Label: URSODIOL tablet


- NDC Code(s): 64380-918-06, 64380-919-06
- Packager: Strides Pharma Science Limited
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated February 1, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use URSODIOL TABLETS safely and effectively. See full prescribing information for URSODIOL TABLETS.
URSODIOL tablets, for oral use
Initial U.S. Approval: 1997INDICATIONS AND USAGE
Ursodiol tablets, 250 mg and 500 mg are bile acids indicated for the treatment of patients with primary biliary cholangitis. (1) (1)
DOSAGE AND ADMINISTRATION
- Recommended adult dosage:13-15 mg/kg/day administered in two to four divided doses with food. (2.1)
- Scored ursodiol tablets, 500 mg:scored tablet can be broken in halves to provide recommended dosage. (2.3, 16)
DOSAGE FORMS AND STRENGTHS
- Ursodiol: 250 mg tablet (3)
- Ursodiol: 500 mg scored tablet (3)
CONTRAINDICATIONS
Patients with complete biliary obstruction and known hypersensitivity or intolerance to ursodiol or any of the components of the formulation. (4) (4)
WARNINGS AND PRECAUTIONS
- Abnormal Liver Function Tests:Liver function tests (γ-GT, alkaline phosphatase, AST, ALT) and bilirubin level should be monitored. Treatment discontinuation should be considered if parameters increase to a level considered clinically significant in patients with stable historical liver function test levels. Caution should be exercised to maintain patients' bile flow. (5.1)
- Enteroliths in Patients with Risk for Intestinal Stenosis or Stasis:Monitor for obstructive gastrointestinal symptoms; if symptoms occur, hold ursodiol until a clinical evaluation has been conducted. (5.2)
ADVERSE REACTIONS
Most common adverse reactions reported with the use of ursodiol during worldwide postmarketing and clinical experience (≥1%) are, in alphabetical order: abdominal discomfort, abdominal pain, alopecia, diarrhea, nausea, pruritus, and rash. (6) (6)
To report SUSPECTED ADVERSE REACTIONS, contact Strides Pharma Inc. at 1-877-244-9825 or go to www.strides.com or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)
DRUG INTERACTIONS
- Bile Acid Sequestering Agents:May interfere with the action of ursodiol by reducing its absorption. (7.1)
- Aluminum-based Antacids:May interfere with the action of ursodiol by reducing its absorption. (7.2)
- Drugs that alter the metabolism of lipids or induce cholestasis may interfere with the action of ursodiol. (7.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 2/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Liver Function Tests
2.3 Scoring the Ursodiol Tablets, 500 mg
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Abnormal Liver Function Tests
5.2 Enteroliths in Patients with Risk for Intestinal Stenosis or Stasis
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Bile Acid Sequestering Agents
7.2 Aluminum-based Antacids
7.3 Drugs Affecting Lipid Metabolism
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Efficacy of Ursodeoxycholic Acid Administered at 13 to 15 mg/kg/day in 3 or 4 Divided Doses to PBC Patients
14.2 Efficacy of Ursodiol Administered at 14 mg/kg/day as a Once Daily Dose to PBC Patients
14.3 Efficacy of Ursodiol Tablets, 250 mg Administered in Twice a Day Versus Four Times a Day Divided Dosing Schedules to PBC Patients
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Ursodiol Tablets, USP 250 mg
16.2 Ursodiol Tablets, USP 500 mg
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
The recommended adult dosage for ursodiol tablets, 250 mg and 500 mg in the treatment of PBC is 13-15 mg/kg/day administered in two to four divided doses with food. Dosing regimen should be adjusted according to each patient's need at the discretion of the physician.
2.2 Liver Function Tests
Liver function tests (γ-GT, alkaline phosphatase, AST, ALT) and bilirubin levels should be monitored every month for three months after start of therapy, and every six months thereafter [see Warnings and Precautions (5.1)].
2.3 Scoring the Ursodiol Tablets, 500 mg
The Ursodiol 500 mg scored tablet can be broken in halves to provide recommended dosage.
