Label: LOPREEZA- estradiol/norethindrone acetate tablet, film coated




- NDC Code(s): 69238-1610-6
- Packager: Amneal Pharmaceuticals NY LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated July 25, 2018
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use LOPREEZA safely and effectively. See full prescribing information for LOPREEZA.
LOPREEZA® (estradiol/norethindrone acetate) tablets, for oral use
Initial U.S. Approval: 1998
WARNING: CARDIOVASCULAR DISORDERS, BREAST CANCER, ENDOMETRIAL CANCER AND PROBABLE DEMENTIA
See full prescribing information for complete boxed warning
Estrogen Plus Progestin Therapy
- Estrogen plus progestin therapy should not be used for the prevention of cardiovascular disease or dementia (5.1, 5.3)
- The Women’s Health Initiative (WHI) estrogen plus progestin substudy reported increased risks of deep vein thrombosis (DVT), pulmonary embolism (PE), stroke and myocardial infarction (MI) (5.1)
- The WHI estrogen plus progestin substudy reported increased risks of invasive breast cancer (5.2)
-
The WHI Memory Study (WHIMS) estrogen plus progestin ancillary study of WHI reported an increased risk of probable dementia in postmenopausal women 65 years of age and older (5.3)
Estrogen-Alone Therapy
- There is an increased risk of endometrial cancer in a woman with a uterus who use unopposed estrogens (5.2)
- Estrogen-alone therapy should not be used for the prevention of cardiovascular disease or dementia (5.2, 5.3)
- The WHI estrogen-alone substudy reported increased risks of stroke and DVT (5.1)
- The WHIMS estrogen-alone ancillary study of WHI reported an increased risk of probable dementia in postmenopausal women 65 years of age and older (5.3)
RECENT MAJOR CHANGES
Warnings and Precautions, Malignant Neoplasm (5.2) 07/2017
INDICATIONS AND USAGE
Lopreezais an estrogen and progestin combination indicated in a woman with a uterus for:
Lopreeza 1 mg/0.5 mg and 0.5 mg/0.1 mg are indicated in a woman with a uterus for:
DOSAGE AND ADMINISTRATION
- One tablet to be taken once daily (2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
- Undiagnosed abnormal genital bleeding (4)
- Known, suspected, or history of breast cancer (4, 5.2)
- Known or suspected estrogen-dependent neoplasia (4, 5.2)
- Active DVT, PE, or history of these conditions (4, 5.1)
- Active arterial thromboembolic disease (for example, stroke and MI), or a history of these conditions (4, 5.1)
- Known anaphylactic reaction or angioedema or hypersensitivity to Lopreeza (4)
- Known liver impairment or disease (4, 5.10)
- Known protein C, protein S, or antithrombin deficiency, or other known thrombophilic disorders (4)
- Known or suspected pregnancy(4, 8.1)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Most common adverse reactions (incidence ≥ 5 percent) are back pain, headache, pain in the extremity, nausea, diarrhea, gastroenteritis, insomnia, emotional lability, upper respiratory tract infection, sinusitis, nasopharyngitis, weight increase, breast pain, post-menopausal bleeding, uterine fibroid vaginal hemorrhage, ovarian cyst, endometrial thickening, viral infection, moniliasis genital, and accidental injury. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Amneal Pharmaceuticals at 1-877-835-5472 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Inducers and/or inhibitors of CYP3A4 may affect estrogen drug metabolism (7.1)
USE IN SPECIFIC POPULATIONS
- Nursing Mothers: Estrogen administration to nursing women has been shown to decrease the quantity and quality of breast milk (8.3)
- Geriatric Use: An increase risk of probable dementia in women over 65 years of age was reported in the Women’s Health Initiative Memory ancillary studies of the Women’s Health Initiative (5.3, 8.5)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
See 17 for PATIENT COUNSELING INFORMATION and PATIENT COUNSELING INFORMATION.
Revised: 7/2018
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CARDIOVASCULAR DISORDERS, BREAST CANCER, ENDOMETRIAL CANCER AND PROBABLE DEMENTIA
1 INDICATIONS AND USAGE
1.2 Treatment of Moderate to Severe Symptoms of Vulvar and Vaginal Atrophy due to Menopause
1.3 Prevention of Postmenopausal Osteoporosis
2 DOSAGE AND ADMINISTRATION
2.1 Treatment of Moderate to Severe Vasomotor Symptoms due to Menopause
2.2 Treatment of Moderate to Severe Symptoms of Vulvar and Vaginal Atrophy due to Menopause
2.3 Prevention of Postmenopausal Osteoporosis
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Disorders
5.2 Malignant Neoplasms
5.3 Probable Dementia
5.4 Gallbladder Disease
5.5 Hypercalcemia
5.6 Vision Abnormalities
5.7 Addition of a Progestin When a Woman Has Not Had a Hysterectomy
5.8 Elevated Blood Pressure
5.9 Hypertriglyceridemia
5.10 Hepatic Impairment and/or Past History of Cholestatic Jaundice
5.11 Hypothyroidism
5.12 Fluid Retention
5.13 Hypocalcemia
5.14 Exacerbation of Endometriosis
5.15 Hereditary Angioedema
5.16 Exacerbation of Other Conditions
5.17 Laboratory Tests
5.18 Drug-Laboratory Test Interactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Metabolic Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Effects on Vasomotor Symptoms
14.2 Effects on the Endometrium
14.3 Effects on Uterine Bleeding or Spotting
14.4 Effects on Bone Mineral Density
14.5 Women’s Health Initiative Studies
14.6 Women’s Health Initiative Memory Study
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
17.1 Abnormal Vaginal Bleeding
17.2 Possible Serious Adverse Reactions with Estrogen Plus Progestin Therapy
17.3 Possible Less Serious but Common Adverse Reactions with Estrogen Plus Progestin Therapy
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CARDIOVASCULAR DISORDERS, BREAST CANCER, ENDOMETRIAL CANCER AND PROBABLE DEMENTIA
Estrogen Plus Progestin Therapy
Cardiovascular Disorders and Probable Dementia
Estrogen plus progestin therapy should not be used for the prevention of cardiovascular disease or dementia [see Warnings and Precautions (5.1, 5.3), and Clinical Studies (14.5, 14.6)].
The Women’s Health Initiative (WHI) estrogen plus progestin substudy reported increased risks of deep vein thrombosis (DVT), pulmonary embolism (PE), stroke and myocardial infarction (MI) in postmenopausal women (50 to 79 years of age) during 5.6 years of treatment with daily oral conjugated estrogen (CE) [0.625 mg] combined with medroxyprogesterone acetate (MPA) [2.5 mg], relative to placebo [see Warnings and Precautions (5.1), and Clinical Studies (14.5)].
The WHI Memory Study (WHIMS) estrogen plus progestin ancillary study of the WHI reported an increased risk of developing probable dementia in postmenopausal women 65 years of age or older during 4 years of treatment with daily CE (0.625 mg) combined with MPA (2.5 mg), relative to placebo. It is unknown whether this finding applies to younger postmenopausal women [see Warnings and Precautions (5.3), Use in Specific Populations (8.5), and Clinical Studies (14.6)].
