Label: amphotec- Amphotericin B injection, lipid complex
-
Contains inactivated NDC Code(s)
NDC Code(s): 64116-021-01, 64116-025-01 - Packager: InterMune, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated February 27, 2006
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- N/A - Section Title Not Found In Database
-
DESCRIPTION
AMPHOTEC® is a sterile, pyrogen-free, lyophilized powder for reconstitution and intravenous (IV) administration. AMPHOTEC consists of a 1:1 (molar ratio) complex of amphotericin B and cholesteryl sulfate. Upon reconstitution, AMPHOTEC forms a colloidal dispersion of microscopic disc-shaped particles.
Note: Liposomal encapsulation or incorporation into a lipid complex can substantially affect a drug’s functional properties relative to those of the unencapsulated drug or non-lipid associated drug. In addition, different liposomal or lipid-complex products with a common active ingredient may vary from one another in the chemical composition and physical form of the lipid component. Such differences may affect the functional properties of these drug products.
Amphotericin B is an antifungal polyene antibiotic produced by a strain of Streptomyces nodosus.
Amphotericin B, which is the established name for [1R (1R*,3S*,5R*,6R*,9R*,11R*, 15S*,16R*,17R*,18S*,19E,21E,23E,25E,27E,29E,31E,33R*,35S*,36R*,37S*)]-33-[(3-Amino-3,6-dideoxy-ß-D-mannopyranosyl)oxy]-1,3,5,6,9,11,17,37-octahydroxy-15,16,18-trimethyl-13-oxo-14,39-dioxabicyclo[33.3.1] nonatriaconta-19,21,23,25,27, 29,31-heptaene-36-carboxylic acid, has the following structure:
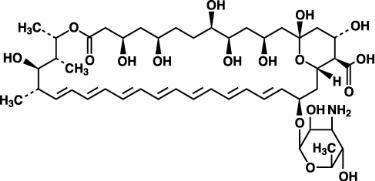
The molecular formula of the drug is C47H73NO17; its molecular weight is 924.10.
AMPHOTEC is available in 50 mg and 100 mg single dose vials. Each 50 mg single dose vial contains amphotericin B, 50 mg; sodium cholesteryl sulfate, 26.4 mg; tromethamine, 5.64 mg; disodium edetate dihydrate, 0.372 mg; lactose monohydrate, 950 mg; and hydrochloric acid, qs, as a sterile, nonpyrogenic, lyophilized powder. Each 100 mg single dose vial contains amphotericin B, 100 mg; sodium cholesteryl sulfate, 52.8 mg; tromethamine, 11.28 mg; disodium edetate dihydrate, 0.744 mg; lactose monohydrate, 1900 mg; and hydrochloric acid, qs, as a sterile, nonpyrogenic, lyophilized powder.
-
MICROBIOLOGY
Mechanism of Action
The active ingredient of AMPHOTEC, amphotericin B, is a polyene antibiotic that acts by binding to sterols (primarily ergosterol) in cell membranes of sensitive fungi, with subsequent leakage of intracellular contents and cell death due to changes in membrane permeability. Amphotericin B also binds to the sterols (primarily cholesterol) in mammalian cell membranes, which is believed to account for its toxicity in animals and humans.
Activity in vitro and in vivo
AMPHOTEC is active in vitro against Aspergillus and Candida species. One hundred and twelve clinical isolates of four different Aspergillus species and 88 clinical isolates of five different Candida species were tested, with a majority of minimum inhibitory concentrations (MICs) < 1 μg/mL. AMPHOTEC is also active in vitro against other fungi. In vitro, AMPHOTEC is fungistatic or fungicidal, depending upon the concentration of the drug and the susceptibility of the fungal organism. However, standardized techniques for susceptibility testing for antifungal agents have not been established, and results of susceptibility studies do not necessarily correlate with clinical outcome.
AMPHOTEC is active in murine models against Aspergillus fumigatus, Candida albicans, Coccidioides immitis and Cryptococcus neoformans, and in an immunosuppressed rabbit model of aspergillosis in which endpoints were prolonged survival of infected animals and clearance of microorganisms from target organ(s). AMPHOTEC also was active in a hamster model of visceral leishmaniasis, a disease caused by infection of macrophages of the mononuclear phagocytic system by a protozoal parasite of the genus Leishmania. In this hamster model the endpoints were also prolonged survival of infected animals and clearance of microorganisms from target organ(s).