To break ursodiol 500 mg scored tablet easily, place the tablet on a flat surface with the scored section on top. Hold the tablet with your thumbs placed close to the scored part of the tablet (groove). Then apply gentle pressure and snap the tablet segments apart (segments breaking incorrectly should not be used). The segments should be washed down unchewed, with water, keeping the segments in the mouth can reveal a bitter taste. Due to the bitter taste, segments should be stored separately from whole tablets. [see How Supplied/ Storage and Handling (16)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Abnormal Liver Function Tests
Liver function tests (γ-GT, alkaline phosphatase, AST, ALT) and bilirubin levels should be monitored every month for three months after start of therapy, and every six months thereafter. This monitoring will allow the early detection of a possible deterioration of the hepatic function. Treatment discontinuation should be considered if the above parameters increase to a level considered clinically significant in patients with stable historical liver function test levels.
Caution has to be exercised to maintain the bile flow of the patients taking ursodiol.
5.2 Enteroliths in Patients with Risk for Intestinal Stenosis or Stasis
There have been rare postmarketing reports of ursodiol-treated patients who developed enteroliths (bezoars) resulting in obstructive symptoms that required surgical intervention. These patients had medical conditions that predisposed them to intestinal stenosis or stasis (e.g., surgical enteroanastomoses, Crohn's disease). If a patient presents with obstructive gastrointestinal symptoms, hold ursodiol until a clinical evaluation has been conducted.
-
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
The following table summarizes the adverse reactions observed in two placebo-controlled clinical trials.
ADVERSE REACTIONS
VISIT AT 12 MONTHS
VISIT AT 24 MONTHS
UDCA
n (%)
Placebo
n (%)
UDCA
n (%)
Placebo
n (%)
Diarrhea
---
---
1 (1.32)
---
Elevated creatinine
---
---
1 (1.32)
---
Elevated blood glucose
1 (1.18)
---
1 (1.32)
---
Leukopenia
---
---
2 (2.63)
---
Peptic ulcer
---
---
1 (1.32)
---
Skin rash
---
---
2 (2.63)
---
Thrombocytopenia
---
---
1 (1.32)
---
Note: Those adverse reactions occurring at the same or higher incidence in the placebo as in the UDCA group have been deleted from this table (this includes diarrhea and thrombocytopenia at 12 months, nausea/vomiting, fever and other toxicity).
UDCA = Ursodeoxycholic acid = Ursodiol
In a randomized, cross-over study in sixty PBC patients, seven patients (11.6%) reported nine adverse reactions: abdominal pain and asthenia (1 patient), nausea (3 patients), dyspepsia (2 patients) and anorexia and esophagitis (1 patient each). One patient on the twice a day regimen (total dose 1,000 mg) withdrew due to nausea. All of these nine adverse reactions except esophagitis were observed with the twice a day regimen at a total daily dose of 1,000 mg or greater. However, an adverse reaction may occur at any dose.
6.2 Postmarketing Experience
The following adverse reactions, presented by system organ class in alphabetical order, have been identified during post approval use of ursodiol. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Gastrointestinal disorders: abdominal discomfort, abdominal pain, enteroliths (bezoars), constipation, diarrhea, dyspepsia, nausea, vomiting.
- General disorders and administration site conditions: malaise, peripheral edema, pyrexia.
- Hepatobiliary disorders:jaundice (or aggravation of pre-existing jaundice).
- Immune System Disorders: Drug hypersensitivity to include facial edema, urticaria, angioedema and laryngeal edema.
- Abnormal Laboratory Tests:ALT increased, AST increased, blood alkaline phosphatase increased, blood bilirubin increased, γ-GT increased, hepatic enzyme increased, liver function test abnormal, transaminases increased.
- Musculoskeletal and connective tissue disorders: myalgia.
- Nervous system disorders: dizziness, headache.
- Respiratory, thoracic and mediastinal disorders: cough.
- Skin and subcutaneous tissue disorder: alopecia, pruritus, rash.
