Label: IRINOTECAN HYDROCHLORIDE injection



- NDC Code(s): 0143-9583-01
- Packager: Hikma Pharmaceuticals USA Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated June 28, 2020
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use irinotecan hydrochloride injection safely and effectively. See full prescribing information for Irinotecan Hydrochloride Injection, USP.
Irinotecan Hydrochloride Injection, USP
Initial U.S. Approval: 1996WARNING: DIARRHEA and MYELOSUPPRESSION
See full prescribing information for complete boxed warning.-
Early and late forms of diarrhea can occur. Early diarrhea may be accompanied by cholinergic symptoms which may be prevented or ameliorated by atropine. Late diarrhea can be life threatening and should be treated promptly with loperamide. Monitor patients with diarrhea and give fluid and electrolytes as needed. Institute antibiotic therapy if patients develop ileus, fever, or severe neutropenia. Interrupt irinotecan hydrochloride injection and reduce subsequent doses if severe diarrhea occurs. (2.1, 5.1)
-
Severe myelosuppression may occur. (5.2)
RECENT MAJOR CHANGES
Warnings and Precautions, Embryo-Fetal Toxicity (5.9) 01/2020
INDICATIONS AND USAGE
Irinotecan Hydrochloride Injection, USP is a topoisomerase inhibitor indicated for patients with metastatic carcinoma of the colon or rectum whose disease has recurred or progressed following initial fluorouracil-based therapy. (1)
DOSAGE AND ADMINISTRATION
- Colorectal cancer single agent regimen 1: irinotecan hydrochloride injection 125 mg/m2 intravenous infusion over 90 minutes on days 1, 8, 15, 22 then 2-week rest. (2.1)
- Colorectal cancer single agent regimen 2: irinotecan hydrochloride injection 350 mg/m2 intravenous infusion over 90 minutes on day 1 every 3 weeks. (2.1)
DOSAGE FORMS AND STRENGTHS
Irinotecan hydrochloride injection is available in two single-dose sizes: (3)
- 2 mL-fill vial containing 40 mg irinotecan hydrochloride injection
- 5 mL-fill vial containing 100 mg irinotecan hydrochloride injection
CONTRAINDICATIONS
- Hypersensitivity to irinotecan hydrochloride injection or its excipients (4)
WARNINGS AND PRECAUTIONS
- Diarrhea and cholinergic reactions: Early diarrhea (occurring during or shortly after infusion of irinotecan hydrochloride injection) is usually transient and may be accompanied by cholinergic symptoms. Consider prophylactic or therapeutic administration of 0.25 mg to 1 mg of intravenous or subcutaneous atropine (unless clinically contraindicated). Late diarrhea (generally occurring more than 24 hours after administration of irinotecan hydrochloride injection) can occur. Monitor and replace fluid and electrolytes. Treat with loperamide. Use antibiotic support for ileus and fever. Interrupt irinotecan hydrochloride injection and reduce subsequent doses if severe diarrhea occurs. (5.1)
- Myelosuppression: Manage promptly with antibiotic support. Interrupt irinotecan hydrochloride injection and reduce subsequent doses if necessary. (5.2)
- Patients with Reduced UGT1A1 Activity: Individuals who are homozygous for the UGT1A1*28 allele are at increased risk for neutropenia following initiation of irinotecan hydrochloride injection treatment. (5.3)
- Hypersensitivity: Hypersensitivity reactions including severe anaphylactic or anaphylactoid reactions have been observed. Discontinue irinotecan hydrochloride injection if this occurs. (5.4)
- Renal Impairment/Renal Failure: Rare cases of renal impairment and acute renal failure have been identified, usually in patients who became volume depleted from severe vomiting and/or diarrhea. (5.5)
- Pulmonary Toxicity: Interstitial Pulmonary Disease (IPD)-like events, including fatalities, have occurred. Interrupt for new or progressive dysnpnea, cough, and fever pending evaluation. If IPD diagnosed, discontinue and institute appropriate treatment as needed. (5.6)
- Toxicity of the 5 Day Regimen: Irinotecan hydrochloride injection should not be used in combination with a regimen of 5-FU/LV administered for 4–5 consecutive days every 4 weeks outside of a clinical study. (5.7)
-
Embryo-Fetal Toxicity: Irinotecan hydrochloride injection can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. Advise male patients with female partners of reproductive potential to use condoms. (5.9, 8.1, 8.3)
- Patients with Hepatic Impairment: In clinical trials, irinotecan hydrochloride injection has not been administered to patients with serum bilirubin > 2.0 mg/dL, or transaminases > 3 times ULN if no liver metastases, or transaminases > 5 times ULN if liver metastases. With the weekly dosage schedule, patients with total bilirubin levels 1.0–2.0 mg/dL had greater likelihood of grade 3–4 neutropenia. (5.10)
ADVERSE REACTIONS
Common adverse reactions (≥30%) observed in single agent therapy clinical studies are: nausea, vomiting, abdominal pain, diarrhea, constipation, anorexia, neutropenia, leukopenia (including lymphocytopenia), anemia, asthenia, fever, body weight decreasing, alopecia. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Hikma Pharmaceuticals USA Inc. at 1-877-845-0689 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong CYP3A4 Inducers: Do not administer strong CYP3A4 inducers with Irinotecan Hydrochloride Injection. (7.2)
- Strong CYP3A4 Inhibitors: Do not administer strong CYP3A4 inhibitors with Irinotecan Hydrochloride Injection. (7.3)
USE IN SPECIFIC POPULATIONS
-
Lactation: Advise not to breastfeed. (8.2)
- Geriatric Use: Closely monitor patients greater than 65 years of age because of a greater risk of early and late diarrhea in this population. (8.5)
- Patients with Renal Impairment: Use caution and do not use in patients on dialysis. (8.6)
- Patients with Hepatic Impairment: Use caution. (5.10, 8.7, 12.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 6/2020
-
Early and late forms of diarrhea can occur. Early diarrhea may be accompanied by cholinergic symptoms which may be prevented or ameliorated by atropine. Late diarrhea can be life threatening and should be treated promptly with loperamide. Monitor patients with diarrhea and give fluid and electrolytes as needed. Institute antibiotic therapy if patients develop ileus, fever, or severe neutropenia. Interrupt irinotecan hydrochloride injection and reduce subsequent doses if severe diarrhea occurs. (2.1, 5.1)
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DIARRHEA AND MYELOSUPPRESSION
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Colorectal Single Agent Regimens 1 and 2
2.2 Dosage in Patients with Reduced UGT1A1 Activity
2.3 Premedication
2.4 Preparation of Infusion Solution
2.5 Safe Handling
2.6 Extravasation
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea and Cholinergic Reactions
5.2 Myelosuppression
5.3 Patients With Reduced UGT1A1 Activity
5.4 Hypersensitivity
5.5 Renal Impairment/Renal Failure
5.6 Pulmonary Toxicity
5.7 Toxicity of the 5 Day Regimen
5.8 Increased Toxicity in Patients with Performance Status 2
5.9 Embryo-Fetal Toxicity
5.10 Patients with Hepatic Impairment
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 Inducers
7.2 Strong CYP3A4 or UGT1A1 Inhibitors
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Metastatic Colorectal Cancer
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DIARRHEA AND MYELOSUPPRESSION
-
Early and late forms of diarrhea can occur. Early diarrhea may be accompanied by cholinergic symptoms which may be prevented or ameliorated by atropine. Late diarrhea can be life threatening and should be treated promptly with loperamide. Monitor patients with diarrhea and give fluid and electrolytes as needed. Institute antibiotic therapy if patients develop ileus, fever, or severe neutropenia. Interrupt irinotecan hydrochloride injection and reduce subsequent doses if severe diarrhea occurs [see Dosage and Administration (2.1) and Warnings and Precautions (5.1)].
- Severe myelosuppression may occur [see Warnings and Precautions (5.2)].
-
Early and late forms of diarrhea can occur. Early diarrhea may be accompanied by cholinergic symptoms which may be prevented or ameliorated by atropine. Late diarrhea can be life threatening and should be treated promptly with loperamide. Monitor patients with diarrhea and give fluid and electrolytes as needed. Institute antibiotic therapy if patients develop ileus, fever, or severe neutropenia. Interrupt irinotecan hydrochloride injection and reduce subsequent doses if severe diarrhea occurs [see Dosage and Administration (2.1) and Warnings and Precautions (5.1)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Colorectal Single Agent Regimens 1 and 2
Administer irinotecan hydrochloride injection as a 90-minute intravenous infusion. The currently recommended regimens are shown in Table 1.
A reduction in the starting dose by one dose level of irinotecan hydrochloride injection may be considered for patients with any of the following conditions: prior pelvic/abdominal radiotherapy, performance status of 2, or increased bilirubin levels. Dosing for patients with bilirubin >2 mg/dL cannot be recommended because there is insufficient information to recommend a dose in these patients.
Table 1. Single-Agent Regimens of Irinotecan Hydrochloride Injection and Dose Modifications - *
-
Subsequent doses may be adjusted as high as 150 mg/m2 or to as low as 50 mg/m2 in 25 to 50 mg/m2 decrements depending upon individual patient tolerance.
