Label: SIMPONI ARIA- golimumab solution

- NDC Code(s): 57894-350-01, 57894-350-89
- Packager: Janssen Biotech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated September 9, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use SIMPONI ARIA safely and effectively. See full prescribing information for SIMPONI ARIA.
SIMPONI ARIA ®(golimumab) injection, for intravenous use
Initial U.S. Approval: 2009WARNING: SERIOUS INFECTIONS and MALIGNANCY
See full prescribing information for complete boxed warning.
- Serious infections leading to hospitalization or death including tuberculosis (TB), bacterial sepsis, invasive fungal (such as histoplasmosis), and other opportunistic infections have occurred in patients receiving SIMPONI ARIA ( 5.1).
- Discontinue SIMPONI ARIA if a patient develops a serious infection or sepsis ( 5.1).
- Perform test for latent TB; if positive, start treatment for TB prior to starting SIMPONI ARIA ( 5.1).
- Monitor all patients for active TB during treatment, even if initial latent TB test is negative ( 5.1).
- Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, of which SIMPONI ARIA is a member ( 5.2).
INDICATIONS AND USAGE
SIMPONI ARIA is a tumor necrosis factor (TNF) blocker indicated for the treatment of:
- Adult patients with moderately to severely active Rheumatoid Arthritis (RA) in combination with methotrexate ( 1.1)
- Active Psoriatic Arthritis (PsA) in patients 2 years of age and older ( 1.2)
- Adult patients with active Ankylosing Spondylitis (AS) ( 1.3)
- Active polyarticular Juvenile Idiopathic Arthritis (pJIA) in patients 2 years of age and older ( 1.4)
DOSAGE AND ADMINISTRATION
- Adult patients with Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis:
- 2 mg/kg intravenous infusion over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter ( 2.1)
- Pediatric patients with polyarticular Juvenile Idiopathic Arthritis and Psoriatic Arthritis:
- 80 mg/m 2intravenous infusion over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter ( 2.2)
- Dilution of supplied SIMPONI ARIA solution with 0.9% Sodium Chloride Injection, USP is required prior to administration. Alternatively, 0.45% Sodium Chloride Injection, USP can also be used ( 2.4)
DOSAGE FORMS AND STRENGTHS
- Injection: 50 mg/4 mL (12.5 mg/mL) solution in a single-dose vial ( 3)
CONTRAINDICATIONS
- None ( 4)
WARNINGS AND PRECAUTIONS
- Serious Infections: Do not start SIMPONI ARIA during an active infection. If an infection develops, monitor carefully, and stop SIMPONI ARIA if infection becomes serious ( 5.1).
- Invasive Fungal Infections: For patients who develop a systemic illness on SIMPONI ARIA, consider empiric antifungal therapy for those who reside in or travel to regions where mycoses are endemic ( 5.1).
- Hepatitis B Reactivation: Monitor HBV carriers during and several months after therapy. If reactivation occurs, stop SIMPONI ARIA and begin anti-viral therapy ( 5.1).
- Malignancies: More cases of lymphoma have been observed among patients receiving TNF blockers compared with patients in the control groups. Cases of other malignancies have been observed among patients receiving TNF blockers ( 5.2).
- Congestive Heart Failure: Worsening, or new onset, may occur. Stop SIMPONI ARIA if new or worsening symptoms occur ( 5.3).
- Demyelinating Disorders: Exacerbation or new onset may occur ( 5.4).
- Lupus-like Syndrome: Discontinue SIMPONI ARIA if symptoms develop ( 5.5).
- Hypersensitivity Reactions: Serious systemic hypersensitivity reactions including anaphylaxis may occur ( 5.11).
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 3%) are: upper respiratory tract infection, alanine aminotransferase increased, viral infection, aspartate aminotransferase increased, neutrophil count decreased, bronchitis, hypertension, and rash ( 6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-JANSSEN (1-800-526-7736) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SERIOUS INFECTIONS and MALIGNANCY
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis (RA)
1.2 Psoriatic Arthritis (PsA)
1.3 Ankylosing Spondylitis (AS)
1.4 Polyarticular Juvenile Idiopathic Arthritis (pJIA)
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Adults with Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
2.2 Dosage in Pediatric Patients with Polyarticular Juvenile Idiopathic Arthritis and Psoriatic Arthritis
2.3 Evaluation for Tuberculosis and Hepatitis B Prior to Dosage
2.4 Important Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
5.2 Malignancies
5.3 Congestive Heart Failure
5.4 Demyelinating Disorders
5.5 Autoimmunity
5.6 Use with Abatacept
5.7 Use with Anakinra
5.8 Switching Between Biological Disease Modifying Antirheumatic Drugs (DMARDs)
5.9 Hematologic Cytopenias
5.10 Vaccinations/Therapeutic Infectious Agents
5.11 Hypersensitivity Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Methotrexate
7.2 Biologic Products for RA, PsA, AS, and pJIA
7.3 Live Vaccines/Therapeutic Infectious Agents
7.4 Cytochrome P450 Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis
14.2 Psoriatic Arthritis
14.3 Ankylosing Spondylitis
14.4 Polyarticular Juvenile Idiopathic Arthritis (pJIA)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SERIOUS INFECTIONS and MALIGNANCY
SERIOUS INFECTIONS
Patients treated with SIMPONI ARIA are at increased risk for developing serious infections that may lead to hospitalization or death [see Warnings and Precautions (5.1)] . Most patients who developed these infections were taking concomitant immunosuppressants such as methotrexate or corticosteroids.
Discontinue SIMPONI ARIA if a patient develops a serious infection.
Reported infections with TNF blockers, of which SIMPONI ARIA is a member, include:
- Active tuberculosis, including reactivation of latent tuberculosis. Patients with tuberculosis have frequently presented with disseminated or extrapulmonary disease. Test patients for latent tuberculosis before SIMPONI ARIA use and during therapy. Initiate treatment for latent tuberculosis prior to SIMPONI ARIA use.
- Invasive fungal infections including histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis and pneumocystosis. Patients with histoplasmosis or other invasive fungal infections may present with disseminated, rather than localized, disease. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. Consider empiric antifungal therapy in patients at risk for invasive fungal infections who develop severe systemic illness.
- Bacterial, viral, and other infections due to opportunistic pathogens, including Legionella and Listeria.
Consider the risks and benefits of treatment with SIMPONI ARIA prior to initiating therapy in patients with chronic or recurrent infection.
Monitor patients closely for the development of signs and symptoms of infection during and after treatment with SIMPONI ARIA, including the possible development of tuberculosis in patients who tested negative for latent tuberculosis infection prior to initiating therapy [see Warnings and Precautions (5.1)] .
MALIGNANCY
Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF-blockers, of which SIMPONI ARIA is a member [see Warnings and Precautions (5.2)] .
-
1 INDICATIONS AND USAGE
1.1 Rheumatoid Arthritis (RA)
SIMPONI ARIA, in combination with methotrexate (MTX), is indicated for the treatment of adult patients with moderately to severely active rheumatoid arthritis.
1.2 Psoriatic Arthritis (PsA)
SIMPONI ARIA is indicated for the treatment of active psoriatic arthritis in patients 2 years of age and older.
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage in Adults with Rheumatoid Arthritis, Psoriatic Arthritis, and Ankylosing Spondylitis
The SIMPONI ARIA dosage regimen is 2 mg per kg given as an intravenous infusion over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter. Follow the dilution and administration instructions for SIMPONI ARIA [see Dosage and Administration (2.4)] .
For patients with rheumatoid arthritis (RA), SIMPONI ARIA should be given in combination with methotrexate.
The efficacy and safety of switching between intravenous and subcutaneous formulations and routes of administration have not been established.
2.2 Dosage in Pediatric Patients with Polyarticular Juvenile Idiopathic Arthritis and Psoriatic Arthritis
The SIMPONI ARIA dosage regimen, based on body surface area (BSA), is 80 mg/m 2given as an intravenous infusion over 30 minutes at weeks 0 and 4, and every 8 weeks thereafter. Follow the dilution and administration instructions for SIMPONI ARIA [see Dosage and Administration (2.4)] .
2.3 Evaluation for Tuberculosis and Hepatitis B Prior to Dosage
Prior to initiating SIMPONI ARIA and periodically during therapy, evaluate patients for active tuberculosis and test for latent infection [see Warnings and Precautions (5.1)] . Prior to initiating SIMPONI ARIA, test patients for hepatitis B viral infection [see Warnings and Precautions (5.1)] .
2.4 Important Administration Instructions
SIMPONI ARIA solution for intravenous infusion should be diluted by a healthcare professional using aseptic technique as follows:
- Calculate the dosage and the number of SIMPONI ARIA vials needed based on the recommended adult dosage of 2 mg/kg and the patient's weight for RA, PsA and AS. Calculate the dosage and number of SIMPONI ARIA vials needed based on the recommended pediatric dosage of 80 mg/m 2and the patient's body surface area (BSA), for pJIA and pediatric patients with PsA. Each 4 mL vial of SIMPONI ARIA contains 50 mg of golimumab.
- Check that the solution in each vial is colorless to light yellow. The solution may develop a few fine translucent particles, as golimumab is a protein. Do not use if opaque particles, discoloration, or other foreign particles are present.
- Dilute the total volume of the SIMPONI ARIA solution with 0.9% Sodium Chloride Injection, USP to a final volume of 100 mL. For example, this can be accomplished by withdrawing a volume of the 0.9% Sodium Chloride Injection, USP from the 100-mL infusion bag or bottle equal to the total volume of SIMPONI ARIA. Slowly add the total volume of SIMPONI ARIA solution to the 100-mL infusion bag or bottle. Gently mix. Discard any unused solution remaining in the vials. Alternatively, SIMPONI ARIA can be diluted using the same method described above with 0.45% Sodium Chloride Injection, USP.
- Prior to infusion, visually inspect the diluted SIMPONI ARIA solution for particulate matter or discoloration. Do not use if these are present.
- Use only an infusion set with an in-line, sterile, non-pyrogenic, low protein-binding filter (pore size 0.22 micrometer or less).
- Do not infuse SIMPONI ARIA concomitantly in the same intravenous line with other agents. No physical biochemical compatibility studies have been conducted to evaluate the use of SIMPONI ARIA with other intravenous agents in the same intravenous line.
- Infuse the diluted solution over 30 minutes.
- Once diluted, the infusion solution can be stored for up to 4 hours at room temperature.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Serious Infections
Patients treated with SIMPONI ARIA are at increased risk for developing serious infections involving various organ systems and sites that may lead to hospitalization or death.
