Label: GALANTAMINE HYDROBROMIDE capsule, extended release

- NDC Code(s): 43353-984-05
- Packager: Aphena Pharma Solutions - Tennessee, LLC
- This is a repackaged label.
- Source NDC Code(s): 0591-3497
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated June 13, 2016
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
DESCRIPTION
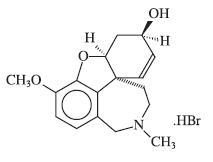
Galantamine hydrobromide is a reversible, competitive acetylcholinesterase inhibitor. Galantamine hydrobromide is known chemically as (4aS,6R,8aS)-4a,5,9,10,11,12-hexahydro-3-methoxy-11-methyl-6H-benzofuro[3a,3,2-ef][2]benzazepin-6-ol hydrobromide. It has a molecular formula of C17H21NO3•HBr and a molecular weight of 368.27. Galantamine hydrobromide is a white to almost white powder and is sparingly soluble in water. The structural formula for galantamine hydrobromide is:
Galantamine hydrobromide extended-release capsules are available in opaque hard gelatin extended-release capsules of 8 mg (white), 16 mg (pink and white), and 24 mg (pink) containing galantamine hydrobromide, equivalent to respectively 8, 16 and 24 mg galantamine base. Inactive ingredients include gelatin, hydroxypropyl cellulose, magnesium stearate, colloidal silicon dioxide and titanium dioxide. The 16 mg capsule and 24 mg capsule also contain red ferric oxide.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Although the etiology of cognitive impairment in Alzheimer’s disease (AD) is not fully understood, it has been reported that acetylcholine-producing neurons degenerate in the brains of patients with Alzheimer’s disease. The degree of this cholinergic loss has been correlated with degree of cognitive impairment and density of amyloid plaques (a neuropathological hallmark of Alzheimer’s disease).
Galantamine, a tertiary alkaloid, is a competitive and reversible inhibitor of acetylcholinesterase. While the precise mechanism of galantamine’s action is unknown, it is postulated to exert its therapeutic effect by enhancing cholinergic function. This is accomplished by increasing the concentration of acetylcholine through reversible inhibition of its hydrolysis by cholinesterase. If this mechanism is correct, galantamine’s effect may lessen as the disease process advances and fewer cholinergic neurons remain functionally intact. There is no evidence that galantamine alters the course of the underlying dementing process.
Pharmacokinetics
Galantamine is well absorbed with absolute oral bioavailability of about 90%. It has a terminal elimination half-life of about 7 hours and pharmacokinetics are linear over the range of 8-32 mg/day.
The maximum inhibition of acetylcholinesterase activity of about 40% was achieved about one hour after a single oral dose of 8 mg galantamine in healthy male subjects.
Absorption and Distribution
Galantamine is rapidly and completely absorbed with time to peak concentration about 1 hour. Bioavailability of the tablet was the same as the bioavailability of an oral solution. Food did not affect the AUC of galantamine but Cmax decreased by 25% and Tmax was delayed by 1.5 hours. The mean volume of distribution of galantamine is 175 L.
The plasma protein binding of galantamine is 18% at therapeutically relevant concentrations. In whole blood, galantamine is mainly distributed to blood cells (52.7%). The blood to plasma concentration ratio of galantamine is 1.2.
Metabolism and Elimination
Galantamine is metabolized by hepatic cytochrome P450 enzymes, glucuronidated, and excreted unchanged in the urine. In vitro studies indicate that cytochrome CYP2D6 and CYP3A4 were the major cytochrome P450 isoenzymes involved in the metabolism of galantamine, and inhibitors of both pathways increase oral bioavailability of galantamine modestly (see PRECAUTIONS, Drug-Drug Interactions). O-demethylation, mediated by CYP2D6 was greater in extensive metabolizers of CYP2D6 than in poor metabolizers. In plasma from both poor and extensive metabolizers, however, unchanged galantamine and its glucuronide accounted for most of the sample radioactivity.
In studies of oral 3H-galantamine, unchanged galantamine and its glucuronide, accounted for most plasma radioactivity in poor and extensive CYP2D6 metabolizers. Up to 8 hours post-dose, unchanged galantamine accounted for 39-77% of the total radioactivity in the plasma, and galantamine glucuronide for 14-24%. By 7 days, 93-99% of the radioactivity had been recovered, with about 95% in urine and about 5% in the feces. Total urinary recovery of unchanged galantamine accounted for, on average, 32% of the dose and that of galantamine glucuronide for another 12% on average.
After i.v. or oral administration, about 20% of the dose was excreted as unchanged galantamine in the urine in 24 hours, representing a renal clearance of about 65 mL/min, about 20-25% of the total plasma clearance of about 300 mL/min.
Galantamine hydrobromide 24 mg extended-release capsules administered once daily under fasting conditions are bioequivalent to galantamine hydrobromide immediate-release tablets 12 mg twice daily with respect to AUC24h and Cmin. The Cmax and Tmax of the extended-release capsules were lower and occurred later, respectively, compared with the immediate-release tablets, with Cmax about 25% lower and median Tmax occurring about 4.5 – 5.0 hours after dosing. Dose-proportionality is observed for galantamine hydrobromide extended-release capsules over the dose range of 8 to 24 mg daily and steady state is achieved within a week. There was no effect of age on the pharmacokinetics of galantamine hydrobromide extended-release capsules. CYP2D6 poor metabolizers had drug exposures that were approximately 50% higher than for extensive metabolizers.
There are no appreciable differences in pharmacokinetic parameters when galantamine hydrobromide extendedrelease capsules are given with food compared to when they are given in the fasted state.
Special Populations
CYP2D6 Poor Metabolizers
Approximately 7% of the normal population has a genetic variation that leads to reduced levels of activity of CYP2D6 isozyme. Such individuals have been referred to as poor metabolizers. After a single oral dose of 4 mg or 8 mg galantamine, CYP2D6 poor metabolizers demonstrated a similar Cmax and about 35% AUC∞ increase of unchanged galantamine compared to extensive metabolizers.
A total of 356 patients with Alzheimer’s disease enrolled in two Phase 3 studies were genotyped with respect to CYP2D6 (n=210 hetero-extensive metabolizers, 126 homo-extensive metabolizers, and 20 poor metabolizers). Population pharmacokinetic analysis indicated that there was a 25% decrease in median clearance in poor metabolizers compared to extensive metabolizers. Dosage adjustment is not necessary in patients identified as poor metabolizers as the dose of drug is individually titrated to tolerability.
Hepatic Impairment:
Following a single 4 mg dose of galantamine tablets, the pharmacokinetics of galantamine in subjects with mild hepatic impairment (n=8; Child-Pugh score of 5-6) were similar to those in healthy subjects. In patients with moderate hepatic impairment (n=8; Child-Pugh score of 7-9), galantamine clearance was decreased by about 25% compared to normal volunteers. Exposure would be expected to increase further with increasing degree of hepatic impairment (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Renal Impairment:
Following a single 8 mg dose of galantamine tablets, AUC increased by 37% and 67% in moderate and severely renal-impaired patients compared to normal volunteers (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Elderly:
Data from clinical trials in patients with Alzheimer’s disease indicate that galantamine concentrations are 30-40% higher than in healthy young subjects.