- †
-
Provided intolerable toxicity does not develop, treatment with additional cycles may be continued indefinitely as long as patients continue to experience clinical benefit.
- ‡
- Subsequent doses may be adjusted as low as 200 mg/m2 in 50 mg/m2 decrements depending upon individual patient tolerance.
Regimen 1
(weekly)*125 mg/m2 intravenous infusion over 90 minutes, days 1,8,15,22 then 2-week rest Starting Dose and Modified Dose Levels†(mg/m2) Starting Dose Dose Level -1 Dose Level -2 125 100 75 Regimen 2
(every 3 weeks)‡350 mg/m2 intravenous infusion over 90 minutes, once every 3 weeks† Starting Dose and Modified Dose Levels (mg/m2) Starting Dose Dose Level -1 Dose Level -2 350 300 250 Dose Modifications
Based on recommended dose-levels described in Table 1, Single-Agent Regimens of Irinotecan Hydrochloride Injection and Dose Modifications, subsequent doses should be adjusted as suggested in Table 2, Recommended Dose Modifications for Single-Agent Schedules. All dose modifications should be based on the worst preceding toxicity.
Table 2. Recommended Dose Modifications For Single-Agent Schedules* A new cycle of therapy should not begin until the granulocyte count has recovered to ≥1500/mm3, and the platelet count has recovered to ≥100,000/mm3, and treatment-related diarrhea is fully resolved. Treatment should be delayed 1 to 2 weeks to allow for recovery from treatment-related toxicities. If the patient has not recovered after a 2-week delay, consideration should be given to discontinuing irinotecan hydrochloride injection. Worst Toxicity
NCI Grade† (Value)
During a Cycle of Therapy At the Start of the Next Cycles of Therapy (After Adequate Recovery), Compared with the Starting Dose in the Previous Cycle* Weekly Weekly Once Every 3 Weeks No toxicity Maintain dose level ↑ 25 mg/m2 up to a maximum dose of 150 mg/m2 Maintain dose level Neutropenia 1 (1500 to 1999/mm3) Maintain dose level Maintain dose level Maintain dose level 2 (1000 to 1499/mm3) ↓ 25 mg/m2 Maintain dose level Maintain dose level 3 (500 to 999/mm3) Omit dose until resolved to ≤ grade 2, then ↓ 25 mg/m2 ↓ 25 mg/m2 ↓ 50 mg/m2 4 (<500/mm3) Omit dose until resolved to ≤ grade 2, then ↓ 50 mg/m2 ↓ 50 mg/m2 ↓ 50 mg/m2 Neutropenic fever Omit dose until resolved, then ↓ 50 mg/m2 when resolved ↓ 50 mg/m2 ↓ 50 mg/m2 Other hematologic toxicities Dose modifications for leukopenia, thrombocytopenia, and anemia during a cycle of therapy and at the start of subsequent cycles of therapy are also based on NCI toxicity criteria and are the same as recommended for neutropenia above. Diarrhea 1 (2–3 stools/day > pretx‡) Maintain dose level Maintain dose level Maintain dose level 2 (4–6 stools/day > pretx) ↓ 25 mg/m2 Maintain dose level Maintain dose level 3 (7–9 stools/day > pretx) Omit dose until resolved to ≤ grade 2, then ↓ 25 mg/m2 ↓ 25 mg/m2 ↓ 50 mg/m2 4 (≥10 stools/day > pretx) Omit dose until resolved to ≤ grade 2 then ↓ 50 mg/m2 ↓ 50 mg/m2 ↓ 50 mg/m2 Other nonhematologic§
toxicities1 Maintain dose level Maintain dose level Maintain dose level 2 ↓ 25 mg/m2 ↓ 25 mg/m2 ↓ 50 mg/m2 3 Omit dose until resolved to ≤ grade 2, then ↓ 25 mg/m2 ↓ 25 mg/m2 ↓ 50 mg/m2 4 Omit dose until resolved to ≤ grade 2, then ↓ 50 mg/m2 ↓ 50 mg/m2 ↓ 50 mg/m2 2.2 Dosage in Patients with Reduced UGT1A1 Activity
When administered in combination with other agents, or as a single-agent, a reduction in the starting dose by at least one level of irinotecan hydrochloride injection should be considered for patients known to be homozygous for the UGT1A1*28 allele [see Dosage and Administration (2.1) and Warnings and Precautions (5.3)]. However, the precise dose reduction in this patient population is not known, and subsequent dose modifications should be considered based on individual patient tolerance to treatment (see Tables 1–2).
2.3 Premedication
It is recommended that patients receive premedication with antiemetic agents. In clinical studies of the weekly dosage schedule, the majority of patients received 10 mg of dexamethasone given in conjunction with another type of antiemetic agent, such as a 5-HT3 blocker (e.g., ondansetron or granisetron). Antiemetic agents should be given on the day of treatment, starting at least 30 minutes before administration of irinotecan hydrochloride injection. Physicians should also consider providing patients with an antiemetic regimen (e.g., prochlorperazine) for subsequent use as needed.
Prophylactic or therapeutic administration of atropine should be considered in patients experiencing cholinergic symptoms.
2.4 Preparation of Infusion Solution
Inspect vial contents for particulate matter and discoloration and repeat inspection when drug product is withdrawn from vial into syringe.
Irinotecan hydrochloride injection 20 mg/mL is intended for single use only and any unused portion should be discarded.
Irinotecan hydrochloride injection must be diluted prior to infusion. Irinotecan hydrochloride injection should be diluted in 5% Dextrose Injection, USP, (preferred) or 0.9% Sodium Chloride Injection, USP, to a final concentration range of 0.12 mg/mL to 2.8 mg/mL. Other drugs should not be added to the infusion solution.
The solution is physically and chemically stable for up to 24 hours at room temperature and in ambient fluorescent lighting. Solutions diluted in 5% Dextrose Injection, USP, and stored at refrigerated temperatures (approximately 2° to 8°C, 36° to 46°F), and protected from light are physically and chemically stable for 48 hours. Refrigeration of admixtures using 0.9% Sodium Chloride Injection, USP, is not recommended due to a low and sporadic incidence of visible particulates. Freezing irinotecan hydrochloride injection and admixtures of irinotecan hydrochloride injection may result in precipitation of the drug and should be avoided.
The irinotecan hydrochloride injection solution should be used immediately after reconstitution as it contains no antibacterial preservative. Because of possible microbial contamination during dilution, it is advisable to use the admixture prepared with 5% Dextrose Injection, USP, within 24 hours if refrigerated (2° to 8°C, 36° to 46°F). In the case of admixtures prepared with 5% Dextrose Injection, USP, or Sodium Chloride Injection, USP, the solutions should be used within 4 hours if kept at room temperature. If reconstitution and dilution are performed under strict aseptic conditions (e.g. on Laminar Air Flow bench), irinotecan hydrochloride injection solution should be used (infusion completed) within 12 hours at room temperature or 24 hours if refrigerated (2° to 8°C, 36° to 46°F).
2.5 Safe Handling
Care should be exercised in the handling and preparation of infusion solutions prepared from irinotecan hydrochloride injection. The use of gloves is recommended. If a solution of irinotecan hydrochloride injection contacts the skin, wash the skin immediately and thoroughly with soap and water. If irinotecan hydrochloride injection contacts the mucous membranes, flush thoroughly with water. Several published guidelines for handling and disposal of anticancer agents are available.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Diarrhea and Cholinergic Reactions
Early diarrhea (occurring during or shortly after infusion of irinotecan hydrochloride injection) is usually transient and infrequently severe. It may be accompanied by cholinergic symptoms of rhinitis, increased salivation, miosis, lacrimation, diaphoresis, flushing, and intestinal hyperperistalsis that can cause abdominal cramping. Bradycardia may also occur. Early diarrhea and other cholinergic symptoms may be prevented or treated. Consider prophylactic or therapeutic administration of 0.25 mg to 1 mg of intravenous or subcutaneous atropine (unless clinically contraindicated). These symptoms are expected to occur more frequently with higher irinotecan doses.
Late diarrhea (generally occurring more than 24 hours after administration of irinotecan hydrochloride injection) can be life threatening since it may be prolonged and may lead to dehydration, electrolyte imbalance, or sepsis. Grade 3–4 late diarrhea occurred in 23–31% of patients receiving weekly dosing. In the clinical studies, the median time to the onset of late diarrhea was 5 days with 3-week dosing and 11 days with weekly dosing. Late diarrhea can be complicated by colitis, ulceration, bleeding, ileus, obstruction, and infection. Cases of megacolon and intestinal perforation have been reported. Patients should have loperamide readily available to begin treatment for late diarrhea. Begin loperamide at the first episode of poorly formed or loose stools or the earliest onset of bowel movements more frequent than normal. One dosage regimen for loperamide is 4 mg at the first onset of late diarrhea and then 2 mg every 2 hours until the patient is diarrhea-free for at least 12 hours. Loperamide is not recommended to be used for more than 48 consecutive hours at these doses, because of the risk of paralytic ileus. During the night, the patient may take 4 mg of loperamide every 4 hours. Monitor and replace fluid and electrolytes. Use antibiotic support for ileus, fever, or severe neutropenia. Subsequent weekly chemotherapy treatments should be delayed in patients until return of pretreatment bowel function for at least 24 hours without anti-diarrhea medication. Patients must not be treated with irinotecan until resolution of the bowel obstruction. If grade 2, 3, or 4 late diarrhea recurs, subsequent doses of irinotecan hydrochloride injection should be decreased [see Dosage and Administration (2)].