-
7 DRUG INTERACTIONS
7.1 Bile Acid Sequestering Agents
Bile acid sequestering agents such as cholestyramine and colestipol may interfere with the action of ursodiol by reducing its absorption.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Available published data on the use of ursodiol in pregnant women derived from randomized controlled trials, observational studies, and case series collected over several decades have not identified a drug-associated risk of major birth defects, miscarriage, or other adverse maternal or fetal outcomes. Most of the reported exposures to ursodiol occurred in the second and third trimester of pregnancy. In animal reproduction studies, ursodiol had no adverse effects on embryo-fetal development when administered at doses greater than human therapeutic doses (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in the clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
No adverse effects on embryo-fetal development were observed with oral administration of ursodiol to pregnant rats and rabbits during organogenesis at doses up to 22 and 7 times, respectively, the maximum recommended human dose (based on body surface area).
8.2 Lactation
Ursodiol is naturally present in human milk. There are no reports of adverse effects of ursodiol on the breastfed child, but the reports are extremely limited. There are no data on the effects of ursodiol on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for ursodiol and any potential adverse effects on the breastfed child from ursodiol or from the underlying maternal condition.
-
10 OVERDOSAGE
There have been no reports of accidental or intentional overdosage with ursodiol. Single oral doses of ursodiol at 10 g/kg in mice and dogs, and 5 g/kg in rats were not lethal. A single oral dose of ursodiol at 1.5 g/kg was lethal in hamsters. Symptoms of acute toxicity were salivation and vomiting in dogs, and ataxia, dyspnea, ptosis, agonal convulsions and coma in hamsters.
-
11 DESCRIPTION
Ursodiol tablets, USP 250 mg is available as a film-coated tablet for oral administration. Ursodiol tablets, USP 500 mg is available as a scored film-coated tablet for oral administration.
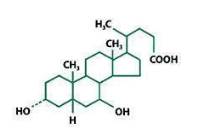
Ursodiol (ursodeoxycholic acid, UDCA) is a naturally occurring bile acid found in small quantities in normal human bile and in larger quantities in the biles of certain species of bears. It is a bitter-tasting white or almost white powder consisting of crystalline particles freely soluble in alcohol and glacial acetic acid, sparingly soluble in chloroform, slightly soluble in ether, acetone, methylene chloride and practically insoluble in water. The chemical name of ursodiol is 3α,7β-dihydroxy-5β-cholan-24-oic (C24H40O4). Ursodiol has a molecular weight of 392.56. Its structure is shown below.
Inactive ingredients: Sodium Starch Glycolate, Povidone, Sodium Lauryl Sulfate, Tromethamine, Microcrystalline Cellulose, Polyethylene Glycol 3350, Magnesium Stearate, Opadry Clear YS-1-7006, Hypromellose, Macrogol/PEG 400, Macrogol/PEG 8000, Purified Water.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ursodiol, a naturally occurring hydrophilic bile acid, derived from cholesterol, is present as a minor fraction of the total human bile acid pool. Oral administration of ursodiol increases this fraction in a dose related manner, to become the major biliary acid, replacing/displacing toxic concentrations of endogenous hydrophobic bile acids that tend to accumulate in cholestatic liver disease.
In addition to the replacement and displacement of toxic bile acids, other mechanisms of action include cytoprotection of the injured bile duct epithelial cells (cholangiocytes) against toxic effects of bile acids, inhibition of apoptosis of hepatocytes, immunomodulatory effects, and stimulation of bile secretion by hepatocytes and cholangiocytes.
12.2 Pharmacodynamics
Lithocholic acid, when administered chronically to animals, causes cholestatic liver injury that may lead to death from liver failure in certain species unable to form sulfate conjugates. Ursodiol is 7-dehydroxylated more slowly than chenodiol. For equimolar doses of ursodiol and chenodiol, steady state levels of lithocholic acid in biliary bile acids are lower during ursodiol administration than with chenodiol administration. Humans and chimpanzees can sulfate lithocholic acid. Although liver injury has not been associated with ursodiol therapy, a reduced capacity to sulfate may exist in some individuals.