Opportunistic infections due to bacterial, mycobacterial, invasive fungal, viral, or parasitic organisms including aspergillosis, blastomycosis, candidiasis, coccidioidomycosis, histoplasmosis, legionellosis, listeriosis, pneumocystosis, and tuberculosis have been reported with TNF-blockers. Patients have frequently presented with disseminated rather than localized disease. The concomitant use of a TNF-blocker and abatacept or anakinra was associated with a higher risk of serious infections; therefore, the concomitant use of SIMPONI ARIA and these biologic products is not recommended [see Warnings and Precautions (5.6, 5.7)and Drug Interactions (7.2)] .
Treatment with SIMPONI ARIA should not be initiated in patients with an active infection, including clinically important localized infections. Patients greater than 65 years of age, patients with co-morbid conditions and/or patients taking concomitant immunosuppressants such as corticosteroids or methotrexate may be at greater risk of infection. Consider the risks and benefits of treatment prior to initiating SIMPONI ARIA in patients:
- with chronic or recurrent infection;
- who have been exposed to tuberculosis;
- with a history of an opportunistic infection;
- who have resided or traveled in areas of endemic tuberculosis or endemic mycoses, such as histoplasmosis, coccidioidomycosis, or blastomycosis; or
- with underlying conditions that may predispose them to infection.
Monitoring
Closely monitor patients for the development of signs and symptoms of infection during and after treatment with SIMPONI ARIA. Discontinue SIMPONI ARIA if a patient develops a serious infection, an opportunistic infection, or sepsis. For patients who develop a new infection during treatment with SIMPONI ARIA, perform a prompt and complete diagnostic workup appropriate for an immunocompromised patient and initiate appropriate antimicrobial therapy and closely monitor them.
Tuberculosis
Cases of reactivation of tuberculosis or new tuberculosis infections have been observed in patients receiving TNF-blockers, including patients who have previously received treatment for latent or active tuberculosis. Evaluate patients for tuberculosis risk factors and test for latent infection prior to initiating SIMPONI ARIA and periodically during therapy.
Treatment of latent tuberculosis infection prior to therapy with TNF-blockers has been shown to reduce the risk of tuberculosis reactivation during therapy. Prior to initiating SIMPONI ARIA, assess if treatment for latent tuberculosis is needed; An induration of 5 mm or greater is a positive tuberculin skin test, even for patients previously vaccinated with Bacille Calmette-Guerin (BCG).
Consider anti-tuberculosis therapy prior to initiation of SIMPONI ARIA in patients with a past history of latent or active tuberculosis in whom an adequate course of treatment cannot be confirmed, and for patients with a negative test for latent tuberculosis but having risk factors for tuberculosis infection. Consultation with a physician with expertise in the treatment of tuberculosis is recommended to aid in the decision whether initiating anti-tuberculosis therapy is appropriate for an individual patient.
Cases of active tuberculosis have occurred in patients treated with the subcutaneous formulation of golimumab during and after treatment for latent tuberculosis. Monitor patients for the development of signs and symptoms of tuberculosis including patients who tested negative for latent tuberculosis infection prior to initiating therapy, patients who are on treatment for latent tuberculosis, or patients who were previously treated for tuberculosis infection.
Consider tuberculosis in the differential diagnosis in patients who develop a new infection during SIMPONI ARIA treatment, especially in patients who have previously or recently traveled to countries with a high prevalence of tuberculosis, or who have had close contact with a person with active tuberculosis.
Invasive Fungal Infections
If patients develop a serious systemic illness and they reside or travel in regions where mycoses are endemic, consider invasive fungal infection in the differential diagnosis. Consider appropriate empiric antifungal therapy and take into account both the risk for severe fungal infection and the risks of antifungal therapy while a diagnostic workup is being performed. Antigen and antibody testing for histoplasmosis may be negative in some patients with active infection. To aid in the management of such patients, consider consultation with a physician with expertise in the diagnosis and treatment of invasive fungal infections.
Hepatitis B Virus Reactivation
The use of TNF-blockers, of which SIMPONI ARIA is a member, has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic hepatitis B carriers (i.e., surface antigen positive). In some instances, HBV reactivation occurring in conjunction with TNF-blocker therapy has been fatal. The majority of these reports have occurred in patients who received concomitant immunosuppressants.
All patients should be tested for HBV infection before initiating TNF-blocker therapy. For patients who test positive for hepatitis B surface antigen, consultation with a physician with expertise in the treatment of hepatitis B is recommended before initiating TNF-blocker therapy. The risks and benefits of treatment should be considered prior to prescribing TNF-blockers, including SIMPONI ARIA, to patients who are carriers of HBV. Adequate data are not available on whether antiviral therapy can reduce the risk of HBV reactivation in HBV carriers who are treated with TNF-blockers. Patients who are carriers of HBV and require treatment with TNF-blockers should be closely monitored for clinical and laboratory signs of active HBV infection throughout therapy and for several months following termination of therapy.
In patients who develop HBV reactivation, TNF-blockers should be stopped and antiviral therapy with appropriate supportive treatment should be initiated. The safety of resuming TNF-blockers after HBV reactivation has been controlled is not known. Therefore, prescribers should exercise caution when considering resumption of TNF-blockers in this situation and monitor patients closely.
5.2 Malignancies
Malignancies in Pediatric Patients
Malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with TNF-blocking agents (initiation of therapy ≤ 18 years of age), including golimumab. Approximately half the cases were lymphomas, including Hodgkin's and non-Hodgkin's lymphoma. The other cases represented a variety of malignancies, including rare malignancies that are usually associated with immunosuppression, and malignancies that are not usually observed in children and adolescents. The malignancies occurred after a median of 30 months (range 1 to 84 months) after the first dose of TNF-blocker therapy. Most of the patients were receiving concomitant immunosuppressants. Most cases were reported postmarketing and are derived from a variety of sources, including registries and spontaneous postmarketing reports.
Malignancies in Adult Patients
The risks and benefits of TNF-blocker treatment including SIMPONI ARIA should be considered prior to initiating therapy in patients with a known malignancy other than a successfully treated non-melanoma skin cancer (NMSC) or when considering continuing a TNF-blocker in patients who develop a malignancy.
In the controlled portions of clinical trials of TNF-blockers including the subcutaneous formulation of golimumab more cases of lymphoma have been observed among patients receiving anti-TNF treatment compared with patients in the control groups. Patients with RA and other chronic inflammatory diseases, particularly patients with highly active disease and/or chronic exposure to immunosuppressant therapies, may be at higher risk (up to several fold) than the general population for the development of lymphoma, even in the absence of TNF-blocking therapy. Cases of acute and chronic leukemia have been reported with TNF-blocker use, including SIMPONI ARIA, in rheumatoid arthritis and other indications. Even in the absence of TNF-blocker therapy, patients with rheumatoid arthritis may be at a higher risk (approximately 2-fold) than the general population for the development of leukemia.
Rare postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL) have been reported in patients treated with TNF-blocking agents. This rare type of T-cell lymphoma has a very aggressive disease course and is usually fatal. Nearly all of the reported TNF-blocker associated cases have occurred in patients with Crohn's disease or ulcerative colitis. The majority were in adolescent and young adult males. Almost all these patients had received treatment with azathioprine (AZA) or 6-mercaptopurine (6–MP) concomitantly with a TNF-blocker at or prior to diagnosis. A risk for the development for hepatosplenic T-cell lymphoma in patients treated with TNF-blockers cannot be excluded.
Melanoma and Merkel cell carcinoma have been reported in patients treated with TNF-blocking agents, including SIMPONI ARIA. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
In controlled trials of other TNF-blockers in patients at higher risk for malignancies (e.g., patients with chronic obstructive pulmonary disease [COPD], patients with Wegener's granulomatosis treated with concomitant cyclophosphamide) a greater portion of malignancies occurred in the TNF-blocker group compared to the controlled group. In an exploratory clinical trial evaluating the use of the subcutaneous formulation of golimumab in patients with severe persistent asthma, more patients treated with golimumab reported malignancies compared with control patients. The significance of this finding is unknown.
During the controlled portion of the Phase 3 trial in RA for SIMPONI ARIA, the incidence of malignancies other than lymphoma and NMSC per 100-patient-years of follow-up was 0.56 (95% CI: 0.01, 3.11) in the SIMPONI ARIA group compared with an incidence of 0 (95% CI: 0.00, 3.79) in the placebo group.
5.3 Congestive Heart Failure
Cases of worsening congestive heart failure (CHF) and new onset CHF have been reported with TNF-blockers, including SIMPONI ARIA. Some cases had a fatal outcome. In several exploratory trials of other TNF-blockers in the treatment of CHF, there were greater proportions of TNF-blocker treated patients who had CHF exacerbations requiring hospitalization or increased mortality. SIMPONI ARIA has not been studied in patients with a history of CHF and SIMPONI ARIA should be used with caution in patients with CHF. If a decision is made to administer SIMPONI ARIA to patients with CHF, these patients should be closely monitored during therapy, and SIMPONI ARIA should be discontinued if new or worsening symptoms of CHF appear.
5.4 Demyelinating Disorders
Use of TNF-blockers, including SIMPONI ARIA, has been associated with rare cases of new onset or exacerbation of central nervous system (CNS) demyelinating disorders, including multiple sclerosis (MS), and peripheral demyelinating disorders, including Guillain-Barré syndrome. Cases of central demyelination, MS, optic neuritis, and peripheral demyelinating polyneuropathy have rarely been reported in patients treated with golimumab. Prescribers should exercise caution in considering the use of TNF-blockers, including SIMPONI ARIA, in patients with central or peripheral nervous system demyelinating disorders. Discontinuation of SIMPONI ARIA should be considered if these disorders develop.
5.5 Autoimmunity
Treatment with TNF blockers, including SIMPONI ARIA, may result in the formation of antinuclear antibodies (ANA). Rarely, treatment with TNF blockers, may result in the development of a lupus-like syndrome [see Adverse Reactions (6.1)] . If a patient develops symptoms suggestive of a lupus-like syndrome following treatment with SIMPONI ARIA, treatment should be discontinued.
5.6 Use with Abatacept
In controlled trials, the concurrent administration of another TNF-blocker and abatacept was associated with a greater proportion of serious infections than the use of a TNF-blocker alone; and the combination therapy, compared to the use of a TNF-blocker alone, has not demonstrated improved clinical benefit in the treatment of RA. Therefore, the combination of TNF-blockers, including SIMPONI ARIA, and abatacept is not recommended [see Drug Interactions (7.2)] .
