolide-treated rodents and human risk, there are no human data to substantiate this conclusion.
Pergolide was evaluated for mutagenic potential in a battery of tests that included an Ames bacterial mutation assay, a DNA repair assay in cultured rat hepatocytes, an in vitro mammalian cell gene mutation assay in cultured L5178Y cells, and a determination of chromosome alteration in bone marrow cells of Chinese hamsters. A weak mutagenic response was noted in the mammalian cell gene mutation assay only after metabolic activation with rat liver microsomes. No mutagenic effects were obtained in the 2 other in vitro assays and in the in vivo assay. The relevance of these findings in humans is unknown.
A fertility study in male and female mice showed that fertility was maintained at 0.6 and 1.7 mg/kg/day but decreased at 5.6 mg/kg/day. Prolactin has been reported to be involved in stimulating and maintaining progesterone levels required for implantation in mice and, therefore, the impaired fertility at the high dose may have occurred because of depressed prolactin levels.
Usage in Pregnancy − Pregnancy Category B
Reproduction studies were conducted in mice at doses of 5, 16, and 45 mg/kg/day and in rabbits at doses of 2, 6, and 16 mg/kg/day. The highest doses tested in mice and rabbits were 375 and 133 times the 6 mg/day maximum human dose administered in controlled clinical trials. In these studies, there was no evidence of harm to the fetus due to pergolide.
There are, however, no adequate and well-controlled studies in pregnant women. Among women who received pergolide for endocrine disorders in premarketing studies, there were 33 pregnancies that resulted in healthy babies and 6 pregnancies that resulted in congenital abnormalities (3 major, 3 minor); a causal relationship has not been established. Because human data are limited and because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
It is not known whether this drug is excreted in human milk. The pharmacologic action of pergolide suggests that it may interfere with lactation. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions to pergolide in nursing infants, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Geriatric Use
Of the total number of subjects in clinical studies of pergolide, 78 were 65 and over. There were no apparent differences in efficacy between these subjects and younger subjects. There was an increased incidence of confusion, somnolence, and peripheral edema in patients 65 and over. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
-
ADVERSE REACTIONS
Commonly Observed— In premarketing clinical trials, the most commonly observed adverse events associated with use of pergolide which were not seen at an equivalent incidence among placebo-treated patients were: nervous system complaints, including dyskinesia, hallucinations, somnolence, insomnia; digestive complaints, including nausea, constipation, diarrhea, dyspepsia; and respiratory system complaints, including rhinitis.
Associated With Discontinuation of Treatment— Twenty-seven percent (27%) of approximately 1200 patients receiving pergolide for treatment of Parkinson's disease in premarketing clinical trials in the US and Canada discontinued treatment due to adverse events. The events most commonly causing discontinuation were related to the nervous system (15.5%), primarily hallucinations (7.8%) and confusion (1.8%).
Fatalities —seeWARNINGS
Incidence in Controlled Clinical Trials— The table that follows enumerates adverse events that occurred at a frequency of 1% or more among patients taking pergolide who participated in the premarketing controlled clinical trials comparing pergolide with placebo. In a double-blind, controlled study of 6 months’ duration, patients with Parkinson’s disease were continued on l-dopa/carbidopa and were randomly assigned to receive either pergolide or placebo as additional therapy.
The prescriber should be aware that these figures cannot be used to predict the incidence of side effects in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contribution of drug and nondrug factors to the side-effect incidence rate in the population studied.