12.3 Pharmacokinetics
Ursodiol (UDCA) is normally present as a minor fraction of the total bile acids in humans (about 5%). Following oral administration, the majority of ursodiol is absorbed by passive diffusion and its absorption is incomplete. Once absorbed, ursodiol undergoes hepatic extraction to the extent of about 50% in the absence of liver disease. As the severity of liver disease increases, the extent of extraction decreases. In the liver, ursodiol is conjugated with glycine or taurine, then secreted into bile. These conjugates of ursodiol are absorbed in the small intestine by passive and active mechanisms. The conjugates can also be deconjugated in the ileum by intestinal enzymes, leading to the formation of free ursodiol that can be reabsorbed and reconjugated in the liver. Nonabsorbed ursodiol passes into the colon where it is mostly 7-dehydroxylated to lithocholic acid. Some ursodiol is epimerized to chenodiol (CDCA) via a 7-oxo intermediate. Chenodiol also undergoes 7-dehydroxylation to form lithocholic acid. These metabolites are poorly soluble and excreted in the feces. A small portion of lithocholic acid is reabsorbed, conjugated in the liver with glycine, or taurine and sulfated at the 3 position. The resulting sulfated lithocholic acid conjugates are excreted in bile and then lost in feces.
In healthy subjects, at least 70% of ursodiol (unconjugated) is bound to plasma protein. No information is available on the binding of conjugated ursodiol to plasma protein in healthy subjects or PBC patients. Its volume of distribution has not been determined, but is expected to be small since the drug is mostly distributed in the bile and small intestine. Ursodiol is excreted primarily in the feces. With treatment, urinary excretion increases, but remains less than 1% except in severe cholestatic liver disease.
During chronic administration of ursodiol, it becomes a major biliary and plasma bile acid. At a chronic dose of 13 to 15 mg/kg/day, ursodiol constitutes 30-50% of biliary and plasma bile acids.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In two 24-month oral carcinogenicity studies in mice, ursodiol at doses up to 1,000 mg/kg/day (3,000 mg/m2/day) was not tumorigenic. Based on body surface area, for a 50 kg person of average height (1.46 m2 body surface area), this dose represents 5.4 times the recommended maximum clinical dose of 15 mg/kg/day (555 mg/m2/day).
In a two-year oral carcinogenicity study in Fischer 344 rats, ursodiol at doses up to 300 mg/kg/day (1,800 mg/m2/day, 3.2 times the recommended maximum human dose based on body surface area) was not tumorigenic.
In a life-span (126-138 weeks) oral carcinogenicity study, Sprague-Dawley rats were treated with doses of 33 to 300 mg/kg/day, 0.4 to 3.2 times the recommended maximum human dose based on body surface area. Ursodiol produced a significantly (p≤0.5, Fisher's exact test) increased incidence of pheochromocytomas of the adrenal medulla in females of the highest dose group.
In 103-week oral carcinogenicity studies of lithocholic acid, a metabolite of ursodiol, doses up to 250 mg/kg/day in mice and 500 mg/kg/day in rats did not produce any tumors. In a 78-week rat study, intrarectal instillation of lithocholic acid (1 mg/kg/day) for 13 months did not produce colorectal tumors. A tumor-promoting effect was observed when it was administered after a single intrarectal dose of a known carcinogen N-methyl-N'-nitro-N-nitrosoguanidine. On the other hand, in a 32-week rat study, ursodiol at a daily dose of 240 mg/kg (1,440 mg/m2, 2.6 times the maximum recommended human dose based on body surface area) suppressed the colonic carcinogenic effect of another known carcinogen azoxymethane.
Ursodiol was not genotoxic in the Ames test, the mouse lymphoma cell (L5178Y, TK+/-) forward mutation test, the human lymphocyte sister chromatid exchange test, the mouse spermatogonia chromosome aberration test, the Chinese hamster micronucleus test and the Chinese hamster bone marrow cell chromosome aberration test.
Ursodiol at oral doses of up to 2,700 mg/kg/day (16,200 mg/m2/day, 29 times the recommended maximum human dose based on body surface area) was found to have no effect on fertility and reproductive performance of male and female rats.