Gender and Race:
No specific pharmacokinetic study was conducted to investigate the effect of gender and race on the disposition of galantamine hydrobromide, but a population pharmacokinetic analysis indicates (n= 539 males and 550 females) that galantamine clearance is about 20% lower in females than in males (explained by lower body weight in females) and race (n= 1029 White, 24 Black, 13 Asian and 23 other) did not affect the clearance of galantamine hydrobromide.
Drug-Drug Interactions
(see also PRECAUTIONS, Drug-Drug Interactions)
Multiple metabolic pathways and renal excretion are involved in the elimination of galantamine so no single pathway appears predominant. Based on in vitro studies, CYP2D6 and CYP3A4 were the major enzymes involved in the metabolism of galantamine. CYP2D6 was involved in the formation of O-desmethyl-galantamine, whereas CYP3A4 mediated the formation of galantamine-N-oxide. Galantamine is also glucuronidated and excreted unchanged in urine.
(A) Effect of Other Drugs on the Metabolism of Galantamine Hydrobromide:
Drugs that are potent inhibitors for CYP2D6 or CYP3A4 may increase the AUC of galantamine. Multiple dose pharmacokinetic studies demonstrated that the AUC of galantamine increased 30% and 40%, respectively, during coadministration of ketoconazole and paroxetine. As co-administered with erythromycin, another CYP3A4 inhibitor, the galantamine AUC increased only 10%. Population PK analysis with a database of 852 patients with Alzheimer’s disease showed that the clearance of galantamine was decreased about 25-33% by concurrent administration of amitriptyline (n = 17), fluoxetine (n = 48), fluvoxamine (n = 14), and quinidine (n = 7), known inhibitors of CYP2D6.
Concurrent administration of H2-antagonists demonstrated that ranitidine did not affect the pharmacokinetics of galantamine, and cimetidine increased the galantamine AUC by approximately 16%.
A multiple dose pharmacokinetic study with concurrent administration of memantine, an N-methyl-D-aspartate (NMDA) receptor antagonist, demonstrated that co-administration of memantine in a dose of 10 mg BID did not affect the pharmacokinetic profile of galantamine (16 mg daily) at steady state.
(B) Effect of Galantamine Hydrobromide on the Metabolism of Other Drugs:
In vitro studies show that galantamine did not inhibit the metabolic pathways catalyzed by CYP1A2, CYP2A6, CYP3A4, CYP4A, CYP2C, CYP2D6 and CYP2E1. This indicated that the inhibitory potential of galantamine towards the major forms of cytochrome P450 is very low. Multiple doses of galantamine (24 mg/day) had no effect on the pharmacokinetics of digoxin and warfarin (R- and S- forms). Galantamine had no effect on the increased prothrombin time induced by warfarin.
-
CLINICAL TRIALS
The effectiveness of galantamine hydrobromide as a treatment for Alzheimer’s disease is demonstrated by the results of 5 randomized, double-blind, placebo-controlled clinical investigations in patients with probable Alzheimer’s disease, 4 with the tablet and 1 with the extended-release capsule [diagnosed by NINCDS-ADRDA criteria, with Mini-Mental State Examination scores that were ≥ 10 and ≤ 24]. Doses studied with the tablet formulation were 8-32 mg/day given as twice daily doses. In 3 of the 4 studies with the tablet, patients were started on a low dose of 8 mg, then titrated weekly by 8 mg/day to 24 or 32 mg as assigned. In the fourth study (USA 4-week Dose-Escalation Fixed-Dose Study) dose escalation of 8 mg/day occurred over 4 week intervals. The mean age of patients participating in these 4 galantamine hydrobromide trials was 75 years with a range of 41 to 100. Approximately 62% of patients were women and 38% were men. The racial distribution was White 94%, Black 3% and other races 3%. Two other studies examined a three times daily dosing regimen; these also showed or suggested benefit but did not suggest an advantage over twice daily dosing.
Study Outcome Measures:
In each study, the primary effectiveness of galantamine hydrobromide immediate-release tablets was evaluated using a dual outcome assessment strategy as measured by the Alzheimer’s Disease Assessment Scale (ADAS-cog) and the Clinician’s Interview Based Impression of Change that required the use of caregiver information (CIBIC-plus).
The ability of galantamine hydrobromide to improve cognitive performance was assessed with the cognitive subscale of the Alzheimer’s Disease Assessment Scale (ADAS-cog), a multi-item instrument that has been extensively validated in longitudinal cohorts of Alzheimer’s disease patients. The ADAS-cog examines selected aspects of cognitive performance including elements of memory, orientation, attention, reasoning, language and praxis. The ADAS-cog scoring range is from 0 to 70, with higher scores indicating greater cognitive impairment. Elderly normal adults may score as low as 0 or 1, but it is not unusual for non-demented adults to score slightly higher. The patients recruited as participants in each study using the tablet formulation had mean scores on ADAS-cog of approximately 27 units, with a range from 5 to 69. Experience gained in longitudinal studies of ambulatory patients with mild to moderate Alzheimer’s disease suggests that they gain 6 to 12 units a year on the ADAS-cog. Lesser degrees of change, however, are seen in patients with very mild or very advanced disease because the ADAS-cog is not uniformly sensitive to change over the course of the disease. The annualized rate of decline in the placebo patients participating in galantamine hydrobromide trials was approximately 4.5 units per year.
The ability of galantamine hydrobromide to produce an overall clinical effect was assessed using a Clinician’s Interview Based Impression of Change that required the use of caregiver information, the CIBIC-plus. The CIBIC-plus is not a single instrument and is not a standardized instrument like the ADAS-cog. Clinical trials for investigational drugs have used a variety of CIBIC formats, each different in terms of depth and structure. As such, results from a CIBIC-plus reflect clinical experience from the trial or trials in which it was used and cannot be compared directly with the results of CIBIC-plus evaluations from other clinical trials. The CIBIC-plus used in the trials was a semi-structured instrument based on a comprehensive evaluation at baseline and subsequent time-points of 4 major areas of patient function: general, cognitive, behavioral and activities of daily living. It represents the assessment of a skilled clinician based on his/her observation at an interview with the patient, in combination with information supplied by a caregiver familiar with the behavior of the patient over the interval rated. The CIBIC-plus is scored as a seven point categorical rating, ranging from a score of 1, indicating “markedly improved,” to a score of 4, indicating “no change” to a score of 7, indicating “marked worsening.” The CIBIC-plus has not been systematically compared directly to assessments not using information from caregivers (CIBIC) or other global methods.
Immediate-Release Tablets
U.S. Twenty-One Week Fixed-Dose Study
In a study of 21 weeks duration, 978 patients were randomized to doses of 8, 16, or 24 mg of immediate-release galantamine hydrobromide per day, or to placebo, each given in 2 divided doses. Treatment was initiated at 8 mg/day for all patients randomized to galantamine hydrobromide, and increased by 8 mg/day every 4 weeks. Therefore, the maximum titration phase was 8 weeks and the minimum maintenance phase was 13 weeks (in patients randomized to 24 mg/day of galantamine hydrobromide).
