Label: ZALEPLON capsule

- NDC Code(s): 43063-505-30
- Packager: PD-Rx Pharmaceuticals, Inc.
- This is a repackaged label.
- Source NDC Code(s): 65862-214
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: CIV
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated September 26, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
WARNING: COMPLEX SLEEP BEHAVIORS
Complex sleep behaviors including sleep-walking, sleep-driving, and engaging in other activities while not fully awake may occur following use of zaleplon. Some of these events may result in serious injuries, including death. Discontinue zaleplon immediately if a patient experiences a complex sleep behavior (see CONTRAINDICATIONS and Complex Sleep Behaviors under WARNINGS) .
-
DESCRIPTION
Zaleplon is a nonbenzodiazepine hypnotic from the pyrazolopyrimidine class. The chemical name of zaleplon is N-[3-(3-cyanopyrazolo[1,5-a]pyrimidin-7-yl)phenyl]-N-ethylacetamide. Its molecular formula is C 17H 15N 5O, and its molecular weight is 305.34. The structural formula is shown below.
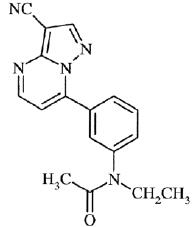
Zaleplon USP is a white to light pale yellow, crystalline powder that is practically insoluble in water and sparingly soluble in alcohol or propylene glycol. Its partition coefficient in octanol/water is constant (log PC = 1.23) over the pH range of 1 to 7.
Zaleplon capsules USP contain zaleplon USP as the active ingredient. Inactive ingredients consist of microcrystalline cellulose, lactose monohydrate, pregelatinized starch (maize), sodium lauryl sulfate, colloidal silicon dioxide, and stearic acid. The empty hard gelatin capsule shells contain FD&C Blue #1, FD&C Green #3, FD&C Yellow #5, titanium dioxide, and gelatin. The capsules are printed with edible ink containing black iron oxide and shellac.
Meets the USP Dissolution Test - 2. -
CLINICAL PHARMACOLOGY
Pharmacodynamics and Mechanism of Action
While zaleplon is a hypnotic agent with a chemical structure unrelated to benzodiazepines, barbiturates, or other drugs with known hypnotic properties, it interacts with the gamma-aminobutyric acid-benzodiazepine (GABA-BZ) receptor complex. Subunit modulation of the GABA-BZ receptor chloride channel macromolecular complex is hypothesized to be responsible for some of the pharmacological properties of benzodiazepines, which include sedative, anxiolytic, muscle relaxant, and anticonvulsive effects in animal models.
Other nonclinical studies have also shown that zaleplon binds selectively to the brain omega-1 receptor situated on the alpha subunit of the GABA A/chloride ion channel receptor complex and potentiates t-butyl-bicyclophosphorothionate (TBPS) binding. Studies of binding of zaleplon to recombinant GABA A receptors (α 1β 1γ 2 [omega-1] and α 2β 1γ 2 [omega-2]) have shown that zaleplon has a low affinity for these receptors, with preferential binding to the omega-1 receptor.Pharmacokinetics
The pharmacokinetics of zaleplon have been investigated in more than 500 healthy subjects (young and elderly), nursing mothers, and patients with hepatic disease or renal disease. In healthy subjects, the pharmacokinetic profile has been examined after single doses of up to 60 mg and once-daily administration at 15 mg and 30 mg for 10 days. Zaleplon was rapidly absorbed with a time to peak concentration (t max) of approximately 1 hour and a terminal-phase elimination half-life (t 1/2) of approximately 1 hour. Zaleplon does not accumulate with once-daily administration and its pharmacokinetics are dose proportional in the therapeutic range.
Absorption
Zaleplon is rapidly and almost completely absorbed following oral administration. Peak plasma concentrations are attained within approximately 1 hour after oral administration. Although zaleplon is well absorbed, its absolute bioavailability is approximately 30% because it undergoes significant presystemic metabolism.
Distribution
Zaleplon is a lipophilic compound with a volume of distribution of approximately 1.4 L/kg following intravenous (IV) administration, indicating substantial distribution into extravascular tissues. The in vitro plasma protein binding is approximately 60% ± 15% and is independent of zaleplon concentration over the range of 10 ng/mL to 1000 ng/mL. This suggests that zaleplon disposition should not be sensitive to alterations in protein binding. The blood to plasma ratio for zaleplon is approximately 1, indicating that zaleplon is uniformly distributed throughout the blood with no extensive distribution into red blood cells.
Metabolism
After oral administration, zaleplon is extensively metabolized, with less than 1% of the dose excreted unchanged in urine. Zaleplon is primarily metabolized by aldehyde oxidase to form 5-oxo-zaleplon. Zaleplon is metabolized to a lesser extent by cytochrome P450 (CYP) 3A4 to form desethylzaleplon, which is quickly converted, presumably by aldehyde oxidase, to 5-oxo-desethylzaleplon. These oxidative metabolites are then converted to glucuronides and eliminated in urine. All of zaleplon’s metabolites are pharmacologically inactive.
Elimination
After either oral or IV administration, zaleplon is rapidly eliminated with a mean t ½ of approximately 1 hour. The oral-dose plasma clearance of zaleplon is about 3 L/h/kg and the IV zaleplon plasma clearance is approximately 1 L/h/kg. Assuming normal hepatic blood flow and negligible renal clearance of zaleplon, the estimated hepatic extraction ratio of zaleplon is approximately 0.7, indicating that zaleplon is subject to high first-pass metabolism.
After administration of a radiolabeled dose of zaleplon, 70% of the administered dose is recovered in urine within 48 hours (71% recovered within 6 days), almost all as zaleplon metabolites and their glucuronides. An additional 17% is recovered in feces within 6 days, most as 5-oxo-zaleplon.Effect of Food
In healthy adults a high-fat/heavy meal prolonged the absorption of zaleplon compared to the fasted state, delaying t max by approximately 2 hours and reducing C max by approximately 35%. Zaleplon AUC and elimination half-life were not significantly affected. These results suggest that the effects of zaleplon on sleep onset may be reduced if it is taken with or immediately after a high-fat/heavy meal.
Special Populations
Age
The pharmacokinetics of zaleplon have been investigated in three studies with elderly men and women ranging in age from 65 to 85 years. The pharmacokinetics of zaleplon in elderly subjects, including those over 75 years of age, are not significantly different from those in young healthy subjects.
Gender
There is no significant difference in the pharmacokinetics of zaleplon in men and women.
Race
The pharmacokinetics of zaleplon have been studied in Japanese subjects as representative of Asian populations. For this group, C max and AUC were increased 37% and 64%, respectively. This finding can likely be attributed to differences in body weight, or alternatively, may represent differences in enzyme activities resulting from differences in diet, environment, or other factors. The effects of race on pharmacokinetic characteristics in other ethnic groups have not been well characterized.
Hepatic impairment
Zaleplon is metabolized primarily by the liver and undergoes significant presystemic metabolism. Consequently, the oral clearance of zaleplon was reduced by 70% and 87% in compensated and decompensated cirrhotic patients, respectively, leading to marked increases in mean C max and AUC (up to 4-fold and 7-fold in compensated and decompensated patients, respectively), in comparison with healthy subjects. The dose of zaleplon should therefore be reduced in patients with mild to moderate hepatic impairment (see DOSAGE AND ADMINISTRATION). Zaleplon is not recommended for use in patients with severe hepatic impairment.