-
14 CLINICAL STUDIES
14.1 Efficacy of Ursodeoxycholic Acid Administered at 13 to 15 mg/kg/day in 3 or 4 Divided Doses to PBC Patients
A U.S., multicenter, randomized, double-blind, placebo-controlled study was conducted to evaluate the efficacy of ursodeoxycholic acid at a dose of 13 to 15 mg/kg/day, administered in 3 or 4 divided doses in 180 patients with PBC (78% received four times a day dosage). Upon completion of the double-blind portion, all patients entered an open-label active treatment extension phase.
Treatment failure, the main efficacy endpoint measured during this study, was defined as death, need for liver transplantation, histologic progression by two stages or to cirrhosis, development of varices, ascites or encephalopathy, marked worsening of fatigue or pruritus, inability to tolerate the drug, doubling of serum bilirubin and voluntary withdrawal. After two years of double-blind treatment, the incidence of treatment failure was significantly (p<0.01) reduced in the ursodiol tablets, 250 mg group (20 of 86 (23%) as compared to the placebo group (40 of 86 (47%). Time to treatment failure, which excluded doubling of serum bilirubin and voluntary withdrawal, was also significantly (p<0.001) delayed in the ursodiol tablets, 250 mg treated group (n=86, 803.8±24.9 d vs. 641.1±24.4 d for the placebo group (n=86) on average) regardless of either histologic stage or baseline bilirubin levels (>1.8 or ≤1.8 mg/dL).
Using a definition of treatment failure, which excluded doubling of serum bilirubin and voluntary withdrawal, time to treatment failure was significantly delayed in the ursodiol tablets, 250 mg group. In comparison with placebo, treatment with ursodiol tablets, 250 mg resulted in a significant improvement in the following serum hepatic biochemistries when compared to baseline: total bilirubin, SGOT, alkaline phosphatase and IgM.
14.2 Efficacy of Ursodiol Administered at 14 mg/kg/day as a Once Daily Dose to PBC Patients
A second study conducted in Canada randomized 222 PBC patients to ursodiol, 14 mg/kg/day or placebo, administered as a once daily dose in a double-blind manner during a two-year period. At two years, a statistically significant (p<0.001) difference between the two treatments (n=106 for the ursodiol tablets, 250 mg group and n=106 for the placebo group), in favor of ursodiol, was demonstrated in the following: reduction in the proportion of patients exhibiting a more than 50% increase in serum bilirubin; median percent decrease in bilirubin (-17.12% for the ursodiol tablets, 250 mg group vs. +20.00% for the placebo group), transaminases (-40.54% for the ursodiol tablets, 250 mg group vs. +5.71% for the placebo group) and alkaline phosphatase (-47.61% for the ursodiol tablets, 250 mg group vs. -5.69% for the placebo group); incidence of treatment failure; and time to treatment failure. The definition of treatment failure included: discontinuing the study for any reason; a total serum bilirubin level greater than or equal to 1.5 mg/dl or increasing to a level equal to or greater than two times the baseline level; and the development of ascites or encephalopathy. Evaluation of patients at 4 years or longer was inadequate due to the high drop-out rate (n=10 withdrew from the ursodiol tablets, 250 mg group vs. n=15 from the placebo group) and small number of patients. Therefore, death, need for liver transplantation, histological progression by two stages or to cirrhosis, development of varices, ascites or encephalopathy, marked worsening of fatigue or pruritus, inability to tolerate the drug, doubling of serum bilirubin and voluntary withdrawal were not assessed.