5.7 Use with Anakinra
Concurrent administration of anakinra (an interleukin-1 antagonist) and another TNF-blocker was associated with a greater portion of serious infections and neutropenia and no additional benefits compared with the TNF-blocker alone. Therefore, the combination of anakinra with TNF-blockers, including SIMPONI ARIA, is not recommended [see Drug Interactions (7.2)] .
5.8 Switching Between Biological Disease Modifying Antirheumatic Drugs (DMARDs)
Care should be taken when switching from one biologic product to another biologic product since overlapping biological activity may further increase the risk of infection.
5.9 Hematologic Cytopenias
There have been reports of pancytopenia, leukopenia, neutropenia, agranulocytosis, aplastic anemia, and thrombocytopenia in patients receiving golimumab. Caution should be exercised when using TNF-blockers, including SIMPONI ARIA, in patients who have or have had significant cytopenias.
5.10 Vaccinations/Therapeutic Infectious Agents
Live Vaccines
Avoid live vaccines in patients treated with SIMPONI ARIA. In patients receiving anti-TNF therapy, limited data are available on the response to live vaccination, or on the secondary transmission of infection by live vaccines. Use of live vaccines could result in clinical infections, including disseminated infections.
Administration of live vaccines to infants exposed to SIMPONI ARIA in uterois not recommended for 6 months following the mother's last SIMPONI ARIA infusion during pregnancy [see Drug Interactions (7.3)and Use in Specific Populations (8.1)] .
Whenever possible update immunizations prior to initiation of treatment with SIMPONI ARIA following current immunization guidelines for patients receiving immunosuppressive agents. Advise patients to discuss with the physician before seeking any immunizations.
Therapeutic Infectious Agents
Other uses of therapeutic infectious agents such as live attenuated bacteria (e.g., BCG bladder instillation for the treatment of cancer) could result in clinical infections, including disseminated infections. It is recommended that therapeutic infectious agents not be given concurrently with SIMPONI ARIA.
5.11 Hypersensitivity Reactions
In postmarketing experience, serious systemic hypersensitivity reactions (including anaphylaxis) have been reported following administration of the subcutaneous and intravenous formulations of golimumab including SIMPONI ARIA. Hypersensitivity reactions including hives, pruritus, dyspnea, and nausea, were reported during infusion and generally within an hour after infusion. Some of these reactions occurred after the first administration of golimumab. If an anaphylactic or other serious allergic reaction occurs, administration of SIMPONI ARIA should be discontinued immediately and appropriate therapy instituted.
-
6 ADVERSE REACTIONS
The most serious adverse reactions were:
- Serious Infections [see Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The safety data described below are based on one, randomized, double-blind, controlled Phase 3 trial in patients with RA receiving SIMPONI ARIA by intravenous infusion (Trial RA). The protocol included provisions for patients taking placebo to receive treatment with SIMPONI ARIA at Week 16 or Week 24 either by patient response (based on uncontrolled disease activity) or by design, so that adverse events cannot always be unambiguously attributed to a given treatment. Comparisons between placebo and SIMPONI ARIA were based on the first 24 weeks of exposure.
Trial RA included 197 control-treated patients and 463 SIMPONI ARIA-treated patients (which includes control-treated patients who switched to SIMPONI ARIA at Week 16). The proportion of patients who discontinued treatment due to adverse reactions in the controlled phase of Trial RA through Week 24 was 3.5% for SIMPONI ARIA-treated patients and 0.5% for placebo-treated patients. Upper respiratory tract infection was the most common adverse reaction reported in the trial through Week 24 occurring in 6.5% of SIMPONI ARIA-treated patients as compared with 7.6% of control-treated patients, respectively.
Infections
Serious infections observed in SIMPONI ARIA-treated patients included sepsis, pneumonia, cellulitis, abscess, opportunistic infections, tuberculosis (TB), and invasive fungal infections. Cases of TB included pulmonary and extrapulmonary TB. The majority of the TB cases occurred in countries with a high incidence rate of TB [see Warnings and Precautions (5.1)] .
In the controlled phase of Trial RA through Week 24, infections were observed in 27% of SIMPONI ARIA-treated patients compared with 24% of control-treated patients, and serious infections were observed in 0.9% of SIMPONI ARIA-treated patients and 0.0% of control-treated patients. Through Week 24, the incidence of serious infections per 100 patient-years of follow-up was 2.2 (95% CI 0.61, 5.71) for the SIMPONI ARIA group, and 0 (0.00, 3.79) for the placebo group. In the controlled and uncontrolled portions of Trial RA, 958 total patient-years of follow-up with a median follow-up of approximately 92 weeks, the incidence per 100 patient-years of all serious infections was 4.07 (95% CI: 2.90, 5.57) in patients receiving SIMPONI ARIA [see Warnings and Precautions (5.1)] . In the controlled and uncontrolled portions of Trial RA, in SIMPONI ARIA-treated patients, the incidence of active TB per 100 patient-years was 0.31 (95% CI: 0.06; 0.92) and the incidence of other opportunistic infections per 100 patient-years was 0.42 (95% CI: 0.11, 1.07).
Malignancies
One case of malignancy other than lymphoma and NMSC with SIMPONI ARIA was reported through Week 24 during the controlled phase of Trial RA. In the controlled and uncontrolled portions through approximately 92 weeks, the incidence of malignancies per 100 patient-years, other than lymphoma and NMSC, in SIMPONI ARIA-treated patients was 0.31 (95% CI: 0.06, 0.92) and the incidence of NMSC was 0.1 (95% CI: 0.00, 0.58).
Liver Enzyme Elevations
There have been reports of severe hepatic reactions including acute liver failure in patients receiving TNF-blockers.
In the controlled phase of Trial RA, through Week 24, ALT elevations ≥ 5 × ULN occurred in 0.8% of SIMPONI ARIA-treated patients and 0% of control-treated patients and ALT elevations ≥ 3 × ULN occurred in 2.3% of SIMPONI ARIA-treated patients and 2.5% of control-treated patients.
In the controlled phase of Trial PsA, through Week 24, ALT elevations ≥ 5 × ULN occurred in 1.7% of SIMPONI ARIA-treated patients and <1% of placebo-treated patients, and ALT elevations ≥ 3 × ULN to < 5 × ULN occurred in 2.9% of SIMPONI ARIA-treated patients and <1% of placebo-treated patients.
Since many of the patients in the Phase 3 trials were also taking medications that cause liver enzyme elevations (e.g., nonsteroidal anti-inflammatory drugs [NSAIDs], MTX, or isoniazid prophylaxis), the relationship between SIMPONI ARIA and liver enzyme elevation is not clear.
Autoimmune Disorders and Autoantibodies
At Week 20 in Trial RA, 17% of SIMPONI ARIA-treated patients and 13% of control patients were newly antinuclear antibody (ANA)-positive. Of these patients, one SIMPONI ARIA-treated patient and no control-treated patients had newly positive anti-dsDNA antibodies [see Warnings and Precautions (5.5)] .
Administration Reactions
In the controlled phase of Trial RA through Week 24, 1.1% of SIMPONI ARIA infusions were associated with an infusion reaction compared with 0.2% of infusions in the control group. The most common infusion reaction in SIMPONI ARIA-treated patients was rash. No serious infusion reactions were reported.
Other Adverse Reactions
Table 1 summarizes the adverse drug reactions that occurred at a rate of at least 1% in the SIMPONI ARIA + MTX group with a higher incidence than in the placebo + MTX group during the controlled period of Trial RA through Week 24.
Table 1: Adverse Drug Reactions Reported by ≥ 1% of SIMPONI ARIA-Treated Patients and with a Higher Incidence than Placebo-Treated Patients in Trial RA through Week 24 Placebo + MTX SIMPONI ARIA + MTX Patients treated 197 463 Adverse Reaction Infections and infestations Upper respiratory tract infection (such as upper respiratory tract infection, nasopharyngitis, pharyngitis, laryngitis, and rhinitis) 12% 13% Viral infections (such as influenza and herpes) 3% 4% Bacterial infections 0% 1% Bronchitis 1% 3% Vascular disorders Hypertension 2% 3% Skin and subcutaneous disorders Rash 1% 3% General disorders and administration site conditions Pyrexia 1% 2% Blood and lymphatic disorders Leukopenia 0% 1% Other and Less Common Clinical Trial Adverse Drug Reactions
Adverse drug reactions that do not appear in Table 1 or that occurred < 1% in SIMPONI ARIA-treated patients during Trial RA through Week 24 that do not appear in the Warnings and Precautions section included the following events listed by system organ class:
Infections and infestations:Superficial fungal infection, sinusitis, abscess, lower respiratory tract infection (pneumonia), pyelonephritis
Investigations:Alanine aminotransferase (ALT) increased, aspartate aminotransferase (AST) increased, neutrophil count decreased
Nervous system disorders:Dizziness, paresthesia
Gastrointestinal disorders:Constipation
Psoriatic Arthritis
Trial PsA evaluated 480 patients [see Clinical Studies (14.2)] . The adverse reactions were similar to those observed in patients with RA, with the exception of psoriasis (new onset or worsening, palmar/plantar and pustular), which occurred in <1% of SIMPONI ARIA-treated patients. The incidence of the adverse reactions reported in Trial PsA were similar to Trial RA with the exceptions of higher incidence in SIMPONI ARIA for ALT increased (7.9% vs. 2.1% in placebo), AST increased (5.4% vs. 2.1% in placebo), and neutrophil count decreased (4.6% vs. 2.1% in placebo).
Ankylosing Spondylitis
Trial AS evaluated 208 patients [see Clinical Studies (14.3)] . The adverse reactions were similar to those reported in patients with RA, with the exception of the higher incidence of ALT increased, which occurred in 2.9% of SIMPONI ARIA-treated patients compared with none of the placebo-treated patients.
Pediatric Patients with Polyarticular Juvenile Idiopathic Arthritis and Psoriatic Arthritis
Trial pJIA evaluated 127 patients with JIA with active polyarthritis [see Use in Specific Populations (8.4)and Clinical Studies (14.4)]. The adverse reactions observed were consistent with the established safety profile of SIMPONI ARIA in adult patients with RA and PsA.
6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to golimumab in the trials described below with the incidence of antibodies in other trials or to other products may be misleading.