Renal impairment
Because renal excretion of unchanged zaleplon accounts for less than 1% of the administered dose, the pharmacokinetics of zaleplon are not altered in patients with renal insufficiency. No dose adjustment is necessary in patients with mild to moderate renal impairment. Zaleplon has not been adequately studied in patients with severe renal impairment.Drug-Drug Interactions
Because zaleplon is primarily metabolized by aldehyde oxidase, and to a lesser extent by CYP3A4, inhibitors of these enzymes might be expected to decrease zaleplon’s clearance and inducers of these enzymes might be expected to increase its clearance. Zaleplon has been shown to have minimal effects on the kinetics of warfarin (both R- and S- forms), imipramine, ethanol, ibuprofen, diphenhydramine, thioridazine, and digoxin. However, the effects of zaleplon on inhibition of enzymes involved in the metabolism of other drugs have not been studied (see Drug Interactions under PRECAUTIONS).
Clinical Trials
Controlled Trials Supporting Effectiveness
Zaleplon (typically administered in doses of 5 mg, 10 mg, or 20 mg) has been studied in patients with chronic insomnia (n = 3,435) in 12 placebo- and active-drug-controlled trials. Three of the trials were in elderly patients (n = 1,019). It has also been studied in transient insomnia (n = 264). Because of its very short half-life, studies focused on decreasing sleep latency, with less attention to duration of sleep and number of awakenings, for which consistent differences from placebo were not demonstrated. Studies were also carried out to examine the time course of effects on memory and psychomotor function, and to examine withdrawal phenomena.
Transient Insomnia
Normal adults experiencing transient insomnia during the first night in a sleep laboratory were evaluated in a double-blind, parallel-group trial comparing the effects of two doses of zaleplon (5 mg and 10 mg) with placebo. Zaleplon 10 mg, but not 5 mg, was superior to placebo in decreasing latency to persistent sleep (LPS), a polysomnographic measure of time to onset of sleep.
Chronic Insomnia
Non-elderly patients
Adult outpatients with chronic insomnia were evaluated in three double-blind, parallel-group outpatient studies, one of 2 weeks duration and two of 4 weeks duration, that compared the effects of zaleplon at doses of 5 mg (in two studies), 10 mg, and 20 mg with placebo on a subjective measure of time to sleep onset (TSO). Zaleplon 10 mg and 20 mg were consistently superior to placebo for TSO, generally for the full duration of all three studies. Although both doses were effective, the effect was greater and more consistent for the 20 mg dose. The 5 mg dose was less consistently effective than were the 10 mg and 20 mg doses. Sleep latency with zaleplon 10 mg and 20 mg was on the order of 10 to 20 minutes (15% to 30%) less than with placebo in these studies.
Adult outpatients with chronic insomnia were evaluated in six double-blind, parallel-group sleep laboratory studies that varied in duration from a single night up to 35 nights. Overall, these studies demonstrated a superiority of zaleplon 10 mg and 20 mg over placebo in reducing LPS on the first 2 nights of treatment. At later time points in 5-, 14-, and 28-night studies, a reduction in LPS from baseline was observed for all treatment groups, including the placebo group, and thus, a significant difference between zaleplon and placebo was not seen beyond 2 nights. In a 35-night study, zaleplon 10 mg was significantly more effective than placebo in reducing LPS at the primary efficacy endpoint on nights 29 and 30.
Elderly patients
Elderly outpatients with chronic insomnia were evaluated in two 2-week, double-blind, parallel-group outpatient studies that compared the effects of zaleplon 5 mg and 10 mg with placebo on a subjective measure of time to sleep onset (TSO). Zaleplon at both doses was superior to placebo on TSO, generally for the full duration of both studies, with an effect size generally similar to that seen in younger persons. The 10 mg dose tended to have a greater effect in reducing TSO.
Elderly outpatients with chronic insomnia were also evaluated in a 2-night sleep laboratory study involving doses of 5 mg and 10 mg. Both 5 mg and 10 mg doses of zaleplon were superior to placebo in reducing latency to persistent sleep (LPS).
Generally in these studies, there was a slight increase in sleep duration, compared to baseline, for all treatment groups, including placebo, and thus, a significant difference from placebo on sleep duration was not demonstrated.Studies Pertinent to Safety Concerns for Sedative/Hypnotic Drugs
Memory Impairment
Studies involving the exposure of normal subjects to single fixed doses of zaleplon (10 mg or 20 mg) with structured assessments of short-term memory at fixed times after dosing (e.g., 1, 2, 3, 4, 5, 8, and 10 hours) generally revealed the expected impairment of short-term memory at 1 hour, the time of peak exposure to zaleplon, for both doses, with a tendency for the effect to be greater after 20 mg. Consistent with the rapid clearance of zaleplon, memory impairment was no longer present as early as 2 hours post dosing in one study, and in none of the studies after 3 to 4 hours. Nevertheless, spontaneous reporting of adverse events in larger premarketing clinical trials revealed a difference between zaleplon and placebo in the risk of next-day amnesia (3% vs 1%), and an apparent dose-dependency for this event (see ADVERSE REACTIONS).
Sedative/Psychomotor Effects
Studies involving the exposure of normal subjects to single fixed doses of zaleplon (10 mg or 20 mg) with structured assessments of sedation and psychomotor function (e.g., reaction time and subjective ratings of alertness) at fixed times after dosing (e.g., 1, 2, 3, 4, 5, 8, and 10 hours) generally revealed the expected sedation and impairment of psychomotor function at 1 hour, the time of peak exposure to zaleplon, for both doses. Consistent with the rapid clearance of zaleplon, impairment of psychomotor function was no longer present as early as 2 hours post dosing in one study, and in none of the studies after 3 to 4 hours. Spontaneous reporting of adverse events in larger premarketing clinical trials did not suggest a difference between zaleplon and placebo in the risk of next-day somnolence (see ADVERSE REACTIONS).
Withdrawal-Emergent Anxiety and Insomnia
During nightly use for an extended period, pharmacodynamic tolerance or adaptation to some effects of hypnotics may develop. If the drug has a short elimination half-life, it is possible that a relative deficiency of the drug or its active metabolites (i.e., in relationship to the receptor site) may occur at some point in the interval between each night’s use. This sequence of events is believed to be responsible for two clinical findings reported to occur after several weeks of nightly use of other rapidly eliminated hypnotics: increased wakefulness during the last quarter of the night and the appearance of increased signs of daytime anxiety.
Zaleplon has a short half-life and no active metabolites. At the primary efficacy endpoint (nights 29 and 30) in a 35-night sleep laboratory study, polysomnographic recordings showed that wakefulness was not significantly longer with zaleplon than with placebo during the last quarter of the night. No increase in the signs of daytime anxiety was observed in clinical trials with zaleplon. In two sleep laboratory studies involving 14- and 28-nightly doses of zaleplon (5 mg and 10 mg in one study and 10 mg and 20 mg in the second) and structured assessments of daytime anxiety, no increases in daytime anxiety were detected. Similarly, in a pooled analysis (all the parallel-group, placebo-controlled studies) of spontaneously reported daytime anxiety, no difference was observed between zaleplon and placebo.