Drug Resistance
Variants with reduced susceptibility to amphotericin B have been isolated from several fungal species after serial passage in cell culture media containing the drug and from some patients receiving prolonged therapy with amphotericin B deoxycholate. Although the relevance of drug resistance to clinical outcome has not been established, fungal organisms that are resistant to amphotericin B may also be resistant to AMPHOTEC.
-
CLINICAL PHARMACOLOGY
Pharmacokinetics
The pharmacokinetics of amphotericin B, administered as AMPHOTEC, were studied in 51 bone marrow transplant patients with systemic fungal infections. The median (range) age and weight of those patients were 32 (3 to 52) years and 69.5 (14 to 116) kg, respectively. AMPHOTEC doses ranged from 0.5 to 8.0 mg/kg/day. The assay used in this study to measure amphotericin B in plasma does not distinguish amphotericin B that is complexed with cholesteryl sulfate from uncomplexed amphotericin B. A population modeling approach was used to estimate pharmacokinetic parameters (see table). The pharmacokinetics of amphotericin B, administered as AMPHOTEC, were best described by an open, two compartment structural model. The pharmacokinetics of amphotericin B, administered as AMPHOTEC, were nonlinear. Steady state volume of distribution (Vss) and total plasma clearance (CLt) increased with escalating doses, resulting in less than proportional increases in plasma concentration over a dose range of 0.5 to 8.0 mg/kg/day. The increased volume of distribution probably reflected uptake by tissues. The covariates of body weight and dose level accounted for a substantial portion of the variability of the pharmacokinetic estimates between patients. The unexplained variability in clearance was 26%. Based on the population model developed for these patients, pharmacokinetic parameters were predicted for two doses of AMPHOTEC and are provided in the following table:
Predicted Pharmacokinetic Parameters of Amphotericin B after Administration of Multiple Doses of AMPHOTEC [a] AMPHOTEC (mg/kg/day) Mean Pharmacokinetic Parameter [b] 3 4 [a] Data obtained using population modeling in 51 bone marrow transplant patients. The modeling assumes amphotericin B pharmacokinetics after administration of AMPHOTEC is best described by a 2-compartment model. Infusion rate = 1 mg/kg/hour. [b] Definitions: Vss - Volume of distribution at steady state, CLt - Total plasma clearance, Cmax - Maximum plasma concentration achieved at the end of an infusion, AUCss – Area under the plasma concentration time curve at steady-state. Vss (L/kg) 3.8 4.1 CLt (L/h/kg) 0.105 0.112 Distribution Half-Life (minutes) 3.5 3.5 Elimination Half-Life (hours) 27.5 28.2 Cmax (μg/mL) 2.6 2.9 AUCss (μg/mL•h) 29 36 In addition, the pharmacokinetics of amphotericin B, administered as amphotericin B deoxycholate, were studied in 15 patients in whom amphotericin B deoxycholate was administered for the treatment of aspergillus infections or empirical therapy. The median (range) age and weight for these patients were 21 (4 to 66) years and 60 (19 to 117) kg, respectively. A population modeling approach was used to estimate the pharmacokinetic parameters. The pharmacokinetics of amphotericin B, administered as amphotericin B deoxycholate, was best described as an open, two-compartment model with linear elimination.
The predicted pharmacokinetic parameters are provided in the following table:
Predicted Pharmacokinetic Parameters of Amphotericin B after Administration of Multiple Doses of 1 mg/kg Amphotericin B Deoxycholate [a] Mean Pharmacokinetic Parameter [b] Values [a] Data obtained using population modeling in 15 patients in whom amphotericin B deoxycholate was administered for treatment of aspergillus infection or empiric therapy. The modeling assumes amphotericin B pharmacokinetics after administration of amphotericin B deoxycholate are best described by a 2-compartment model. Infusion rate = 0.25 mg/kg/hour. [b] Definitions: Vss - Volume of distribution at steady state, CLt - Total plasma clearance, Cmax - Maximum plasma concentration achieved at the end of an infusion, AUCss - Area under the plasma concentration time curve at steady-state. Vss (L/kg) 1.1 CLt (L/h/kg) 0.028 Distribution Half-Life (minutes) 38 Elimination Half-Life (hours) 39 Cmax (μg/mL) 2.9 AUCss (μg/mL•h) 36 An analytical assay that is able to distinguish between amphotericin B in the AMPHOTEC complex and amphotericin B which is not complexed to cholesteryl sulfate was used to analyze samples from a study of 25 patients who were either immunocompromised with aspergillosis or both febrile and neutropenic. Following a 1 mg/kg/hour infusion, 25 ± 18% (mean ± SD) of the total amphotericin B concentration measured in plasma was in the AMPHOTEC complex, dropping to 9.3 ± 7.9% at 1 hour and 7.5 ± 9.3% at 24 hours after the end of the infusion.