Using an enzyme immunoassay (EIA) method, antibodies to golimumab were detected in 13 (3%) golimumab-treated patients following IV administration of SIMPONI ARIA in combination with MTX through Week 24 of Trial RA, of which all were neutralizing antibodies.
A drug-tolerant enzyme immunoassay (drug-tolerant EIA) method for detecting antibodies to golimumab was developed and validated. This method is approximately 16-fold more sensitive than the original EIA method with less interference from golimumab in serum. Through approximately 6 months, the incidence of antibodies to golimumab with the drug-tolerant EIA method for Trials RA, PsA, AS, and pJIA was 21%, 19%, 19% and 31%, respectively. Where tested, approximately one-third to one-half were neutralizing.
Patients with RA, PsA, AS and pJIA who developed antibodies to golimumab generally had lower trough steady-state serum concentrations of golimumab [see Clinical Pharmacology (12.3)].
6.3 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of golimumab. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to golimumab exposure:
General Disorders and Administration Site Conditions:Infusion-related reactions [see Warnings and Precautions (5.11)]
Neoplasm benign and malignant: Melanoma, Merkel cell carcinoma [see Warnings and Precautions (5.2)]
Immune system disorders: Serious systemic hypersensitivity reactions (including anaphylactic reaction) [see Warnings and Precautions (5.11)] , sarcoidosis
Respiratory, thoracic and mediastinal disorders: Interstitial lung disease
Skin and subcutaneous tissue disorders: Skin exfoliation, lichenoid reactions, bullous skin reactions
-
7 DRUG INTERACTIONS
7.1 Methotrexate
SIMPONI ARIA should be used with MTX for the treatment of RA [see Clinical Studies (14.1)] . Following IV administration, concomitant administration of methotrexate decreases the clearance of SIMPONI ARIA by approximately 9% based on population pharmacokinetics (PK) analysis. In addition, concomitant administration of methotrexate decreases the SIMPONI ARIA clearance by reducing the development of antibodies to golimumab.
7.2 Biologic Products for RA, PsA, AS, and pJIA
An increased risk of serious infections has been seen in clinical RA studies of other TNF-blockers used in combination with anakinra or abatacept, with no added benefit; therefore, use of SIMPONI ARIA with other biologic products, including abatacept or anakinra, is not recommended [see Warnings and Precautions (5.6and 5.7)] . A higher rate of serious infections has also been observed in RA patients treated with rituximab who received subsequent treatment with a TNF-blocker. The concomitant use of SIMPONI ARIA with biologics approved to treat RA, PsA, AS, and pJIA is not recommended because of the possibility of an increased risk of infection.
7.3 Live Vaccines/Therapeutic Infectious Agents
Live vaccines should not be given concurrently with SIMPONI ARIA [see Warnings and Precautions (5.10)] .
Therapeutic infectious agents should not be given concurrently with SIMPONI ARIA [see Warnings and Precautions (5.10)] .
Infants born to women treated with SIMPONI ARIA during their pregnancy may be at increased risk of infection for up to 6 months. Administration of live vaccines to infants exposed to SIMPONI ARIA in uterois not recommended for 6 months following the mother's last SIMPONI ARIA infusion during pregnancy [see Warnings and Precautions (5.10), Use in Specific Populations (8.1)] .
7.4 Cytochrome P450 Substrates
The formation of CYP450 enzymes may be suppressed by increased levels of cytokines (e.g., TNFα) during chronic inflammation. Therefore, it is expected that for a molecule that antagonizes cytokine activity, such as golimumab, the formation of CYP450 enzymes could be normalized. Upon initiation or discontinuation of SIMPONI ARIA in patients being treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin) or drug concentration (e.g., cyclosporine or theophylline) is recommended and the individual dose of the drug product may be adjusted as needed.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from postmarketing case reports with golimumab use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. An observational study of northern European births observed similar unadjusted rates of major birth defects in infants exposed in utero to golimumab compared to no treatment or non-biologic systemic therapy. However, this study had important limitations ( see Data).
Monoclonal antibodies, such as golimumab, are transported across the placenta during the third trimester of pregnancy and may affect immune response in the in uteroexposed infant. There are clinical considerations for the use of SIMPONI ARIA in pregnant women (see Clinical Considerations) . In an animal reproductive study, golimumab administered by the subcutaneous route to pregnant monkeys, during the period of organogenesis, at doses that produced exposures approximately 200 times the maximum recommended human dose (MRHD) had no adverse fetal effects.
All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. The estimated background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risks of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and of miscarriage is 15–20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Golimumab crosses the placenta during pregnancy. Another TNF-blocking monoclonal antibody administered during pregnancy was detected for up to 6 months in the serum of infants. Consequently, these infants may be at increased risk of infection. Administration of live vaccines to infants exposed to SIMPONI ARIA in uterois not recommended for 6 months following the mother's last SIMPONI ARIA infusion during pregnancy [see Warnings and Precautions (5.10)and Drug Interactions (7.3)] .
Data
Human Data
An observational, exposure-based, cohort study based on data from the Swedish, Danish, and Finnish Medical Birth Registers conducted between 2006–2020 (Sweden and Denmark) and 2006–2019 (Finland) compared the risk of major birth defects in 134 live-born infants exposed to golimumab (116 from women treated for rheumatic conditions, 18 from women treated for ulcerative colitis) to no treatment or nonbiologic systemic therapy. The unadjusted rate of major birth defects in infants exposed in uterowas similar across all groups. However, this study had important limitations such as a small number of pregnant women exposed to golimumab, a wide exposure ascertainment window, and incomplete risk adjustment for potential confounders.
Animal Data
In an embryofetal developmental toxicology study in which pregnant cynomolgus monkeys were treated with golimumab during the period of organogenesis from gestation days (GD) 20 to 51, exposures up to 200 times greater than the exposure at the MRHD (on an area under the curve (AUC) basis with maternal subcutaneous doses up to 50 mg/kg twice weekly) produced no evidence of fetal malformations or embryotoxicity. There was no evidence of maternal toxicity. Umbilical cord blood samples collected at the end of the second trimester showed that fetuses were exposed to golimumab during gestation.
In a pre- and postnatal developmental study in which pregnant cynomolgus monkeys were treated with golimumab from gestation day 50 to postpartum day 33, maximal drug concentrations up to 33 times greater than that found with the MRHD (on a maximum blood concentration (C max) basis at steady-state with maternal subcutaneous doses up to 50 mg/kg twice weekly) were not associated with any evidence of developmental defects in infants. There was no evidence of maternal toxicity. Golimumab was present in fetal serum at the end of the second trimester and in neonatal serum from the time of birth and for up to 6 months postpartum.
8.2 Lactation
Risk Summary
There is no information regarding the presence of SIMPONI ARIA in human milk, the effects on breastfed infants, or the effects on milk production. Maternal IgG is known to be present in human milk. Golimumab is present in the milk of lactating cynomolgus monkeys (see Data) . If golimumab is transferred into human milk, the effects of local exposure in the gastrointestinal tract and potential limited systemic exposure in the infant to golimumab are unknown. The developmental and health benefits of breast-feeding should be considered along with the mother's clinical need for SIMPONI ARIA and any potential adverse effects on the breast-fed infants from SIMPONI ARIA, or from the underlying maternal condition.
8.4 Pediatric Use
Safety and effectiveness of SIMPONI ARIA for active polyarticular juvenile idiopathic arthritis and PsA have been established in pediatric patients 2 years and older.
Use of SIMPONI ARIA in these age groups is supported by evidence from adequate and well-controlled studies of SIMPONI ARIA in adults with RA and PsA, pharmacokinetic data from adult patients with RA and PsA and pediatric patients with JIA with active polyarthritis, and safety data from a clinical study in 127 pediatric patients 2 to < 18 years of age with JIA with active polyarthritis. The observed pre-dose (trough) concentrations are generally comparable between adults with RA and PsA and pediatric patients with JIA with active polyarthritis, and the PK exposure is expected to be comparable between adult PsA and pediatric patients with PsA [see Adverse Reactions (6.1), Clinical Pharmacology (12.3)and Clinical Studies (14.2, 14.4)] .
Malignancies, some fatal, have been reported among children, adolescents, and young adults who received treatment with golimumab and other TNF-blocking agents [see Warnings and Precautions (5.2)].
The safety and effectiveness in pediatric patients below the age of 2 years have not been established in pJIA or in PsA. The safety and effectiveness of SIMPONI ARIA in pediatric patients with conditions other than pJIA and PsA have not been established.
8.5 Geriatric Use
In Trial RA, the number of patients ages 65 or older was too small to make comparisons with younger SIMPONI ARIA-treated patients. Because there is a higher incidence of infections in the geriatric population in general, caution should be used in treating geriatric patients with SIMPONI ARIA.
- 10 OVERDOSAGE
-
11 DESCRIPTION
Golimumab is a human IgG1қ monoclonal antibody specific for human tumor necrosis factor alpha (TNFα) that exhibits multiple glycoforms with molecular masses of approximately 150 to 151 kilodaltons. Golimumab was created using genetically engineered mice immunized with human TNF, resulting in an antibody with human-derived antibody variable and constant regions. Golimumab is produced by a recombinant cell line cultured by continuous perfusion and is purified by a series of steps that includes measures to inactivate and remove viruses.
The SIMPONI ARIA ®(golimumab) Injection is a sterile solution of the golimumab antibody supplied in a 4-mL glass vial for intravenous infusion.
SIMPONI ARIA is a preservative-free, colorless to light yellow solution with a pH of approximately 5.5. SIMPONI ARIA is not made with natural rubber latex. Each 4-mL vial of SIMPONI ARIA contains 50 mg golimumab, L-histidine (1.14 mg), L-histidine monohydrochloride monohydrate (6.42 mg), polysorbate 80 (0.6 mg), sorbitol (180 mg), and water for injection.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Golimumab is a human monoclonal antibody that binds to both the soluble and transmembrane bioactive forms of human TNFα. This interaction prevents the binding of TNFα to its receptors, thereby inhibiting the biological activity of TNFα (a cytokine protein). There was no evidence of the golimumab antibody binding to other TNF superfamily ligands; in particular, the golimumab antibody did not bind or neutralize human lymphotoxin. Golimumab did not lyse human monocytes expressing transmembrane TNF in the presence of complement or effector cells.
Elevated TNFα levels in the blood, synovium, and joints have been implicated in the pathophysiology of several chronic inflammatory diseases such as rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis. TNFα is an important mediator of the articular inflammation that is characteristic of these diseases. Golimumab modulated the in vitrobiological effects mediated by TNF in several bioassays, including the expression of adhesion proteins responsible for leukocyte infiltration (E-selectin, ICAM-1 and VCAM-1) and the secretion of proinflammatory cytokines (IL-6, IL-8, G-CSF and GM-CSF). The clinical relevance of these findings is unknown.