Rebound insomnia, defined as a dose-dependent temporary worsening in sleep parameters (latency, total sleep time, and number of awakenings) compared to baseline following discontinuation of treatment, is observed with short- and intermediate-acting hypnotics. Rebound insomnia following discontinuation of zaleplon relative to baseline was examined at both nights 1 and 2 following discontinuation in three sleep laboratory studies (14, 28, and 35 nights) and five outpatient studies utilizing patient diaries (14 and 28 nights). Overall, the data suggest that rebound insomnia may be dose dependent. At 20 mg, there appeared to be both objective (polysomnographic) and subjective (diary) evidence of rebound insomnia on the first night after discontinuation of treatment with zaleplon. At 5 mg and 10 mg, there was no objective and minimal subjective evidence of rebound insomnia on the first night after discontinuation of treatment with zaleplon. At all doses, the rebound effect appeared to resolve by the second night following withdrawal. In the 35-night study, there was a worsening in sleep on the first night off for both the 10 mg and 20 mg groups compared to placebo, but not to baseline. This discontinuation-emergent effect was mild, had the characteristics of the return of the symptoms of chronic insomnia, and appeared to resolve by the second night after zaleplon discontinuation.
Other Withdrawal-Emergent Phenomena
The potential for other withdrawal phenomena was also assessed in 14- to 28-night studies, including both the sleep laboratory studies and the outpatient studies, and in open-label studies of 6- and 12-month durations. The Benzodiazepine Withdrawal Symptom Questionnaire was used in several of these studies, both at baseline and then during days 1 and 2 following discontinuation. Withdrawal was operationally defined as the emergence of 3 or more new symptoms after discontinuation. Zaleplon was not distinguishable from placebo at doses of 5 mg, 10 mg, or 20 mg on this measure, nor was zaleplon distinguishable from placebo on spontaneously reported withdrawal-emergent adverse events. There were no instances of withdrawal delirium, withdrawal associated hallucinations, or any other manifestations of severe sedative/hypnotic withdrawal. -
INDICATIONS AND USAGE
Zaleplon capsules are indicated for the short-term treatment of insomnia. Zaleplon capsules have been shown to decrease the time to sleep onset for up to 30 days in controlled clinical studies (see Clinical Trials under CLINICAL PHARMACOLOGY). They have not been shown to increase total sleep time or decrease the number of awakenings.
The clinical trials performed in support of efficacy ranged from a single night to 5 weeks in duration. The final formal assessments of sleep latency were performed at the end of treatment. -
CONTRAINDICATIONS
Zaleplon capsules are contraindicated in patients:
- who have experienced complex sleep behaviors after taking zaleplon capsules (see WARNINGS).
- with hypersensitivity to zaleplon or any excipients in the formulation (see PRECAUTIONS) .
-
WARNINGS
Complex Sleep Behaviors
Complex sleep behaviors including sleep-walking, sleep-driving, and engaging in other activities while not fully awake may occur following the first or any subsequent use of zaleplon. Patients can be seriously injured or injure others during complex sleep behaviors. Such injuries may result in a fatal outcome. Other complex sleep behaviors (e.g., preparing and eating food, making phone calls, or having sex) have also been reported. Patients usually do not remember these events. Post-marketing reports have shown that complex sleep behaviors may occur with zaleplon alone at recommended dosages, with or without the concomitant use of alcohol or other central nervous system (CNS) depressants.
CNS-Depressant Effects and Next-Day Impairment
Zaleplon, like other hypnotics, has CNS-depressant effects. Because of the rapid onset of action, zaleplon should only be ingested immediately prior to going to bed or after the patient has gone to bed and has experienced difficulty falling asleep.
Coadministration with other CNS depressants (e.g., benzodiazepines, opioids, tricyclic antidepressants, alcohol) increases the risk of CNS depression. Dosage adjustments of zaleplon and of other concomitant CNS depressants may be necessary when zaleplon is administered with such agents because of the potentially additive effects. The use of zaleplon with other sedative-hypnotics at bedtime or the middle of the night is not recommended (see DOSAGE AND ADMINISTRATION).
The risk of next-day psychomotor impairment, including impaired driving, is increased if zaleplon is taken with less than a full night of sleep remaining (7 to 8 hours); if a higher than the recommended dose is taken; if coadministered with other CNS depressants or alcohol; or if coadministered with other drugs that increase the blood levels of zaleplon. Patients should be warned against driving and other activities requiring complete mental alertness if zaleplon is taken in these circumstances (see DOSAGE AND ADMINISTRATION and Clinical Trials under CLINICAL PHARMACOLOGY).
Vehicle drivers and machine operators should be warned that, as with other hypnotics, there may be a possible risk of adverse reactions including drowsiness, prolonged reaction time, dizziness, sleepiness, blurred/double vision, reduced alertness, and impaired driving the morning after therapy. In order to minimize this risk a full night of sleep (7 to 8 hours) is recommended.
Because zaleplon can cause drowsiness and a decreased level of consciousness, patients, particularly the elderly, are at higher risk of falls.
Need to Evaluate for Co-morbid Diagnoses
Because sleep disturbances may be the presenting manifestation of a physical and/or psychiatric disorder, symptomatic treatment of insomnia should be initiated only after a careful evaluation of the patient. The failure of insomnia to remit after 7 to 10 days of treatment may indicate the presence of a primary psychiatric and/or medical illness that should be evaluated.
Worsening of insomnia or the emergence of new thinking or behavior abnormalities may be the consequence of an unrecognized psychiatric or physical disorder. Such findings have emerged during the course of treatment with sedative/hypnotic drugs, including zaleplon. Because some of the important adverse effects of zaleplon appear to be dose-related, it is important to use the lowest possible effective dose, especially in the elderly (see DOSAGE AND ADMINISTRATION).
Severe Anaphylactic and Anaphylactoid Reactions
Rare cases of angioedema involving the tongue, glottis or larynx have been reported in patients after taking the first or subsequent doses of sedative-hypnotics, including zaleplon. Some patients have had additional symptoms such as dyspnea, throat closing, or nausea and vomiting that suggest anaphylaxis. Some patients have required medical therapy in the emergency department. If angioedema involves the tongue, glottis or larynx, airway obstruction may occur and be fatal. Patients who develop angioedema after treatment with zaleplon should not be rechallenged with the drug.
Abnormal Thinking and Behavioral Changes
A variety of abnormal thinking and behavior changes have been reported to occur in association with the use of sedative/hypnotics. Some of these changes may be characterized by decreased inhibition (e.g., aggressiveness and extroversion that seem out of character), similar to effects produced by alcohol and other CNS depressants. Other reported behavioral changes have included bizarre behavior, agitation, hallucinations, and depersonalization. Amnesia and other neuropsychiatric symptoms may occur unpredictably.
It can rarely be determined with certainty whether a particular instance of the abnormal behaviors listed above is drug induced, spontaneous in origin, or a result of an underlying psychiatric or physical disorder. Nonetheless, the emergence of any new behavioral sign or symptom of concern requires careful and immediate evaluation.
Withdrawal Effects
Following rapid dose decrease or abrupt discontinuation of the use of sedative/hypnotics, there have been reports of signs and symptoms similar to those associated with withdrawal from other CNS-depressant drugs (see DRUG ABUSE AND DEPENDENCE).
-
PRECAUTIONS
General
Timing of Drug Administration
Zaleplon should be taken immediately before bedtime or after the patient has gone to bed and has experienced difficulty falling asleep. As with all sedative/hypnotics, taking zaleplon while still up and about may result in short-term memory impairment, hallucinations, impaired coordination, dizziness, and lightheadedness.