Pharmacokinetics in Special Populations
A population modeling approach was used to assess the effect of renal function, hepatic function, and age on the pharmacokinetics of AMPHOTEC in 51 patients receiving bone marrow transplants as described earlier.
Renal Impairment: The pharmacokinetics of amphotericin B, administered as AMPHOTEC, were not related to baseline serum creatinine clearance in the population studied; the median (range) creatinine clearance for this population was 74.0 (range: 35 - 202) mL/min/70 kg. The effect of more severe renal impairment on the pharmacokinetics of AMPHOTEC has not been studied.
Hepatic Impairment: The pharmacokinetics of amphotericin B, administered as AMPHOTEC, were not related to baseline liver function, as determined by liver enzymes and total bilirubin. For the population tested, the mean ± SD values for AST and total Bilirubin were 59.4 ± 70.0 IU/mL and 3.5 ± 3.7 mg/dL, respectively. The effect of more severe hepatic impairment on the pharmacokinetics of AMPHOTEC has not been studied.
Age: The pharmacokinetics of amphotericin B, administered as AMPHOTEC, were not related to the age of the patient. The median (range) age for the population in this study was 32 (3 to 52) years.
- INDICATIONS AND USAGE
-
DESCRIPTION OF CLINICAL STUDIES
Clinical Studies in Aspergillosis
Data from 161 patients with proven or probable aspergillus infection were pooled from 5 non-comparative open label studies, one of which included emergency use patients. The patients were treated with AMPHOTEC because of failure to respond to amphotericin B deoxycholate (n=49), development of nephrotoxicity while receiving amphotericin B deoxycholate (n=62), preexisting renal impairment (n=25), or other reasons (n=25).
The median age of these 161 patients (92 males and 69 females) was 41 years (range 2 months to 85 years). For the 155 patients with baseline neutrophil data, 33 patients (21%) had neutrophil counts of < 500/mm3. The underlying diseases included bone marrow transplant, 69 (43%); hematological malignancy, 51(32%); solid organ transplant, 25 (15%); solid tumor, 3 (2%); and other diagnoses, 13 (8%) including surgery, 4; HIV infection, 3; immunosuppression for autoimmune disease, 3; diabetes, 2; and no known underlying disease, 1. Pulmonary involvement was the primary infection site, 118 patients (73%), followed by sinus, 14 (9%), CNS, 9 (6%), skin/wound, 9 (6%), and others, 10 (6%) including 3 with bone involvement, 2 with hepatic involvement, 2 with disseminated disease and 1 each with endocarditis, ophthalmitis, otitis, and involvement of the hard palate. The 49 patients enrolled due to failure to respond to amphotericin B had received amphotericin B deoxycholate prior to AMPHOTEC for ≤ 7 days (11 patients), 8 - 14 days (16 patients), and > 14 days (22 patients).
Patients were defined by their physicians as being refractory to amphotericin B deoxycholate therapy based on overall clinical judgment after receiving either a minimum of 7 days of amphotericin B deoxycholate or a minimum total dose of 15 mg/kg of amphotericin B deoxycholate. Nephrotoxicity was defined as a serum creatinine that had doubled from baseline, increased by ≥ 1.5 mg/dL or increased to ≥ 2.0 mg/dL. Preexisting renal impairment was defined as a serum creatinine that had increased to ≥ 2.0 mg/dL due to reasons other than amphotericin B deoxycholate administration.
Classifications of diagnosis and response were based on the definitions previously developed by the Mycoses Study Group.1 A retrospective response analysis was conducted in which a “complete response” was defined as resolution of all attributable symptoms, signs, and radiographic abnormalities present at enrollment, and a “partial response” was defined as major improvement of the abovementioned parameters. The total number of responders was the sum of the number of “complete” and “partial” responses.
Of the 161 patients, 80 were considered evaluable for response. Eighty-one (81) were excluded on the basis of inadequate diagnosis, confounding factors, or receiving ≤ 4 doses of AMPHOTEC. In the evaluable patients, the median daily dose was 4 mg/kg/day (range 0.73 - 7.5 mg/kg/day) and the cumulative median dose was 6.3 g (range 0.36 - 34.4 grams). Median duration of treatment was 24 days (range 5 - 129 days).