12.2 Pharmacodynamics
Following treatment with SIMPONI ARIA in patients with RA, decreases from baseline were observed in tissue inhibitor of metalloproteinase-1 (TIMP-1), matrix metalloproteinase-1 (MMP-1), matrix metalloproteinase-3 (MMP-3), resistin, interleukin-6 (IL-6), macrophage inflammatory protein-1 (MIP-1b), vascular endothelial growth factor (VEGF), serum amyloid A (SAA), S100A12, and high sensitivity C-Reactive protein (hsCRP). Conversely, increases from baseline were observed in tartrate-resistant acid phosphatase (TRAP-5b). The clinical relevance of this information is not known.
12.3 Pharmacokinetics
Golimumab exhibited approximately dose-proportional pharmacokinetics in patients with RA over the dose range of 0.1 to 10.0 mg/kg following a single intravenous dose.
Absorption
Following a single intravenous administration of 2 mg/kg SIMPONI ARIA, a mean C maxof 44.4 ± 11.3 mcg/mL was observed in patients with RA. Data directly comparing 2 mg/kg intravenous administration and 50 mg subcutaneous administration are not available.
Distribution
Following a single intravenous administration of 2 mg/kg SIMPONI ARIA, the mean volume of distribution was estimated to be 115 ± 19 mL/kg in healthy subjects, and 151 ± 61 mL/kg in patients with RA. The volume of distribution of golimumab may indicate that golimumab is distributed primarily in the circulatory system with limited extravascular distribution.
Elimination
Following a single intravenous administration of 2 mg/kg SIMPONI ARIA, the systemic clearance of golimumab was estimated to be 6.9 ± 2.0 mL/day/kg in healthy subjects and 7.6 ± 2.0 mL/day/kg in patients with RA. The mean terminal half-life was estimated to be 12 ± 3 days in healthy subjects and the mean terminal half-life in RA patients was 14 ± 4 days.
Multiple Doses
When 2 mg/kg SIMPONI ARIA was administered intravenously to patients with RA at weeks 0, 4 and every 8 weeks thereafter, serum concentrations reached steady-state by Week 12. With concomitant use of MTX, treatment with 2 mg/kg SIMPONI ARIA every 8 weeks resulted in a mean steady-state trough serum concentration of approximately 0.4 ± 0.4 mcg/mL in patients with active RA. The mean steady-state trough serum concentration in patients with PsA was 0.7 ± 0.6 mcg/mL. The mean steady-state trough serum concentration in patients with AS was 0.8 ± 0.6 mcg/mL.
Patients with RA, PsA and AS who developed antibodies to golimumab generally had lower trough steady-state serum concentrations of golimumab [see Adverse Reactions (6.2)].
Specific Populations
No formal study of the effect of renal or hepatic impairment on the PK of golimumab was conducted.
Body Weight
Following intravenous administration, patients with higher body weight tended to have slightly higher serum golimumab concentrations than patients with lower body weights when golimumab was administered on a mg/kg (body weight) basis. However, based on population PK analysis, there were no clinically relevant differences in golimumab exposure following intravenous administration of 2 mg/kg SIMPONI ARIA in adult patients across a range of different body weights.
Pediatrics
When 80 mg/m 2SIMPONI ARIA was administered intravenously to patients with JIA with active polyarthritis at weeks 0, 4 and every 8 weeks thereafter, serum concentrations reached steady-state by Week 12. With concomitant use of MTX, treatment with 80 mg/m 2SIMPONI ARIA resulted in a mean steady-state trough serum golimumab concentration of approximately 0.5 ± 0.4 mcg/mL and a mean steady-state AUC of 425 ± 125 mcg∙day/mL in patients with JIA with active polyarthritis. Overall, the observed steady-state golimumab trough concentrations in patients with JIA with active polyarthritis were within the range of those observed for adult RA and PsA after administration of SIMPONI ARIA.
Consistent with the intravenous data in adult patients with RA, population pharmacokinetic analyses for intravenous SIMPONI ARIA in pJIA revealed that there were no clinically relevant differences in golimumab exposure following intravenous administration of 80 mg/m 2SIMPONI ARIA in pediatric patients across a range of age and different body weights. The immune response effect on golimumab clearance in patients with JIA with active polyarthritis was comparable to adults with RA.
Drug Interaction Studies
Specific drug interaction studies have not been conducted with SIMPONI ARIA.
Population PK analysis indicated that concomitant use of MTX, NSAIDs, oral corticosteroids, or sulfasalazine (SSZ) did not significantly influence the clearance of golimumab following IV administration.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term animal studies of golimumab have not been conducted to evaluate its carcinogenic potential. Mutagenicity studies have not been conducted with golimumab. A fertility study conducted in mice using an analogous anti-mouse TNFα antibody administered by the intravenous route at doses up to 40 mg/kg once per week showed no impairment of fertility.
-
14 CLINICAL STUDIES
14.1 Rheumatoid Arthritis
The efficacy and safety of SIMPONI ARIA were evaluated in one multicenter, randomized, double-blind, controlled trial (Trial RA, NCT00973479) in 592 patients ≥ 18 years of age with moderately to severely active RA despite concurrent MTX therapy and had not previously been treated with a biologic TNF-blocker. Patients were diagnosed according to the American College of Rheumatology (ACR) criteria, at least 3 months prior to administration of study agent and were required to have at least 6 swollen and 6 tender joints. Patients were randomized to receive either SIMPONI ARIA 2 mg/kg (N=395) or placebo (N=197) over a 30-minute intravenous infusion at Weeks 0, 4 and every 8 weeks thereafter in addition to their weekly maintenance MTX dose (15–25 mg). All patients receiving placebo + MTX received SIMPONI ARIA + MTX after Week 24, but the trial remained blinded until all patients had completed 108 weeks of treatment. Efficacy data were collected and analyzed through Week 52. Patients were allowed to continue stable doses of concomitant low dose corticosteroids (equivalent to ≤ 10 mg of prednisone a day) and/or NSAIDs. The use of other DMARDs including cytotoxic agents or other biologics was prohibited.
The primary endpoint in Trial RA was the percentage of patients achieving an ACR 20 response at Week 14. In Trial RA, the majority of subjects were women (82%) and were Caucasian (80%) with a median age of 52 years and a median weight of 70 kg. Median disease duration was 4.7 years, and 50% of the patients used at least one DMARD other than MTX in the past. At baseline, 81% of patients received concomitant NSAIDs and 81% of patients received low-dose corticosteroids (equivalent to ≤ 10 mg of prednisone a day). The median baseline DAS28-CRP was 5.9 and the median van der Heijde-Sharp score at baseline was 28.5.
Clinical Response
A greater percentage of patients treated with the combination of SIMPONI ARIA + MTX achieved ACR 20 at Week 14 and ACR 50 at Week 24 versus patients treated with the placebo + MTX as shown in Table 2. The percent of patients achieving ACR 20 responses by visit for Trial RA is shown in Figure 1.
Table 2: Trial RA – Proportion of Patients with an ACR Response Trial RA
Active RA, despite MTXPlacebo + MTX SIMPONI ARIA + MTX 95% CI * N † 197 395 ACR 20 Week 14 25% 59% 25.9, 41.4 Week 24 32% 63% 23.3, 39.4 ACR 50 Week 14 9% 30% 15.3, 27.2 Week 24 13% 35% 15.1, 28.4 ACR 70 Week 14 3% 12% 5.3, 13.4 Week 24 4% 18% 8.8, 18.1 Figure 1: Trial RA – Percent of Patients Achieving ACR 20 Response Over Time: Randomized Patients 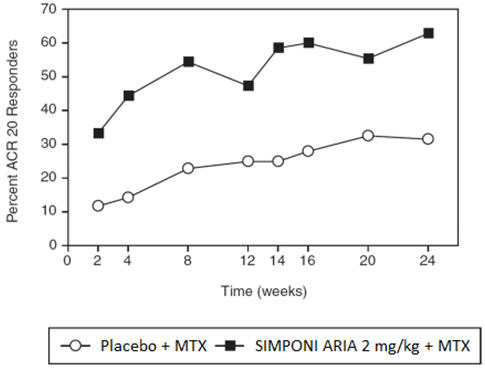
The analysis is based on the intent-to-treat population. Last observation carried forward was performed for missing data. Patients who discontinued treatment due to lack of efficacy were counted as non-responders, as were patients who started prohibited medication or failed to achieve at least a 10% improvement in joint counts at Week 16. The improvement in all components of the ACR response criteria for the SIMPONI ARIA + MTX group was greater compared to the placebo + MTX group in Trial RA as shown in Table 3.
Table 3: Trial RA ─ Components of ACR Response at Week 14 Trial RA
Active RA, despite MTXPlacebo + MTX SIMPONI ARIA + MTX Note: All values are means. - *
- N reflects randomized patients; actual number of patients evaluable for each endpoint may vary.
N * 197 395 Number of swollen joints (0–66) Baseline 15 15 Week 14 11 6 Number of tender joints (0–68) Baseline 26 26 Week 14 20 13 Patient's assessment of pain (0–10) Baseline 6.5 6.5 Week 14 5.6 3.9 Patient's global assessment of disease activity (0–10) Baseline 6.5 6.5 Week 14 5.5 4.0 Physician's global assessment of disease activity (0–10) Baseline 6.3 6.2 Week 14 4.9 3.1 HAQ score (0–3) Baseline 1.6 1.6 Week 14 1.4 1.1 CRP (mg/dL) (0–1) Baseline 2.2 2.8 Week 14 1.8 0.9 At Week 14, a greater proportion of patients treated with SIMPONI ARIA + MTX achieved a low level of disease activity as measured by a DAS28-CRP less than 2.6 compared with the placebo + MTX group (15% compared to 5%; 95% CI for difference [6.3%, 15.5%]).
Radiographic Response
In Trial RA, structural joint damage was assessed radiographically and expressed as a change in van der Heijde-Modified Sharp Score (vdH-S) and its components, the erosion score and Joint Space Narrowing (JSN) score, at Week 24 compared to baseline. The SIMPONI ARIA + MTX treatment group inhibited the progression of structural damage compared with placebo + MTX, as assessed by total vdH-S score as shown in Table 4.