Breast Cancer
The WHI estrogen plus progestin substudy also demonstrated an increased risk of invasive breast cancer [see Warnings and Precautions (5.2), and Clinical Studies (14.5)].
In the absence of comparable data, these risks should be assumed to be similar for other doses of CE and MPA and other combinations and dosage forms of estrogens and progestins.
Estrogens with or without progestins should be prescribed at the lowest effective doses and for the shortest duration consistent with treatment goals and risks for the individual woman.
Estrogen-Alone Therapy
Endometrial Cancer
There is an increased risk of endometrial cancer in a woman with a uterus who uses unopposed estrogens. Adding a progestin to estrogen therapy has been shown to reduce the risk of endometrial hyperplasia, which may be a precursor to endometrial cancer. Adequate diagnostic measures, including directed or random endometrial sampling when indicated, should be undertaken to rule out malignancy in postmenopausal women with undiagnosed persistent or recurring abnormal genital bleeding [see Warnings and Precautions (5.2)].
Cardiovascular Disorders and Probable Dementia
Estrogen-alone therapy should not be used for the prevention of cardiovascular disease or dementia [see Warnings and Precautions (5.1, 5.3), and Clinical Studies (14.5, 14.6)].
The WHI estrogen-alone substudy reported increased risks of stroke and DVT in postmenopausal women (50 to 79 years of age) during 7.1 years of treatment with daily oral CE (0.625 mg)-alone, relative to placebo [see Warnings and Precautions (5.1), and Clinical Studies (14.5)].
The WHIMS estrogen-alone ancillary study of the WHI reported an increased risk of developing probable dementia in postmenopausal women 65 years of age or older during 5.2 years of treatment with daily CE (0.625 mg)-alone, relative to placebo. It is unknown whether this finding applies to younger postmenopausal women [see Warnings and Precautions (5.3), Use in Specific Populations (8.5), and Clinical Studies (14.6)].
In the absence of comparable data, these risks should be assumed to be similar for other doses of CE and other dosage forms of estrogens.
Estrogens with or without progestins should be prescribed at the lowest effective doses and for the shortest duration consistent with treatment
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
Use of estrogen-alone, or in combination with a progestin, should be with the lowest effective dose and for the shortest duration consistent with treatment goals and risks for the individual woman. Postmenopausal women should be re-evaluated periodically as clinically appropriate to determine if treatment is still necessary.
2.1 Treatment of Moderate to Severe Vasomotor Symptoms due to Menopause
Lopreeza therapy consists of a single tablet to be taken once daily for the treatment of moderate to severe vasomotor symptoms due to menopause.
- Lopreeza 1 mg/0.5 mg
- Lopreeza 0.5 mg/0.1 mg
-
3 DOSAGE FORMS AND STRENGTHS
Lopreeza tablets are available in two strengths:
- Each tablet of Lopreeza 1 mg/ 0.5 mg contains 1 mg of estradiol and 0.5 mg of norethindrone acetate. The tablets are white, round, biconvex, film-coated tablets debossed with “ALH” on the one side and plain on the other side.
- Each tablet of Lopreeza 0.5 mg/ 0.1 mg contains 0.5 mg of estradiol and 0.1 mg of norethindrone acetate. The tablets are white, round, biconvex, film-coated tablets debossed with “ALL” on the one side and plain on the other side.
-
4 CONTRAINDICATIONS
Lopreeza is contraindicated in women with any of the following conditions:
- Undiagnosed abnormal genital bleeding
- Known, suspected, or history of breast cancer
- Known, past or suspected estrogen-dependent neoplasia
- Active DVT, PE, or history of these conditions
- Active arterial thromboembolic disease (for example stroke and MI), or a history of these conditions
- Known anaphylactic reaction or angioedema or hypersensitivity to Lopreeza
- Known liver impairment or disease
- Known protein C, protein S, or antithrombin deficiency, or other known thrombophilic disorders
- Known or suspected pregnancy
-
5 WARNINGS AND PRECAUTIONS
5.1 Cardiovascular Disorders
An increased risk of PE, DVT, stroke and MI has been reported with estrogen plus progestin therapy. An increased risk of stroke and DVT has been reported with estrogen-alone therapy. Should any of these occur or be suspected, estrogen with or without progestin therapy should be discontinued immediately.
Risk factors for arterial vascular disease (for example, hypertension, diabetes mellitus, tobacco use, hypercholesterolemia, and obesity) and/or venous thromboembolism (VTE) (for example, personal history or family history of VTE, obesity, and systemic lupus erythematosus) should be managed appropriately.
Stroke
In the WHI estrogen plus progestin substudy, a statistically significant increased risk of stroke was reported in women 50 to 79 years of age receiving daily CE (0.625 mg) plus MPA (2.5 mg) compared to women in the same age group receiving placebo (33 versus 25 per 10,000 women-years) [see Clinical Studies (14.5)]. The increase in risk was demonstrated after the first year and persisted.1 Should a stroke occur or be suspected, estrogen plus progestin therapy should be discontinued immediately.
In the WHI estrogen-alone substudy, a statistically significant increased risk of stroke was reported in women 50 to 79 years of age receiving daily CE (0.625 mg)-alone compared to women in the same age group receiving placebo (45 versus 33 per 10,000 women-years). The increase in risk was demonstrated in year 1 and persisted [see Clinical Studies (14.5)]. Should a stroke occur or be suspected, estrogen-alone therapy should be discontinued immediately.
Subgroup analyses of women 50 to 59 years of age suggest no increased risk of stroke for those women receiving CE (0.625 mg)-alone versus those receiving placebo (18 versus 21 per 10,000 women-years). 1
Coronary Heart Disease
In the WHI estrogen plus progestin substudy, there was a statistically non-significant increase risk of coronary heart disease (CHD) events (defined as nonfatal MI, silent MI, or CHD death) reported in women receiving daily CE (0.625 mg) plus MPA (2.5 mg) compared to women receiving placebo (41 versus 34 per 10,000 women-years). 1 An increase in relative risk was demonstrated in year 1, and a trend toward decreasing relative risk was reported in years 2 through 5 [see Clinical Studies (14.5)].
In the WHI estrogen-alone substudy, no overall effect on CHD events was reported in women receiving estrogen-alone compared to placebo 2 [see Clinical Studies (14.5)].
Subgroup analyses of women 50 to 59 years of age suggest a statistically non-significant reduction in CHD events (CE [0.625 mg]-alone compared to placebo) in women with less than 10 years since menopause (8 versus 16 per 10,000 women-years). 1
In postmenopausal women with documented heart disease (n=2,763), average 66.7 years of age, in a controlled clinical trial of secondary prevention of cardiovascular disease (Heart and Estrogen/Progestin Replacement Study [HERS]), treatment with daily CE (0.625 mg plus MPA (2.5 mg) demonstrated no cardiovascular benefit. During an average follow-up of 4.1 years, treatment with CE plus MPA did not reduce the overall rate of CHD events in postmenopausal women with established CHD. There were more CHD events in the CE plus MPA-treated group than in the placebo group in year 1, but not during the subsequent years. Two thousand, three hundred and twenty‑one (2,321) women from the original HERS trial agreed to participate in an open label extension of HERS, HERS II. Average follow-up in HERS II was an additional 2.7 years, for a total of 6.8 years overall. Rates of CHD events were comparable among women in the CE plus MPA group and the placebo group in HERS, HERS II, and overall.