Response Rates for Evaluable Patients Patient Group (n) Complete Response Partial Response Total Responders
[a]Response
Rate[a] Total responders = Complete responses + Partial responses. [b] Defined, based on overall clinical judgment, after receiving a minimum of 7 days of amphotericin B deoxycholate or a minimum total dose of 15 mg/kg of amphotericin B deoxycholate. [c] Defined as a serum creatinine that had doubled from baseline or increased by ≥ 1.5 mg/dL or increased to ≥ 2.0 mg/dL. [d] Defined as a serum creatinine that had increased to ≥ 2.0 mg/dL due to reasons other than amphotericin B deoxycholate. Amphotericin B deoxycholate failure (28) [b] 3 9 12 43% Nephrotoxicity (36) [c] 5 12 17 47% Preexisting renal impairment (16) [d] 1 7 8 50% Total (80) 9 28 37 46% There is no directly comparable control group for the patients described in the above table to be certain whether similar patients would have responded had amphotericin B deoxycholate therapy been continued. A randomized study comparing AMPHOTEC with amphotericin B deoxycholate for therapy of invasive aspergillosis is currently undergoing analysis.
Renal Function
Patients with Renal Dysfunction at Baseline
The subset of patients with aspergillosis from the above five noncomparative open label studies, who initiated treatment with AMPHOTEC when their serum creatinine was ≥ 2.0 mg/dL (n = 47) experienced a mean decline in serum creatinine during treatment. In part, this decline may be attributed to patient dropout over time from this group. A historical control group was selected by reviewing medical charts of patients from January 1990 to June 1994 at 6 medical centers ( M.D. Anderson Cancer Center, Fred Hutchinson Cancer Research Center, H. Lee Moffitt Cancer Center, University of Pittsburgh, Memorial Sloan-Kettering Cancer Center, and Bone Marrow Transplant Program at Emory University). The mean change in serum creatinine was evaluated for similar cohorts of patients from this historical control group, with the baseline for assessing change being the day each patient’s serum creatinine reached ≥ 2.0 mg/dL. As shown in the figure, serum creatinine levels were lower during treatment with AMPHOTEC when compared to the serum creatinine levels of amphotericin B deoxycholate patients in the historical control group. There is no directly comparable group to be certain whether this decline is significantly better than the results of serum creatinine levels in patients who had continued on amphotericin B deoxycholate. Since these data were obtained from two separate studies, no statistical testing of the differences between these two groups was performed.
Changes in Mean Serum Creatinine Over Time in Patients with
Aspergillosis and Baseline Serum Creatinine ≥ 2.0 mg/dL [a]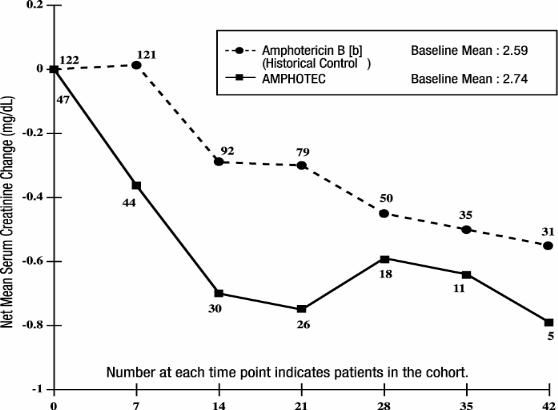
[a] These curves do not represent the clinical course of a given patient, but that of an openlabel cohort of patients.
[b] Administered as amphotericin B deoxycholate.
Patients with Normal Renal Function at Baseline
In a randomized, double-blind, multicenter study, 213 febrile neutropenic patients were given empirically either 4 mg/kg/day of AMPHOTEC or 0.8 mg/kg/day of amphotericin B deoxycholate for a maximum of 14 days. This study was primarily designed to compare the safety profiles of these two treatments. NOTE: AMPHOTEC is NOT approved for empirical treatment in febrile neutropenic patients.
In the above study, patients had largely normal renal function at baseline; median serum creatinine levels were 0.8 mg/dL for both treatment groups. The mean change in serum creatinine was evaluated for patients with baseline creatinine ≤ 1.5 mg/dL. As shown in the graph, patients in both treatment groups showed an increase in serum creatinine while on study, however AMPHOTEC patients experienced significantly less creatinine increase at each time point.