Incidence of Treatment-Emergent Adverse Experiences in the Placebo-Controlled Clinical Trial Percentage of Patients Reporting Events Pergolide Placebo Body system/Adverse Event* N=189 N=187 *Events reported by at least 1% of patients receiving pergolide are included. Body as a Whole Pain 7.0 2.1 Abdominal pain 5.8 2.1 Injury, accident 5.8 7.0 Headache 5.3 6.4 Asthenia 4.2 4.8 Chest pain 3.7 2.1 Flu syndrome 3.2 2.1 Neck pain 2.7 1.6 Back pain 1.6 2.1 Surgical procedure 1.6 <1 Chills 1.1 0 Face edema 1.1 0 Infection 1.1 0 Cardiovascular Postural hypotension 9.0 7.0 Vasodilatation 3.2 <1 Palpitation 2.1 <1 Hypotension 2.1 <1 Syncope 2.1 1.1 Hypertension 1.6 1.1 Arrhythmia 1.1 <1 Myocardial infarction 1.1 <1 Digestive Nausea 24.3 12.8 Constipation 10.6 5.9 Diarrhea 6.4 2.7 Dyspepsia 6.4 2.1 Anorexia 4.8 2.7 Dry mouth 3.7 <1 Vomitting 2.7 1.6 Hemic and Lymphatic Anemia 1.1 <1 Metabolic and Nutritional Peripheral edema 7.4 4.3 Edema 1.6 0 Weight gain 1.6 0 Musuloskeletal Arthralgia 1.6 2.1 Bursitis 1.6 <1 Myalgia 1.1 <1 Twitching 1.1 0 Nervous System Dyskinesia 62.4 24.6 Dizziness 19.1 13.9 Hallucinations 13.8 3.2 Dystonia 11.6 8.0 Confusion 11.1 9.6 Somnolence 10.1 3.7 Insomnia 7.9 3.2 Anxiety 6.4 4.3 Tremor 4.2 7.5 Depression 3.2 5.4 Abnormal dreams 2.7 4.3 Personality disorder 2.1 <1 Psychosis 2.1 0 Abnormal gait 1.6 1.6 Akathisia 1.6 0 Extrapyramidal syndrome 1.6 1.1 Incoordination 1.6 <1 Paresthesia 1.6 3.2 Akinesia 1.1 1.1 Hypertonia 1.1 0 Neuralgia 1.1 <1 Speech disorder 1.1 1.6 Respiratory system Rhinitis 12.2 5.4 Dyspnea 4.8 1.1 Epistaxis 1.6 <1 Hiccup 1.1 0 Skin and Appendages Rash 3.2 2.1 Sweating 2.1 2.7 Special Senses Abnormal vision 5.8 5.4 Diplopia 2.1 0 Taste perversion 1.6 0 Eye disorder 1.1 0 Urogenital System Urinary frequency 2.7 6.4 Urinary tract infection 2.7 3.7 Hematuria 1.1 <1 Events Observed During the Premarketing Evaluation of Permax— This section reports event frequencies evaluated as of October 1988 for adverse events occurring in a group of approximately 1800 patients who took multiple doses of pergolide. The conditions and duration of exposure to pergolide varied greatly, involving well-controlled studies as well as experience in open and uncontrolled clinical settings. In the absence of appropriate controls in some of the studies, a causal relationship between these events and treatment with pergolide cannot be determined.
The following enumeration by organ system describes events in terms of their relative frequency of reporting in the data base. Events of major clinical importance are also described in the Warnings and Precautions sections.
The following definitions of frequency are used: frequent adverse events are defined as those occurring in at least 1/100 patients; infrequent adverse events are those occurring in 1/100 to 1/1000 patients; rare events are those occurring in fewer than 1/1000 patients.
Body as a Whole—Frequent: headache, asthenia, accidental injury, pain, abdominal pain, chest pain, back pain, flu syndrome, neck pain, fever; Infrequent: facial edema, chills, enlarged abdomen, malaise, neoplasm, hernia, pelvic pain, sepsis, cellulitis, moniliasis, abscess, jaw pain, hypothermia; Rare: acute abdominal syndrome, LE syndrome.
Cardiovascular System—Frequent: postural hypotension, syncope, hypertension, palpitations, vasodilatations, congestive heart failure; Infrequent: myocardial infarction, tachycardia, heart arrest, abnormal electrocardiogram, angina pectoris, thrombophlebitis, bradycardia, ventricular extrasystoles, cerebrovascular accident, ventricular tachycardia, cerebral ischemia, atrial fibrillation, varicose vein, pulmonary embolus, AV block, shock; Rare: vasculitis, pulmonary hypertension, pericarditis, migraine, heart block, cerebral hemorrhage.
Digestive System—Frequent: nausea, vomiting, dyspepsia, diarrhea, constipation, dry mouth, dysphagia; Infrequent: flatulence, abnormal liver function tests, increased appetite, salivary gland enlargement, thirst, gastroenteritis, gastritis, periodontal abscess, intestinal obstruction, nausea and vomiting, gingivitis, esophagitis, cholelithiasis, tooth caries, hepatitis, stomach ulcer, melena, hepatomegaly, hematemesis, eructation; Rare: sialadenitis, peptic ulcer, pancreatitis, jaundice, glossitis, fecal incontinence, duodenitis, colitis, cholecystitis, aphthous stomatitis, esophageal ulcer.
Endocrine System—Infrequent: hypothyroidism, adenoma, diabetes mellitus, ADH inappropriate; Rare: endocrine disorder, thyroid adenoma.
Hemic and Lymphatic System—Frequent: anemia; Infrequent: leukopenia, lymphadenopathy, leukocytosis, thrombocytopenia, petechia, megaloblastic anemia, cyanosis; Rare: purpura, lymphocytosis, eosinophilia, thrombocythemia, acute lymphoblastic leukemia, polycythemia, splenomegaly.