Avoid diuretics or laxatives in patients with diarrhea.
5.2 Myelosuppression
Irinotecan Hydrochloride Injection can cause severe myelosuppression. Bacterial, viral, and fungal infections have occurred in patients treated with Irinotecan Hydrochloride Injection.
Deaths due to sepsis following severe neutropenia have been reported in patients treated with irinotecan hydrochloride injection. In the clinical studies evaluating the weekly dosage schedule, neutropenic fever (concurrent NCI grade 4 neutropenia and fever of grade 2 or greater) occurred in 3% of the patients; 6% of patients received G-CSF for the treatment of neutropenia. Manage febrile neutropenia promptly with antibiotic support [see Warnings and Precautions (5.1)]. Hold irinotecan hydrochloride injection if neutropenic fever occurs or if the absolute neutrophil count drops <1000/mm3. After recovery to an absolute neutrophil count ≥1000/mm3, subsequent doses of irinotecan hydrochloride injection should be reduced [see Dosage and Administration (2)].
When evaluated in the trials of weekly administration, the frequency of grade 3 and 4 neutropenia was higher in patients who received previous pelvic/abdominal irradiation than in those who had not received such irradiation (48% [13/27] versus 24% [67/277]; p=0.04). Patients who have previously received pelvic/abdominal irradiation are at increased risk of severe myelosuppression following the administration of irinotecan hydrochloride injection. Based on sparse available data, the concurrent administration of irinotecan hydrochloride injection with irradiation is not recommended.
Patients with baseline serum total bilirubin levels of 1.0 mg/dL or more also had a greater likelihood of experiencing first-cycle grade 3 or 4 neutropenia than those with bilirubin levels that were less than 1.0 mg/dL (50% [19/38] versus 18% [47/266]; p<0.001). Patients with deficient glucuronidation of bilirubin, such as those with Gilbert's syndrome, may be at greater risk of myelosuppression when receiving therapy with irinotecan hydrochloride injection.
5.3 Patients With Reduced UGT1A1 Activity
Individuals who are homozygous for the UGT1A1*28 allele (UGT1A1 7/7 genotype) are at increased risk for neutropenia following initiation of irinotecan hydrochloride injection treatment.
In a study of 66 patients who received single-agent irinotecan hydrochloride injection (350 mg/m2 once-every-3-weeks), the incidence of grade 4 neutropenia in patients homozygous for the UGT1A1*28 allele was 50%, and in patients heterozygous for this allele (UGT1A1 6/7 genotype) the incidence was 12.5%. No grade 4 neutropenia was observed in patients homozygous for the wild-type allele (UGT1A1 6/6 genotype).
When administered in combination with other agents or as a single-agent, a reduction in the starting dose by at least one level of irinotecan hydrochloride injection should be considered for patients known to be homozygous for the UGT1A1*28 allele. However, the precise dose reduction in this patient population is not known and subsequent dose modifications should be considered based on individual patient tolerance to treatment [see Dosage and Administration (2)].
UGT1A1 Testing
A laboratory test is available to determine the UGT1A1 status of patients. Testing can detect the UGT1A1 6/6, 6/7 and 7/7 genotypes.5.4 Hypersensitivity
Hypersensitivity reactions including severe anaphylactic or anaphylactoid reactions have been observed. Discontinue irinotecan hydrochloride injection if anaphylactic reaction occurs.
5.5 Renal Impairment/Renal Failure
Renal impairment and acute renal failure have been identified, usually in patients who became volume depleted from severe vomiting and/or diarrhea.
5.6 Pulmonary Toxicity
Interstitial Pulmonary Disease (IPD)-like events, including fatalities, have occurred in patients receiving irinotecan (in combination with 5-FU/LV and as monotherapy). Risk factors include pre-existing lung disease, use of pneumotoxic drugs, radiation therapy, and colony stimulating factors. Patients with risk factors should be closely monitored for respiratory symptoms before and during irinotecan hydrochloride injection therapy. In Japanese studies, a reticulonodular pattern on chest x-ray was observed in a small percentage of patients. New or progressive, dyspnea, cough, and fever should prompt interruption of chemotherapy, pending diagnostic evaluation. If IPD is diagnosed, irinotecan hydrochloride injection and other chemotherapy should be discontinued and appropriate treatment instituted as needed [see Adverse Reactions (6.1)].
5.7 Toxicity of the 5 Day Regimen
Outside of a well-designed clinical study, irinotecan hydrochloride injection should not be used in combination with a regimen of 5-FU/LV administered for 4–5 consecutive days every 4 weeks because of reports of increased toxicity, including toxic deaths. Irinotecan hydrochloride injection should be used as recommended in Table 2 [see Dosage and Administration (2)].
5.8 Increased Toxicity in Patients with Performance Status 2
In the clinical trials, higher rates of hospitalization, neutropenic fever, thromboembolism, first-cycle treatment discontinuation, and early deaths were observed in patients with a baseline performance status of 2 than in patients with a baseline performance status of 0 or 1.
5.9 Embryo-Fetal Toxicity
Based on its mechanism of action and findings in animals, irinotecan hydrochloride injection can cause fetal harm when administered to a pregnant woman. In animal studies, intravenous administration of irinotecan during the period of organogenesis resulted in embryofetal mortality and teratogenicity in pregnant animals at exposures lower than the human exposure based on area under the curve (AUC) at the clinical dose of 125 mg/m2. Advise pregnant women of the potential risk to a fetus.
Advise female patients of reproductive potential to avoid becoming pregnant and to use highly effective contraception during treatment with irinotecan hydrochloride injection and for 6 months after the final dose. Advise male patients with female partners of reproductive potential to use condoms during treatment and for 3 months after the final dose of irinotecan hydrochloride injection [see Use in Specific Populations (8.1), (8.3) and Nonclinical Toxicology (13.1)].
5.10 Patients with Hepatic Impairment
The use of irinotecan hydrochloride injection in patients with significant hepatic impairment has not been established. In clinical trials of either dosing schedule, irinotecan was not administered to patients with serum bilirubin >2.0 mg/dL, or transaminase >3 times the upper limit of normal if no liver metastasis, or transaminase >5 times the upper limit of normal with liver metastasis. In clinical trials of the weekly dosage schedule, patients with modestly elevated baseline serum total bilirubin levels (1.0 to 2.0 mg/dL) had a significantly greater likelihood of experiencing first-cycle, grade 3 or 4 neutropenia than those with bilirubin levels that were less than 1.0 mg/dL (50% [19/38] versus 18% [47/226]; p<0.001) [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
-
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Common adverse reactions (≥30%) observed in single agent therapy clinical studies are: nausea, vomiting, abdominal pain, diarrhea, constipation, anorexia, neutropenia, leukopenia (including lymphocytopenia), anemia, asthenia, fever, body weight decreasing, and alopecia.
Second-Line Single-Agent Therapy
Weekly Dosage Schedule
In three clinical studies evaluating the weekly dosage schedule, 304 patients with metastatic carcinoma of the colon or rectum that had recurred or progressed following 5-FU-based therapy were treated with irinotecan hydrochloride injection. Seventeen of the patients died within 30 days of the administration of irinotecan hydrochloride injection; in five cases (1.6%, 5/304), the deaths were potentially drug-related. One of the patients died of neutropenic sepsis without fever. Neutropenic fever occurred in nine (3.0%) other patients; these patients recovered with supportive care.One hundred nineteen (39.1%) of the 304 patients were hospitalized because of adverse events; 81 (26.6%) patients were hospitalized for events judged to be related to administration of irinotecan hydrochloride injection. The primary reasons for drug-related hospitalization were diarrhea, with or without nausea and/or vomiting (18.4%); neutropenia/leukopenia, with or without diarrhea and/or fever (8.2%); and nausea and/or vomiting (4.9%).
The first dose of at least one cycle of irinotecan hydrochloride injection was reduced for 67% of patients who began the studies at the 125-mg/m2 starting dose. Within-cycle dose reductions were required for 32% of the cycles initiated at the 125-mg/m2 dose level. The most common reasons for dose reduction were late diarrhea, neutropenia, and leukopenia. Thirteen (4.3%) patients discontinued treatment with irinotecan hydrochloride injection because of adverse events. The adverse events in Table 3 are based on the experience of the 304 patients enrolled in the three studies described in Clinical Studies (14.1).