Changes in Mean Serum Creatinine Over Time in Patients with Febrile
Neutropenia, and Baseline Serum Creatinine ≤ 1.5 mg/dL [a]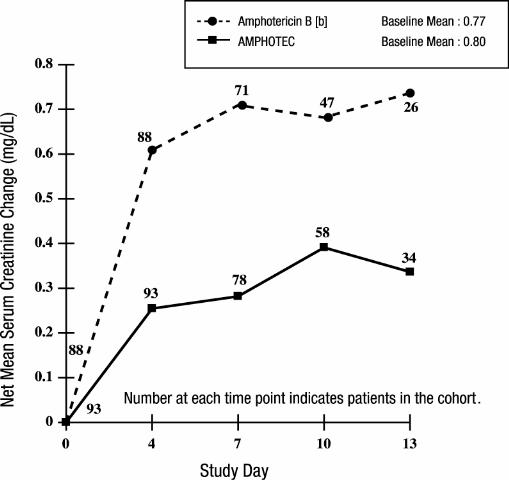
[a] These curves do not represent the clinical course of a given patient, but that of a cohort of patients.
[b] Administered as amphotericin B deoxycholate.
Hypokalemia
In the same empiric study, significantly more amphotericin B deoxycholate patients had at least one laboratory result of serum potassium < 3.0 mEq/L at least one time in the study compared with AMPHOTEC patients (23% vs. 7%), although concomitant supplemental potassium was allowed in the study design. Both groups received approximately equal amounts of potassium supplementation.
- CONTRAINDICATIONS
-
WARNINGS
Anaphylaxis has been reported with amphotericin B deoxycholate and other amphotericin B-containing drugs. Immediate treatment of anaphylaxis or anaphylactoid reactions is required. Epinephrine, oxygen, intravenous steroids, and airway management should be administered as indicated. If severe respiratory distress occurs, the infusion should be immediately discontinued. The patient should not receive further infusions of AMPHOTEC.
-
PRECAUTIONS
General
AMPHOTEC should be administered intravenously. Acute infusionrelated reactions including fever, chills, hypoxia, hypotension, nausea, or tachypnea, may occur 1 to 3 hours after starting intravenous infusion. These reactions are usually more severe or more frequent with the initial doses of AMPHOTEC and usually diminish with subsequent doses. Acute infusion-related reactions can be managed by pretreatment with antihistamines and corticosteroids and/or by reducing the rate of infusion and by prompt administration of antihistamines and corticosteroids. (See ADVERSE REACTIONS).
Rapid intravenous infusion should be avoided.
Laboratory Tests
Particularly tests of renal and hepatic function, serum electrolytes, complete blood count and prothrombin time should be monitored as medically indicated.
Drug Interactions
No formal drug interaction studies have been conducted with AMPHOTEC®. When administered concomitantly, the following drugs are known to interact with amphotericin B; therefore the following drugs may interact with AMPHOTEC.
Antineoplastic Agents
Concurrent use of antineoplastic agents and amphotericin B may enhance the potential for renal toxicity, bronchospasm, and hypotension. Caution is urged when antineoplastic agents are given concomitantly with AMPHOTEC.
Corticosteroids and Corticotropin (ACTH)
Concurrent use of corticosteroids and corticotropin (ACTH) with amphotericin B may potentiate hypokalemia which could predispose the patient to cardiac dysfunction. If corticosteroids or corticotropin are used concomitantly with AMPHOTEC, serum electrolytes and cardiac function should be monitored.
Cyclosporine and Tacrolimus
In the same randomized, double-blind, empiric trial to compare AMPHOTEC and amphotericin B deoxycholate, patients with normal baseline serum creatinine were prospectively enrolled into four strata: adults receiving cyclosporine or tacrolimus (n=89); or pediatric patients (< 16 years old) receiving cyclosporine or tacrolimus (n=15); adults not receiving cyclosporine or tacrolimus (n=75); or pediatric patients not receiving cyclosporine or tacrolimus (n=34). Patients were assessed for renal toxicity defined as either a doubling or an increase of 1.0 mg/dL or more from baseline serum creatinine, or ≥ 50% decrease from baseline calculated creatinine clearance. Adults and pediatric patients receiving cyclosporine or tacrolimus in addition to AMPHOTEC had a significantly lower rate of renal toxicity (31%, 16/51), compared to the amphotericin B deoxycholate patients receiving cyclosporine or tacrolimus (68%, 34/50). In the adults and pediatric patients not receiving cyclosporine or tacrolimus, only 8% (4/51) of the AMPHOTEC patients experienced renal toxicity compared to 35% (17/49) of the amphotericin B deoxycholate patients.
Digitalis Glycosides
Concurrent use of amphotericin B may induce hypokalemia and may potentiate digitalis toxicity. If digitalis glycosides are administered concomitantly with AMPHOTEC, serum potassium levels should be closely monitored.