Venous Thromboembolism
In the WHI estrogen plus progestin substudy, a statistically significant 2-fold greater rate of VTE (DVT and PE), was reported in women receiving daily CE (0.625 mg) plus MPA (2.5 mg) compared to women receiving placebo (35 versus 17 per 10,000 women-years). Statistically significant increases in risk for both DVT (26 versus 13 per 10,000 women-years) and PE (18 versus 8 per 10,000 women-years) were also demonstrated. The increase in VTE risk was demonstrated during the first year and persisted 3[see Clinical Studies (14.5)]. Should a VTE occur or be suspected, estrogen plus progestin therapy should be discontinued immediately.
In the WHI estrogen-alone substudy, the risk of VTE was increased for women receiving daily CE
(0.625 mg)-alone compared to placebo (30 versus 22 per 10,000 women-years), although only the increased risk of DVT reached statistical significance (23 versus 15 per 10,000 women-years). The increase in VTE risk was demonstrated during the first 2 years 4 [see Clinical Studies (14.5)]. Should a VTE occur or be suspected, estrogen-alone therapy should be discontinued immediately.
If feasible, estrogens should be discontinued at least 4 to 6 weeks before surgery of the type associated with an increased risk of thromboembolism, or during periods of prolonged immobilization.
5.2 Malignant Neoplasms
Breast Cancer
The most important randomized clinical trial providing information about breast cancer in estrogen plus progestin users is the WHI substudy of daily CE (0.625 mg) plus MPA (2.5 mg). After a mean follow-up of 5.6 years, the estrogen plus progestin substudy reported an increased risk of invasive breast cancer in women who took daily CE plus MPA. In this substudy, prior use of estrogen-alone or estrogen plus progestin therapy was reported by 26 percent of the women. The relative risk of invasive breast cancer was 1.24, and the absolute risk was 41 versus 33 cases per 10,000 women-years, for CE plus MPA compared with placebo [see Clinical Studies (14.5)]. Among women who reported prior use of hormone therapy, the relative risk of invasive breast cancer was 1.86, and the absolute risk was 46 versus 25 cases per 10,000 women-years, for CE plus MPA compared with placebo. Among women who reported no prior use of hormone therapy, the relative risk of invasive breast cancer was 1.09, and the absolute risk was 40 versus 36 cases per 10,000 women-years, for CE plus MPA compared with placebo. In the same substudy, invasive breast cancers were larger, were more likely to be node positive, and were diagnosed at a more advanced stage in the CE (0.625 mg) plus MPA (2.5 mg) group compared with the placebo group. Metastatic disease was rare, with no apparent difference between the two groups. Other prognostic factors, such as histologic subtype, grade and hormone receptor status did not differ between the groups 5[see Clinical Studies (14.5)].
The most important randomized clinical trial providing information about breast cancer in estrogen-alone users is the WHI substudy of daily CE (0.625 mg)-alone. In the WHI estrogen-alone substudy, after an average follow‑up of 7.1 years, daily CE-alone was not associated with an increased risk of invasive breast cancer [relative risk (RR) 0.80 6[see Clinical Studies (14.5)].
Consistent with the WHI clinical trials, observational studies have also reported an increased risk of breast cancer for estrogen plus progestin therapy, and a smaller increased risk for estrogen-alone therapy, after several years of use. The risk increased with duration of use, and appeared to return to baseline over about 5 years after stopping treatment (only the observational studies have substantial data on risk after stopping). Observational studies also suggest that the risk of breast cancer was greater, and became apparent earlier, with estrogen plus progestin therapy as compared to estrogen-alone therapy. However, these studies have not found significant variation in the risk of breast cancer among different estrogen plus progestin combinations, doses, or routes of administration.
The use of estrogen-alone and estrogen plus progestin has been reported to result in an increase in abnormal mammograms requiring further evaluation.
In a one-year trial among 1,176 women who received either unopposed 1 mg estradiol or a combination of 1 mg estradiol plus one of three different doses of NETA (0.1, 0.25, 0.5 mg), seven new cases of breast cancer were diagnosed, two of which occurred among the group of 295 women treated with Lopreeza 1 mg/0.5 mg and two of which occurred among the group of 294 women treated with 1 mg estradiol/0.1 mg NETA.
All women should receive yearly breast examinations by a healthcare provider and perform monthly breast self-examinations. In addition, mammography examinations should be scheduled based on patient age, risk factors, and prior mammogram results.
Endometrial Cancer
Endometrial hyperplasia (a possible precursor of endometrial cancer) has been reported to occur at a rate of approximately 1 percent or less with Lopreeza.
An increased risk of endometrial cancer has been reported with the use of unopposed estrogen therapy in a woman with a uterus. The reported endometrial cancer risk among unopposed estrogen users is about 2 to 12 times greater than in nonusers, and appears dependent on duration of treatment and on estrogen dose. Most studies show no significant increased risk associated with use of estrogens for less than 1 year. The greatest risk appears to be associated with prolonged use, with increased risks of 15- to 24‑fold for 5 to 10 years or more. This risk has been shown to persist for at least 8 to 15 years after estrogen therapy is discontinued.
Clinical surveillance of all women using estrogen-alone or estrogen plus progestin therapy is important. Adequate diagnostic measures, including directed or random endometrial sampling when indicated, should be undertaken to rule out malignancy in postmenopausal women with undiagnosed persistent or recurring abnormal genital bleeding.
There is no evidence that the use of natural estrogens results in a different endometrial risk profile than synthetic estrogens of equivalent estrogen dose. Adding a progestin to estrogen therapy in postmenopausal women has been shown to reduce the risk of endometrial hyperplasia, which may be a precursor to endometrial cancer.
Ovarian Cancer
The WHI estrogen plus progestin substudy reported a statistically non-significant increased risk of ovarian cancer. After an average follow-up of 5.6 years, the relative risk for ovarian cancer for CE plus MPA versus placebo was 1.58 (95 percent CI, 0.77-3.24]. The absolute risk for CE plus MPA versus placebo was 4 versus 3 cases per 10,000 women-years.7
A meta-analysis of 17 prospective and 35 retrospective epidemiology studies found that women who used hormonal therapy for menopausal symptoms had an increased risk for ovarian cancer. The primary analysis, using case-control comparisons, included 12,110 cancer cases from the 17 prospective studies. The relative risks associated with current use of hormonal therapy was 1.41 (95% confidence interval [CI] 1.32 to 1.50); there was no difference in the risk estimates by duration of the exposure (less than 5 years [median of 3 years] vs. greater than 5 years [median of 10 years] of use before the cancer diagnosis). The relative risk associated with combined current and recent use (discontinued use within 5 years before cancer diagnosis) was 1.37 (95% CI 1.27-1.48), and the elevated risk was significant for both estrogen-alone and estrogen plus progestin products. The exact duration of hormone therapy use associated with an increased risk of ovarian cancer, however, is unknown.
5.3 Probable Dementia
In the WHIMS estrogen plus progestin ancillary study of WHI, a population of 4,532 postmenopausal women 65 to 79 years of age was randomized to daily CE (0.625 mg) plus MPA (2.5 mg) or placebo.