Effects on the ADAS-cog:
Figure 1 illustrates the time course for the change from baseline in ADAS-cog scores for all four dose groups over the 21 weeks of the study. At 21 weeks of treatment, the mean differences in the ADAS-cog change scores for the galantamine hydrobromide-treated patients compared to the patients on placebo were 1.7, 3.3, and 3.6 units for the 8, 16 and 24 mg/day treatments, respectively. The 16 mg/day and 24 mg/day treatments were statistically significantly superior to placebo and to the 8 mg/day treatment. There was no statistically significant difference between the 16 mg/day and 24 mg/day dose groups.
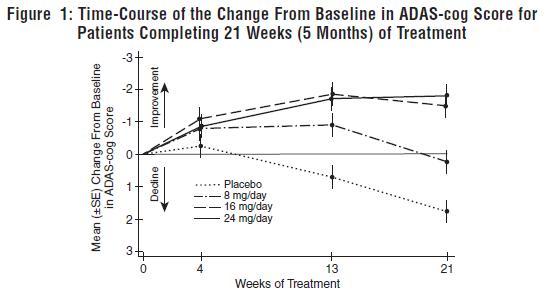
Figure 2 illustrates the cumulative percentages of patients from each of the four treatment groups who had attained at least the measure of improvement in ADAS-cog score shown on the X-axis. Three change scores (10-point, 7-point and 4-point reductions) and no change in score from baseline have been identified for illustrative purposes, and the percent of patients in each group achieving that result is shown in the inset table.
The curves demonstrate that both patients assigned to galantamine and placebo have a wide range of responses, but that the galantamine hydrobromide groups are more likely to show the greater improvements.
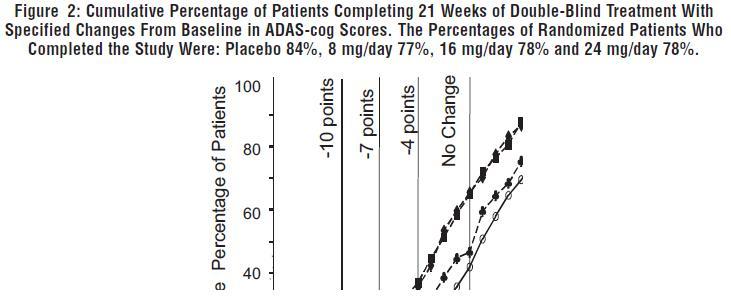
Change in ADAS-cog Treatment -10 -7 -4 0 Placebo 3.6% 7.6% 19.6% 41.8% 8 mg/day 5.9% 13.9% 25.7% 46.5% 16 mg/day 7.2% 15.9% 35.6% 65.4% 24 mg/day 10.4% 22.3% 37.0% 64.9% Effects on the CIBIC-plus:
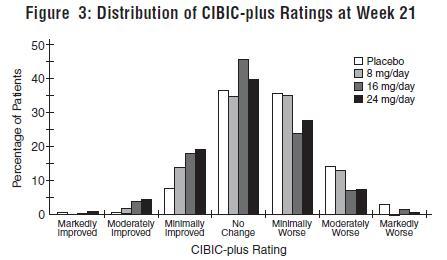
Figure 3 is a histogram of the percentage distribution of CIBIC-plus scores attained by patients assigned to each of the four treatment groups who completed 21 weeks of treatment. The galantamine hydrobromide-placebo differences for these groups of patients in mean rating were 0.15, 0.41 and 0.44 units for the 8, 16 and 24 mg/day treatments, respectively. The 16 mg/day and 24 mg/day treatments were statistically significantly superior to placebo. The differences vs. the 8 mg/day treatment for the 16 and 24 mg/day treatments were 0.26 and 0.29, respectively. There were no statistically significant differences between the 16 mg/day and 24 mg/day dose groups.
U.S. Twenty-Six Week Fixed-Dose Study
In a study of 26 weeks duration, 636 patients were randomized to either a dose of 24 mg or 32 mg of immediate-release galantamine hydrobromide per day, or to placebo, each given in two divided doses. The 26-week study was divided into a 3-week dose titration phase and a 23-week maintenance phase.
Effects on the ADAS-cog:
Figure 4 illustrates the time course for the change from baseline in ADAS-cog scores for all three dose groups over the 26 weeks of the study. At 26 weeks of treatment, the mean differences in the ADAS-cog change scores for the galantamine hydrobromide treated patients compared to the patients on placebo were 3.9 and 3.8 units for the 24 mg/day and 32 mg/day treatments, respectively. Both treatments were statistically significantly superior to placebo, but were not significantly different from each other.
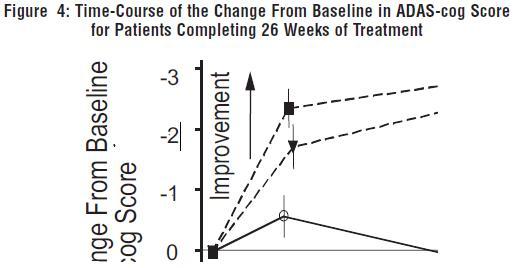
Figure 5 illustrates the cumulative percentages of patients from each of the three treatment groups who had attained at least the measure of improvement in ADAS-cog score shown on the X-axis. Three change scores (10-point, 7-point and 4-point reductions) and no change in score from baseline have been identified for illustrative purposes, and the percent of patients in each group achieving that result is shown in the inset table.
The curves demonstrate that both patients assigned to galantamine hydrobromide and placebo have a wide range of responses, but that the galantamine hydrobromide groups are more likely to show the greater improvements. A curve for an effective treatment would be shifted to the left of the curve for placebo, while an ineffective or deleterious treatment would be superimposed upon, or shifted to the right of the curve for placebo, respectively.
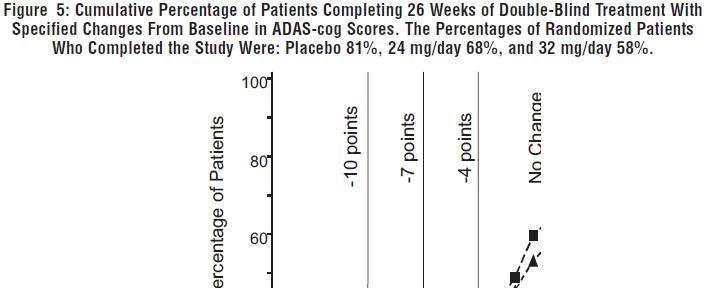
Change in ADAS-cog Treatment -10 -7 -4 0 Placebo 2.1% 5.7% 16.6% 43.9% 24 mg/day 7.6% 18.3% 33.6% 64.1% 32 mg/day 11.1% 19.7% 33.3% 58.1% Effects on the CIBIC-plus:
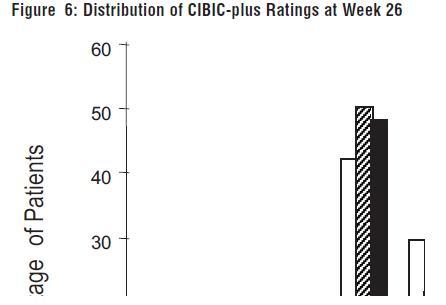
Figure 6 is a histogram of the percentage distribution of CIBIC-plus scores attained by patients assigned to each of the three treatment groups who completed 26 weeks of treatment. The mean galantamine hydrobromide-placebo differences for these groups of patients in the mean rating were 0.28 and 0.29 units for 24 and 32 mg/day of galantamine hydrobromide, respectively. The mean ratings for both groups were statistically significantly superior to placebo, but were not significantly different from each other.