Use in the Elderly and/or Debilitated Patients
Impaired motor and/or cognitive performance after repeated exposure or unusual sensitivity to sedative/hypnotic drugs is a concern in the treatment of elderly and/or debilitated patients. A dose of 5 mg is recommended for elderly patients to decrease the possibility of side effects (see DOSAGE AND ADMINISTRATION). Elderly and/or debilitated patients should be monitored closely.
Use in Patients with Concomitant Illness
Clinical experience with zaleplon in patients with concomitant systemic illness is limited. Zaleplon should be used with caution in patients with diseases or conditions that could affect metabolism or hemodynamic responses.
Although preliminary studies did not reveal respiratory depressant effects at hypnotic doses of zaleplon in normal subjects, caution should be observed if zaleplon is prescribed to patients with compromised respiratory function, because sedative/hypnotics have the capacity to depress respiratory drive. Controlled trials of acute administration of zaleplon 10 mg in patients with mild to moderate chronic obstructive pulmonary disease or moderate obstructive sleep apnea showed no evidence of alterations in blood gases or apnea/hypopnea index, respectively. However, patients with compromised respiration due to preexisting illness should be monitored carefully.
The dose of zaleplon should be reduced to 5 mg in patients with mild to moderate hepatic impairment (see DOSAGE AND ADMINISTRATION). It is not recommended for use in patients with severe hepatic impairment.
No dose adjustment is necessary in patients with mild to moderate renal impairment. Zaleplon has not been adequately studied in patients with severe renal impairment.Use in Patients with Depression
In primarily depressed patients treated with sedative-hypnotics, worsening of depression, including suicidal thoughts and actions (including completed suicides), have been reported. As with other sedative/hypnotic drugs, zaleplon should be administered with caution to patients exhibiting signs or symptoms of depression. Suicidal tendencies may be present in such patients and protective measures may be required. Intentional overdosage is more common in this group of patients (see OVERDOSAGE); therefore, the least amount of drug that is feasible should be prescribed for the patient at any one time.
Allergic-Type Reactions FD&C Yellow # 5
This product contains FD&C Yellow #5 (tartrazine) which may cause allergic-type reactions (including bronchial asthma) in certain susceptible persons. Although the overall incidence of FD&C Yellow #5 (tartrazine) sensitivity in the general population is low, it is frequently seen in patients who also have aspirin hypersensitivity.
Information for Patients
A patient Medication Guide is also available for zaleplon. The prescriber or health professional should instruct patients, their families, and their caregivers to read the Medication Guide and should assist them in understanding its contents. Patients should be given the opportunity to discuss the contents of the Medication Guide and to obtain answers to any questions that they may have.
Instruct patients and their families that zaleplon may cause complex sleep behaviors, including sleep-walking, sleep-driving, preparing and eating food, making phone calls, or having sex while not being fully awake. Serious injuries and death have occurred during complex sleep behavior episodes. Tell patients to discontinue zaleplon and notify their healthcare provider immediately if they develop any of these symptoms (see BOXED WARNING and WARNINGS).
Advise patients that increased drowsiness and decreased consciousness may increase the risk of falls in some patients (see WARNINGS).Drug Interactions
As with all drugs, the potential exists for interaction with other drugs by a variety of mechanisms.
CNS-Active Drugs
Ethanol
Zaleplon 10 mg potentiated the CNS-impairing effects of ethanol 0.75 g/kg on balance testing and reaction time for 1 hour after ethanol administration and on the digit symbol substitution test (DSST), symbol copying test, and the variability component of the divided attention test for 2.5 hours after ethanol administration. The potentiation resulted from a CNS pharmacodynamic interaction; zaleplon did not affect the pharmacokinetics of ethanol.
Imipramine
Coadministration of single doses of zaleplon 20 mg and imipramine 75 mg produced additive effects on decreased alertness and impaired psychomotor performance for 2 to 4 hours after administration. The interaction was pharmacodynamic with no alteration of the pharmacokinetics of either drug.
Paroxetine
Coadministration of a single dose of zaleplon 20 mg and paroxetine 20 mg daily for 7 days did not produce any interaction on psychomotor performance. Additionally, paroxetine did not alter the pharmacokinetics of zaleplon, reflecting the absence of a role of CYP2D6 in zaleplon’s metabolism.
Thioridazine
Coadministration of single doses of zaleplon 20 mg and thioridazine 50 mg produced additive effects on decreased alertness and impaired psychomotor performance for 2 to 4 hours after administration. The interaction was pharmacodynamic with no alteration of the pharmacokinetics of either drug.
Venlafaxine
Coadministration of a single dose of zaleplon 10 mg and multiple doses of venlafaxine ER (extended release) 150 mg did not result in any significant changes in the pharmacokinetics of either zaleplon or venlafaxine. In addition, there was no pharmacodynamic interaction as a result of coadministration of zaleplon and venlafaxine ER.
Promethazine
Coadministration of a single dose of zaleplon and promethazine (10 and 25 mg, respectively) resulted in a 15% decrease in maximal plasma concentrations of zaleplon, but no change in the area under the plasma concentration-time curve. However, the pharmacodynamics of coadministration of zaleplon and promethazine have not been evaluated. Caution should be exercised when these 2 agents are coadministered.Drugs That Induce CYP3A4
Rifampin
CYP3A4 is ordinarily a minor metabolizing enzyme of zaleplon. Multiple-dose administration of the potent CYP3A4 inducer rifampin (600 mg every 24 hours, q24h, for 14 days), however, reduced zaleplon C max and AUC by approximately 80%. The coadministration of a potent CYP3A4 enzyme inducer, although not posing a safety concern, thus could lead to ineffectiveness of zaleplon. An alternative non-CYP3A4 substrate hypnotic agent may be considered in patients taking CYP3A4 inducers such as rifampin, phenytoin, carbamazepine, and phenobarbital.Drugs That Inhibit CYP3A4
CYP3A4 is a minor metabolic pathway for the elimination of zaleplon because the sum of desethylzaleplon (formed via CYP3A4 in vitro) and its metabolites, 5-oxo-desethylzaleplon and 5-oxo-desethylzaleplon glucuronide, account for only 9% of the urinary recovery of a zaleplon dose. Coadministration of single, oral doses of zaleplon with erythromycin (10 mg and 800 mg respectively), a strong, selective CYP3A4 inhibitor, produced a 34% increase in zaleplon’s maximal plasma concentrations and a 20% increase in the area under the plasma concentration-time curve. The magnitude of interaction with multiple doses of erythromycin is unknown. Other strong selective CYP3A4 inhibitors such as ketoconazole can also be expected to increase the exposure of zaleplon. A routine dosage adjustment of zaleplon is not considered necessary.
Drugs That Inhibit Aldehyde Oxidase
The aldehyde oxidase enzyme system is less well studied than the cytochrome P450 enzyme system.
Diphenhydramine: Diphenhydramine is reported to be a weak inhibitor of aldehyde oxidase in rat liver, but its inhibitory effects in human liver are not known. There is no pharmacokinetic interaction between zaleplon and diphenhydramine following the administration of a single dose (10 mg and 50 mg, respectively) of each drug. However, because both of these compounds have CNS effects, an additive pharmacodynamic effect is possible.Drugs That Inhibit Both Aldehyde Oxidase and CYP3A4
Cimetidine: Cimetidine inhibits both aldehyde oxidase ( in vitro) and CYP3A4 ( in vitro and in vivo), the primary and secondary enzymes, respectively, responsible for zaleplon metabolism. Concomitant administration of zaleplon (10 mg) and cimetidine (800 mg) produced an 85% increase in the mean C max and AUC of zaleplon. An initial dose of 5 mg should be given to patients who are concomitantly being treated with cimetidine (see DOSAGE AND ADMINISTRATION).