Table 3. Adverse Events Occurring in >10% of 304 Previously Treated Patients with Metastatic Carcinoma of the Colon or Rectum* - *
- Severity of adverse events based on NCI CTC (version 1.0)
- †
- Occurring > 24 hours after administration of irinotecan hydrochloride injection
- ‡
- Occurring ≤24 hours after administration of irinotecan hydrochloride injection
- §
- Primarily upper respiratory infections
- ¶
- Not applicable; complete hair loss = NCI grade 2
% of Patients Reporting Body System & Event NCI Grades 1–4 NCI Grades 3 & 4 GASTROINTESTINAL Diarrhea (late)† 88 31 7–9 stools/day (grade 3) - (16) ≥10 stools/day (grade 4) - (14) Nausea 86 17 Vomiting 67 12 Anorexia 55 6 Diarrhea (early)‡ 51 8 Constipation 30 2 Flatulence 12 0 Stomatitis 12 1 Dyspepsia 10 0 HEMATOLOGIC Leukopenia 63 28 Anemia 60 7 Neutropenia 54 26 500 to <1000/mm3 (grade 3) - (15) <500/mm3 (grade 4) - (12) BODY AS A WHOLE Asthenia 76 12 Abdominal cramping/pain 57 16 Fever 45 1 Pain 24 2 Headache 17 1 Back pain 14 2 Chills 14 0 Minor infection§ 14 0 Edema 10 1 Abdominal enlargement 10 0 METABOLIC AND NUTRITIONAL ↓ Body weight 30 1 Dehydration 15 4 ↑ Alkaline phosphatase 13 4 ↑ SGOT 10 1 DERMATOLOGIC Alopecia 60 NA¶ Sweating 16 0 Rash 13 1 RESPIRATORY Dyspnea 22 4 ↑ Coughing 17 0 Rhinitis 16 0 NEUROLOGIC Insomnia 19 0 Dizziness 15 0 CARDIOVASCULAR Vasodilation (flushing) 11 0 Once-Every-3-Week Dosage Schedule
A total of 535 patients with metastatic colorectal cancer whose disease had recurred or progressed following prior 5-FU therapy participated in the two phase 3 studies: 316 received irinotecan, 129 received 5-FU, and 90 received best supportive care. Eleven (3.5%) patients treated with irinotecan died within 30 days of treatment. In three cases (1%, 3/316), the deaths were potentially related to irinotecan treatment and were attributed to neutropenic infection, grade 4 diarrhea, and asthenia, respectively. One (0.8%, 1/129) patient treated with 5-FU died within 30 days of treatment; this death was attributed to grade 4 diarrhea.Hospitalizations due to serious adverse events occurred at least once in 60% (188/316) of patients who received irinotecan, 63% (57/90) who received best supportive care, and 39% (50/129) who received 5-FU-based therapy. Eight percent of patients treated with irinotecan and 7% treated with 5-FU-based therapy discontinued treatment due to adverse events.
Of the 316 patients treated with irinotecan, the most clinically significant adverse events (all grades, 1–4) were diarrhea (84%), alopecia (72%), nausea (70%), vomiting (62%), cholinergic symptoms (47%), and neutropenia (30%). Table 4 lists the grade 3 and 4 adverse events reported in the patients enrolled to all treatment arms of the two studies described in Clinical Studies (14.1).
Table 4. Percent Of Patients Experiencing Grade 3 & 4 Adverse Events In Comparative Studies Of Once-Every-3-Week Irinotecan Therapy* - *
- Severity of adverse events based on NCI CTC (version 1.0)
- †
- BSC = best supportive care
- ‡
- Hepatic includes events such as ascites and jaundice
- §
- Cutaneous signs include events such as rash
- ¶
- Respiratory includes events such as dyspnea and cough
- #
-
Neurologic includes events such as somnolence
- Þ
- Cardiovascular includes events such as dysrhythmias, ischemia, and mechanical cardiac dysfunction
- ß
-
Other includes events such as accidental injury, hepatomegaly, syncope, vertigo, and weight loss
Adverse EventStudy 1 Study 2 Irinotecan
N=189BSC †
N=90Irinotecan
N=1275-FU
N=129TOTAL Grade 3/4 Adverse Events 79 67 69 54 GASTROINTESTINAL Diarrhea 22 6 22 11 Vomiting 14 8 14 5 Nausea 14 3 11 4 Abdominal pain 14 16 9 8 Constipation 10 8 8 6 Anorexia 5 7 6 4 Mucositis 2 1 2 5 HEMATOLOGIC Leukopenia/Neutropenia 22 0 14 2 Anemia 7 6 6 3 Hemorrhage 5 3 1 3 Thrombocytopenia 1 0 4 2 Infection without grade 3/4 neutropenia 8 3 1 4 with grade 3/4 neutropenia 1 0 2 0 Fever without grade 3/4 neutropenia 2 1 2 0 with grade 3/4 neutropenia 2 0 4 2 BODY AS A WHOLE Pain 19 22 17 13 Asthenia 15 19 13 12 METABOLIC AND NUTRITIONAL Hepatic‡ 9 7 9 6 DERMATOLOGIC Hand and foot syndrome 0 0 0 5 Cutaneous signs§ 2 0 1 3 RESPIRATORY¶ 10 8 5 7 NEUROLOGIC# 12 13 9 4 CARDIOVASCULARÞ 9 3 4 2 OTHERß 32 28 12 14 The incidence of akathisia in clinical trials of the weekly dosage schedule was greater (8.5%, 4/47 patients) when prochlorperazine was administered on the same day as irinotecan hydrochloride injection than when these drugs were given on separate days (1.3%, 1/80 patients). The 8.5% incidence of akathisia, however, is within the range reported for use of prochlorperazine when given as a premedication for other chemotherapies.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of irinotecan hydrochloride injection. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Myocardial ischemic events have been observed following irinotecan hydrochloride injection therapy. Thromboembolic events have been observed in patients receiving irinotecan hydrochloride injection.
Symptomatic pancreatitis, asymptomatic pancreatic enzyme elevation have been reported. Increases in serum levels of transaminases (i.e., AST and ALT) in the absence of progressive liver metastasis have been observed.
Hyponatremia, mostly with diarrhea and vomiting, has been reported.
Transient dysarthria has been reported in patients treated with irinotecan hydrochloride injection; in some cases, the event was attributed to the cholinergic syndrome observed during or shortly after infusion of irinotecan.
Interaction between irinotecan hydrochloride injection and neuromuscular blocking agents cannot be ruled out. Irinotecan has anticholinesterase activity, which may prolong the neuromuscular blocking effects of suxamethonium and the neuromuscular blockade of non-depolarizing drugs may be antagonized.
Infections: fungal and viral infections have been reported.
-
7 DRUG INTERACTIONS
7.1 Strong CYP3A4 Inducers
Exposure to irinotecan or its active metabolite SN-38 is substantially reduced in adult and pediatric patients concomitantly receiving the CYP3A4 enzyme-inducing anticonvulsants phenytoin, phenobarbital, carbamazepine, or St. John’s wort. The appropriate starting dose for patients taking these or other strong inducers such as rifampin and rifabutin has not been defined. Consider substituting non-enzyme inducing therapies at least 2 weeks prior to initiation of irinotecan hydrochloride injection therapy. Do not administer strong CYP3A4 inducers with irinotecan hydrochloride injection unless there are no therapeutic alternatives.
7.2 Strong CYP3A4 or UGT1A1 Inhibitors
Irinotecan and its active metabolite, SN-38, are metabolized via the human cytochrome P450 3A4 isoenzyme (CYP3A4) and uridine diphosphate-glucuronosyl transferase 1A1 (UGT1A1), respectively, [see Clinical Pharmacology (12.3)]. Patients receiving concomitant ketoconazole, a CYP3A4 and UGT1A1 inhibitor, have increased exposure to irinotecan and its active metabolite SN-38. Coadministration of irinotecan hydrochloride injection with other inhibitors of CYP3A4 (e.g., clarithromycin, indinavir, itraconazole, lopinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telaprevir, voriconazole) or UGT1A1 (e.g., atazanavir, gemfibrozil, indinavir) may increase systemic exposure to irinotecan or SN-38. Discontinue strong CYP3A4 inhibitors at least 1 week prior to starting irinotecan hydrochloride injection therapy. Do not administer strong CYP3A4 or UGT1A1 inhibitors with irinotecan hydrochloride injection unless there are no therapeutic alternatives.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action, irinotecan hydrochloride injection can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)].
Available postmarketing and published data reporting the use of irinotecan hydrochloride injection in pregnant women, are insufficient and confounded by the concomitant use of other cytotoxic drugs, to evaluate for any drug-associated risk for major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal studies, intravenous administration of irinotecan to rats and rabbits during the period of organogenesis resulted in embryofetal mortality and teratogenicity in pregnant animals at exposures lower than the human exposure based on AUC at the clinical dose of 125 mg/m2 (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal DataRadioactivity related to 14C-irinotecan crosses the placenta of rats following intravenous administration. Intravenous administration of irinotecan to rats at a dose of 6 mg/kg/day (approximately 0.2 times the clinical exposure (AUC) at the 125 mg/m2 dose based on exposure data from a separate rat study) during the period of organogenesis resulted in increased post-implantation loss and decreased numbers of live fetuses; at doses ≥ 1.2 mg/kg/day (approximately 0.03 times the clinical exposure (AUC) at the 125 mg/m2 dose based on exposure data from a separate rat study) there were increases in a variety of external, visceral, and skeletal abnormalities. Administration of irinotecan to pregnant rabbits at a dose of 6 mg/kg (approximately half of the clinical dose of 125 mg/m2 based on BSA) resulted in similar findings to those in rats, with increased post-implantation loss, decreased live fetuses, and increased external, visceral, and skeletal abnormalities.