International Twenty-Six Week Fixed-Dose Study
In a study of 26 weeks duration identical in design to the USA 26-Week Fixed-Dose Study, 653 patients were randomized to either a dose of 24 mg or 32 mg of immediate-release galantamine hydrobromide per day, or to placebo, each given in two divided doses. The 26-week study was divided into a 3-week dose titration phase and a 23-week maintenance phase.
Effects on the ADAS-cog:
Figure 7 illustrates the time course for the change from baseline in ADAS-cog scores for all three dose groups over the 26 weeks of the study. At 26 weeks of treatment, the mean differences in the ADAS-cog change scores for the galantamine hydrobromide-treated patients compared to the patients on placebo were 3.1 and 4.1 units for the 24 mg/day and 32 mg/day treatments, respectively. Both treatments were statistically significantly superior to placebo, but were not significantly different from each other.
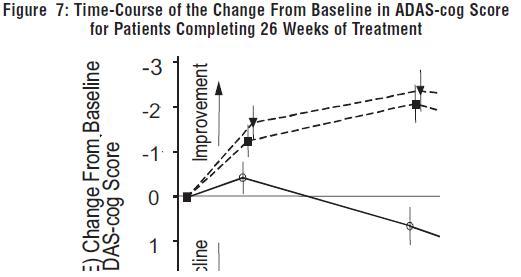
Figure 8 illustrates the cumulative percentages of patients from each of the three treatment groups who had attained at least the measure of improvement in ADAS-cog score shown on the X-axis. Three change scores (10-point, 7-point and 4-point reductions) and no change in score from baseline have been identified for illustrative purposes, and the percent of patients in each group achieving that result is shown in the inset table.
The curves demonstrate that both patients assigned to galantamine hydrobromide and placebo have a wide range of responses, but that the galantamine hydrobromide groups are more likely to show the greater improvements.
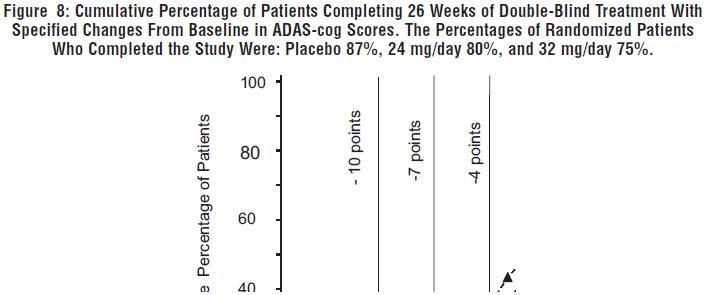
Change in ADAS-cog Treatment -10 -7 -4 0 Placebo 1.2% 5.8% 15.2% 39.8% 24 mg/day 4.5% 15.4% 30.8% 65.4% 32 mg/day 7.9% 19.7% 34.9% 63.8% Effects on the CIBIC-plus:
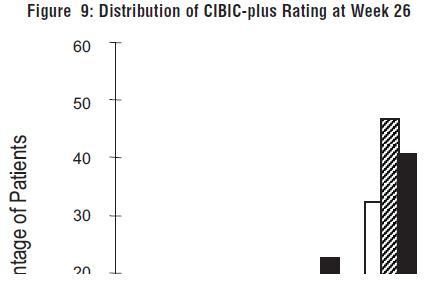
Figure 9 is a histogram of the percentage distribution of CIBIC-plus scores attained by patients assigned to each of the three treatment groups who completed 26 weeks of treatment. The mean galantamine hydrobromide-placebo differences for these groups of patients in the mean rating of change from baseline were 0.34 and 0.47 for 24 and 32 mg/day of galantamine hydrobromide, respectively. The mean ratings for the galantamine hydrobromide groups were statistically significantly superior to placebo, but were not significantly different from each other.
International Thirteen-Week Flexible-Dose Study
In a study of 13 weeks duration, 386 patients were randomized to either a flexible dose of 24-32 mg/day of immediate-release galantamine hydrobromide or to placebo, each given in two divided doses. The 13-week study was divided into a 3-week dose titration phase and a 10-week maintenance phase. The patients in the active treatment arm of the study were maintained at either 24 mg/day or 32 mg/day at the discretion of the investigator.
Effects on the ADAS-cog:
Figure 10 illustrates the time course for the change from baseline in ADAS-cog scores for both dose groups over the 13 weeks of the study. At 13 weeks of treatment, the mean difference in the ADAS-cog change scores for the treated patients compared to the patients on placebo was 1.9. Galantamine hydrobromide at a dose of 24-32 mg/day was statistically significantly superior to placebo.
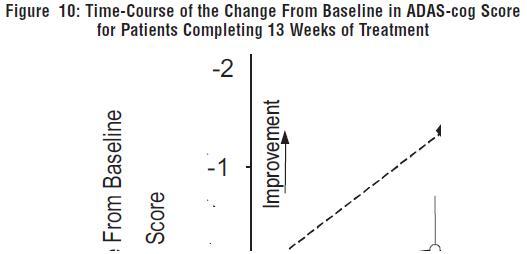
Figure 11 illustrates the cumulative percentages of patients from each of the two treatment groups who had attained at least the measure of improvement in ADAS-cog score shown on the X-axis. Three change scores (10-point, 7-point and 4-point reductions) and no change in score from baseline have been identified for illustrative purposes, and the percent of patients in each group achieving that result is shown in the inset table.
The curves demonstrate that both patients assigned to galantamine hydrobromide and placebo have a wide range of responses, but that the galantamine hydrobromide group is more likely to show the greater improvement.
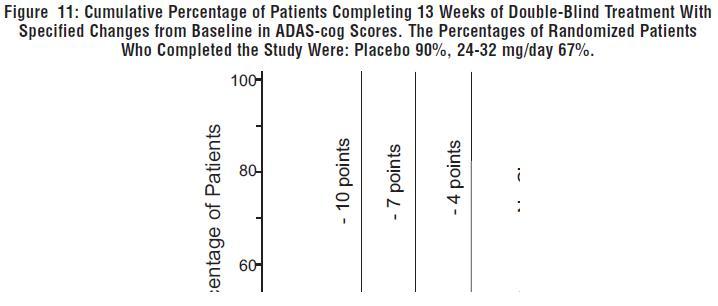
Change in ADAS-cog Treatment -10 -7 -4 0 Placebo 1.9% 5.6% 19.4% 50.0% 24 or 32 mg/day 7.1% 18.8% 32.9% 65.3% Effects on the CIBIC-plus:
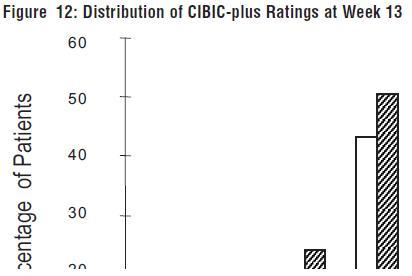
Figure 12 is a histogram of the percentage distribution of CIBIC-plus scores attained by patients assigned to each of the two treatment groups who completed 13 weeks of treatment. The mean galantamine hydrobromide-placebo differences for the group of patients in the mean rating of change from baseline was 0.37 units. The mean rating for the 24-32 mg/day group was statistically significantly superior to placebo.