Flucytosine
Concurrent use of flucytosine with amphotericin B containing preparations may increase the toxicity of flucytosine by possibly increasing its cellular uptake and/or impairing its renal excretion. Caution is urged when flucytosine is given concomitantly with AMPHOTEC.
Imidazoles (e.g., ketoconazole, miconazole, clotrimazole, fluconazole, etc.)
Antagonism between amphotericin B and imidazole derivatives such as miconazole and ketoconazole which inhibit ergosterol synthesis, has been reported in both in vitro and in vivo animal studies. The clinical significance of these findings has not been determined.
Other Nephrotoxic Medications
Concurrent use of amphotericin B and agents such as aminoglycosides and pentamidine may enhance the potential for drug-induced renal toxicity. Caution is urged if aminoglycosides or pentamidine are used concomitantly with AMPHOTEC. Intensive monitoring of renal function is recommended in patients requiring any combination of nephrotoxic medications.
Carcinogenesis, Mutagenesis and Impairment of Fertility
No long-term studies in animals have been performed with AMPHOTEC or amphotericin B deoxycholate to evaluate carcinogenic potential. AMPHOTEC and/or amphotericin B deoxycholate were not mutagenic in vitro with and without an exogenous mammalian microsomal metabolic activation system when assayed in the Salmonella reverse mutation assay, the CHO chromosomal aberration assay and the mouse lymphoma forward mutation assay. AMPHOTEC was also negative in vivo in the mouse bone marrow micronucleus assay. No studies have been conducted to determine if AMPHOTEC affects fertility or if it produces adverse effects when administered peri- or post-natally in animals. In multiple dose toxicity studies of up to 13 weeks in rats at doses up to 0.5 times the recommended human dose and in dogs at doses up to 0.4 times the recommended human dose (based on body surface area), ovarian and testicular histology were unaffected.
Pregnancy
Teratogenic Effects. Pregnancy Category B:
There are no reports of pregnant women having been treated with AMPHOTEC. Reproductive studies with AMPHOTEC in rats at doses up to 0.4 times the recommended human dose and in rabbits at doses up to 1.1 times the recommended human dose have revealed no evidence of harm to the fetus due to treatment with AMPHOTEC. Because animal reproduction studies are not always predictive of human response and because adequate and well controlled studies have not been conducted in pregnant women, AMPHOTEC should be used during pregnancy only if the anticipated benefit to the patient outweighs the potential risk to the fetus.
Nursing Mothers
It is not known whether AMPHOTEC is excreted in milk. Because of the potential for serious adverse reactions in nursing infants from amphotericin B, a decision should be made to discontinue nursing or discontinue treatment with AMPHOTEC, taking into account the importance of the drug to the mother.
Pediatric Use
Ninety-seven pediatric patients with systemic fungal infections have been treated with AMPHOTEC, at daily doses (mg/kg) similar to those given to adults. No unexpected adverse events have been reported. In the same empiric, multicenter trial, pediatric patients (< 16 years) treated with AMPHOTEC had significantly less renal toxicity than amphotericin B deoxycholate patients. Only 12% (3/25) of pediatric patients treated with AMPHOTEC developed nephrotoxicity compared to 52% (11/21) of pediatric patients receiving amphotericin B deoxycholate. Renal toxicity defined as either a doubling or an increase of 1.0 mg/dL or more from baseline serum creatinine, or ≥ 50% decrease from baseline calculated creatinine clearance.
-
ADVERSE REACTIONS
The following adverse events are based on the experience of 572 AMPHOTEC patients from 5 open studies of patients with systemic fungal infections, of whom 526 were treated with a daily dose of 3 - 6 mg/kg. Additionally, comparative adverse event data from 150 AMPHOTEC (4 or 6 mg/kg/day) and 146 amphotericin B deoxycholate (0.8 or 1 mg/kg/day) patients in prospectively randomized doubleblinded studies of empiric treatment of febrile and neutropenic patients or treatment of aspergillosis are also provided.
Infusion-related adverse events: Infusion-related adverse events (1 to 3 hours after starting intravenous infusion) occurred most frequently in association with the first infusion of AMPHOTEC. Their frequency and severity decreased with subsequent dosing. Based on the combined non-comparative studies, 35% (197/569) of the patients reported chills or chills and fever, possibly or probably related to AMPHOTEC, on the first day of dosing, compared to 14% (58/422) by the seventh dose. In the comparative studies, a similar decreasing trend was noted for AMPHOTEC and amphotericin B deoxycholate.