Drugs Highly Bound to Plasma Protein
Zaleplon is not highly bound to plasma proteins (fraction bound 60%±15%); therefore, the disposition of zaleplon is not expected to be sensitive to alterations in protein binding. In addition, administration of zaleplon to a patient taking another drug that is highly protein bound should not cause transient increase in free concentrations of the other drug.
Drugs with a Narrow Therapeutic Index
Digoxin: Zaleplon (10 mg) did not affect the pharmacokinetic or pharmacodynamic profile of digoxin (0.375 mg q24h for 8 days).
Warfarin: Multiple oral doses of zaleplon (20 mg q24h for 13 days) did not affect the pharmacokinetics of warfarin (R+)- or (S-)-enantiomers or the pharmacodynamics (prothrombin time) following a single 25 mg oral dose of warfarin.Drugs That Alter Renal Excretion
Ibuprofen: Ibuprofen is known to affect renal function and, consequently, alter the renal excretion of other drugs. There was no apparent pharmacokinetic interaction between zaleplon and ibuprofen following single dose administration (10 mg and 600 mg, respectively) of each drug. This was expected because zaleplon is primarily metabolized and renal excretion of unchanged zaleplon accounts for less than 1% of the administered dose.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Lifetime carcinogenicity studies of zaleplon were conducted in mice and rats. Mice received doses of 25 mg/kg/day, 50 mg/kg/day, 100 mg/kg/day, and 200 mg/kg/day in the diet for two years. These doses are equivalent to 6 to 49 times the maximum recommended human dose (MRHD) of 20 mg on a mg/m 2 basis. There was a significant increase in the incidence of hepatocellular adenomas in female mice in the high dose group. Rats received doses of 1 mg/kg/day, 10 mg/kg/day, and 20 mg/kg/day in the diet for two years. These doses are equivalent to 0.5 to 10 times the maximum recommended human dose (MRHD) of 20 mg on a mg/m 2 basis. Zaleplon was not carcinogenic in rats.
Mutagenesis
Zaleplon was clastogenic, both in the presence and absence of metabolic activation, causing structural and numerical aberrations (polyploidy and endoreduplication), when tested for chromosomal aberrations in the in vitro Chinese hamster ovary cell assay. In the in vitro human lymphocyte assay, zaleplon caused numerical, but not structural, aberrations only in the presence of metabolic activation at the highest concentrations tested. In other in vitro assays, zaleplon was not mutagenic in the Ames bacterial gene mutation assay or the Chinese hamster ovary HGPRT gene mutation assay. Zaleplon was not clastogenic in two in vivo assays, the mouse bone marrow micronucleus assay and the rat bone marrow chromosomal aberration assay, and did not cause DNA damage in the rat hepatocyte unscheduled DNA synthesis assay.
Impairment of Fertility
In a fertility and reproductive performance study in rats, mortality and decreased fertility were associated with administration of an oral dose of zaleplon of 100 mg/kg/day to males and females prior to and during mating. This dose is equivalent to 49 times the maximum recommended human dose (MRHD) of 20 mg on a mg/m 2 basis. Follow-up studies indicated that impaired fertility was due to an effect on the female.
Pregnancy
In embryofetal development studies in rats and rabbits, oral administration of up to 100 mg/kg/day and 50 mg/kg/day, respectively, to pregnant animals throughout organogenesis produced no evidence of teratogenicity. These doses are equivalent to 49 (rat) and 48 (rabbit) times the maximum recommended human dose (MRHD) of 20 mg on a mg/m 2 basis. In rats, pre- and postnatal growth was reduced in the offspring of dams receiving 100 mg/kg/day. This dose was also maternally toxic, as evidenced by clinical signs and decreased maternal body weight gain during gestation. The no-effect dose for rat offspring growth reduction was 10 mg/kg (a dose equivalent to 5 times the MRHD of 20 mg on a mg/m 2 basis). No adverse effects on embryofetal development were observed in rabbits at the doses examined.
In a pre- and postnatal development study in rats, increased stillbirth and postnatal mortality, and decreased growth and physical development, were observed in the offspring of females treated with doses of 7 mg/kg/day or greater during the latter part of gestation and throughout lactation. There was no evidence of maternal toxicity at this dose. The no-effect dose for offspring development was 1 mg/kg/day (a dose equivalent to 0.5 times the MRHD of 20 mg on a mg/m 2 basis). When the adverse effects on offspring viability and growth were examined in a cross-fostering study, they appeared to result from both in utero and lactational exposure to the drug.
There are no studies of zaleplon in pregnant women; therefore, zaleplon capsules are not recommended for use in women during pregnancy.Nursing Mothers
A study in lactating mothers indicated that the clearance and half-life of zaleplon is similar to that in young normal subjects. A small amount of zaleplon is excreted in breast milk, with the highest excreted amount occurring during a feeding at approximately 1 hour after zaleplon administration. Since the small amount of the drug from breast milk may result in potentially important concentrations in infants, and because the effects of zaleplon on a nursing infant are not known, it is recommended that nursing mothers not take zaleplon.
Pediatric Use
The safety and effectiveness of zaleplon in pediatric patients have not been established.
Geriatric Use
A total of 628 patients in double-blind, placebo-controlled, parallel-group clinical trials who received zaleplon were at least 65 years of age; of these, 311 received 5 mg and 317 received 10 mg. In both sleep laboratory and outpatient studies, elderly patients with insomnia responded to a 5 mg dose with a reduced sleep latency, and thus 5 mg is the recommended dose in this population. During short-term treatment (14 night studies) of elderly patients with zaleplon, no adverse event with a frequency of at least 1% occurred at a significantly higher rate with either 5 mg or 10 mg zaleplon than with placebo.
-
ADVERSE REACTIONS
The premarketing development program for zaleplon included zaleplon exposures in patients and/or normal subjects from 2 different groups of studies: approximately 900 normal subjects in clinical pharmacology/pharmacokinetic studies; and approximately 2,900 exposures from patients in placebo-controlled clinical effectiveness studies, corresponding to approximately 450 patient exposure years. The conditions and duration of treatment with zaleplon varied greatly and included (in overlapping categories) open-label and double-blind phases of studies, inpatients and outpatients, and short-term or longer-term exposure. Adverse reactions were assessed by collecting adverse events, results of physical examinations, vital signs, weights, laboratory analyses, and ECGs.
Adverse events during exposure were obtained primarily by general inquiry and recorded by clinical investigators using terminology of their own choosing. Consequently, it is not possible to provide a meaningful estimate of the proportion of individuals experiencing adverse events without first grouping similar types of events into a smaller number of standardized event categories. In the tables and tabulations that follow, COSTART terminology has been used to classify reported adverse events.
The stated frequencies of adverse events represent the proportion of individuals who experienced, at least once, a treatment-emergent adverse event of the type listed. An event was considered treatment-emergent if it occurred for the first time or worsened while receiving therapy following baseline evaluation.Adverse Findings Observed in Short-Term, Placebo-Controlled Trials
Adverse Events Associated With Discontinuation of Treatment
In premarketing placebo-controlled, parallel-group phase 2 and phase 3 clinical trials, 3.1% of 744 patients who received placebo and 3.7% of 2,149 patients who received zaleplon discontinued treatment because of an adverse clinical event. This difference was not statistically significant. No event that resulted in discontinuation occurred at a rate of ≥ 1%.