Irinotecan administered to rat dams for the period following organogenesis through weaning at doses of 6 mg/kg/day caused decreased learning ability and decreased female body weights in the offspring.
8.2 Lactation
Risk Summary
Irinotecan and its metabolites are present in human milk. There is no information regarding the effects of irinotecan on the breastfed infant, or on milk production. Because of the potential for serious adverse reactions from irinotecan hydrochloride injection in the breastfed child, advise lactating women not to breastfeed during treatment with irinotecan hydrochloride injection and for 7 days after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status in female patients of reproductive potential prior to initiating irinotecan hydrochloride injection.
Contraception
Irinotecan hydrochloride injection can cause fetal harm when administered to a pregnant woman.
Females
Advise female patients of reproductive potential to use effective contraception during treatment and for 6 months after the final dose of irinotecan hydrochloride injection [see Use in Specific Populations (8.1) and Nonclinical Toxicology (13.1)].
Males
Due to the potential for genotoxicity, advise male patients with female partners of reproductive potential to use condoms during treatment and for 3 months after the final dose of irinotecan hydrochloride injection [see Nonclinical Toxicology (13.1)].
Infertility
Females
Based on postmarketing reports, female fertility may be impaired by treatment with irinotecan hydrochloride injection. Menstrual dysfunction has been reported following irinotecan hydrochloride injection administration.
Males
Based on findings from animal studies, male fertility may be impaired by treatment with irinotecan hydrochloride injection [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The effectiveness of irinotecan in pediatric patients has not been established. Results from two open-label, single arm studies were evaluated. One hundred and seventy children with refractory solid tumors were enrolled in one phase 2 trial in which 50 mg/m2 of irinotecan was infused for 5 consecutive days every 3 weeks. Grade 3–4 neutropenia was experienced by 54 (31.8%) patients. Neutropenia was complicated by fever in 15 (8.8%) patients. Grade 3–4 diarrhea was observed in 35 (20.6%) patients. This adverse event profile was comparable to that observed in adults. In the second phase 2 trial of 21 children with previously untreated rhabdomyosarcoma, 20 mg/m2 of irinotecan was infused for 5 consecutive days on weeks 0, 1, 3 and 4. This single agent therapy was followed by multimodal therapy. Accrual to the single agent irinotecan phase was halted due to the high rate (28.6%) of progressive disease and the early deaths (14%). The adverse event profile was different in this study from that observed in adults; the most significant grade 3 or 4 adverse events were dehydration experienced by 6 patients (28.6%) associated with severe hypokalemia in 5 patients (23.8%) and hyponatremia in 3 patients (14.3%); in addition Grade 3–4 infection was reported in 5 patients (23.8%) (across all courses of therapy and irrespective of causal relationship).
Pharmacokinetic parameters for irinotecan and SN-38 were determined in 2 pediatric solid-tumor trials at dose levels of 50 mg/m2 (60-min infusion, n=48) and 125 mg/m2 (90-min infusion, n=6). Irinotecan clearance (mean ± S.D.) was 17.3 ± 6.7 L/h/m2 for the 50 mg/m2 dose and 16.2 ± 4.6 L/h/m2 for the 125 mg/m2 dose, which is comparable to that in adults.
Dose-normalized SN-38 AUC values were comparable between adults and children. Minimal accumulation of irinotecan and SN-38 was observed in children on daily dosing regimens [daily × 5 every 3 weeks or (daily × 5) × 2 weeks every 3 weeks].
8.5 Geriatric Use
Patients greater than 65 years of age should be closely monitored because of a greater risk of early and late diarrhea in this population [see Clinical Pharmacology (12.3) and Adverse Reactions (6.1)]. The starting dose of irinotecan hydrochloride injection in patients 70 years and older for the once-every-3-week-dosage schedule should be 300 mg/m2[see Clinical Pharmacology (12.3) and Dosage and Administration (2)].
The frequency of grade 3 and 4 late diarrhea by age was significantly greater in patients ≥65 years than in patients <65 years (40% [53/133] versus 23% [40/171]; p=0.002). In another study of 183 patients treated on the weekly schedule, the frequency of grade 3 or 4 late diarrhea in patients ≥65 years of age was 28.6% [26/91] and in patients <65 years of age was 23.9% [22/92].
8.6 Renal Impairment
The influence of renal impairment on the pharmacokinetics of irinotecan has not been evaluated. Therefore, use caution in patients with impaired renal function. Irinotecan hydrochloride injection is not recommended for use in patients on dialysis.
8.7 Hepatic Impairment
Irinotecan clearance is diminished in patients with hepatic impairment while exposure to the active metabolite SN-38 is increased relative to that in patients with normal hepatic function. The magnitude of these effects is proportional to the degree of liver impairment as measured by elevations in total bilirubin and transaminase concentrations. Therefore, use caution when administering irinotecan hydrochloride injection to patients with hepatic impairment. The tolerability of irinotecan in patients with hepatic dysfunction (bilirubin greater than 2 mg/dl) has not been assessed sufficiently, and no recommendations for dosing can be made [see Dosage and Administration (2), Warnings and Precautions (5.10) and Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
In U.S. phase 1 trials, single doses of up to 345 mg/m2 of irinotecan were administered to patients with various cancers. Single doses of up to 750 mg/m2 of irinotecan have been given in non-U.S. trials. The adverse events in these patients were similar to those reported with the recommended dosage and regimen. There have been reports of overdosage at doses up to approximately twice the recommended therapeutic dose, which may be fatal. The most significant adverse reactions reported were severe neutropenia and severe diarrhea. There is no known antidote for overdosage of irinotecan hydrochloride injection. Maximum supportive care should be instituted to prevent dehydration due to diarrhea and to treat any infectious complications.
-
11 DESCRIPTION
Irinotecan Hydrochloride Injection, USP is an antineoplastic agent of the topoisomerase I inhibitor class.
Irinotecan Hydrochloride Injection, USP is supplied as a sterile, pale yellow, clear, aqueous solution. Each milliliter of solution contains 20 mg of irinotecan hydrochloride (on the basis of the trihydrate salt), 45 mg of sorbitol, NF, and 0.9 mg of lactic acid, USP. The pH of the solution has been adjusted to 3.5 (range, 3.0 to 3.8) with sodium hydroxide or hydrochloric acid. Irinotecan Hydrochloride Injection, USP is intended for dilution with 5% Dextrose Injection, USP (D5W), or 0.9% Sodium Chloride Injection, USP, prior to intravenous infusion. The preferred diluent is 5% Dextrose Injection, USP.
Irinotecan hydrochloride is a semisynthetic derivative of camptothecin, an alkaloid extract from plants such as Camptotheca acuminata or is chemically synthesized.
The chemical name is (S)-4,11-diethyl-3,4,12,14-tetrahydro-4-hydroxy-3,14-dioxo1H-pyrano[3',4':6,7]-indolizino[1,2-b]quinolin-9-yl-[1,4'bipiperidine]-1'-carboxylate, monohydrochloride, trihydrate. Its empirical formula is C33H38N4O6•HCl•3H2O and molecular weight is 677.19. It is slightly soluble in water and organic solvents. Its structural formula is as follows:
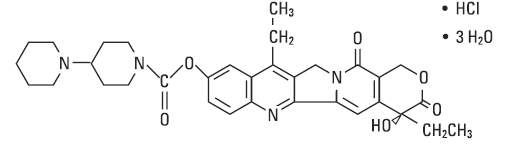
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Irinotecan is a derivative of camptothecin. Camptothecins interact specifically with the enzyme topoisomerase I, which relieves torsional strain in DNA by inducing reversible single-strand breaks. Irinotecan and its active metabolite SN-38 bind to the topoisomerase I-DNA complex and prevent religation of these single-strand breaks. Current research suggests that the cytotoxicity of irinotecan is due to double-strand DNA damage produced during DNA synthesis when replication enzymes interact with the ternary complex formed by topoisomerase I, DNA, and either irinotecan or SN-38. Mammalian cells cannot efficiently repair these double-strand breaks.
12.2 Pharmacodynamics
Irinotecan serves as a water-soluble precursor of the lipophilic metabolite SN-38. SN-38 is formed from irinotecan by carboxylesterase-mediated cleavage of the carbamate bond between the camptothecin moiety and the dipiperidino side chain. SN-38 is approximately 1000 times as potent as irinotecan as an inhibitor of topoisomerase I purified from human and rodent tumor cell lines. In vitro cytotoxicity assays show that the potency of SN-38 relative to irinotecan varies from 2-to 2000-fold; however, the plasma area under the concentration versus time curve (AUC) values for SN-38 are 2% to 8% of irinotecan and SN-38 is 95% bound to plasma proteins compared to approximately 50% bound to plasma proteins for irinotecan [see Clinical Pharmacology (12.3)]. The precise contribution of SN-38 to the activity of irinotecan hydrochloride injection is thus unknown. Both irinotecan and SN-38 exist in an active lactone form and an inactive hydroxy acid anion form. A pH-dependent equilibrium exists between the two forms such that an acid pH promotes the formation of the lactone, while a more basic pH favors the hydroxy acid anion form.