Adverse events that were considered to be possibly or probably related to AMPHOTEC and that occurred in 5% or more of the patients are summarized in the table below:
Summary of Probably and Possibly Related Adverse Events Reported by ≥ 5% of AMPHOTEC Patients Non-Comparative Studies Comparative Studies [a]
Adverse
EventAMPHOTEC
(n=572)
%AMPHOTEC
Aspergillosis
Patients
(n=161)
%AMPHOTEC
(n=150)
%Amphotericin B
Deoxycholate
(n=146)
%[a] From AMPHOTEC (4 or 6 mg/kg/day) and amphotericin B deoxycholate (0.8 or 1 mg/kg/day) patients in prospectively randomized double-blinded studies of empiric treatment of febrile and neutropenic patients or treatment of first-line aspergillosis, respectively. [b] Includes patients with “kidney function abnormal” which was associated with an increase in creatinine. Body as a Whole Chills 50 55 77 56 Fever 33 34 55 47 Headache 5 8 4 3 Chills and fever 3 3 7 2 Cardiovascular System Hypotension 10 9 12 5 Tachycardia 10 12 9 5 Hypertension 7 9 7 6 Digestive System Nausea 8 12 7 7 Nausea and vomiting 7 11 4 7 Vomiting 6 8 11 8 Liver function test abnormal 4 4 11 8 Hemic and Lymphatic System Thrombocytopenia 6 7 1 1 Metabolic/Nutritional Disorders Creatinine
increased [b]12 12 21 34 Hypokalemia 8 7 26 29 Hypomagnesemia 4 7 6 11 Hyperbilirubinemia 3 2 19 17 Alkaline phosphatase increased 3 3 7 8 Hyperglycemia 1 1 6 9 Respiratory System Dyspnea 5 4 9 4 Hypoxia 5 6 9 5 Additionally, the following adverse events also occurred in 5% or more of AMPHOTEC patients; however, the causal relationship of these adverse events is uncertain:
General (body as a whole)
Abdomen enlarged, abdominal pain, back pain, chest pain, face edema, injection site inflammation, mucous membrane disorder, pain, sepsis
Metabolic and Nutritional Disorders
Edema, generalized edema, hypocalcemia, hypophosphatemia, peripheral edema, weight gain
Respiratory System
Apnea, asthma, cough increased, epistaxis, hyperventilation, lung disorder, rhinitis
Urogenital
Hematuria
The following adverse events occurred in 1% to less than 5% of AMPHOTEC patients. The causal association between these adverse events and AMPHOTEC is uncertain.
General (body as a whole)
Accidental injury, allergic reaction, asthenia, death, hypothermia, immune system disorder, infection, injection site pain, injection site reaction, neck pain
Cardiovascular System
Arrhythmia, atrial fibrillation, bradycardia, congestive heart failure, heart arrest, phlebitis, shock, supraventricular tachycardia, syncope, vasodilatation, venoocclusive liver disease, ventricular extrasystoles
Digestive System
Anorexia, bloody diarrhea, constipation, dyspepsia, fecal incontinence, gamma glutamyl transpeptidase increased, gastrointestinal disorder, gastrointestinal hemorrhage, gingivitis, glossitis, hepatic failure, melena, mouth ulceration, oral moniliasis, rectal disorder
Hemic and Lymphatic System
Ecchymosis, fibrinogen increased, hypochromic anemia, leukocytosis, leukopenia, petechia, thromboplastin decreased
Metabolic and Nutritional Disorders
Acidosis, BUN increased, dehydration, hyponatremia, hyperkalemia, hyperlipemia, hypernatremia, hypervolemia, hypoglycemia, hypoproteinemia, lactic dehydrogenase increased, AST (SGOT) increased, ALT (SGPT) increased, weight loss
Nervous System
Agitation, anxiety, convulsion, depression, hallucinations, hypertonia, nervousness, neuropathy, paresthesia, psychosis, speech disorder, stupor
Respiratory System
Hemoptysis, lung edema, pharyngitis, pleural effusion, respiratory disorder, sinusitis
- OVERDOSAGE
-
DOSAGE AND ADMINISTRATION
The recommended dose for adults and pediatric patients is 3 - 4 mg/kg as required, once a day.
AMPHOTEC, reconstituted in Sterile Water for Injection, is administered diluted in 5% Dextrose for Injection by intravenous infusion at a rate of 1 mg/kg/hour. A test dose immediately preceding the first dose is advisable when commencing all new courses of treatment. A small amount of drug (e.g., 10 mL of the final preparation containing between 1.6 to 8.3 mg) should be infused over 15 to 30 minutes and the patient carefully observed for the next 30 minutes.