14.3 Efficacy of Ursodiol Tablets, 250 mg Administered in Twice a Day Versus Four Times a Day Divided Dosing Schedules to PBC Patients
A randomized, two-period crossover study in fifty PBC patients compared efficacy of ursodiol tablets, 250 mg in twice a day versus four times a day divided dosing schedules in 50 patients for 6 months in each crossover period. Mean percent changes from baseline in liver test results and Mayo risk score (n=46) and serum enrichment with UDCA (n=34) were not statistically significant with any dosage at any time interval. This study demonstrated that UDCA (13 to 15 mg/kg/day) given twice a day is equally effective to UDCA given four times a day. In addition, ursodiol tablets, 250 mg was given as a single versus three times a day dosing schedules in 10 patients. Due to the small number of patients in this arm of the study, it was not possible to conduct statistical comparisons between these regimens.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 Ursodiol Tablets, USP 250 mg
Each ursodiol tablet USP, 250 mg white to off white, elliptical shaped, biconvex film-coated tablet, debossed with "250" on one side and plain on the other side, contains 250 mg of ursodiol. Available in bottles of 100 tablets (NDC 64380-918-06).
16.2 Ursodiol Tablets, USP 500 mg
Each ursodiol tablet USP, 500 mg white to off white, elliptical shaped, biconvex film-coated tablet, debossed with "500" on one side and scored on the other side, contains 500 mg of ursodiol. Available in bottles of 100 tablets (NDC 64380-919-06).
Store at 20°C to 25°C (68°F to 77°F). Dispense in a tight container using a child-resistant closure.
Half-tablets (scored ursodiol tablet USP, 500 mg tablets broken in half) maintain acceptable quality for up to 28 days when stored in the current packaging (bottles) at 20°C to 25°C (68°F to 77°F). Due to the bitter taste, the halved segments should be stored separately from the whole tablets [see Dosage and Administration (2.2)].
-
17 PATIENT COUNSELING INFORMATION
Enteroliths in Patients with Risk for Intestinal Stenosis or Stasis
Advise patients or their caretaker(s) to notify their healthcare provider if they experience obstructive gastrointestinal symptoms due to the risk of enteroliths [see Warnings and Precautions (5.2)].
Drug Interactions
Patients should be informed that absorption of ursodiol tablets, 250 mg and 500 mg may be reduced if they are taking bile acid sequestering agents, such as cholestyramine and colestipol, aluminum-based antacids, or drugs known to alter the metabolism of cholesterol [see Drug Interactions (7)].
Rx only
Manufactured by:
Strides Pharma Science Ltd.
Bengaluru – 562106, India.
Distributed by:
Strides Pharma Inc.
East Brunswick, NJ 08816.
Revised: 02/2023
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
URSODIOL
ursodiol tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64380-918 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength URSODIOL (UNII: 724L30Y2QR) (URSODIOL - UNII:724L30Y2QR) URSODIOL 250 mg Inactive Ingredients Ingredient Name Strength POVIDONE K30 (UNII: U725QWY32X) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TROMETHAMINE (UNII: 023C2WHX2V) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) Product Characteristics Color WHITE (White to off white) Score no score Shape OVAL (elliptical) Size 13mm Flavor Imprint Code 250 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64380-918-06 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/08/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA213504 02/08/2021 URSODIOL
ursodiol tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64380-919 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength URSODIOL (UNII: 724L30Y2QR) (URSODIOL - UNII:724L30Y2QR) URSODIOL 500 mg Inactive Ingredients Ingredient Name Strength POVIDONE K30 (UNII: U725QWY32X) SODIUM LAURYL SULFATE (UNII: 368GB5141J) TROMETHAMINE (UNII: 023C2WHX2V) HYPROMELLOSE 2910 (6 MPA.S) (UNII: 0WZ8WG20P6) MAGNESIUM STEARATE (UNII: 70097M6I30) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POLYETHYLENE GLYCOL 8000 (UNII: Q662QK8M3B) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) Product Characteristics Color WHITE (White to off white) Score 2 pieces Shape OVAL (elliptical) Size 17mm Flavor Imprint Code 500 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64380-919-06 100 in 1 BOTTLE; Type 0: Not a Combination Product 02/08/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA213504 02/08/2021 Labeler - Strides Pharma Science Limited (650738743) Registrant - Strides Pharma Global Pte. Ltd. (659220961) Establishment Name Address ID/FEI Business Operations Strides Pharma Science Limited 918513263 ANALYSIS(64380-918, 64380-919) , MANUFACTURE(64380-918, 64380-919) , PACK(64380-918, 64380-919)