Metabolic and Nutritional System—Frequent: peripheral edema, weight loss, weight gain; Infrequent: dehydration, hypokalemia, hypoglycemia, iron deficiency anemia, hyperglycemia, gout, hypercholesteremia; Rare: electrolyte imbalance, cachexia, acidosis, hyperuricemia.
Musculoskeletal System—Frequent: twitching, myalgia, arthralgia; Infrequent: bone pain, tenosynovitis, myositis, bone sarcoma, arthritis; Rare: osteoporosis, muscle atrophy, osteomyelitis.
Nervous System—Frequent: dyskinesia, dizziness, hallucinations, confusion, somnolence, insomnia, dystonia, paresthesia, depression, anxiety, tremor, akinesia, extrapyramidal syndrome, abnormal gait, abnormal dreams, incoordination, psychosis, personality disorder, nervousness, choreoathetosis, amnesia, paranoid reaction, abnormal thinking; Infrequent: akathisia, neuropathy, neuralgia, hypertonia, delusions, convulsion, libido increased, euphoria, emotional lability, libido decreased, vertigo, myoclonus, coma, apathy, paralysis, neurosis, hyperkinesia, ataxia, acute brain syndrome, torticollis, meningitis, manic reaction, hypokinesia, hostility, agitation, hypotonia; Rare: stupor, neuritis, intracranial hypertension, hemiplegia, facial paralysis, brain edema, myelitis, hallucinations and confusion after abrupt discontinuation.
Respiratory System—Frequent: rhinitis, dyspnea, pneumonia, pharyngitis, cough increased; Infrequent: epistaxis, hiccup, sinusitis, bronchitis, voice alteration, hemoptysis, asthma, lung edema, pleural effusion, laryngitis, emphysema, apnea, hyperventilation; Rare: pneumothorax, lung fibrosis, larynx edema, hypoxia, hypoventilation, hemothorax, carcinoma of lung.
Skin and Appendages System—Frequent: sweating, rash; Infrequent: skin discoloration, pruritus, acne, skin ulcer, alopecia, dry skin, skin carcinoma, seborrhea, hirsutism, herpes simplex, eczema, fungal dermatitis, herpes zoster; Rare: vesiculobullous rash, subcutaneous nodule, skin nodule, skin benign neoplasm, lichenoid dermatitis.
Special Senses System—Frequent: abnormal vision, diplopia; Infrequent: otitis media, conjunctivitis, tinnitus, deafness, taste perversion, ear pain, eye pain, glaucoma, eye hemorrhage, photophobia, visual field defect; Rare: blindness, cataract, retinal detachment, retinal vascular disorder.
Urogenital System—Frequent: urinary tract infection, urinary frequency, urinary incontinence, hematuria, dysmenorrhea; Infrequent: dysuria, breast pain, menorrhagia, impotence, cystitis, urinary retention, abortion, vaginal hemorrhage, vaginitis, priapism, kidney calculus, fibrocystic breast, lactation, uterine hemorrhage, urolithiasis, salpingitis, pyuria, metrorrhagia, menopause, kidney failure, breast carcinoma, cervical carcinoma; Rare: amenorrhea, bladder carcinoma, breast engorgement, epididymitis, hypogonadism, leukorrhea, nephrosis, pyelonephritis, urethral pain, uricaciduria, withdrawal bleeding.
Postintroduction Reports— Voluntary reports of adverse events temporally associated with pergolide that have been received since market introduction and which may have no causal relationship with the drug, include the following: neuroleptic malignant syndrome and Raynaud’s phenomenon.
-
OVERDOSAGE
There is no clinical experience with massive overdosage. The largest overdose involved a young hospitalized adult patient who was not being treated with pergolide but who intentionally took 60 mg of the drug. He experienced vomiting, hypotension, and agitation. Another patient receiving a daily dosage of 7 mg of pergolide unintentionally took 19 mg/day for 3 days, after which his vital signs were normal but he experienced severe hallucinations. Within 36 hours of resumption of the prescribed dosage level, the hallucinations stopped. One patient unintentionally took 14 mg/day for 23 days instead of her prescribed 1.4 mg/day dosage. She experienced severe involuntary movements and tingling in her arms and legs. Another patient who inadvertently received 7 mg instead of the prescribed 0.7 mg experienced palpitations, hypotension, and ventricular extrasystoles. The highest total daily dose (prescribed for several patients with refractory Parkinson's disease) has exceeded 30 mg.
Symptoms— Animal studies indicate that the manifestations of overdosage in man might include nausea, vomiting, convulsions, decreased blood pressure, and CNS stimulation. The oral median lethal doses in mice and rats were 54 and 15 mg/kg respectively.