Age, Gender and Race:
Patient’s age, gender, or race did not predict clinical outcome of treatment.
Extended-Release Capsules
The efficacy of galantamine hydrobromide extended-release capsules was studied in a randomized, double-blind, placebo-controlled trial which was 6 months in duration, and had an initial 4-week dose-escalation phase. In this trial, patients were assigned to one of 3 treatment groups: galantamine hydrobromide extended-release capsules in a flexible dose of 16 to 24 mg once daily; immediate-release galantamine hydrobromide tablets in a flexible dose of 8 to 12 mg twice daily; and placebo. The primary efficacy measures in this study were the ADAS-cog and CIBIC-plus. On the protocol-specified primary efficacy analysis at Month 6, a statistically significant improvement favoring galantamine hydrobromide extended-release capsules over placebo was seen for the ADAS-cog, but not for the CIBIC-plus. Galantamine hydrobromide extended-release capsules showed a statistically significant improvement when compared with placebo on the Alzheimer’s Disease Cooperative Study-Activities of Daily Living (ADCS-ADL) scale, a measure of function, and a secondary efficacy measure in this study. The effects of both galantamine hydrobromide extended-release capsules and the galantamie hydrobromide immediate-release tablets on the ADAS-cog, CIBIC-plus, and ADCS-ADL were similar in this study.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Anesthesia
Galantamine, as a cholinesterase inhibitor, is likely to exaggerate the neuromuscular blocking effects of succinylcholine-type and similar neuromuscular blocking agents during anesthesia.
Cardiovascular Conditions
Because of their pharmacological action, cholinesterase inhibitors have vagotonic effects on the sinoatrial and atrioventricular nodes, leading to bradycardia and AV block. These actions may be particularly important to patients with supraventricular cardiac conduction disorders or to patients taking other drugs concomitantly that significantly slow heart rate. Postmarketing surveillance of marketed anticholinesterase inhibitors has shown, however, that bradycardia and all types of heart block have been reported in patients both with and without known underlying cardiac conduction abnormalities. Therefore all patients should be considered at risk for adverse effects on cardiac conduction.
In randomized controlled trials, bradycardia was reported more frequently in galantamine-treated patients than in placebo-treated patients, but was rarely severe and rarely led to treatment discontinuation. The overall frequency of this event was 2-3% for galantamine doses up to 24 mg/day compared with <1% for placebo. No increased incidence of heart block was observed at the recommended doses.
Patients treated with galantamine up to 24 mg/day using the recommended dosing schedule showed a dose-related increase in risk of syncope (placebo 0.7% [2/286]; 4 mg BID 0.4% [3/692]; 8 mg BID 1.3% [7/552]; 12 mg BID 2.2% [6/273]).
Gastrointestinal Conditions
Through their primary action, cholinomimetics may be expected to increase gastric acid secretion due to increased cholinergic activity. Therefore, patients should be monitored closely for symptoms of active or occult gastrointestinal bleeding, especially those with an increased risk for developing ulcers, e.g., those with a history of ulcer disease or patients using concurrent nonsteroidal anti-inflammatory drugs (NSAIDS). Clinical studies of galantamine hydrobromide have shown no increase, relative to placebo, in the incidence of either peptic ulcer disease or gastrointestinal bleeding.
Galantamine hydrobromide, as a predictable consequence of its pharmacological properties, has been shown to produce nausea, vomiting, diarrhea, anorexia, and weight loss (see ADVERSE REACTIONS).
Genitourinary
Although this was not observed in clinical trials with galantamine hydrobromide, cholinomimetics may cause bladder outflow obstruction.
Neurological Conditions
Seizures: Cholinesterase inhibitors are believed to have some potential to cause generalized convulsions. However, seizure activity may also be a manifestation of Alzheimer’s disease. In clinical trials, there was no increase in the incidence of convulsions with galantamine hydrobromide, compared to placebo.
Pulmonary Conditions
Because of its cholinomimetic action, galantamine should be prescribed with care to patients with a history of severe asthma or obstructive pulmonary disease.
-
PRECAUTIONS
Information for Patients and Caregivers:
Caregivers should be instructed about the recommended dosage and administration of galantamine hydrobromide extended-release capsules. Galantamine hydrobromide extended-release capsules should be administered once daily in the morning, preferably with food (although not required). Dose escalation (dose increases) should follow a minimum of four weeks at prior dose.
Patients and caregivers should be advised that the most frequent adverse events associated with use of the drug can be minimized by following the recommended dosage and administration.
Patients and caregivers should be advised to ensure adequate fluid intake during treatment. If therapy has been interrupted for several days or longer, the patient should be restarted at the lowest dose and the dose escalated to the current dose.
Deaths in Subjects with Mild Cognitive Impairment (MCI)
In two randomized placebo controlled trials of 2 years duration in subjects with mild cognitive impairment (MCI), a total of 13 subjects on galantamine hydrobromide (n=1026) and 1 subject on placebo (n=1022) died. The deaths were due to various causes which could be expected in an elderly population; about half of the galantamine hydrobromide deaths appeared to result from various vascular causes (myocardial infarction, stroke, and sudden death).
Although the difference in mortality between galantamine hydrobromide and placebo-treated groups in these two studies was significant, the results are highly discrepant with other studies of galantamine hydrobromide. Specifically, in these two MCI studies, the mortality rate in the placebo-treated subjects was markedly lower than the rate in placebo-treated patients in trials of galantamine hydrobromide in Alzheimer’s disease or other dementias (0.7 per 1000 person years compared to 22-61 per 1000 person years, respectively). Although the mortality rate in the galantamine hydrobromide-treated MCI subjects was also lower than that observed in galantamine hydrobromide-treated patients in Alzheimer’s disease and other dementia trials (10.2 per 1000 person years compared to 23-31 per 1000 person years, respectively), the relative difference was much less. When the Alzheimer’s disease and other dementia studies were pooled (n=6000), the mortality rate in the placebo group numerically exceeded that in the galantamine hydrobromide group. Furthermore, in the MCI studies, no subjects in the placebo group died after 6 months, a highly unexpected finding in this population.
Individuals with mild cognitive impairment demonstrate isolated memory impairment greater than expected for their age and education, but do not meet current diagnostic criteria for Alzheimer’s disease.
Special Populations
Hepatic Impairment
In patients with moderately impaired hepatic function, dose titration should proceed cautiously (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION). The use of galantamine hydrobromide in patients with severe hepatic impairment is not recommended.