The infusion time may be shortened to a minimum of 2 hours for patients who show no evidence of intolerance or infusion-related reactions. If the patient experiences acute reactions or cannot tolerate the infusion volume, the infusion time may be extended.
Directions for reconstitution and preparation of infusion admixture
AMPHOTEC must be reconstituted by addition of Sterile Water for Injection. Using sterile syringe and a 20-gauge needle, rapidly add the following volumes to the vial to provide a liquid containing 5 mg of amphotericin B per mL. Shake gently by hand, rotating the vial until all solids have dissolved. Note that the fluid may be opalescent or clear.
50 mg/vial add 10 mL Sterile Water for Injection 100 mg/vial add 20 mL Sterile Water for Injection For infusion, further dilute the reconstituted liquid to a final concentration of approximately 0.6 mg/mL (range 0.16 mg/mL to 0.83 mg/mL). The following table provides dilution recommendations:
Dose of
AMPHOTECVolume of Reconstituted
AMPHOTECInfusion Bag Size for
5% Dextrose for Injection10 – 35 mg 2 – 7 mL 50 mL 35 – 70 mg 7 – 14 mL 100 mL 70 – 175 mg 14 – 35 mL 250 mL 175 – 350 mg 35 – 70 mL 500 mL 350 – 1000 mg 70 – 200 mL 1000 mL Do not reconstitute the lyophilized powder with saline or dextrose solutions, or admix the reconstituted liquid with saline or electrolytes.
The use of any solution other than those recommended, or the presence of a bacteriostatic agent (e.g., benzyl alcohol) in the solution may cause precipitation of AMPHOTEC. Do not filter or use an in-line filter with AMPHOTEC.
Do not mix the infusion admixture with other drugs. If administered through an existing intravenous line, flush with 5% Dextrose for Injection prior to, and following, infusion of AMPHOTEC, otherwise administer via a separate line.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration whenever solution and container permit. Do not use if a precipitate or foreign matter is present, or if the seal is not intact. Strict aseptic technique always should be observed during reconstitution and dilution since no preservatives are present in the lyophilized drug or in the solutions used for reconstitution and dilution.
After reconstitution, the drug should be refrigerated at 2-8°C (36-46°F) and used within 24 hours. Do not freeze. After further dilution with 5% Dextrose for Injection, the infusion should be stored in a refrigerator (2-8°C) and used within 24 hours. Partially used vials should be discarded.
-
HOW SUPPLIED
AMPHOTEC® (Amphotericin B) Cholesteryl Sulfate Complex for Injection is a sterile lyophilized powder supplied in single use glass vials. Each vial is individually packaged.
AMPHOTEC 50 mg in 20 mL vial (NDC 64116-025-01)
AMPHOTEC 100 mg in 50 mL vial (NDC 64116-021-01)
STORAGE
Store unopened vials of AMPHOTEC at 15-30°C (59-86°F).
AMPHOTEC should be retained in the carton until time of use.
Manufactured by:
Ben Venue Laboratories, Inc., Bedford, OH 44146, USADistributed by:
InterMune, Inc., Brisbane, CA 94005U.S. Patent Numbers 4,822,777; 5,032,582; 5,194,266; 5,077,057.
Rx Only
- REFERENCES
-
INGREDIENTS AND APPEARANCE
AMPHOTEC
amphotericin b injection, lipid complexProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64116-025 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength amphotericin B (UNII: 7XU7A7DROE) (amphotericin B - UNII:7XU7A7DROE) 50 mg in 10 mL Inactive Ingredients Ingredient Name Strength sodium cholesteryl sulfate () 26.4 mg in 10 mL tromethamine (UNII: 023C2WHX2V) 5.64 mg in 10 mL disodium edetate dihydrate () 0.372 mg in 10 mL lactose monohydrate (UNII: EWQ57Q8I5X) 950 mg in 10 mL hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64116-025-01 10 mL in 1 VIAL, SINGLE-USE AMPHOTEC
amphotericin b injection, lipid complexProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64116-021 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength amphotericin B (UNII: 7XU7A7DROE) (amphotericin B - UNII:7XU7A7DROE) 100 mg in 20 mL Inactive Ingredients Ingredient Name Strength sodium cholesteryl sulfate () 52.8 mg in 20 mL tromethamine (UNII: 023C2WHX2V) 11.28 mg in 20 mL disodium edetate dihydrate () 0.744 mg in 20 mL lactose monohydrate (UNII: EWQ57Q8I5X) 1900 mg in 20 mL hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64116-021-01 20 mL in 1 VIAL, SINGLE-USE Labeler - InterMune, Inc.