After an average follow-up of 4 years, 40 women in the CE plus MPA and 21 women in the placebo group were diagnosed with probable dementia. The relative risk of probable dementia for the CE plus MPA versus placebo was 2.05 (95 percent CI, 1.21-3.48). The absolute risk of probable dementia for CE plus MPA versus placebo was 45 versus 22 cases per 10,000 women-years 8 [see Use in Specific Populations (8.5), and Clinical Studies (14.6)].
In the WHIMS estrogen-alone ancillary study of WHI, a population of 2,947 hysterectomized women 65 to 79 years of age was randomized to daily CE (0.625 mg)-alone or placebo. After an average follow-up of 5.2 years, 28 women in the estrogen-alone group and 19 women in the placebo group were diagnosed with probable dementia. The relative risk of probable dementia for CE-alone versus placebo was 1.49 (95 percent CI, 0.83-2.66). The absolute risk of probable dementia for CE-alone versus placebo was 37 versus 25 cases per 10,000 women-years 8 [see Use in Specific Populations (8.5), and Clinical Studies (14.6)].
When data from the two populations in the WHIMS estrogen-alone and estrogen plus progestin ancillary studies were pooled as planned in the WHIMS protocol, the reported overall relative risk of probable dementia was 1.76 (95 percent CI, 1.19-2.60). Since both ancillary studies were conducted in women 65 to 79 years of age, it is unknown whether these findings apply to younger postmenopausal women 8 [see Use in Specific Populations (8.5), and Clinical Studies (14.6)].
5.4 Gallbladder Disease
A 2- to 4-fold increase in the risk of gallbladder disease requiring surgery in postmenopausal women receiving estrogens has been reported.
5.5 Hypercalcemia
Estrogen administration may lead to severe hypercalcemia in patients with breast cancer and bone metastases. If hypercalcemia occurs, use of the drug should be stopped and appropriate measures taken to reduce the serum calcium level.
5.6 Vision Abnormalities
Retinal vascular thrombosis has been reported in patients receiving estrogens. Discontinue medication pending examination if there is a sudden partial or complete loss of vision, or a sudden onset of proptosis, diplopia, or migraine. If examination reveals papilledema or retinal vascular lesions, estrogens should be permanently discontinued.
5.7 Addition of a Progestin When a Woman Has Not Had a Hysterectomy
Studies of the addition of a progestin for 10 or more days of a cycle of estrogen administration, or daily with estrogen in a continuous regimen, have reported a lowered incidence of endometrial hyperplasia than would be induced by estrogen treatment alone. Endometrial hyperplasia may be a precursor to endometrial cancer.
There are, however, possible risks that may be associated with the use of progestins with estrogens compared to estrogen-alone regimens. These include an increased risk of breast cancer.
5.8 Elevated Blood Pressure
In a small number of case reports, substantial increases in blood pressure have been attributed to idiosyncratic reactions to estrogens. In a large, randomized, placebo-controlled clinical trial, a generalized effect of estrogen therapy on blood pressure was not seen.
5.9 Hypertriglyceridemia
In women with pre-existing hypertriglyceridemia, estrogen therapy may be associated with elevations of plasma triglycerides leading to pancreatitis. Consider discontinuation of treatment if pancreatitis occurs.
5.10 Hepatic Impairment and/or Past History of Cholestatic Jaundice
Estrogens may be poorly metabolized in women with impaired liver function. For women with a history of cholestatic jaundice associated with past estrogen use or with pregnancy, caution should be exercised, and in the case of recurrence, medication should be discontinued.
5.11 Hypothyroidism
Estrogen administration leads to increased thyroid-binding globulin (TBG) levels. Women with normal thyroid function can compensate for the increased TBG by making more thyroid hormone, thus maintaining free T4 and T3 serum concentrations in the normal range. Women dependent on thyroid hormone replacement therapy who are also receiving estrogen may require increased doses of their thyroid replacement therapy. These women should have their thyroid function monitored to maintain their free thyroid hormone levels in an acceptable range.
5.12 Fluid Retention
Estrogens plus progestins may cause some degree of fluid retention. Women with conditions that might be influenced by this factor, such as a cardiac or renal impairment, warrant careful observation when estrogens plus progestins are prescribed.
5.13 Hypocalcemia
Estrogen therapy should be used with caution in women with hypoparathyroidism as estrogen-induced hypocalcemia may occur.
5.14 Exacerbation of Endometriosis
A few cases of malignant transformation of residual endometrial implants have been reported in women treated post-hysterectomy with estrogen-alone therapy. For women known to have residual endometriosis post-hysterectomy, the addition of progestin should be considered.
5.15 Hereditary Angioedema
Exogenous estrogens may exacerbate symptoms of angioedema in women with hereditary angioedema.
5.16 Exacerbation of Other Conditions
Estrogen therapy may cause an exacerbation of asthma, diabetes mellitus, epilepsy, migraine, porphyria, systemic lupus erythematosus, and hepatic hemangiomas and should be used with caution in women with these conditions.
5.17 Laboratory Tests
Serum follicle stimulating hormone (FSH) and estradiol levels have not been shown to be useful in the management of moderate to severe vasomotor symptoms and moderate to severe symptoms of vulvar and vaginal atrophy.
5.18 Drug-Laboratory Test Interactions
Accelerated prothrombin time, partial thromboplastin time, and platelet aggregation time; increased platelet count; increased factors II, VII antigen, VIII coagulant activity, IX, X, XII, VII-X complex, and beta-thromboglobulin; decreased levels of anti-factor Xa and antithrombin III, decreased antithrombin III activity, increased levels of fibrinogen and fibrinogen activity; increased plasminogen antigen and activity.
Increased TBG levels leading to increased circulating total thyroid hormone levels as measured by protein-bound iodine (PBI), T4 levels (by column or by radioimmunoassay), or T3 levels by radioimmunoassay. T3 resin uptake is decreased, reflecting the elevated TBG. Free T4 and free T3 concentrations are unaltered. Women on thyroid replacement therapy may require higher doses of thyroid hormone.
Other binding proteins may be elevated in serum, for example, corticosteroid binding globulin (CBG), sex hormone-binding globulin (SHBG), leading to increased total circulating corticosteroids and sex steroids, respectively. Free hormone concentrations, such as testosterone and estradiol, may be decreased. Other plasma proteins may be increased (angiotensinogen/rennin substrate, alpha-1 antitrypsin, ceruloplasmin).
Increased plasma high-density lipoprotein (HDL) and HDL2 cholesterol subfraction concentration, reduced
low-density lipoprotein (LDL) cholesterol concentration, increased triglyceride levels.
Impaired glucose tolerance.
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- Cardiovascular Disorders [see Boxed Warning, Warnings and Precautions (5.1)]
- Malignant Neoplasms [see Boxed Warning, Warnings and Precautions, (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse reactions reported with Lopreeza 1 mg/0.5 mg by investigators during clinical trials regardless of causality assessment are shown in Table 1.