Renal Impairment
In patients with moderately impaired renal function, dose titration should proceed cautiously (see CLINICAL PHARMACOLOGY and DOSAGE AND ADMINISTRATION). In patients with severely impaired renal function (CLcr < 9 mL/min) the use of galantamine hydrobromide is not recommended.
Drug-Drug Interactions
(see also CLINICAL PHARMACOLOGY, Drug-Drug Interactions)
Use With Anticholinergics
Galantamine hydrobromide has the potential to interfere with the activity of anticholinergic medications.
Use With Cholinomimetics and Other Cholinesterase Inhibitors
A synergistic effect is expected when cholinesterase inhibitors are given concurrently with succinylcholine, other cholinesterase inhibitors, similar neuromuscular blocking agents or cholinergic agonists such as bethanechol.
A) Effect of Other Drugs on Galantamine
In vitro
CYP3A4 and CYP2D6 are the major enzymes involved in the metabolism of galantamine. CYP3A4 mediates the formation of galantamine-N-oxide; CYP2D6 leads to the formation of O-desmethyl-galantamine. Because galantamine is also glucuronidated and excreted unchanged, no single pathway appears predominant.
In vivo
Cimetidine and Ranitidine: Galantamine was administered as a single dose of 4 mg on day 2 of a 3-day treatment with either cimetidine (800 mg daily) or ranitidine (300 mg daily). Cimetidine increased the bioavailability of galantamine by approximately 16%. Ranitidine had no effect on the PK of galantamine.
Ketoconazole: Ketoconazole, a strong inhibitor of CYP3A4 and an inhibitor of CYP2D6, at a dose of 200 mg BID for 4 days, increased the AUC of galantamine by 30%.
Erythromycin: Erythromycin, a moderate inhibitor of CYP3A4 at a dose of 500 mg QID for 4 days, affected the AUC of galantamine minimally (10% increase).
Paroxetine: Paroxetine, a strong inhibitor of CYP2D6, at 20 mg/day for 16 days, increased the oral bioavailability of galantamine by about 40%.
Memantine: Memantine, an N-methyl-D-aspartate receptor antagonist, at a dose of 10 mg BID, had no effect on the pharmacokinetics of galantamine (16 mg/day) at steady state.
B) Effect of Galantamine on Other Drugs
In vitro
Galantamine did not inhibit the metabolic pathways catalyzed by CYP1A2, CYP2A6, CYP3A4, CYP4A, CYP2C, CYP2D6 or CYP2E1. This indicates that the inhibitory potential of galantamine towards the major forms of cytochrome P450 is very low.
In vivo
Warfarin: Galantamine at 24 mg/day had no effect on the pharmacokinetics of R- and S-warfarin (25 mg single dose) or on the prothrombin time. The protein binding of warfarin was unaffected by galantamine.
Digoxin: Galantamine at 24 mg/day had no effect on the steady-state pharmacokinetics of digoxin (0.375 mg once daily) when they were coadministered. In this study, however, one healthy subject was hospitalized for 2nd and 3rd degree heart block and bradycardia.
Carcinogenesis, Mutagenesis and Impairment of Fertility
In a 24-month oral carcinogenicity study in rats, a slight increase in endometrial adenocarcinomas was observed at 10 mg/kg/day (4 times the Maximum Recommended Human Dose [MRHD] on a mg/m2 basis or 6 times on an exposure [AUC] basis) and 30 mg/kg/day (12 times MRHD on a mg/m2 basis or 19 times on an AUC basis). No increase in neoplastic changes was observed in females at 2.5 mg/kg/day (equivalent to the MRHD on a mg/m2 basis or 2 times on an AUC basis) or in males up to the highest dose tested of 30 mg/kg/day (12 times the MRHD on a mg/m2 and AUC basis).
Galantamine was not carcinogenic in a 6-month oral carcinogenicity study in transgenic (P 53-deficient) mice up to 20 mg/kg/day, or in a 24-month oral carcinogenicity study in male and female mice up to 10 mg/kg/day (2 times the MRHD on a mg/m2 basis and equivalent on an AUC basis).
Galantamine produced no evidence of genotoxic potential when evaluated in the in vitro Ames S. typhimurium or E. coli reverse mutation assay, in vitro mouse lymphoma assay, in vivo micronucleus test in mice, or in vitro chromosome aberration assay in Chinese hamster ovary cells.
No impairment of fertility was seen in rats given up to 16 mg/kg/day (7 times the MRHD on a mg/m2 basis) for 14 days prior to mating in females and for 60 days prior to mating in males.
Pregnancy
Pregnancy Category B: In a study in which rats were dosed from day 14 (females) or day 60 (males) prior to mating through the period of organogenesis, a slightly increased incidence of skeletal variations was observed at doses of 8 mg/kg/day (3 times the Maximum Recommended Human Dose [MRHD] on a mg/m2 basis) and 16 mg/kg/day. In a study in which pregnant rats were dosed from the beginning of organogenesis through day 21 post-partum, pup weights were decreased at 8 and 16 mg/kg/day, but no adverse effects on other postnatal developmental parameters were seen. The doses causing the above effects in rats produced slight maternal toxicity. No major malformations were caused in rats given up to 16 mg/kg/day. No drug related teratogenic effects were observed in rabbits given up to 40 mg/kg/day (32 times the MRHD on a mg/m2 basis) during the period of organogenesis.
There are no adequate and well-controlled studies of galantamine hydrobromide in pregnant women. Galantamine hydrobromide should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
-
ADVERSE REACTIONS
Pre-Marketing Clinical Trial Experience:
The specific adverse event data described in this section are based on studies of the immediate-release tablet formulation. In clinical trials, once-daily treatment with galantamine hydrobromide extended-release capsules was well tolerated and adverse events were similar to those seen with galantamine hydrobromide immediate-release tablets.
Adverse Events Leading to Discontinuation:
In two large scale, placebo-controlled trials of 6 months duration in which patients were titrated weekly from 8 to 16 to 24, and to 32 mg/day, the risk of discontinuation because of an adverse event in the galantamine group exceeded that in the placebo group by about threefold. In contrast, in a 5-month trial with escalation of the dose by 8 mg/day every 4 weeks, the overall risk of discontinuation because of an adverse event was 7%, 7%, and 10% for the placebo, galantamine 16 mg/day, and galantamine 24 mg/day groups, respectively, with gastrointestinal adverse effects the principle reason for discontinuing galantamine. Table 1 shows the most frequent adverse events leading to discontinuation in this study.
Table 1: Most Frequent Adverse Events Leading to Discontinuation in a Placebo-Controlled, Double-Blind Trial With a 4-Week Dose Escalation Schedule 4-Week Escalation Adverse Event Placebo
N=28616 mg/day
N=27924 mg/day
N=273Nausea <1% 2% 4% Vomiting 0% 1% 3% Anorexia <1% 1% <1% Dizziness <1% 2% 1% Syncope 0% 0% 1% Adverse Events Reported in Controlled Trials:
The reported adverse events in trials using galantamine hydrobromide immediate-release tablets reflect experience gained under closely monitored conditions in a highly selected patient population. In actual practice or in other clinical trials, these frequency estimates may not apply, as the conditions of use, reporting behavior and the types of patients treated may differ.