Adverse Events Occurring at an Incidence of 1% or More Among Zaleplon 20 mg-Treated Patients
Table 1 enumerates the incidence of treatment-emergent adverse events for a pool of three 28-night and one 35-night placebo-controlled studies of zaleplon at doses of 5 mg or 10 mg and 20 mg. The table includes only those events that occurred in 1% or more of patients treated with zaleplon 20 mg and that had a higher incidence in patients treated with zaleplon 20 mg than in placebo-treated patients.
The prescriber should be aware that these figures cannot be used to predict the incidence of adverse events in the course of usual medical practice where patient characteristics and other factors differ from those which prevailed in the clinical trials. Similarly, the cited frequencies cannot be compared with figures obtained from other clinical investigations involving different treatments, uses, and investigators. The cited figures, however, do provide the prescribing physician with some basis for estimating the relative contribution of drug and non-drug factors to the adverse event incidence rate in the population studied.
Table 1 Incidence (%) of Treatment-Emergent Adverse Events in Long-Term (28 and 35 Nights) Placebo-Controlled Clinical Trials of Zaleplon a Body System
Preferred TermPlacebo
(n = 344)Zaleplon
5 mg or 10 mg
(n = 569)Zaleplon
20 mg
(n = 297)a) Events for which the incidence for zaleplon 20 mg-treated patients was at least 1% and greater than the incidence among placebo-treated patients. Incidence greater than 1% has been rounded to the nearest whole number.
Body as a whole
Abdominal pain
3
6
6
Asthenia
5
5
7
Headache
35
30
42
Malaise
<1
<1
2
Photosensitivity reaction
<1
<1
1
Digestive system
Anorexia
<1
<1
2
Colitis
0
0
1
Nausea
7
6
8
Metabolic and nutritional
Peripheral edema
<1
<1
1
Nervous system
Amnesia
1
2
4
Confusion
<1
<1
1
Depersonalization
<1
<1
2
Dizziness
7
7
9
Hallucinations
<1
<1
1
Hypertonia
<1
1
1
Hypesthesia
<1
<1
2
Paresthesia
1
3
3
Somnolence
4
5
6
Tremor
1
2
2
Vertigo
<1
<1
1
Respiratory system
Epistaxis
<1
<1
1
Special senses
Abnormal vision
<1
<1
2
Ear pain
0
<1
1
Eye pain
2
4
3
Hyperacusis
<1
1
2
Parosmia
<1
<1
2
Urogenital system
Dysmenorrhea
2
3
4
Other Adverse Events Observed During the Premarketing Evaluation of Zaleplon
Listed below are COSTART terms that reflect treatment-emergent adverse events as defined in the introduction to the ADVERSE REACTIONS section. These events were reported by patients treated with zaleplon at doses in a range of 5 mg/day to 20 mg/day during premarketing phase 2 and phase 3 clinical trials throughout the United States, Canada, and Europe, including approximately 2,900 patients. All reported events are included except those already listed in Table 1 or elsewhere in labeling, those events for which a drug cause was remote, and those event terms that were so general as to be uninformative. It is important to emphasize that although the events reported occurred during treatment with zaleplon, they were not necessarily caused by it.
Events are further categorized by body system and listed in order of decreasing frequency according to the following definitions: frequent adverse events are those occurring on one or more occasions in at least 1/100 patients; infrequent adverse events are those occurring in less than 1/100 patients but at least 1/1,000 patients; rare events are those occurring in fewer than 1/1,000 patients.
Body as a whole - Frequent: back pain, chest pain, fever; Infrequent: chest pain substernal, chills, face edema, generalized edema, hangover effect, neck rigidity.
Cardiovascular system - Frequent: migraine; Infrequent: angina pectoris, bundle branch block, hypertension, hypotension, palpitation, syncope, tachycardia, vasodilatation, ventricular extrasystoles; Rare: bigeminy, cerebral ischemia, cyanosis, pericardial effusion, postural hypotension, pulmonary embolus, sinus bradycardia, thrombophlebitis, ventricular tachycardia.
Digestive system - Frequent: constipation, dry mouth, dyspepsia; Infrequent: eructation, esophagitis, flatulence, gastritis, gastroenteritis, gingivitis, glossitis, increased appetite, melena, mouth ulceration, rectal hemorrhage, stomatitis; Rare: aphthous stomatitis, biliary pain, bruxism, cardiospasm, cheilitis, cholelithiasis, duodenal ulcer, dysphagia, enteritis, gum hemorrhage, increased salivation, intestinal obstruction, abnormal liver function tests, peptic ulcer, tongue discoloration, tongue edema, ulcerative stomatitis.
Endocrine system - Rare: diabetes mellitus, goiter, hypothyroidism.
Hemic and lymphatic system - Infrequent: anemia, ecchymosis, lymphadenopathy; Rare: eosinophilia, leukocytosis, lymphocytosis, purpura.
Metabolic and nutritional - Infrequent: edema, gout, hypercholesteremia, thirst, weight gain; Rare: bilirubinemia, hyperglycemia, hyperuricemia, hypoglycemia, hypoglycemic reaction, ketosis, lactose intolerance, AST (SGOT) increased, ALT (SGPT) increased, weight loss.
Musculoskeletal system - Frequent: arthralgia, arthritis, myalgia; Infrequent: arthrosis, bursitis, joint disorder (mainly swelling, stiffness, and pain), myasthenia, tenosynovitis; Rare: myositis, osteoporosis.
Nervous system - Frequent: anxiety, depression, nervousness, thinking abnormal (mainly difficulty concentrating); Infrequent: abnormal gait, agitation, apathy, ataxia, circumoral paresthesia, emotional lability, euphoria, hyperesthesia, hyperkinesia, hypotonia, incoordination, insomnia, libido decreased, neuralgia, nystagmus; Rare: CNS stimulation, delusions, dysarthria, dystonia, facial paralysis, hostility, hypokinesia, myoclonus, neuropathy, psychomotor retardation, ptosis, reflexes decreased, reflexes increased, sleep talking, sleep walking, slurred speech, stupor, trismus.
Respiratory system - Frequent: bronchitis; Infrequent: asthma, dyspnea, laryngitis, pneumonia, snoring, voice alteration; Rare: apnea, hiccup, hyperventilation, pleural effusion, sputum increased.
Skin and appendages - Frequent: pruritus, rash; Infrequent: acne, alopecia, contact dermatitis, dry skin, eczema, maculopapular rash, skin hypertrophy, sweating, urticaria, vesiculobullous rash; Rare: melanosis, psoriasis, pustular rash, skin discoloration.
Special senses - Frequent: conjunctivitis, taste perversion; Infrequent: diplopia, dry eyes, photophobia, tinnitus, watery eyes; Rare: abnormality of accommodation, blepharitis, cataract specified, corneal erosion, deafness, eye hemorrhage, glaucoma, labyrinthitis, retinal detachment, taste loss, visual field defect.
Urogenital system - Infrequent: bladder pain, breast pain, cystitis, decreased urine stream, dysuria, hematuria, impotence, kidney calculus, kidney pain, menorrhagia, metrorrhagia, urinary frequency, urinary incontinence, urinary urgency, vaginitis; Rare: albuminuria, delayed menstrual period, leukorrhea, menopause, urethritis, urinary retention, vaginal hemorrhage. -
DRUG ABUSE AND DEPENDENCE
Controlled Substance Class
Zaleplon is classified as a Schedule IV controlled substance by federal regulation.