Treatment— To obtain up-to-date information about the treatment of overdose, a good resource is your certified Regional Poison Control Center. Telephone numbers of certified poison control centers are listed in the Physicians’ Desk Reference (PDR). In managing overdosage, consider the possibility of multiple drug overdoses, interaction among drugs, and unusual drug kinetics in your patient.
Management of overdosage may require supportive measures to maintain arterial blood pressure. Cardiac function should be monitored; an antiarrhythmic agent may be necessary. If signs of CNS stimulation are present, a phenothiazine or other butyrophenone neuroleptic agent may be indicated; the efficacy of such drugs in reversing the effects of overdose has not been assessed.
Protect the patient’s airway and support ventilation and perfusion. Meticulously monitor and maintain, within acceptable limits, the patient’s vital signs, blood gases, serum electrolytes, etc. Absorption of drugs from the gastrointestinal tract may be decreased by giving activated charcoal, which, in many cases, is more effective than emesis or lavage; consider charcoal instead of or in addition to gastric emptying. Repeated doses of charcoal over time may hasten elimination of some drugs that have been absorbed. Safeguard the patient’s airway when employing gastric emptying or charcoal.
There is no experience with dialysis or hemoperfusion, and these procedures are unlikely to be of benefit.
-
DOSAGE AND ADMINISTRATION
Administration of Permax should be initiated with a daily dosage of 0.05 mg for the first 2 days. The dosage should then be gradually increased by 0.1 or 0.15 mg/day every third day over the next 12 days of therapy. The dosage may then be increased by 0.25 mg/day every third day until an optimal therapeutic dosage is achieved.
Permax is usually administered in divided doses 3 times per day. During dosage titration, the dosage of concurrent l-dopa/carbidopa may be cautiously decreased.
In clinical studies, the mean therapeutic daily dosage of Permax was 3 mg/day. The average concurrent daily dosage of l-dopa/carbidopa (expressed as l-dopa) was approximately 650 mg/day. The efficacy of Permax at doses above 5 mg/day has not been systematically evaluated. Doses of pergolide above 5 mg/day are not recommended (seeWARNINGS).
-
HOW SUPPLIED
Tablets (modified rectangle shape, scored):
0.05 mg, ivory, debossed with A 024, in bottles of 30 (UC5336) — NDC 0187-0839-01
0.25 mg, green, debossed with A 025, in bottles of 100 (UC5337) — NDC 0187-0840-02
1 mg, pink, debossed with A 026, in bottles of 100 (UC5338) — NDC 0187-0841-02
Store at 25°C (77°F); excursions permitted to 15°C-30°C (59°F-86°F) [see USP Controlled Room Temperature].
PERMAX is a registered trademark of Eli Lilly and Company, and licensed in the US to Valeant Pharmaceuticals International.
Literature revised August 18, 2004
Manufactured for:
Valeant Pharmaceuticals International
3300 Hyland Ave.
Costa Mesa, CA 92626 U.S.A.Part Number 3083900EX00
PRINTED IN USA -
INGREDIENTS AND APPEARANCE
PERMAX
pergolide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0187-0839 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength pergolide (UNII: 24MJ822NZ9) (pergolide - UNII:24MJ822NZ9) 0.05 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium () iron oxide () lactose () magnesium stearate (UNII: 70097M6I30) povidone () L-methionine () Product Characteristics Color WHITE (ivory) Score 2 pieces Shape RECTANGLE Size 12mm Flavor Imprint Code A;024 Contains Coating false Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0187-0839-01 30 in 1 BOTTLE PERMAX
pergolide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0187-0840 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength pergolide (UNII: 24MJ822NZ9) (pergolide - UNII:24MJ822NZ9) 0.25 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium () iron oxide () lactose () magnesium stearate (UNII: 70097M6I30) povidone () FD&C Blue No. 2 () Product Characteristics Color GREEN Score 2 pieces Shape RECTANGLE Size 12mm Flavor Imprint Code A;025 Contains Coating false Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0187-0840-02 100 in 1 BOTTLE PERMAX
pergolide tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0187-0841 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength pergolide (UNII: 24MJ822NZ9) (pergolide - UNII:24MJ822NZ9) 1 mg Inactive Ingredients Ingredient Name Strength croscarmellose sodium () iron oxide () lactose () magnesium stearate (UNII: 70097M6I30) povidone () Product Characteristics Color PINK Score 2 pieces Shape RECTANGLE Size 12mm Flavor Imprint Code A;026 Contains Coating false Symbol false Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0187-0841-02 100 in 1 BOTTLE Labeler - Valeant Pharmaceuticals, Inc.