Table 4: Trial RA – Radiographic Change From Baseline at Week 24 Placebo + MTX
(N=197) *SIMPONI ARIA + MTX
(N=395) *,†Mean Mean Change Total vdH-S Score 1.1 0.03 ‡ Change Erosion Score 0.5 -0.1 Change JSN Score 0.6 0.1 At Week 24, a greater proportion of patients in the SIMPONI ARIA + MTX group (71%) had no progression of structural damage (change in the total vdH-S score ≤ 0), compared to 57% of patients in the placebo + MTX group. At Week 52, the mean change from baseline in total vdH-S score was 1.2 in patients originally randomized to placebo + MTX who crossed over to SIMPONI ARIA + MTX at Week 16 or Week 24, and 0.1 in patients originally randomized to SIMPONI ARIA + MTX who remained on active treatment.
Physical Function Response in Patients with RA
Physical function was assessed by the disability index of the Health Assessment Questionnaire (HAQ-DI). At Week 14, the SIMPONI ARIA + MTX group showed greater mean improvement in the HAQ-DI compared with placebo + MTX (0.5 compared to 0.2; 95% CI for difference [0.2, 0.4]).
Other Health-Related Outcomes
General health status was assessed by the 36-item Short Form Health Survey (SF-36). In Trial RA, patients receiving SIMPONI ARIA + MTX demonstrated greater improvement from baseline compared with placebo + MTX in physical component summary (PCS), mental component summary (MCS) scores and in all 8 domains of the SF-36.
Fatigue was assessed by the Functional Assessment of Chronic Illness Therapy-Fatigue score (FACIT-F) in Trial RA. Treatment with SIMPONI ARIA resulted in improvement in fatigue as measured by FACIT-F.
14.2 Psoriatic Arthritis
The efficacy and safety of SIMPONI ARIA were evaluated in a multicenter, randomized, double-blind, placebo-controlled trial in 480 patients ≥ 18 years of age with active psoriatic arthritis despite NSAID or DMARD therapy (Trial PsA, NCT02181673). Previous treatment with a biologic was not allowed. Patients in this trial had a diagnosis of PsA for at least six months and had symptoms of active disease [≥5 swollen joints and ≥5 tender joints and a CRP level of ≥ 0.6 mg/dL]. Patients were randomized to either receive SIMPONI ARIA 2 mg/kg (N=241) or placebo (N=239) as a 30-minute intravenous infusion at Weeks 0, 4, 12 and 20. All patients on placebo received SIMPONI ARIA at Week 24, Week 28 and every 8 weeks thereafter through Week 52. Patients in the SIMPONI ARIA treatment group continued to receive SIMPONI ARIA infusions at Week 28 and every 8 weeks through Week 52.
Patients were allowed to continue stable doses of MTX, NSAIDs, and low dose oral corticosteroids (equivalent to ≤ 10 mg of prednisone per day) during the trial. The use of other DMARDs including cytotoxic agents or other biologics was prohibited.
The primary endpoint was the percentage of patients achieving an ACR 20 response at Week 14.
Patients with each subtype of PsA were enrolled, including polyarticular arthritis with absence of rheumatoid nodules (44%), asymmetric peripheral arthritis (19%), distal interphalangeal joint involvement (8.1%), spondylitis with peripheral arthritis (25%), and arthritis mutilans (4.8%). The median duration of PsA disease was 3.5 years, 86% of patients had previously used MTX, and 35% of patients received at least one other DMARD in the past. At baseline, 76% and 54% of the patients had enthesitis and dactylitis, respectively. The median total modified vdH-S score at baseline was 15.5. During the trial, concomitant medications used included MTX (70%), oral corticosteroids (28%), and NSAIDs (71%).
Clinical Response
In Trial PsA, SIMPONI ARIA treatment, compared with placebo, resulted in a significant improvement in signs and symptoms as demonstrated by the percentage of patients with an ACR 20 response at Week 14 (see Table 5). Similar ACR 20 responses at Week 24 were observed in patients with different PsA subtypes. ACR 20 responses observed in the SIMPONI ARIA-treated groups were similar in patients who were or were not receiving concomitant MTX.
Table 5: Trial PsA –Percentage of Patients with ACR Responses at Weeks 14 and 24 Placebo SIMPONI ARIA Difference from placebo
(95% CI)(N *=239) (N *=241) Note: The analysis is based on the intent-to-treat population. Last observation carried forward was performed for partial missing data and non-responder imputation for completely missing data. Patients who discontinued treatment due to lack of efficacy were imputed as non-responders, as were patients who started prohibited medication, increased corticosteroids or MTX, or failed to achieve at least a 5% improvement in joint counts at Week 16 and received a concomitant medication intervention (corticosteroids, MTX or NSAIDs). ACR 20 response Week 14 22% 75% 53% †
(46, 61)Week 24 24% 77% 53%
(45, 60)ACR 50 response Week 14 6.3% 44% 37%
(30, 44)Week 24 6.3% 54% 47%
(40, 54)ACR 70 response Week 14 2.1% 25% 22%
(17, 28)Week 24 3.3% 33% 29%
(23, 36)The percentage of patients achieving ACR20 responses by visit through Week 24 for Trial PsA is shown in Figure 2.
Figure 2: Trial PsA - Percentage of Patients Achieving ACR20 Response Through Week 24 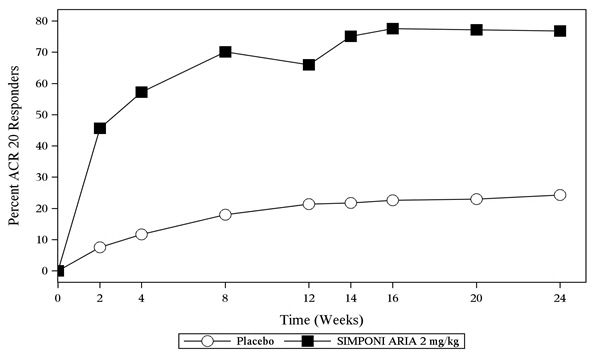
The analysis is based on the intent-to-treat population. Last observation carried forward was performed for partial missing data and non-responder imputation for completely missing data. Patients who discontinued treatment due to lack of efficacy were imputed as non-responders, as were patients who started prohibited medication, increased corticosteroids or MTX, or failed to achieve at least a 5% improvement in joint counts at Week 16 and received a concomitant medication intervention (corticosteroids, MTX or NSAIDs). Table 6 shows the improvement in the individual components of the ACR response criteria for the SIMPONI ARIA and placebo groups in Trial PsA.
Table 6: Trial PsA – Mean Changes in ACR Components at Week 14 Placebo
N *=239SIMPONI ARIA
N *=241Baseline Week 14 change from baseline Baseline Week 14 change from baseline Note: All values are means. ACR Components No. of Swollen Joints (0–66) 14 -2.9 14 -11 Number of Tender Joints (0–68) 26 -4.2 25 -15 Patient's assessment of Pain (0–100 mm) 64 -11 63 -31 Patient Global Assessment (0–100 mm) 63 -11 65 -32 Physician Global Assessment (0–100 mm) 64 -13 62 -39 Disability Index (HAQ) (0–3) † 1.3 -0.13 1.3 -0.60 hsCRP (mg/L) 20 -2.9 19 -16 Patients with enthesitis at baseline were evaluated for mean improvement using the Leeds Enthesitis Index (LEI) on a scale of 0–6. SIMPONI ARIA-treated patients showed a significantly greater improvement in enthesitis, with a mean reduction of 1.8 as compared with a mean reduction in placebo-treated patients of 0.8 at Week 14. Patients with dactylitis at baseline were evaluated for mean improvement on a scale of 0–60. SIMPONI ARIA-treated patients showed a significantly greater improvement, with a mean reduction of 7.8 compared with a mean reduction of 2.8 in placebo-treated patients at Week 14.
Radiographic Response
In Trial PsA, structural joint damage was assessed radiographically and expressed as a change from baseline at Week 24 in total modified vdH-S score and its components, the erosion score and JSN score. SIMPONI ARIA inhibited the progression of structural damage compared with placebo, as assessed by total modified vdH-S score as shown in Table 7.
Table 7: Trial PsA – Radiographic Change From Baseline at Week 24 Placebo
N *=237SIMPONI ARIA
N *=237Difference from placebo
(95% CI)Mean Mean Note: All values are means. - *
- N reflects randomized patients evaluable for radiographic assessment.
Change Total Modified vdH-S Score 2.0 -0.4 -2.3
(-2.9, -1.7)At Week 24, a greater proportion of patients in the SIMPONI ARIA group (72%) had no progression of structural damage (change in the total modified vdH-S score ≤ 0), compared to 43% of patients in the placebo group.
Physical Function and Responses
Improvement in physical function as assessed by the Health Assessment Questionnaire Disability Index (HAQ-DI) demonstrated that the proportion of patients who achieved clinically meaningful improvement of ≥ 0.3 in HAQ-DI score from baseline was greater in the SIMPONI ARIA-treated group compared to placebo at Week 14 (69% compared to 32%).
Other Health Related Outcomes
General health status was assessed by the 36-item Short Form Health Survey (SF-36). In Trial PsA, patients receiving SIMPONI ARIA demonstrated greater improvement from baseline compared with placebo in physical component summary, mental component summary scores and in all 8 domains of the SF-36.
Fatigue was assessed by the Functional Assessment of Chronic Illness Therapy-Fatigue score (FACIT-F) in Trial PsA. Treatment with SIMPONI ARIA resulted in improvement in fatigue as measured by FACIT-F.
Treatment of Pediatric Patients
The efficacy of SIMPONI ARIA in pediatric patients with PsA is based on the pharmacokinetic exposure and extrapolation of the established efficacy of SIMPONI ARIA in adult PsA patients in Trial PsA [see Use in Specific Populations (8.4), Clinical Pharmacology (12.3), Clinical Studies (14.4)].