TABLE 1
ALL TREATMENT-EMERGENT ADVERSE REACTIONS REGARDLESS OF RELATIONSHIP REPORTED AT A FREQUENCY OF ³ 5 PERCENT WITH LOPREEZA 1 MG/0.5 MG
Endometrial
Hyperplasia Study
(12-Months)
Vasomotor
Symptoms Study
(3-Months)
Osteoporosis
Study
(2-Years)
Lopreeza
1 mg/0.5 mg
1 mg E2
Lopreeza
1 mg/0.5 mg
Placebo
Lopreeza
1 mg/0.5 mg
Placebo
(n=295)
(n=296)
(n=29)
(n=34)
(n=47)
(n=48)
Body as a Whole
Back Pain
6%
5%
3%
3%
6%
4%
Headache
16%
16%
17%
18%
11%
6%
Digestive System
Nausea
3%
5%
10%
0%
11%
0%
Gastroenteritis
2%
2%
0%
0%
6%
4%
Nervous System
Insomnia
6%
4%
3%
3%
0%
8%
Emotional Lability
1%
1%
0%
0%
6%
0%
Respiratory System
Upper Respiratory
Tract Infection
18%
15%
10%
6%
15%
19%
Sinusitis
7%
11%
7%
0%
15%
10%
Metabolic and Nutritional
Weight Increase
0%
0%
0%
0%
9%
6%
Urogenital System
Breast Pain
24%
10%
21%
0%
17%
8%
Post-Menopausal Bleeding
5%
15%
10%
3%
11%
0%
Uterine Fibroid
5%
4%
0%
0%
4%
8%
Ovarian Cyst
3%
2%
7%
0%
0%
8%
Resistance Mechanism
Infection Viral
4%
6%
0%
3%
6%
6%
Moniliasis Genital
4%
7%
0%
0%
6%
0%
Secondary Terms
Injury Accidental
4%
3%
3%
0%
17%*
4%*
Other Events
2%
3%
3%
0%
6%
4%
* including one upper extremity fracture in each group
Adverse reactions reported with Lopreeza 0.5 mg/0.1 mg by investigators during clinical trials regardless of causality assessment are shown in Table 2.
TABLE 2
ALL TREATMENT-EMERGENT ADVERSE REACTIONS REGARDLESS OF RELATIONSHIP REPORTED AT A FREQUENCY OF ³ 5 PERCENT WITH LOPREEZA 0.5 MG/0.1 MG
Lopreeza
0.5 mg/0.1 mg
Placebo
(n=194)
(n=200)
Body as a Whole
Back Pain
10%
4%
Headache
22%
19%
Pain in extremity
5%
4%
Digestive System
Nausea
5%
4%
Diarrhea
6%
6%
Respiratory System
Nasopharyngitis
21%
18%
Urogenital System
Endometrial thickening
10%
4%
Vaginal hemorrhage
26%
12%
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of Lopreeza. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Genitourinary System
Changes in vaginal bleeding pattern and abnormal withdrawal bleeding or flow; breakthrough bleeding; spotting; dysmenorrhea, increase in size of uterine leiomyomata; vaginitis, including vaginal candidiasis; change in amount of cervical secretion; changes in cervical ectropion; pre-menstrual-like syndrome; cystitis-like syndrome; ovarian cancer; endometrial hyperplasia; endometrial cancer.
Breast
Tenderness, enlargement, pain, nipple discharge, galactorrhea; fibrocystic breast changes; breast cancer.
Cardiovascular
Deep and superficial venous thrombosis; pulmonary embolism; thrombophlebitis; myocardial infarction, stroke; increase in blood pressure.
Gastrointestinal
Nausea, vomiting; changes in appetite; cholestatic jaundice; abdominal pain/cramps, flatulence, bloating; increased incidence of gallbladder disease and pancreatitis.
Skin
Chloasma or melasma that may persist when drug is discontinued; erythema multiforme; erythema nodosum; hemorrhagic eruption; loss of scalp hair; seborrhea; hirsutism; itching; skin rash; pruritus.
Eyes
Retinal vascular thrombosis, intolerance to contact lenses.
Central Nervous System
Headache; migraine; dizziness; mental depression; chorea; insomnia; nervousness; mood disturbances; irritability; exacerbation of epilepsy; dementia.
Miscellaneous
Increase or decrease in weight; edema; leg cramps; changes in libido; fatigue; exacerbation of asthma; increased triglycerides; hypersensitivity; anaphylactoid/anaphylactic reactions.
-
7 DRUG INTERACTIONS
Co-administration of estradiol with norethindrone acetate did not elicit any apparent influence on the pharmacokinetics of norethindrone acetate. Similarly, no relevant interaction of norethindrone acetate on the pharmacokinetics of estradiol was found within the NETA dose range investigated in a single dose study.
7.1 Metabolic Interactions
Estradiol
In-vitro and in-vivo studies have shown that estrogens are metabolized partially by cytochrome P450 3A4 (CYP3A4). Therefore, inducers or inhibitors of CYP3A4 may affect estrogen drug metabolism. Inducers of CYP3A4 such as St. John’s wort (Hypericum perforatum) preparations, phenobarbital, carbamazepine, and rifampin may reduce plasma concentrations of estrogens, possibly resulting in a decrease in therapeutic effects and/or changes in the uterine bleeding profile. Inhibitors of CYP3A4 such as erythromycin, clarithromycin, ketoconazole, itraconazole, ritonavir and grapefruit juice may increase plasma concentrations of estrogens and result in side effects.
Norethindrone Acetate
Drugs or herbal products that induce or inhibit cytochrome P-450 enzymes, including CYP3A4, may decrease or increase the serum concentrations of norethindrone.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Lopreeza should not be used during pregnancy [see Contraindications (4)]. There appears to be little or no increased risk of birth defects in children born to women who have used estrogens and progestins as an oral contraceptive inadvertently during early pregnancy.
8.3 Nursing Mothers
Lopreeza should not be used during lactation. Estrogen administration to nursing women has been shown to decrease the quantity and quality of the breast milk. Detectable amounts of estrogen and progestin have been identified in the breast milk of women receiving estrogen plus progestin therapy. Caution should be exercised when Lopreeza is administered to a nursing woman.
8.4 Pediatric Use
Lopreeza is not indicated in children. Clinical studies have not been conducted in the pediatric population.
8.5 Geriatric Use
There have not been sufficient numbers of geriatric women involved in clinical studies utilizing Lopreeza to determine whether those over 65 years of age differ from younger subjects in their response to Lopreeza.
The Women’s Health Initiative Studies
In the WHI estrogen plus progestin substudy (daily CE [0.625 mg] plus MPA [2.5 mg] versus placebo), there was a higher relative risk of nonfatal stroke and invasive breast cancer in women greater than 65 years of age [see Clinical Studies (14.5)].
In the WHI estrogen-alone substudy (daily CE [0.625 mg]-alone versus placebo), there was a higher relative risk of stroke in women greater than 65 years of age [see Clinical Studies (14.5)].
The Women’s Health Initiative Memory Study
In the WHIMS ancillary studies of postmenopausal women 65 to 79 years of age, there was an increased risk of developing probable dementia in women receiving estrogen plus progestin or estrogen-alone when compared to placebo. It is unknown whether this finding applies to younger postmenopausal women [see Warnings and Precautions (5.3), and Clinical Studies (14.6)].