Abuse, Dependence, and Tolerance
Abuse and addiction are separate and distinct from physical dependence and tolerance. Abuse is characterized by misuse of the drug for non-medical purposes, often in combination with other psychoactive substances.
Physical dependence is a state of adaption that is manifested by a specific withdrawal syndrome that can be produced by abrupt cessation, rapid dose reduction, decreasing blood level of the drug and/or administration of an antagonist. Tolerance is a state of adaptation in which exposure to a drug induces changes that result in a diminution of one or more of the drug’s effects over time. Tolerance may occur to both the desired and undesired effects of drugs and may develop at different rates for different effects.
Addiction is a primary, chronic, neurobiological disease with genetic, psychosocial, and environmental factors influencing its development and manifestations. It is characterized by behaviors that include one or more of the following: impaired control over drug use, compulsive use, continued use despite harm, and craving. Drug addiction is a treatable disease, utilizing a multidisciplinary approach, but relapse is common.
Abuse
Two studies assessed the abuse liability of zaleplon at doses of 25 mg, 50 mg, and 75 mg in subjects with known histories of sedative drug abuse. The results of these studies indicate that zaleplon has an abuse potential similar to benzodiazepine and benzodiazepine-like hypnotics.
Dependence
The potential for developing physical dependence on zaleplon and a subsequent withdrawal syndrome was assessed in controlled studies of 14-, 28-, and 35-night durations and in open-label studies of 6- and 12-month durations by examining for the emergence of rebound insomnia following drug discontinuation. Some patients (mostly those treated with 20 mg) experienced a mild rebound insomnia on the first night following withdrawal that appeared to be resolved by the second night. The use of the Benzodiazepine Withdrawal Symptom Questionnaire and examination of any other withdrawal-emergent events did not detect any other evidence for a withdrawal syndrome following abrupt discontinuation of zaleplon therapy in pre-marketing studies.
However, available data cannot provide a reliable estimate of the incidence of dependence during treatment at recommended doses of zaleplon. Other sedative/hypnotics have been associated with various signs and symptoms following abrupt discontinuation, ranging from mild dysphoria and insomnia to a withdrawal syndrome that may include abdominal and muscle cramps, vomiting, sweating, tremors, and convulsions. Seizures have been observed in two patients, one of which had a prior seizure, in clinical trials with zaleplon. Seizures and death have been seen following the withdrawal of zaleplon from animals at doses many times higher than those proposed for human use. Because individuals with a history of addiction to, or abuse of, drugs or alcohol are at risk of habituation and dependence, they should be under careful surveillance when receiving zaleplon or any other hypnotic.
Tolerance
Possible tolerance to the hypnotic effects of zaleplon 10 mg and 20 mg was assessed by evaluating time to sleep onset for zaleplon compared with placebo in two 28-night placebo-controlled studies and latency to persistent sleep in one 35-night placebo-controlled study where tolerance was evaluated on nights 29 and 30. No development of tolerance to zaleplon was observed for time to sleep onset over 4 weeks.
-
OVERDOSAGE
Signs and Symptoms
Signs and symptoms of overdose effects of CNS depressants can be expected to present as exaggerations of the pharmacological effects noted in preclinical testing. Overdose is usually manifested by degrees of central nervous system depression ranging from drowsiness to coma. In mild cases, symptoms include drowsiness, mental confusion, and lethargy; in more serious cases, symptoms may include ataxia, hypotonia, hypotension, respiratory depression, rarely coma, and very rarely death.
Loss of consciousness, in addition to signs and symptoms consistent with CNS depressants as described above, have been reported following zaleplon overdose. Individuals have fully recovered from zaleplon overdoses of greater than 200 mg (10 times the maximum recommended dose of zaleplon). Rare instances of fatal outcomes following overdose with zaleplon, most often associated with overdose of additional CNS depressants, have been reported.Recommended Treatment
General symptomatic and supportive measures should be used along with immediate gastric lavage where appropriate. Intravenous fluids should be administered as needed. Animal studies suggest that flumazenil is an antagonist to zaleplon. However, there is no pre-marketing clinical experience with the use of flumazenil as an antidote to a zaleplon overdose. As in all cases of drug overdose, respiration, pulse, blood pressure, and other appropriate signs should be monitored and general supportive measures employed. Hypotension and CNS depression should be monitored and treated by appropriate medical intervention.
-
DOSAGE AND ADMINISTRATION
The dose of zaleplon should be individualized. The recommended dose of zaleplon for most nonelderly adults is 10 mg. For certain low weight individuals, 5 mg may be a sufficient dose. Although the risk of certain adverse events associated with the use of zaleplon appears to be dose dependent, the 20 mg dose has been shown to be adequately tolerated and may be considered for the occasional patient who does not benefit from a trial of a lower dose. Doses above 20 mg have not been adequately evaluated and are not recommended.
Zaleplon should be taken immediately before bedtime or after the patient has gone to bed and has experienced difficulty falling asleep (see PRECAUTIONS). Taking zaleplon with or immediately after a heavy, high-fat meal results in slower absorption and would be expected to reduce the effect of zaleplon on sleep latency (see Pharmacokinetics under CLINICAL PHARMACOLOGY).Special Populations
Elderly patients and debilitated patients appear to be more sensitive to the effects of hypnotics, and respond to 5 mg of zaleplon. The recommended dose for these patients is therefore 5 mg. Doses over 10 mg are not recommended.
Hepatic insufficiency
Patients with mild to moderate hepatic impairment should be treated with zaleplon 5 mg because clearance is reduced in this population. Zaleplon is not recommended for use in patients with severe hepatic impairment.
Renal insufficiency
No dose adjustment is necessary in patients with mild to moderate renal impairment. Zaleplon has not been adequately studied in patients with severe renal impairment.
An initial dose of 5 mg should be given to patients concomitantly taking cimetidine because zaleplon clearance is reduced in this population (see Drug Interactions under PRECAUTIONS). -
HOW SUPPLIED
Zaleplon Capsules USP, 5 mg are Opaque green/opaque pale green colored, size ‘4’ hard gelatin capsule filled with white to off-white powder and imprinted with ‘E19’ on opaque green cap and ‘5 mg’ on opaque pale green body with black ink.
Bottles of 30 NDC 43063-505-30
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [See USP Controlled Room Temperature]. Dispense in a light-resistant container as defined in the USP.
Dispense with Medication Guide available at:www.aurobindousa.com/medication-guides
-
MEDICATION GUIDE
Zaleplon Capsules USP CIV
(zal' e plon)
Rx only
Read this Medication Guide before you start taking zaleplon and each time you get a refill. There may be new information. This Medication Guide does not take the place of talking to your doctor about your medical condition or treatment. You and your doctor should talk about zaleplon when you start taking it and at regular checkups.
What is the most important information I should know about zaleplon?
Zaleplon may cause serious side effects including complex sleep behaviors that may cause serious injury and death. After taking zaleplon, you may get up out of bed while not being fully awake and do an activity that you do not know you are doing (complex sleep behaviors). The next morning, you may not remember that you did anything during the night. These activities may occur with zaleplon whether or not you drink alcohol or take other medicines that make you sleepy.
Reported activities include:- driving a car ("sleep-driving")
- making and eating food
- talking on the phone
- having sex
- sleep-walking
Important:
1. Take zaleplon exactly as prescribed
- Do not take more zaleplon than prescribed.