14.3 Ankylosing Spondylitis
The efficacy and safety of SIMPONI ARIA were evaluated in a multicenter, randomized, double-blind, placebo-controlled trial (Trial AS, NCT02186873) in 208 patients ≥ 18 years of age with active ankylosing spondylitis (AS) and inadequate response or intolerance to NSAIDs. Patients had a diagnosis of definite AS for at least 3 months according to modified New York criteria. Patients had symptoms of active disease [Bath AS Disease Activity Index (BASDAI) ≥ 4, VAS for total back pain of ≥ 4, on scales of 0 to 10 cm (0 to 100 mm), and a hsCRP level of ≥ 0.3 mg/dL (3 mg/L)]. Patients were randomized to receive either SIMPONI ARIA 2 mg/kg (N=105) or placebo (N=103) as a 30-minute intravenous infusion at Weeks 0, 4 and 12. All patients on placebo received SIMPONI ARIA at Week 16, Week 20 and every 8 weeks thereafter through Week 52. Patients in the SIMPONI ARIA treatment group continued to receive SIMPONI ARIA infusions at Week 20 and every 8 weeks through Week 52. Patients were allowed to continue stable doses of concomitant MTX, SSZ, hydroxychloroquine (HCQ), low dose oral corticosteroids (equivalent to ≤ 10 mg of prednisone per day), and/or NSAIDs during the trial. The use of other DMARDs including cytotoxic agents or other biologics was prohibited.
The primary endpoint was the percentage of patients achieving an Assessment in Ankylosing Spondylitis (ASAS) 20 response at Week 16.
In Trial AS, the median duration of AS disease was 2.8 years, median duration of inflammatory back pain was 8 years, 90% were HLA-B27 positive, 8.2% had prior joint surgery or procedure, 5.8% had complete ankylosis of the spine, 14% had received prior therapy with one biologic TNF blocker (other than golimumab) and discontinued for reasons other than lack of efficacy within the first 16 weeks of treatment (primary failure), and 76% received at least one DMARD in the past. During the trial, the use of concomitant medications was NSAIDs (88%), SSZ (38%), corticosteroids (26%), MTX (18%), and HCQ (0.5%).
Clinical Response
In Trial AS, SIMPONI ARIA treatment, compared with placebo, resulted in a significant improvement in signs and symptoms as demonstrated by the percentage of patients with an ASAS 20 response at Week 16 (see Table 8).
Table 8: Trial AS – Percentage of ASAS Responders at Weeks 16 Placebo
N *=103SIMPONI ARIA
N *=105Treatment Difference
(95% CI)Note: The analysis is based on the intent-to-treat population. Last observation carried forward was performed for partial missing data and non-responder imputation for completely missing data. Responders ASAS 20 26% 73% 47% †
(35, 59)ASAS 40 8.7% 48% 39%
(28, 50)The percentage of patients achieving ASAS 20 responses by visit through Week 16 for Trial AS is shown in Figure 3.
Figure 3: Trial AS – Percentage of Patients Achieving an ASAS 20 Response Through Week 16 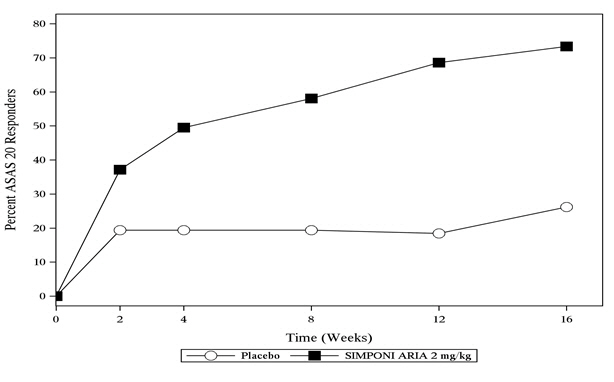
The analysis is based on the intent-to-treat population. Last observation carried forward was performed for partial missing data and non-responder imputation for completely missing data. Table 9 shows the improvement in the components of the ASAS response criteria and other measures of disease activity for the SIMPONI ARIA and placebo groups in Trial AS.
Table 9: Trial AS – Mean Changes in ASAS 20 Components and Other Measures of Disease Activity at Week 16 * Placebo
N *=103SIMPONI ARIA
N *=105Baseline Week 16 change from baseline Baseline Week 16 change from baseline Note: All values are means. - *
- N reflects randomized patients; actual number of patients evaluable for each endpoint may vary.
- †
- Measured on a Visual Analog Scale (VAS) with 0= very well, 100=very poor
- ‡
- Measured on a Visual Analog Scale (VAS) with 0= no pain, 100=most severe pain
- §
- BASFI is Bath Ankylosing Spondylitis Functional Index.
- ¶
- Inflammation is the mean of 2 morning stiffness self-assessments in the BASDAI.
- #
- Bath Ankylosing Spondylitis Metrology Index.
ASAS 20 Response criteria Patient Global Assessment of Disease Activity (0–100 mm) † 71 -8.3 73 -34 Total back pain (0–100 mm) ‡ 73 -12 72 -32 BASFI (0–10) § 6.1 -0.5 6.3 -2.4 Inflammation (0–10) ¶ 7.4 -1.1 7.3 -3.6 BASDAI Score 7.1 -1.1 7.1 -3.1 BASMI # 5.0 -0.1 5.0 -0.4 hsCRP (mg/L) 19 -2.3 20 -17 At Week 16, a greater percentage of patients treated with SIMPONI ARIA achieved a low level of disease activity (<2 [on a scale of 0 to 10 cm] in all four ASAS domains) compared with patients treated with placebo (16.2% vs. 3.9%).
Other Health-Related Outcomes
General health status was assessed by the 36-item Short Form Health Survey (SF-36). In Trial AS, patients receiving SIMPONI ARIA demonstrated greater improvement from baseline compared with placebo in physical component summary and mental component summary scores and in all 8 domains of the SF-36.
SIMPONI ARIA-treated patients showed significant improvement compared with placebo-treated patients in health related quality of life as assessed by the Ankylosing Spondylitis Quality of Life questionnaire (ASQoL).
14.4 Polyarticular Juvenile Idiopathic Arthritis (pJIA)
The efficacy of SIMPONI ARIA in pediatric patients with pJIA is based on the pharmacokinetic exposure and extrapolation of the established efficacy of SIMPONI ARIA in RA patients. Efficacy of SIMPONI ARIA was also assessed in a multicenter, open-label, single-arm study in 127 children (2 to < 18 years of age) with JIA with active polyarthritis despite treatment with MTX for at least 2 months (Trial pJIA, NCT02277444). The polyarticular JIA patient subtypes at study entry included: rheumatoid factor negative (43%), rheumatoid factor positive (35%), enthesitis-related arthritis (9%), oligoarticular extended (6%), juvenile psoriatic arthritis (4%), and systemic JIA without systemic manifestations (3%). All patients received SIMPONI ARIA 80 mg/m 2as an intravenous infusion at Week 0, 4, and every 8 weeks through Week 52. Patients continued stable doses of MTX weekly through Week 28; after Week 28, changes in MTX dose were permitted. Efficacy was assessed as supportive endpoints through Week 52. The efficacy was generally consistent with responses in patients with RA.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
SIMPONI ARIA ®(golimumab) Injection is a colorless to light yellow solution available in packs of 1 vial NDC 57894-350-01.
Storage and Handling
Refrigerate SIMPONI ARIA at 36 ⁰F to 46 ⁰F (2 ⁰C to 8 ⁰C) and protect from light. Keep the product in the original carton to protect from light until the time of use. Do not freeze. Do not shake.
If needed, SIMPONI ARIA may be stored at room temperature up to 77 ºF (25 ºC) for a maximum single period of 30 days in the original carton to protect from light. Once SIMPONI ARIA has been stored at room temperature, do not return the product to the refrigerator. If not used within 30 days at room temperature, discard SIMPONI ARIA.
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Medication Guide).
Advise patients of the potential benefits and risks of SIMPONI ARIA. Instruct patients to read the Medication Guide before starting SIMPONI ARIA therapy and to read it each time the prescription is renewed.
Infections
Inform patients that SIMPONI ARIA may lower the ability of their immune system to fight infections. Instruct the patient of the importance of contacting their doctor if they develop any symptoms of infection, including tuberculosis, invasive fungal infections, and hepatitis B reactivation.
Malignancies
Patients should be counseled about the risk of lymphoma and other malignancies while receiving SIMPONI ARIA. Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
Other Medical Conditions
Advise patients to report any signs of new or worsening medical conditions such as congestive heart failure, demyelinating disorders, autoimmune diseases, liver disease, cytopenias, or psoriasis.
Vaccinations
Inform patients that because SIMPONI ARIA may lower the ability of their immune system to fight infections, they should avoid live vaccines. Inform pregnant patients receiving SIMPONI ARIA that their infants should not receive live vaccines for 6 months following the last infusion of SIMPONI ARIA during pregnancy. Advise patients and infants of women who received SIMPONI ARIA during pregnancy to consult a physician before receiving any immunizations.
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
MEDICATION GUIDE
SIMPONI ARIA ®( SIM-po-nee AHR-ee-uh )
(golimumab)
injection, for intravenous useWhat is the most important information I should know about SIMPONI ARIA? SIMPONI ARIA is a medicine that affects your immune system. SIMPONI ARIA can lower the ability of your immune system to fight infections. Some people have serious infections while receiving SIMPONI ARIA, including tuberculosis (TB), and infections caused by bacteria, fungi, or viruses that spread throughout their body. Some people have died from these serious infections. - Your doctor should test you for TB and hepatitis B before starting SIMPONI ARIA.
- Your doctor should monitor you closely for signs and symptoms of TB during treatment with SIMPONI ARIA.
You should not start receiving SIMPONI ARIA if you have any kind of infection unless your doctor tells you to. Before receiving SIMPONI ARIA, tell your doctor if you: - think you have an infection or have symptoms of an infection such as:
- fever, sweat, or chills
- muscle aches
- cough
- shortness of breath
- blood in phlegm
- weight loss
- warm, red, or painful skin or sores on your body
- diarrhea or stomach pain
- burning when you urinate or urinate more often than normal
- feel very tired
- are being treated for an infection.
- get a lot of infections or have infections that keep coming back.
- have diabetes, HIV, or a weak immune system. People with these conditions have a higher chance for infections.
- have TB, or have been in close contact with someone with TB.
- live, have lived, or traveled to certain parts of the country (such as the Ohio and Mississippi River valleys and the Southwest) where there is an increased chance for getting certain kinds of fungal infections (histoplasmosis, coccidioidomycosis, blastomycosis). These infections may happen or become more severe if you use SIMPONI ARIA. Ask your doctor if you do not know if you have lived in an area where these infections are common.
- have or have had hepatitis B.
- use the medicine ORENCIA (abatacept), KINERET (anakinra), ACTEMRA (tocilizumab) or RITUXAN (rituximab).
After receiving SIMPONI ARIA, call your doctor right away if you have any symptoms of an infection. SIMPONI ARIA can make you more likely to get infections or make worse any infection that you have. Cancer - For children and adults receiving Tumor Necrosis Factor (TNF)-blocker medicines, including SIMPONI ARIA, the chances of getting cancer may increase.