Administration of irinotecan has resulted in antitumor activity in mice bearing cancers of rodent origin and in human carcinoma xenografts of various histological types.
12.3 Pharmacokinetics
After intravenous infusion of irinotecan in humans, irinotecan plasma concentrations decline in a multiexponential manner, with a mean terminal elimination half-life of about 6 to 12 hours. The mean terminal elimination half-life of the active metabolite SN-38 is about 10 to 20 hours. The half-lives of the lactone (active) forms of irinotecan and SN-38 are similar to those of total irinotecan and SN-38, as the lactone and hydroxy acid forms are in equilibrium.
Over the recommended dose range of 50 to 350 mg/m2, the AUC of irinotecan increases linearly with dose; the AUC of SN-38 increases less than proportionally with dose. Maximum concentrations of the active metabolite SN-38 are generally seen within 1 hour following the end of a 90-minute infusion of irinotecan. Pharmacokinetic parameters for irinotecan and SN-38 following a 90-minute infusion of irinotecan at dose levels of 125 and 340 mg/m2 determined in two clinical studies in patients with solid tumors are summarized in Table 5:
Table 5. Summary of Mean (±Standard Deviation) Irinotecan and SN-38 Pharmacokinetic Parameters in Patients with Solid Tumors - *
- Plasma specimens collected for 24 hours following the end of the 90-minute infusion.
- †
- Plasma specimens collected for 48 hours following the end of the 90-minute infusion. Because of the longer collection period, these values provide a more accurate reflection of the terminal elimination half-lives of irinotecan and SN-38.
Dose
(mg/m2)Irinotecan SN-38 Cmax
(ng/mL)AUC0–24
(ng∙h/mL)t1/2
(h)Vz
(L/m2)CL
(L/h/m2)Cmax
(ng/mL)AUC0–24
(ng∙h/mL)t1/2
(h)125
(N=64)1,660
±79710,200
±3,2705.8*
±0.7110
±48.513.3
±6.0126.3
±11.9229
±10810.4
±3.1340
(N=6)3,392
±87420,604
±6,02711.7†
±1.0234
±69.613.9
±4.056.0
±28.2474
±24521.0
±4.3Cmax - Maximum plasma concentration
AUC0–24 - Area under the plasma concentration-time curve from time
0 to 24 hours after the end of the 90-minute infusion
t1/2 - Terminal elimination half-life
Vz - Volume of distribution of terminal elimination phase
CL - Total systemic clearanceDistribution
Irinotecan exhibits moderate plasma protein binding (30% to 68% bound). SN-38 is highly bound to human plasma proteins (approximately 95% bound). The plasma protein to which irinotecan and SN-38 predominantly binds is albumin.
Metabolism
Irinotecan is subject to extensive metabolic conversion by various enzyme systems, including esterases to form the active metabolite SN-38, and UGT1A1 mediating glucuronidation of SN-38 to form the inactive glucuronide metabolite SN-38G. Irinotecan can also undergo CYP3A4-mediated oxidative metabolism to several inactive oxidation products, one of which can be hydrolyzed by carboxylesterase to release SN-38. In vitro studies indicate that irinotecan, SN-38 and another metabolite aminopentane carboxylic acid (APC), do not inhibit cytochrome P-450 isozymes. UGT1A1 activity is reduced in individuals with genetic polymorphisms that lead to reduced enzyme activity such as the UGT1A1*28 polymorphism. Approximately 10% of the North American population is homozygous for the UGT1A1*28 allele (also referred to as UGT1A1 7/7 genotype). In a prospective study, in which irinotecan was administered as a single-agent (350 mg/m2) on a once-every-3-week schedule, patients with the UGT1A1 7/7 genotype had a higher exposure to SN-38 than patients with the wild-type UGT1A1 allele (UGT1A1 6/6 genotype) [see Warnings and Precautions (5.3) and Dosage and Administration (2.2)]. SN-38 glucuronide had 1/50 to 1/100 the activity of SN-38 in cytotoxicity assays using two cell lines in vitro.
Excretion
The disposition of irinotecan has not been fully elucidated in humans. The urinary excretion of irinotecan is 11% to 20%; SN-38, <1%; and SN-38 glucuronide, 3%. The cumulative biliary and urinary excretion of irinotecan and its metabolites (SN-38 and SN-38 glucuronide) over a period of 48 hours following administration of irinotecan in two patients ranged from approximately 25% (100 mg/m2) to 50% (300 mg/m2).
Effect of Age
The pharmacokinetics of irinotecan administered using the weekly schedule was evaluated in a study of 183 patients that was prospectively designed to investigate the effect of age on irinotecan toxicity. Results from this trial indicate that there are no differences in the pharmacokinetics of irinotecan, SN-38, and SN-38 glucuronide in patients <65 years of age compared with patients ≥65 years of age. In a study of 162 patients that was not prospectively designed to investigate the effect of age, small (less than 18%) but statistically significant differences in dose-normalized irinotecan pharmacokinetic parameters in patients <65 years of age compared to patients ≥65 years of age were observed. Although dose-normalized AUC0–24 for SN-38 in patients ≥65 years of age was 11% higher than in patients <65 years of age, this difference was not statistically significant. No change in the starting dose is recommended for geriatric patients receiving the weekly dosage schedule of irinotecan [see Dosage and Administration (2)].
Effect of Hepatic Impairment
Irinotecan clearance is diminished in patients with hepatic impairment while exposure to the active metabolite SN-38 is increased relative to that in patients with normal hepatic function. The magnitude of these effects is proportional to the degree of liver impairment as measured by elevations in total bilirubin and transaminase concentrations. However, the tolerability of irinotecan in patients with hepatic dysfunction (bilirubin greater than 2 mg/dl) has not been assessed sufficiently, and no recommendations for dosing can be made [see Dosage and Administration (2), Warnings and Precautions (5.10) and Use in Specific Populations (8.7)].
Effect of Renal Impairment
The influence of renal impairment on the pharmacokinetics of irinotecan has not been evaluated. Therefore, caution should be undertaken in patients with impaired renal function. Irinotecan Hydrochloride Injection is not recommended for use in patients on dialysis [see Use in Specific Populations (8.6)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term carcinogenicity studies with irinotecan were not conducted. Rats were, however, administered intravenous doses of 2 mg/kg or 25 mg/kg irinotecan once per week for 13 weeks (in separate studies, the 25 mg/kg dose produced an irinotecan Cmax and AUC that were about 7.0 times and 1.3 times the respective values in patients administered 125 mg/m2 weekly) and were then allowed to recover for 91 weeks. Under these conditions, there was a significant linear trend with dose for the incidence of combined uterine horn endometrial stromal polyps and endometrial stromal sarcomas. Irinotecan was clastogenic both in vitro (chromosome aberrations in Chinese hamster ovary cells) and in vivo (micronucleus test in mice). Neither irinotecan nor its active metabolite SN-38 was mutagenic in the in vitro Ames assay.
No significant adverse effects on fertility and general reproductive performance were observed after intravenous administration of irinotecan in doses of up to 6 mg/kg/day to rats and rabbits; however, atrophy of male reproductive organs was observed after multiple daily irinotecan doses both in rodents at 20 mg/kg and in dogs at 0.4 mg/kg. In separate studies in rodents, this dose produced an irinotecan Cmax and AUC about 5 and 1 times, respectively, of the corresponding values in patients administered 125 mg/m2 weekly. In dogs this dose produced an irinotecan Cmax and AUC about one-half and 1/15th, respectively, of the corresponding values in patients administered 125 mg/m2 weekly.
-
14 CLINICAL STUDIES
Irinotecan has been studied in clinical trials as a single agent [see Dosage and Administration (2)]. Weekly and once-every-3-week dosage schedules were used for the single-agent irinotecan studies. Clinical studies of single-agent use are described below.