Since both ancillary studies were conducted in women 65 to 79 years of age, it is unknown whether these findings apply to younger postmenopausal women 8 [see Warnings and Precautions (5.3), and Clinical Studies (14.6)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Lopreeza 1 mg/0.5 mg is a single tablet for oral administration containing 1 mg of estradiol and 0.5 mg of norethindrone acetate and the following excipients: lactose monohydrate, starch (corn), copovidone, talc, magnesium stearate, hypromellose and triacetin.
Lopreeza 0.5 mg/0.1 mg is a single tablet for oral administration containing 0.5 mg of estradiol and 0.1 mg of norethindrone acetate and the following excipients: lactose monohydrate, starch (corn), hydroxypropyl cellulose, talc, magnesium stearate, hypromellose and triacetin.
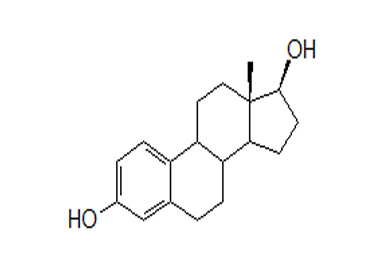
Estradiol (E2), an estrogen, is a white or almost white crystalline powder. Its chemical name is estra-1, 3, 5 (10)-triene-3, 17β-diol hemihydrate with the empirical formula of C18H24O2, ½ H2O and a molecular weight of 281.4. The structural formula of E2 is as follows:
Estradiol
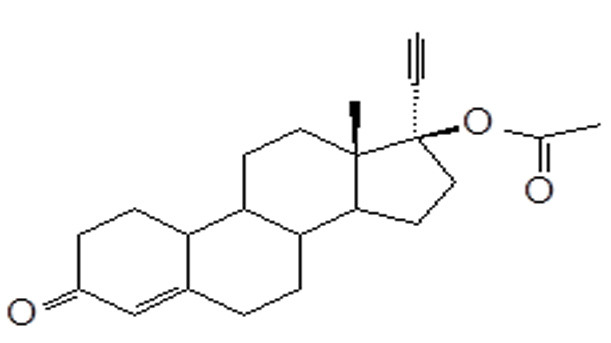
Norethindrone acetate (NETA), a progestin, is a white or yellowish-white crystalline powder. Its chemical name is 17β -acetoxy-19-nor-17α -pregn-4-en-20-yn-3-one with the empirical formula of C22H28O3 and molecular weight of 340.5. The structural formula of NETA is as follows:
Norethindrone Acetate
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Endogenous estrogens are largely responsible for the development and maintenance of the female reproductive system and secondary sexual characteristics. Although circulating estrogens exist in a dynamic equilibrium of metabolic interconversions, estradiol is the principal intracellular human estrogen and is substantially more potent than its metabolites, estrone and estriol, at the receptor level.
The primary source of estrogen in normally cycling adult women is the ovarian follicle, which secretes 70 to 500 mcg of estradiol daily, depending on the phase of the menstrual cycle. After menopause, most endogenous estrogen is produced by conversion of androstenedione, secreted by the adrenal cortex, to estrone in the peripheral tissues. Thus, estrone and the sulfate-conjugated form, estrone sulfate, are the most abundant circulating estrogens in postmenopausal women.
Estrogens act through binding to nuclear receptors in estrogen-responsive tissues. To date, two estrogen receptors have been identified. These vary in proportion from tissue to tissue.
Circulating estrogens modulate the pituitary secretion of the gonadotropins, luteinizing hormone (LH), and FSH through a negative feedback mechanism. Estrogens act to reduce the elevated levels of these hormones seen in postmenopausal women.
Progestin compounds enhance cellular differentiation and generally oppose the actions of estrogens by decreasing estrogen receptor levels, increasing local metabolism of estrogens to less active metabolites, or inducing gene products that blunt cellular responses to estrogen. Progestins exert their effects in target cells by binding to specific progesterone receptors that interact with progesterone response elements in target genes. Progesterone receptors have been identified in the female reproductive tract, breast, pituitary, hypothalamus, and central nervous system.
12.3 Pharmacokinetics
Absorption
Estradiol
Estradiol is absorbed through the gastrointestinal tract. Following oral administration of Lopreeza tablets, peak plasma estradiol concentrations are reached within 5 to 8 hours. The oral bioavailability of estradiol following administration of Lopreeza 1 mg/0.5 mg when compared to a combination oral solution is 53%. Administration of Lopreeza 1 mg/0.5 mg with food did not modify the bioavailability of estradiol.
Norethindrone Acetate
After oral administration, norethindrone acetate is absorbed and transformed to norethindrone. Norethindrone reaches a peak plasma concentration within 0.5 to 1.5 hours after the administration of Lopreeza tablets. The oral bioavailability of norethindrone following administration of Lopreeza 1 mg/0.5 mg when compared to a combination oral solution is 100%. Administration of Lopreeza 1 mg/0.5 mg with food increases norethindrone AUC0-72 by 19% and decreases Cmax by 36%.
The pharmacokinetic parameters of estradiol (E2), estrone (E1), and norethindrone (NET) following oral administration of 1 Lopreeza 1 mg/0.5 mg or 2 Lopreeza 0.5 mg/0.1 mg tablet(s) to healthy postmenopausal women are summarized in Table 3.
TABLE 3
PHARMACOKINETIC PARAMETERS AFTER ADMINISTRATION OF
1 TABLET OF LOPREEZA 1 MG/0.5 MG OR 2 TABLETS OF LOPREEZA 0.5 MG/0.1 MG
TO HEALTHY POSTMENOPAUSAL WOMEN
1 x Lopreeza
1 mg/0.5 mg
(n=24)
2 x Lopreeza
0.5 mg/0.1 mg
(n=24)
Meana (%CV)b Meana (%CV)b Estradiolc (E2) AUC0-t (pg/mL*h) 766.5 (48) 697.3 (53) Cmax (pg/mL) 26.8 (36) 26.5 (37) tmax(h): median (range) 6.0 (0.5-16.0) 6.5 (0.5-16.0) t1/2(h)d 14.0e (29) 14.5f (27) Estronec (E1) AUC0-t (pg/mL*h) 4469.1 (48) 4506.4 (44) Cmax (pg/mL) 195.5 (37) 199.5 (30) tmax(h): median (range) 6.0 (1.0-9.0) 6.0 (2.0-9.0) t1/2(h)d 10.7 (44)g 11.8 (25)g Norethindrone (NET) AUC0-t (pg/mL*h) 21043 (41) 8407.2 (43) Cmax (pg/mL) 5249.5 (47) 2375.4 (41) tmax(h) : median (range) 0.7 (0.7-1.25) 0.8 (0.7-1.3) t1/2(h) 9.8 (32)h 11.4 (36)i AUC = area under the curve, 0 – last quantifiable sample
Cmax = maximum plasma concentration,
tmax = time at maximum plasma concentration,
t1/2 = half-life,
a geometric mean; b geometric % coefficient of variation; c baseline unadjusted data; d baseline unadjusted data; e n=18; f n=16;
g n=13; h n=22; i n=21
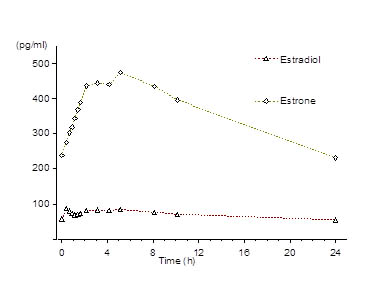
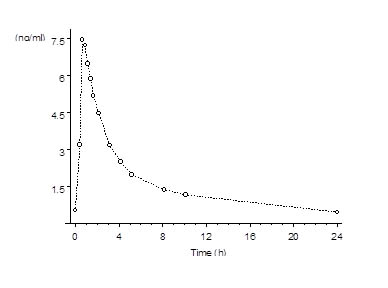
Following continuous dosing with once-daily administration of Lopreeza 1 mg/0.5 mg,serum concentrations of estradiol, estrone, and norethindrone reached steady-state within two weeks with an accumulation of 33 to 47% above concentrations following single dose administration. Unadjusted circulating concentrations of E2, E1, and NET during Lopreeza 1 mg/0.5 mg treatment at steady-state (dosing at time 0) are provided in Figures 1a and 1b.