- There have been cases of unusual cancers in children and teenage patients receiving TNF-blocking agents.
- People with inflammatory diseases, including rheumatoid arthritis (RA), especially those with very active disease, may be more likely to get lymphoma.
- Some people receiving TNF-blockers, like SIMPONI ARIA, developed a rare type of cancer called hepatosplenic T-cell lymphoma. This type of cancer often results in death. Most of these people were male teenagers or young men. Also, most people were being treated for Crohn's disease or ulcerative colitis with a TNF blocker and another medicine called azathioprine or 6-mercaptopurine, (6-MP).
- Some people treated with SIMPONI ARIA developed skin cancer. If any changes in the appearance of your skin or growths on your skin occur during or after your treatment with SIMPONI ARIA, tell your doctor.
- You should see your doctor periodically for skin examinations, especially if you have a history of skin cancer.
What is SIMPONI ARIA? SIMPONI ARIA is a prescription medicine called a TNF-blocker. SIMPONI ARIA is used to treat: - Adults with the medicine methotrexate (MTX) to treat moderately to severely active RA.
- Active psoriatic arthritis (PsA) in people 2 years of age and older.
- Adults with active ankylosing spondylitis (AS).
- Active polyarticular juvenile idiopathic arthritis (pJIA) in people 2 years of age and older.
It is not known if SIMPONI ARIA is safe and effective in children with PsA and pJIA under 2 years of age or in children with conditions other than PsA and pJIA. What should I tell my doctor before starting treatment with SIMPONI ARIA? See " What is the most important information I should know about SIMPONI ARIA?". Before starting SIMPONI ARIA, tell your doctor about all your medical conditions, including if you: - have an infection.
- have or have had lymphoma or any other type of cancer.
- have or have had heart failure.
- have or have had a condition that affects your nervous system, such as multiple sclerosis or Guillain-Barré syndrome.
- have a skin problem called psoriasis.
- have recently received or are scheduled to receive a vaccine. People receiving SIMPONI ARIA should not receive live vaccines or treatment with a weakened bacteria (such as BCG for bladder cancer). People receiving SIMPONI ARIA can receive non-live vaccines.
- have a baby and you were receiving SIMPONI ARIA during your pregnancy. Tell your baby's doctor before your baby receives any vaccine. Your baby may have an increased chance of getting an infection for up to 6 months after birth.
- are pregnant or plan to become pregnant. It is not known if SIMPONI ARIA will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if SIMPONI ARIA passes into your breast milk. You and your doctor should decide if you will receive SIMPONI ARIA or breastfeed.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially, tell your doctor if you: - use ORENCIA (abatacept) or KINERET (anakinra). You should not receive SIMPONI ARIA while you are also receiving ORENCIA (abatacept) or KINERET (anakinra).
- use other TNF-blocker medicines, including REMICADE (infliximab), HUMIRA (adalimumab), ENBREL (etanercept), or CIMZIA (certolizumab pegol).
- receive RITUXAN (rituximab) or ACTEMRA (tocilizumab).
Ask your doctor or pharmacist for a list of these medicines if you are not sure. Keep a list of all your medicines with you to show your doctor and pharmacist each time you get a new medicine. How should I receive SIMPONI ARIA? - SIMPONI ARIA is prepared and given by a healthcare provider through a needle placed in your vein (infusion). The infusion is usually given in your arm and should take 30 minutes.
- Your doctor will decide how much SIMPONI ARIA you will receive. Your usual schedule for receiving SIMPONI ARIA after your first treatment should be:
- 4 weeks after your first treatment
- every 8 weeks after that
- If you miss an appointment to receive SIMPONI ARIA, make another appointment as soon as possible.
What are the possible side effects of SIMPONI ARIA? SIMPONI ARIA can cause serious side effects, including: See " What is the most important information I should know about SIMPONI ARIA?"
Serious Infections.- Some patients have an increased chance of getting serious infections while receiving SIMPONI ARIA. These serious infections include TB and infections caused by viruses, fungi, or bacteria that have spread throughout the body. Some patients die from these infections. If you get an infection while receiving treatment with SIMPONI ARIA your doctor will treat your infection and may need to stop your SIMPONI ARIA treatment. Tell your doctor right away if you have any of the following signs of an infection while receiving or after receiving SIMPONI ARIA:
- a fever
- feel very tired
- have a cough
- have flu-like symptoms
- warm, red, or painful skin
- Your doctor will examine you for TB and perform a test to see if you have TB. If your doctor feels that you are at risk for TB, you may be treated with medicine for TB before you begin treatment with SIMPONI ARIA and during treatment with SIMPONI ARIA. Even if your TB test is negative your doctor should carefully monitor you for TB infections while you are receiving SIMPONI ARIA. People who had a negative TB skin test before receiving SIMPONI ARIA have developed active TB. Tell your doctor if you have any of the following symptoms while receiving or after receiving SIMPONI ARIA:
- cough that does not go away
- low grade fever
- weight loss
- loss of body fat and muscle (wasting)
Hepatitis B infection in people who carry the virus in their blood.If you are a carrier of the hepatitis B virus (a virus that affects the liver), the virus can become active while you use SIMPONI ARIA. Your doctor should do blood tests before you start treatment with SIMPONI ARIA and while you are receiving SIMPONI ARIA. - Tell your doctor if you have any of the following symptoms of a possible hepatitis B infection:
- feel very tired
- dark urine
- skin or eyes look yellow
- little or no appetite
- vomiting
- muscle aches
- clay-colored bowel movements
- fevers
- chills
- stomach discomfort
- skin rash
Heart failure, including new heart failure or worsening of heart failure that you already have can happen in people who use TNF-blocker medicines, including SIMPONI ARIA.If you develop new or worsening heart failure with SIMPONI ARIA, you may need to be treated in a hospital, and it may result in death. - If you have heart failure before starting SIMPONI ARIA, your condition should be watched closely during treatment with SIMPONI ARIA.
- Call your doctor right away if you get new or worsening symptoms of heart failure during treatment with SIMPONI ARIA (such as shortness of breath or swelling of your lower legs or feet, or sudden weight gain).
Nervous System Problems.Rarely, people receiving TNF-blocker medicines, including SIMPONI ARIA, have nervous system problems such as multiple sclerosis or Guillain-Barré syndrome. Tell your doctor right away if you get any of these symptoms: - vision changes
- weakness in your arms or legs
- numbness or tingling in any part of your body
Immune System Problems.Rarely, people receiving TNF-blocker medicines have developed symptoms that are like the symptoms of lupus. Tell your doctor if you have any of these symptoms: - a rash on your cheeks or other parts of the body
- sensitivity to the sun
- new joint or muscle pains
- becoming very tired
- chest pain or shortness of breath
- swelling of the feet, ankles, or legs
Liver Problems.Liver problems can happen in people who receive TNF-blocker medicines, including SIMPONI ARIA. These problems can lead to liver failure and death. Call your doctor right away if you have any of these symptoms: - feel very tired
- skin or eyes look yellow
- poor appetite or vomiting
- pain on the right side of your stomach (abdomen)
Blood Problems.Low blood counts have been seen with SIMPONI ARIA. Your body may not make enough blood cells that help fight infections or help stop bleeding. Symptoms include fever, bruising or bleeding easily, or looking pale. Your doctor will check your blood counts before and during treatment with SIMPONI ARIA. Allergic Reactions.Allergic reactions can happen in people who receive TNF-blocker medicines, including SIMPONI ARIA. Some reactions may be serious and can be life-threatening. Some of these reactions can happen after receiving your first dose of SIMPONI ARIA. Call your doctor right away if you have any of these symptoms of an allergic reaction: - hives
- swollen face
- breathing trouble
- chest pain
The most common side effects of SIMPONI ARIA include: - upper respiratory infection (runny nose, sore throat, and hoarseness or laryngitis)
- abnormal liver tests
- decreased blood cells that fight infection
- viral infections, such as flu and cold sores in the mouth
- bronchitis
- high blood pressure
- rash
These are not all of the possible side effects of SIMPONI ARIA. Tell your doctor about any side effect that bothers you or does not go away. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. General information about the safe and effective use of SIMPONI ARIA. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your doctor or pharmacist for information about SIMPONI ARIA that is written for health professionals. What are the ingredients in SIMPONI ARIA? Active ingredient: golimumab. Inactive ingredients: L-histidine, L-histidine monohydrochloride monohydrate, polysorbate 80, sorbitol, and water for injection. SIMPONI ARIA is preservative-free and is not made with natural rubber latex. Manufactured by: Janssen Biotech, Inc. Horsham, PA 19044, USA US License No. 1864 For patent information: www.janssenpatents.com
© 2017, 2020 Janssen Pharmaceutical CompaniesFor more information go to www.SIMPONIARIA.com or call 1-800-JANSSEN (1-800-526-7736). This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: 9/2020 - PRINCIPAL DISPLAY PANEL - 4 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
SIMPONI ARIA
golimumab solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:57894-350 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength GOLIMUMAB (UNII: 91X1KLU43E) (GOLIMUMAB - UNII:91X1KLU43E) GOLIMUMAB 50 mg in 4 mL Inactive Ingredients Ingredient Name Strength HISTIDINE MONOHYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 6.42 mg in 4 mL HISTIDINE (UNII: 4QD397987E) 1.14 mg in 4 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.6 mg in 4 mL WATER (UNII: 059QF0KO0R) SORBITOL (UNII: 506T60A25R) 180 mg in 4 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-350-01 1 in 1 CARTON 07/19/2013 1 4 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 2 NDC:57894-350-89 1 in 1 CARTON 07/19/2013 2 4 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125433 07/19/2013 Labeler - Janssen Biotech, Inc. (099091753) Establishment Name Address ID/FEI Business Operations Anderson Brecon 053217022 label(57894-350) , pack(57894-350) Establishment Name Address ID/FEI Business Operations Janssen Biologics, B.V. 409612918 api manufacture(57894-350) , analysis(57894-350) Establishment Name Address ID/FEI Business Operations Cilag AG 483237103 manufacture(57894-350) , analysis(57894-350) , label(57894-350) , pack(57894-350) Establishment Name Address ID/FEI Business Operations Simtra US LLC 604719430 manufacture(57894-350) Establishment Name Address ID/FEI Business Operations Janssen Sciences Ireland UC 986030167 api manufacture(57894-350) , analysis(57894-350)