14.1 Metastatic Colorectal Cancer
Second-Line Therapy After 5-FU-Based Treatment
4 Weekly Doses on a 6-Week Cycle: Studies 3, 4, and 5
Data from three open-label, single-agent, clinical studies, involving a total of 304 patients in 59 centers, support the use of irinotecan hydrochloride injection in the treatment of patients with metastatic cancer of the colon or rectum that has recurred or progressed following treatment with 5-FU-based therapy. These studies were designed to evaluate tumor response rate and do not provide information on effects on survival and disease-related symptoms. In each study, irinotecan hydrochloride injection was administered in repeated 6-week cycles consisting of a 90-minute intravenous infusion once weekly for 4 weeks, followed by a 2-week rest period. Starting doses of irinotecan hydrochloride injection in these trials were 100, 125, or 150 mg/m2, but the 150-mg/m2 dose was poorly tolerated (due to high rates of grade 4 late diarrhea and febrile neutropenia). Study 3 enrolled 48 patients and was conducted by a single investigator at several regional hospitals. Study 4 was a multicenter study conducted by the North Central Cancer Treatment Group. All 90 patients enrolled in Study 4 received a starting dose of 125 mg/m2. Study 5 was a multicenter study that enrolled 166 patients from 30 institutions. The initial dose in Study 5 was 125 mg/m2 but was reduced to 100 mg/m2 because the toxicity seen at the 125-mg/m2 dose was perceived to be greater than that seen in previous studies. All patients in these studies had metastatic colorectal cancer, and the majority had disease that recurred or progressed following a 5-FU-based regimen administered for metastatic disease. The results of the individual studies are shown in Table 6.Table 6. Weekly Dosage Schedule: Study Results - *
- Nine patients received 150 mg/m2 as a starting dose; two (22.2%) responded to irinotecan hydrochloride injection.
- †
-
Relative dose intensity for irinotecan hydrochloride injection based on planned dose intensity of 100, 83.3, and 66.7 mg/m2/wk corresponding with 150, 125, and 100 mg/m2 starting doses, respectively.
- ‡
-
Confirmed ≥ 4 to 6 weeks after first evidence of objective response.
Study 3 4 5 Number of Patients 48 90 64 102 Starting Dose (mg/m2/week × 4) 125* 125 125 100 Demographics and Treatment Administration Female/Male (%) 46/54 36/64 50/50 51/49 Median Age in years (range) 63 (29–78) 63 (32–81) 61 (42–84) 64 (25–84) Ethnic Origin (%) White 79 96 81 91 African American 12 4 11 5 Hispanic 8 0 8 2 Oriental/Asian 0 0 0 2 Performance Status (%) 0 60 38 59 44 1 38 48 33 51 2 2 14 8 5 Primary Tumor (%) Colon 100 71 89 87 Rectum 0 29 11 8 Unknown 0 0 0 5 Prior 5-FU Therapy (%) For Metastatic Disease 81 66 73 68 ≤ 6 months after Adjuvant 15 7 27 28 > 6 months after Adjuvant 2 16 0 2 Classification Unknown 2 12 0 3 Prior Pelvic/Abdominal Irradiation (%) Yes 3 29 0 0 Other 0 9 2 4 None 97 62 98 96 Duration of Treatment with irinotecan hydrochloride injection (median, months) 5 4 4 3 Relative Dose Intensity† (median %) 74 67 73 81 Efficacy Confirmed Objective Response Rate (%)‡ 21 13 14 9 (95% CI) (9.3 – 32.3) (6.3 – 20.4) (5.5 – 22.6) (3.3 – 14.3) Time to Response (median, months) 2.6 1.5 2.8 2.8 Response Duration (median, months) 6.4 5.9 5.6 6.4 Survival (median, months) 10.4 8.1 10.7 9.3 1-Year Survival (%) 46 31 45 43 In the intent-to-treat analysis of the pooled data across all three studies, 193 of the 304 patients began therapy at the recommended starting dose of 125 mg/m2. Among these 193 patients, 2 complete and 27 partial responses were observed, for an overall response rate of 15.0% (95% Confidence Interval [CI], 10.0% to 20.1%) at this starting dose. A considerably lower response rate was seen with a starting dose of 100 mg/m2. The majority of responses were observed within the first two cycles of therapy, but responses did occur in later cycles of treatment (one response was observed after the eighth cycle). The median response duration for patients beginning therapy at 125 mg/m2 was 5.8 months (range, 2.6 to 15.1 months). Of the 304 patients treated in the three studies, response rates to irinotecan hydrochloride injection were similar in males and females and among patients older and younger than 65 years. Rates were also similar in patients with cancer of the colon or cancer of the rectum and in patients with single and multiple metastatic sites. The response rate was 18.5% in patients with a performance status of 0 and 8.2% in patients with a performance status of 1 or 2. Patients with a performance status of 3 or 4 have not been studied. Over half of the patients responding to irinotecan hydrochloride injection had not responded to prior 5-FU. Patients who had received previous irradiation to the pelvis responded to irinotecan hydrochloride injection at approximately the same rate as those who had not previously received irradiation.
Once-Every-3-Week Dosage Schedule
Single Arm Study: Study 6
Data from an open-label, single-agent, single-arm, multicenter, clinical study involving a total of 132 patients support a once every-3-week dosage schedule of irinotecan in the treatment of patients with metastatic cancer of the colon or rectum that recurred or progressed following treatment with 5-FU. Patients received a starting dose of 350 mg/m2 given by 30-minute intravenous infusion once every 3 weeks. Among the 132 previously treated patients in this trial, the intent-to-treat response rate was 12.1% (95% CI, 7.0% to 18.1%).Randomized Studies: Studies 7 and 8
Two multicenter, randomized, clinical studies further support the use of irinotecan given by the once-every-3-week dosage schedule in patients with metastatic colorectal cancer whose disease has recurred or progressed following prior 5-FU therapy. In Study 7, second-line irinotecan therapy plus best supportive care was compared with best supportive care alone. In Study 8, second-line irinotecan therapy was compared with infusional 5-FU-based therapy. In both studies, irinotecan was administered intravenously at a starting dose of 350 mg/m2 over 90 minutes once every 3 weeks. The starting dose was 300 mg/m2 for patients who were 70 years and older or who had a performance status of 2. The highest total dose permitted was 700 mg. Dose reductions and/or administration delays were permitted in the event of severe hematologic and/or nonhematologic toxicities while on treatment. Best supportive care was provided to patients in both arms of Study 7 and included antibiotics, analgesics, corticosteroids, transfusions, psychotherapy, or any other symptomatic therapy as clinically indicated. In both studies, concomitant medications such as antiemetics, atropine, and loperamide were given to patients for prophylaxis and/or management of symptoms from treatment. If late diarrhea persisted for greater than 24 hours despite loperamide, a 7-day course of fluoroquinolone antibiotic prophylaxis was given. Patients in the control arm of the Study 8 received one of the following 5-FU regimens: (1) LV, 200 mg/m2 IV over 2 hours; followed by 5-FU, 400 mg/m2 IV bolus; followed by 5-FU, 600 mg/m2 continuous IV infusion over 22 hours on days 1 and 2 every 2 weeks; (2) 5-FU, 250 to 300 mg/m2/day protracted continuous IV infusion until toxicity; (3) 5-FU, 2.6 to 3 g/m2 IV over 24 hours every week for 6 weeks with or without LV, 20 to 500 mg/m2/day every week IV for 6 weeks with 2-week rest between cycles. Patients were to be followed every 3 to 6 weeks for 1 year.A total of 535 patients were randomized in the two studies at 94 centers. The primary endpoint in both studies was survival. The studies demonstrated a significant overall survival advantage for irinotecan compared with best supportive care (p=0.0001) and infusional 5-FU-based therapy (p=0.035) as shown in Figures 1 and 2. In Study 7, median survival for patients treated with irinotecan was 9.2 months compared with 6.5 months for patients receiving best supportive care. In Study 8, median survival for patients treated with irinotecan was 10.8 months compared with 8.5 months for patients receiving infusional 5-FU-based therapy. Multiple regression analyses determined that patients' baseline characteristics also had a significant effect on survival. When adjusted for performance status and other baseline prognostic factors, survival among patients treated with irinotecan remained significantly longer than in the control populations (p=0.001 for Study 7 and p=0.017 for Study 8). Measurements of pain, performance status, and weight loss were collected prospectively in the two studies; however, the plan for the analysis of these data was defined retrospectively. When comparing irinotecan with best supportive care in Study 7, this analysis showed a statistically significant advantage for irinotecan, with longer time to development of pain (6.9 months versus 2.0 months), time to performance status deterioration (5.7 months versus 3.3 months), and time to > 5% weight loss (6.4 months versus 4.2 months). Additionally, 33.3% (33/99) of patients with a baseline performance status of 1 or 2 showed an improvement in performance status when treated with irinotecan versus 11.3% (7/62) of patients receiving best supportive care (p=0.002). Because of the inclusion of patients with non-measurable disease, intent-to-treat response rates could not be assessed.