Figure 1a: Mean Baseline-Uncorrected Estradiol and Estrone Serum Concentration-Time Profiles Following Multiple Doses of Lopreeza 1 mg/0.5 mg (N=24)
Figure 1b: Mean Baseline-Uncorrected Norethindrone Serum Concentration-Time Profile Following Multiple Doses of Lopreeza 1 mg/0.5 mg (N=24)
Distribution
Estradiol
The distribution of exogenous estrogens is similar to that of endogenous estrogens. Estrogens are widely distributed in the body and are generally found in higher concentrations in the sex hormone target organs. Estradiol circulates in the blood bound to SHBG (37%) and to albumin (61%), while only approximately 1 to 2% is unbound.
Norethindrone Acetate
Norethindrone also binds to a similar extent to SHBG (36%) and to albumin (61%).
Metabolism
Estradiol
Exogenous estrogens are metabolized in the same manner as endogenous estrogens. Circulating estrogens exist in a dynamic equilibrium of metabolic interconversions. These transformations take place mainly in the liver. Estradiol is converted reversibly to estrone, and both can be converted to estriol, which is a major urinary metabolite. Estrogens also undergo enterohepatic recirculation via sulfate and glucuronide conjugation in the liver, biliary secretion of conjugates into the intestine, and hydrolysis in the intestine followed by reabsorption. In postmenopausal women, a significant proportion of the circulating estrogens exist as sulfate conjugates, especially estrone sulfate, which serves as a circulating reservoir for the formation of more active estrogens.
Norethindrone Acetate
The most important metabolites of norethindrone are isomers of 5a-dihydro-norethindrone and tetrahydro-norethindrone, which are excreted mainly in the urine as sulfate or glucuronide conjugates.
Excretion
Estradiol
Estradiol, estrone, and estriol are excreted in the urine along with glucuronide and sulfate conjugates. The half-life of estradiol following single dose administration of Lopreeza 1 mg/0.5 mg is 12 to 14 hours.
Norethindrone Acetate
The terminal half-life of norethindrone is about 8 to 11 hours.
Use in Specific Populations
No pharmacokinetic studies were conducted in specific populations, including women with renal or hepatic impairment.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Effects on Vasomotor Symptoms
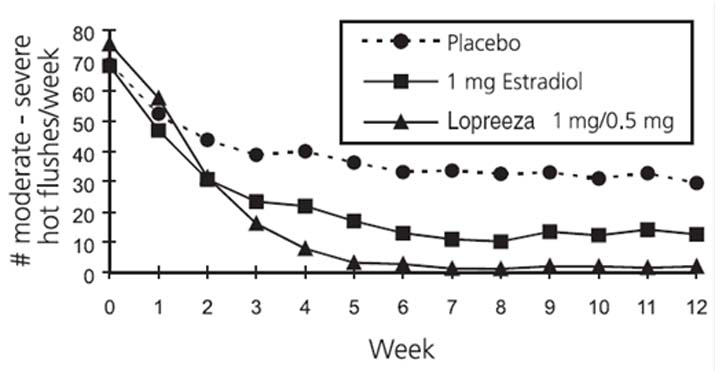
In a 12-week randomized clinical trial involving 92 subjects, Lopreeza 1 mg/0.5 mg was compared to 1 mg of estradiol and to placebo. The mean number and intensity of hot flushes were significantly reduced from baseline to week 4 and 12 in both the Lopreeza 1 mg/0.5 mg and the 1 mg estradiol group compared to placebo (see Figure 2).
Figure 2
Mean Weekly Number of Moderate and Severe Hot
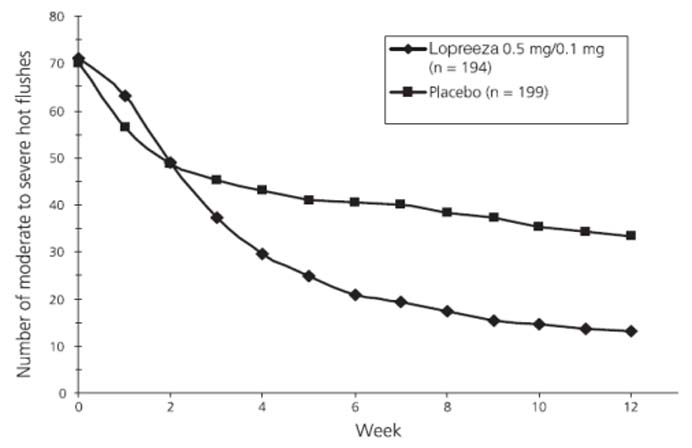
Flushes in a 12-Week StudyIn a study conducted in Europe a total of 577 postmenopausal women were randomly assigned to either Lopreeza 0.5 mg/0.1 mg, 0.5 mg E2/0.25 mg NETA, or placebo for 24 weeks of treatment. The mean number and severity of hot flushes were significantly reduced at week 4 and week 12 in the Lopreeza 0.5 mg/0.1 mg (see Figure 3) and 0.5 mg E2/0.25 mg NETA groups compared to placebo.
Figure 3
Mean Number of Moderate to Severe Hot
Flushes for Weeks 0 Through 12
14.2 Effects on the Endometrium
Lopreeza 1 mg/0.5 mg reduced the incidence of estrogen-induced endometrial hyperplasia at 1 year in a randomized, controlled clinical trial. This trial enrolled 1,176 subjects who were randomized to one of 4 arms: 1 mg estradiol unopposed (n=296), 1 mg E2 + 0.1 mg NETA (n=294), 1 mg E2 + 0.25 mg NETA (n=291), and Lopreeza 1 mg/0.5 mg (n=295). At the end of the study, endometrial biopsy results were available for 988 subjects. The results of the 1 mg estradiol unopposed arm compared to Lopreeza 1 mg/0.5 mg are shown in Table 4.