- Take zaleplon right before you get in bed, not sooner.
2. Do not take zaleplon if you:
- have ever experienced a complex sleep behavior (such as driving a car, making and eating food, talking on the phone or having sex while not fully awake) after taking zaleplon
- drink alcohol
- take other medicines that can make you sleepy. Talk to your doctor about all of your medicines. Your doctor will tell you if you can take zaleplon with your other medicines
- cannot get a full night’s sleep
3. Stop taking zaleplon and call your doctor right away if you find out that you have done any of the above activities after taking zaleplon.
What is zaleplon?
Zaleplon is a sedative-hypnotic (sleep) medicine. Zaleplon is used in adults for the short-term treatment of the symptom of trouble falling asleep from insomnia. Zaleplon does not treat other symptoms of insomnia which include waking up too early in the morning and waking up often during the night.
Zaleplon is not for children.
Zaleplon is a federally controlled substance (C-IV) because it can be abused or lead to dependence. Keep zaleplon in a safe place to prevent misuse and abuse. Selling or giving away zaleplon may harm others, and is against the law. Tell your doctor if you have ever abused or been dependent on alcohol, prescription medicines or street drugs.
Who should not take zaleplon?
Do not take zaleplon capsules if you are allergic to anything in them. See the end of this Medication Guide for a complete list of ingredients in zaleplon capsules.
Zaleplon may not be right for you. Before starting zaleplon, tell your doctor about all of your health conditions, including if you:
- have a history of depression, mental illness, or suicidal thoughts
- have a history of drug or alcohol abuse or addiction
- have kidney or liver disease
- have a lung disease or breathing problems
- are pregnant, planning to become pregnant, or breastfeeding
Tell your doctor about all of the medicines you take including prescription and nonprescription medicines, vitamins and herbal supplements. Medicines can interact, sometimes causing side effects. Do not take zaleplon with other medicines that can make you sleepy.
Know the medicines you take. Keep a list of your medicines with you to show your doctor and pharmacist each time you get a new medicine.
How should I take zaleplon?
- Take zaleplon exactly as prescribed. Do not take more zaleplon than prescribed for you.
- Take zaleplon right before you get into bed. Or you can take zaleplon after you have been in bed and have trouble falling asleep.
- Do not take zaleplon with or right after a meal.
- Do not take zaleplon unless you are able to get a full night’s sleep before you must be active again.
- Call your healthcare provider if your insomnia worsens or is not better within 7 to 10 days. This may mean that there is another condition causing your sleep problem.
- If you take too much zaleplon or overdose, call your doctor or poison control center right away, or get emergency treatment.
What are the possible side effects of zaleplon?
Serious side effects of zaleplon include:
- getting out of bed while not being fully awake and do an activity that you do not know you are doing. (See “What is the most important information I should know about zaleplon?")
- abnormal thoughts and behavior. Symptoms include more outgoing or aggressive behavior than normal, confusion, agitation, hallucinations, worsening of depression, and suicidal thoughts or actions.
- memory loss
- anxiety
- severe allergic reactions. Symptoms include swelling of the tongue or throat, trouble breathing, and nausea and vomiting. Get emergency medical help if you get these symptoms after taking zaleplon.
Call your doctor right away if you have any of the above side effects or any other side effects that worry you while using zaleplon.
Common side effects of zaleplon include:
- drowsiness
- lightheadedness
- dizziness
- “pins and needles” feeling on your skin
- difficulty with coordination
- You may still feel drowsy the next day after taking zaleplon. Do not drive or do other dangerous activities after taking zaleplon until you feel fully awake.
- You may have withdrawal symptoms when you stop taking zaleplon. Withdrawal symptoms include unpleasant feelings, stomach and muscle cramps, vomiting, sweating, shakiness, and rarely seizures. You may also have more trouble sleeping the first few nights after zaleplon is stopped. The problem usually goes away on its own after 1 or 2 nights.
These are not all the side effects of zaleplon. Ask your doctor or pharmacist for more information.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store zaleplon?- Store zaleplon at room temperature between 68° and 77°F (20º to 25ºC).
- Protect from light.
- Keep zaleplon and all medicines out of the reach of children.
General Information about zaleplon
- Medicines are sometimes prescribed for purposes not mentioned in a Medication Guide.
- Do not use zaleplon for a condition for which it was not prescribed.
- Do not give zaleplon to other people, even if they have the same condition. It may harm them and it is against the law.
This Medication Guide summarizes the most important information about zaleplon. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about zaleplon that was written for healthcare professionals.
If you would like more information, contact the Aurobindo Pharma USA, Inc. at 1-866-850-2876.
What are the ingredients in zaleplon capsules?
Active Ingredient: zaleplon
Inactive Ingredients: microcrystalline cellulose, lactose monohydrate, pregelatinized starch (maize), sodium lauryl sulfate, colloidal silicon dioxide, and stearic acid. The empty hard gelatin capsule shells contain FD&C Blue #1, FD&C Green #3, FD&C Yellow #5, titanium dioxide, and gelatin. The capsules are printed with edible ink containing black iron oxide and shellac.
- Zaleplon Capsules USP, 5 mg are Opaque green/opaque pale green colored, size ‘4’ hard gelatin capsule filled with white to off-white powder and imprinted with ‘E19’ on opaque green cap and ‘5 mg’ on opaque pale green body with black ink.
- Zaleplon Capsules USP, 10 mg are Opaque green/opaque light green colored, size ‘4’ hard gelatin capsule filled with white to off-white powder and imprinted with ‘E20’ on opaque green cap and ‘10 mg’ on opaque light green body with black ink.
Dispense with Medication Guide available at: www.aurobindousa.com/medication-guides
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Distributed by:
Aurobindo Pharma USA, Inc.
279 Princeton-Hightstown Road East Windsor, NJ 08520
Manufactured by:
Aurobindo Pharma Limited
Hyderabad-500 032, India
Revised: 10/2021
- PACKAGE LABEL-PRINCIPAL DISPLAY PANEL - 5 mg
-
INGREDIENTS AND APPEARANCE
ZALEPLON
zaleplon capsuleProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:43063-505(NDC:65862-214) Route of Administration ORAL DEA Schedule CIV Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ZALEPLON (UNII: S62U433RMH) (ZALEPLON - UNII:S62U433RMH) ZALEPLON 5 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) STARCH, CORN (UNII: O8232NY3SJ) SODIUM LAURYL SULFATE (UNII: 368GB5141J) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) STEARIC ACID (UNII: 4ELV7Z65AP) FD&C BLUE NO. 1 (UNII: H3R47K3TBD) FD&C GREEN NO. 3 (UNII: 3P3ONR6O1S) FD&C YELLOW NO. 5 (UNII: I753WB2F1M) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) GELATIN, UNSPECIFIED (UNII: 2G86QN327L) FERROSOFERRIC OXIDE (UNII: XM0M87F357) SHELLAC (UNII: 46N107B71O) Product Characteristics Color green (Opaque Green or Opaque Pale Green) Score no score Shape CAPSULE Size 14mm Flavor Imprint Code E19;5;mg Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:43063-505-30 30 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 05/17/2013 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078829 06/06/2008 Labeler - PD-Rx Pharmaceuticals, Inc. (156893695) Registrant - PD-Rx Pharmaceuticals, Inc. (156893695) Establishment Name Address ID/FEI Business Operations PD-Rx Pharmaceuticals, Inc. 156893695 repack(43063-505)