The majority of these adverse events occurred during the dose-escalation period. In those patients who experienced the most frequent adverse event, nausea, the median duration of the nausea was 5-7 days.
Administration of galantamine hydrobromide with food, the use of anti-emetic medication, and ensuring adequate fluid intake may reduce the impact of these events.
The most frequent adverse events, defined as those occurring at a frequency of at least 5% and at least twice the rate on placebo with the recommended maintenance dose of either 16 or 24 mg/day of galantamine hydrobromide under conditions of every 4-week dose-escalation for each dose increment of 8 mg/day, are shown in Table 2. These events were primarily gastrointestinal and tended to be less frequent with the 16 mg/day recommended initial maintenance dose.
Table 2: The Most Frequent Adverse Events in the Placebo-Controlled Trial With Dose Escalation Every 4 Weeks Occurring in at Least 5% of Patients Receiving Galantamine Hydrobromide and at Least Twice the Rate on Placebo.
Adverse EventPlacebo
N=286Galantamine
Hydrobromide
16 mg/day
N=279Galantamine
Hydrobromide
24 mg/day
N=273Nausea 5% 13% 17% Vomiting 1% 6% 10% Diarrhea 6% 12% 6% Anorexia 3% 7% 9% Weight decrease 1% 5% 5% Table 3: The most common adverse events (adverse events occurring with an incidence of at least 2% with galantamine hydrobromide treatment and in which the incidence was greater than with placebo treatment) are listed in Table 3 for four placebo-controlled trials for patients treated with 16 or 24 mg/day of galantamine hydrobromide.
Table 3: Adverse Events Reported in at Least 2% of Patients With Alzheimer’s Disease Administered Galantamine Hydrobromide and at a Frequency Greater Than With Placebo Body System Placebo Galantamine Hydrobromide1 Adverse Event (N=801) (N=1040) 1 Adverse events in patients treated with 16 or 24 mg/day of galantamine hydrobromide in four placebo-controlled trials are included. Body as a whole - general disorders Fatigue 3% 5% Syncope 1% 2% Central & peripheral nervous system disorders Dizziness 6% 9% Headache 5% 8% Tremor 2% 3% Gastrointestinal system disorders Nausea 9% 24% Vomiting 4% 13% Diarrhea 7% 9% Abdominal pain 4% 5% Dyspepsia 2% 5% Heart rate and rhythm disorders Bradycardia 1% 2% Metabolic and nutritional disorders Weight decrease 2% 7% Psychiatric disorders Anorexia 3% 9% Depression 5% 7% Insomnia 4% 5% Somnolence 3% 4% Red blood cell disorders Anemia 2% 3% Respiratory system disorders Rhinitis 3% 4% Urinary system disorders Urinary tract infection 7% 8% Hematuria 2% 3% Adverse events occurring with an incidence of at least 2% in placebo-treated patients that was either equal to or greater than with galantamine hydrobromide treatment were constipation, agitation, confusion, anxiety, hallucination, injury, back pain, peripheral edema, asthenia, chest pain, urinary incontinence, upper respiratory tract infection, bronchitis, coughing, hypertension, fall, and purpura.
There were no important differences in adverse event rates related to dose or sex. There were too few non-Caucasian patients to assess the effects of race on adverse event rates.
No clinically relevant abnormalities in laboratory values were observed.
Other Adverse Events Observed During Clinical Trials
Galantamine hydrobromide immediate-release tablets were administered to 3055 patients with Alzheimer’s disease. A total of 2357 patients received galantamine in placebo-controlled trials and 761 patients with Alzheimer’s disease received galantamine 24 mg/day, the maximum recommended maintenance dose. About 1000 patients received galantamine for at least one year and approximately 200 patients received galantamine for two years.
To establish the rate of adverse events, data from all patients receiving any dose of galantamine in 8 placebo-controlled trials and 6 open-label extension trials were pooled. The methodology to gather and codify these adverse events was standardized across trials, using WHO terminology. All adverse events occurring in approximately 0.1% are included, except for those already listed elsewhere in labeling, WHO terms too general to be informative, or events unlikely to be drug caused. Events are classified by body system and listed using the following definitions: frequent adverse events - those occurring in at least 1/100 patients; infrequent adverse events – those occurring in 1/100 to 1/1000 patients; rare adverse events - those occurring in fewer than 1/1000 patients. These adverse events are not necessarily related to galantamine hydrobromide treatment and in most cases were observed at a similar frequency in placebo-treated patients in the controlled studies. Additional adverse events observed in other clinical trials are also included below.
Body As a Whole – General Disorders: Frequent: chest pain, asthenia, fever, malaise
Cardiovascular System Disorders: Infrequent: postural hypotension, hypotension, dependent edema, cardiac failure, myocardial ischemia or infarction
Central & Peripheral Nervous System Disorders: Infrequent: vertigo, hypertonia, convulsions, involuntary muscle contractions, paresthesia, ataxia, hypokinesia, hyperkinesia, apraxia, aphasia, leg cramps, tinnitus, transient ischemic attack or cerebrovascular accident
Gastrointestinal System Disorders: Frequent: flatulence; Infrequent: gastritis, melena, dysphagia, rectal hemorrhage, dry mouth, saliva increased, diverticulitis, gastroenteritis, hiccup; Rare: esophageal perforation
Heart Rate & Rhythm Disorders: Infrequent: AV block, palpitation, atrial arrhythmias including atrial fibrillation and supraventricular tachycardia, QT prolonged, bundle branch block, T-wave inversion, ventricular tachycardia; Rare: severe bradycardia
Metabolic & Nutritional Disorders: Infrequent: hyperglycemia, alkaline phosphatase increased
Platelet, Bleeding & Clotting Disorders: Infrequent: purpura, epistaxis, thrombocytopenia
Psychiatric Disorders: Infrequent: apathy, paroniria, paranoid reaction, libido increased, delirium; Rare: suicidal ideation, suicide
Urinary System Disorders: Frequent: incontinence; Infrequent: hematuria, micturition frequency, cystitis, urinary retention, nocturia, renal calculi
Post-Marketing Experience:
Other adverse events from post-approval controlled and uncontrolled clinical trials and post-marketing experience observed in patients treated with galantamine hydrobromide include:
Body as a Whole – General Disorders: dehydration (including rare, severe cases leading to renal insufficiency and renal failure)
Psychiatric Disorders: aggression
Gastrointestinal System Disorders: upper and lower GI bleeding
Metabolic & Nutritional Disorders: hypokalemia
These adverse events may or may not be causally related to the drug.
-
OVERDOSAGE
Because strategies for the management of overdose are continually evolving, it is advisable to contact a poison control center to determine the latest recommendations for the management of an overdose of any drug.