Figure 1. Survival
Second-Line Irinotecan vs Best Supportive Care (BSC)
Study 7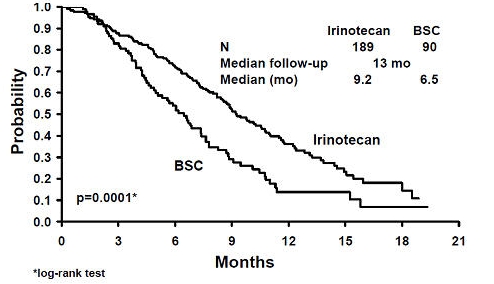
Figure 2. Survival
Second-Line Irinotecan vs Infusion 5-FU
Study 8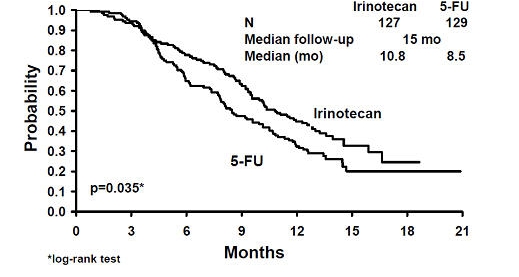
Table 7. Once-Every-3-Week Dosage Schedule: Study Results Study 7 Study 8 Irinotecan BSC* Irinotecan 5-FU Number of patients 189 90 127 129 Demographics and treatment administration Female/Male (%) 32/68 42/58 43/57 35/65 Median age in years (range) 59 (22–75) 62 (34–75) 58 (30–75) 58 (25–75) Performance status (%) 0 47 31 58 54 1 39 46 35 43 2 14 23 8 3 Primary tumor (%) Colon 55 52 57 62 Rectum 45 48 43 38 Prior 5-FU therapy (%) For metastatic disease 70 63 58 68 As adjuvant treatment 30 37 42 32 Prior irradiation (%) 26 27 18 20 Duration of study treatment (median, months) 4.1 -- 4.2 2.8 (Log-rank test) (p=0.02) Relative dose intensity (median %)† 94 -- 95 81 - 99 Survival Survival (median, months) 9.2 6.5 10.8 8.5 (Log-rank test) (p=0.0001) (p=0.035) In the two randomized studies, the EORTC QLQ-C30 instrument was utilized. At the start of each cycle of therapy, patients completed a questionnaire consisting of 30 questions, such as "Did pain interfere with daily activities?" (1 = Not at All, to 4 = Very Much) and "Do you have any trouble taking a long walk?" (Yes or No). The answers from the 30 questions were converted into 15 subscales, that were scored from 0 to 100, and the global health status subscale that was derived from two questions about the patient's sense of general well being in the past week. The results as summarized in Table 8 are based on patients' worst post-baseline scores. In Study 7, a multivariate analysis and univariate analyses of the individual subscales were performed and corrected for multivariate testing. Patients receiving irinotecan reported significantly better results for the global health status, on two of five functional subscales, and on four of nine symptom subscales. As expected, patients receiving irinotecan noted significantly more diarrhea than those receiving best supportive care. In Study 8, the multivariate analysis on all 15 subscales did not indicate a statistically significant difference between irinotecan and infusional 5-FU.
Table 8. EORTC QLQ-C30: Mean Worst Post-Baseline Score* QLQ-C30 Subscale
Study 7
Study 8
Irinotecan
BSC
p-value
Irinotecan
5-FU
p-value
- *
- For the five functional subscales and global health status subscale, higher scores imply better functioning, whereas, on the nine symptom subscales, higher scores imply more severe symptoms. The subscale scores of each patient were collected at each visit until the patient dropped out of the study.
Global health status
47
37
0.03
53
52
0.9
Functional scales
Cognitive
77
68
0.07
79
83
0.9
Emotional
68
64
0.4
64
68
0.9
Social
58
47
0.06
65
67
0.9
Physical
60
40
0.0003
66
66
0.9
Role
53
35
0.02
54
57
0.9
Symptom Scales
Fatigue
51
63
0.03
47
46
0.9
Appetite loss
37
57
0.0007
35
38
0.9
Pain assessment
41
56
0.009
38
34
0.9
Insomnia
39
47
0.3
39
33
0.9
Constipation
28
41
0.03
25
19
0.9
Dyspnea
31
40
0.2
25
24
0.9
Nausea/Vomiting
27
29
0.5
25
16
0.09
Financial impact
22
26
0.5
24
15
0.3
Diarrhea
32
19
0.01
32
22
0.2
-
15 REFERENCES
- NIOSH Alert: Preventing occupational exposures to antineoplastic and other hazardous drugs in healthcare settings. 2004. U.S. Department of Health and Human Services, Public Health Service, Centers for Disease Control and Prevention, National Institute for Occupational Safety and Health, DHHS (NIOSH) Publication No. 2004-165.
- OSHA Technical Manual, TED 1-0.15A, Section VI: Chapter 2. Controlling Occupational Exposure to Hazardous Drugs. OSHA, 1999. http://www.osha.gov/dts/osta/otm/otm_vi/otm_vi_2.html
- American Society of Health-System Pharmacists. ASHP guidelines on handling hazardous drugs. Am J Health-Syst Pharm. 2006; 63:1172–1193.
- Polovich, M., White, J. M., & Kelleher, L.O. (eds.) 2005. Chemotherapy and biotherapy guidelines and recommendations for practice (2nd. ed.) Pittsburgh, PA: Oncology Nursing Society.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Irinotecan Hydrochloride Injection, USP is available in single-dose amber glass vials in the following package sizes:
100 mg/5 mL NDC 0143-9583-01 Store at 20° to 25° C (68° to 77° F). See USP Controlled Room Temperature (excursions permitted to 15° to 30° C (59°to 86° F)). Protect from light. Keep the vial in the carton until the time of use. Protect from freezing.
Inspect the vial for damage and visible signs of leaks before removing from the carton. If damaged, incinerate the unopened package.
-
17 PATIENT COUNSELING INFORMATION
- Patients and caregivers should be informed of gastrointestinal complications, such as nausea, vomiting, abdominal cramping, and diarrhea. Patients should have loperamide readily available to begin treatment for late diarrhea (generally occurring more than 24 hours after administration of irinotecan hydrochloride injection). Begin loperamide at the first episode of poorly formed or loose stools or the earliest onset of bowel movements more frequent than normal. One dosage regimen for loperamide is 4 mg at the first onset of late diarrhea and then 2 mg every 2 hours until the patient is diarrhea-free for at least 12 hours. Loperamide is not recommended to be used for more than 48 consecutive hours at these doses, because of the risk of paralytic ileus. During the night, the patient may take 4 mg of loperamide every 4 hours. Patients should contact their physician if any of the following occur: diarrhea for the first time during treatment; black or bloody stools; symptoms of dehydration such as lightheadedness, dizziness, or faintness; inability to take fluids by mouth due to nausea or vomiting; or inability to get diarrhea under control within 24 hours.
- Patients should be warned about the potential for dizziness or visual disturbances which may occur within 24 hours following the administration of irinotecan hydrochloride injection.
- Explain the significance of routine blood cell counts. Instruct patients to monitor their temperature frequently and immediately report any occurrence of fever or infection.
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.9), Use in Specific Populations (8.1, 8.3), Clinical Pharmacology (12.1) and Nonclinical Toxicology (13.1)]
o Advise pregnant women and females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy.
o Advise females of reproductive potential to use effective contraception during treatment with irinotecan hydrochloride injection and for 6 months after the final dose.
o Advise male patients with female partners of reproductive potential to use condoms during treatment and for 3 months after the final dose of irinotecan hydrochloride injection.
- Lactation
o Advise women not to breastfeed during treatment with irinotecan hydrochloride injection and for at least 7 days after the final dose [see Use in Specific Populations (8.2)]. - Infertility
o Advise females and males of reproductive potential that irinotecan hydrochloride injection may impair fertility [see Use in Specific Populations (8.3)].
- Patients should be alerted to the possibility of alopecia.
- Contains sorbitol.
Manufactured by:
THYMOORGAN PHARMAZIE GmbH
Schiffgraben 23
38690 Goslar
GermanyDistributed by:
Hikma Pharmaceuticals USA Inc.
Berkeley Heights, NJ 07922Novaplus is a registered trademark of Vizient, Inc.
novaplus+
Revised: June 2020
127.207.017/02
-
PRINCIPAL DISPLAY PANEL
NDC 0143-9583-01 Rx ONLY
Irinotecan
HCl Injection, USP
100 mg per 5 mL
(20 mg/mL)
as the trihydrate
For Intravenous use ONLY
MUST BE DILUTED - CYOTOXIC AGENT
5 mL Single Dose Vial

NDC 0143-9583-01 Rx ONLY
Irinotecan
Hydrochloride Injection, USP
100 mg per 5 mL
(20 mg/mL)*
as the trihydrate
For Intravenous use ONLY
MUST BE DILUTED
CYOTOXIC AGENT
1 x 5 mL Single Dose Vial
- SERIALIZATION IMAGE
-
INGREDIENTS AND APPEARANCE
IRINOTECAN HYDROCHLORIDE
irinotecan hydrochloride injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0143-9583 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength IRINOTECAN HYDROCHLORIDE (UNII: 042LAQ1IIS) (IRINOTECAN - UNII:7673326042) IRINOTECAN HYDROCHLORIDE 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength SORBITOL (UNII: 506T60A25R) 45 mg in 1 mL LACTIC ACID (UNII: 33X04XA5AT) 0.9 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0143-9583-01 5 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product 12/20/2010 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA091032 12/20/2010 Labeler - Hikma Pharmaceuticals USA Inc. (001230762) Establishment Name Address ID/FEI Business Operations THYMOORGAN GMBH PHARMAZIE 319029989 analysis(0143-9583) , label(0143-9583) , manufacture(0143-9583) , pack(0143-9583)