As in any case of overdose, general supportive measures should be utilized. Signs and symptoms of significant overdosing of galantamine are predicted to be similar to those of overdosing of other cholinomimetics. These effects generally involve the central nervous system, the parasympathetic nervous system, and the neuromuscular junction. In addition to muscle weakness or fasciculations, some or all of the following signs of cholinergic crisis may develop: severe nausea, vomiting, gastrointestinal cramping, salivation, lacrimation, urination, defecation, sweating, bradycardia, hypotension, respiratory depression, collapse and convulsions. Increasing muscle weakness is a possibility and may result in death if respiratory muscles are involved.
Tertiary anticholinergics such as atropine may be used as an antidote for galantamine hydrobromide overdosage. Intravenous atropine sulfate titrated to effect is recommended at an initial dose of 0.5 to 1.0 mg i.v. with subsequent doses based upon clinical response. Atypical responses in blood pressure and heart rate have been reported with other cholinomimetics when coadministered with quaternary anticholinergics. It is not known whether galantamine hydrobromide and/or its metabolites can be removed by dialysis (hemodialysis, peritoneal dialysis, or hemofiltration). Dose-related signs of toxicity in animals included hypoactivity, tremors, clonic convulsions, salivation, lacrimation, chromodacryorrhea, mucoid feces, and dyspnea.
In one postmarketing report, one patient who had been taking 4 mg of galantamine daily for a week inadvertently ingested eight 4 mg tablets (32 mg total) on a single day. Subsequently, she developed bradycardia, QT prolongation, ventricular tachycardia and torsades de pointes accompanied by a brief loss of consciousness for which she required hospital treatment. Two additional cases of accidental ingestion of 32 mg (nausea, vomiting, and dry mouth; nausea, vomiting, and substernal chest pain) and one of 40 mg (vomiting), resulted in brief hospitalizations for observation with full recovery. One patient who was prescribed 24 mg/day and had a history of hallucinations over the previous two years, mistakenly received 24 mg twice daily for 34 days and developed hallucinations requiring hospitalization. Another patient, who was prescribed 16 mg/day of oral solution, inadvertently ingested 160 mg (40 mL) and experienced sweating, vomiting, bradycardia, and near-syncope one hour later, which necessitated hospital treatment. His symptoms resolved within 24 hours.
-
DOSAGE AND ADMINISTRATION
Galantamine Hydrobromide Extended-Release Capsules
The dosage of galantamine hydrobromide extended-release capsules shown to be effective in a controlled clinical trial is 16-24 mg/day.
The recommended starting dose of galantamine hydrobromide extended-release capsules is 8 mg/day. The dose should be increased to the initial maintenance dose of 16 mg/day after a minimum of 4 weeks. A further increase to 24 mg/day should be attempted after a minimum of 4 weeks at 16 mg/day. Dose increases should be based upon assessment of clinical benefit and tolerability of the previous dose.
Galantamine hydrobromide extended-release capsules should be administered once daily in the morning, preferably with food.
Patients currently being treated with galantamine hydrobromide immediate-release tablets can convert to galantamine hydrobromide extended-release capsules by taking their last dose of galantamine hydrobromide immediate-release tablets in the evening and starting galantamine hydrobromide extended-release capsules once daily treatment the next morning. Converting from galantamine hydrobromide immediate-release tablets to galantamine hydrobromide extended-release capsules should occur at the same total daily dose.
Patients and caregivers should be advised to ensure adequate fluid intake during treatment. If therapy has been interrupted for several days or longer, the patient should be restarted at the lowest dose and the dose escalated to the current dose.
The abrupt withdrawal of galantamine hydrobromide extended-release capsules in those patients who had been receiving doses in the effective range was not associated with an increased frequency of adverse events in comparison with those continuing to receive the same doses of that drug. The beneficial effects of galantamine hydrobromide extended-release capsules are lost, however, when the drug is discontinued.
Doses in Special Populations
Galantamine plasma concentrations may be increased in patients with moderate to severe hepatic impairment. In patients with moderately impaired hepatic function (Child-Pugh score of 7-9), the dose should generally not exceed 16 mg/day. The use of galantamine hydrobromide extended-release capsules in patients with severe hepatic impairment (Child-Pugh score of 10-15) is not recommended.
For patients with moderate renal impairment the dose should generally not exceed 16 mg/day. In patients with severe renal impairment (creatinine clearance < 9 mL/min), the use of galantamine hydrobromide extendedrelease capsules are not recommended.
-
HOW SUPPLIED
Galantamine hydrobromide extended-release capsules contain white to off-white matrix tablets.
8 mg white opaque cap and body, size 1 hard gelatin capsules with inscription “WPI 3496”
16 mg pink opaque cap and white opaque body, size 1 hard gelatin capsules with the inscription “WPI 3497”
24 mg pink opaque cap and body, size 1 hard gelatin capsules with the inscription “WPI 3498”
The capsules are supplied as follows:
8 mg capsules – bottles of 30 NDC 0591-3496-30
8 mg capsules – bottles of 500 NDC 0591-3496-05
16 mg capsules – bottles of 30 NDC 0591-3497-30
16 mg capsules – bottles of 500 NDC 0591-3497-05
24 mg capsules – bottles of 30 NDC 0591-3498-30
24 mg capsules – bottles of 500 NDC 0591-3498-05
Storage and Handling
Galantamine hydrobromide extended-release capsules should be stored at 20° to 25°C (68°-77°F). [See USP controlled room temperature.]
Keep out of reach of children.
Galantamine hydrobromide extended-release capsules are manufactured by:
Watson Laboratories, Inc.
Corona, CA 92880 USADistributed by:
Watson Pharma, Inc.
Corona, CA 92880 USAIssued: June 2008
173479
0608B -
Repackaging Information
Please reference the How Supplied section listed above for a description of individual tablets. This drug product has been received by Aphena Pharma - TN in a manufacturer or distributor packaged configuration and repackaged in full compliance with all applicable cGMP regulations. The package configurations available from Aphena are listed below:
Count 16mg 4200 43353-984-05 Store between 20°-25°C (68°-77°F). See USP Controlled Room Temperature. Dispense in a tight light-resistant container as defined by USP. Keep this and all drugs out of the reach of children.
Repackaged by:

Cookeville, TN 38506
20160613JH - PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
GALANTAMINE HYDROBROMIDE
galantamine hydrobromide capsule, extended releaseProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:43353-984(NDC:0591-3497) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength GALANTAMINE HYDROBROMIDE (UNII: MJ4PTD2VVW) (GALANTAMINE - UNII:0D3Q044KCA) GALANTAMINE 16 mg Inactive Ingredients Ingredient Name Strength GELATIN (UNII: 2G86QN327L) HYDROXYPROPYL CELLULOSE (UNII: RFW2ET671P) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERRIC OXIDE RED (UNII: 1K09F3G675) Product Characteristics Color WHITE (white opaque body) , PINK (pink opaque cap) Score no score Shape CAPSULE Size 19mm Flavor Imprint Code WPI;3497 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:43353-984-05 4200 in 1 BOTTLE; Type 0: Not a Combination Product 11/15/2015 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA079028 12/15/2009 Labeler - Aphena Pharma Solutions - Tennessee, LLC (128385585) Establishment Name Address ID/FEI Business Operations Aphena Pharma Solutions - Tennessee, LLC 128385585 REPACK(43353-984)