TABLE 4
INCIDENCE OF ENDOMETRIAL HYPERPLASIA WITH UNOPPOSED ESTRADIOL AND LOPREEZA 1 MG/0.5 MG IN A 12-MONTH STUDY
1 mg E2
(n=296)Lopreeza
1 mg E2/0.5 mg
NETA
(n=295)1 mg E2/0.25 mg
NETA
(n=291 )1 mg E2/0.1 mg
NETA
(n=294 )No. of subjects with histological evaluation at the end of the study 247 241 251 249 No. (%) of subjects with endometrial hyperplasia at the end of the study 36 (14.6%) 1 (0.4%) 1 (0.4%) 2 (0.8%)
14.3 Effects on Uterine Bleeding or Spotting
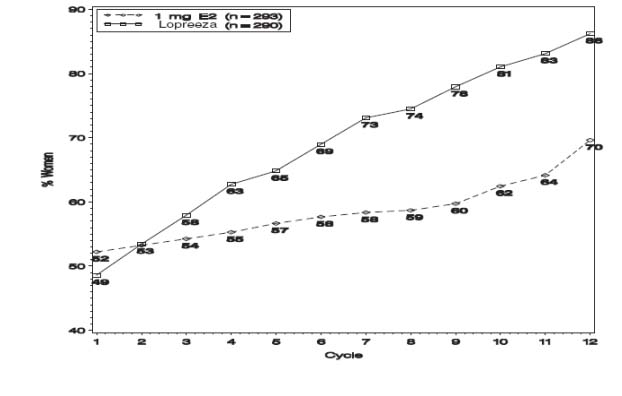
During the initial months of therapy, irregular bleeding or spotting occurred with Lopreeza 1 mg/0.5 mg treatment. However, bleeding tended to decrease over time, and after 12 months of treatment with Lopreeza 1 mg/0.5 mg, about 86 percent of women were amenorrheic (see Figure 4).
Figure 4
Patients Treated with Lopreeza 1 mg/0.5 mg with Cumulative Amenorrhea over Time
Percentage of Women with no Bleeding or Spotting at any Cycle Through Cycle 13
Intent to Treat Population, LOCF
Note: the percentage of patients who were amenorrheic in a given cycle and through cycle 13 is shown. If data were missing, the bleeding value from the last reported day was carried forward (LOCF).
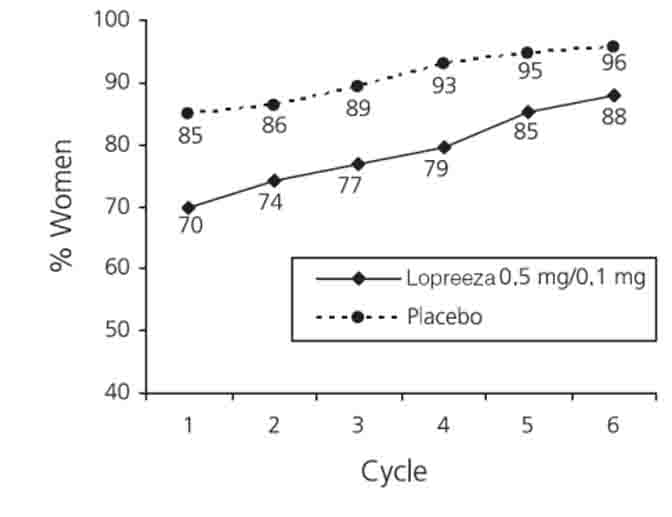
In the clinical trial with Lopreeza 0.5 mg/0.1 mg, 88 percent of women were amenorrheic after 6 months of treatment (See Figure 5).
Figure 5
Patients Treated with Lopreeza 0.5 mg/0.1 mg with
Cumulative Amenorrhea over Time
Percentage of Women with no Bleeding or Spotting at any
Cycle Through Cycle 6, Intent to Treat Population, LOCF
14.4 Effects on Bone Mineral Density
The results of two randomized, multicenter, calcium-supplemented (500 to 1,000 mg per day), placebo-controlled, 2 year clinical trials have shown that Lopreeza 1 mg/0.5 mg and estradiol 0.5 mg are effective in preventing bone loss in postmenopausal women. A total of 462 postmenopausal women with intact uteri and baseline BMD values for lumbar spine within 2 standard deviations of the mean in healthy young women (T-score > -2.0) were enrolled. In a US trial, 327 postmenopausal women (mean time from menopause 2.5 to 3.1 years) with a mean age of 53 years were randomized to 7 groups (0.25 mg, 0.5 mg, and 1 mg of estradiol alone, 1 mg estradiol with 0.25 mg norethindrone acetate, 1 mg estradiol with 0.5 mg norethindrone acetate, and 2 mg estradiol with 1 mg norethindrone acetate, and placebo.) In a European trial (EU trial), 135 postmenopausal women (mean time from menopause 8.4 to 9.3 years) with a mean age of 58 years were randomized to 1 mg estradiolwith 0.25 mg norethindrone acetate, 1 mg estradiol with 0.5 mg norethindrone acetate, andplacebo. Approximately 58 percent and 67 percent of the randomized subjects in the two clinical trials, respectively, completed the two clinical trials. BMD was measured using dual-energy x-ray absorptiometry (DXA).
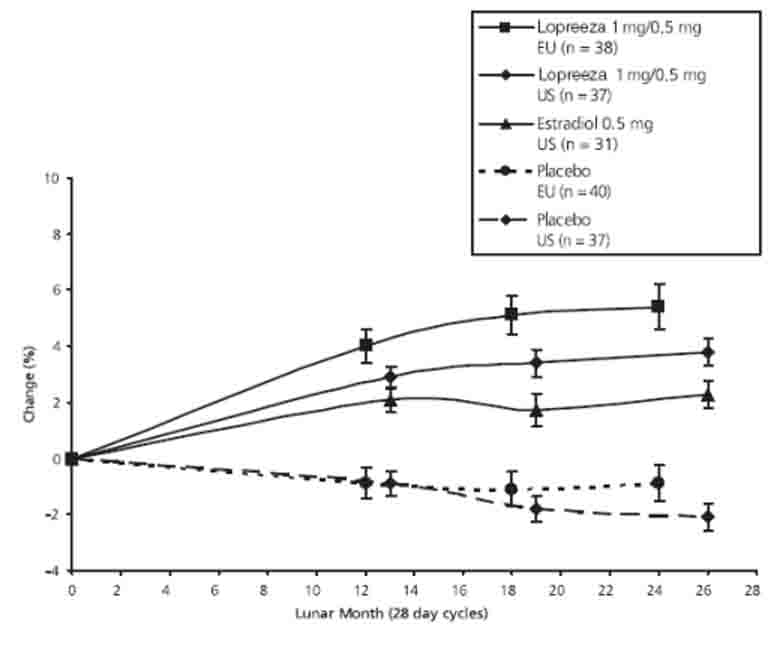
A summary of the results comparing Lopreeza 1 mg/0.5 mg and estradiol 0.5 mg to placebo from the two prevention trials is shown in Table 5.
TABLE 5
PERCENTAGE CHANGE (MEAN ± SD) IN BONE MINERAL DENSITY (BMD) FOR
LOPREEZA 1 MG/0.5 MG AND 0.5 MG E2†
(Intent to Treat Analysis, Last Observation Carried Forward)
US Trial
EU Trial
Placebo
(n=37)0.5 mg E2†
(n=31)
Lopreeza
1 mg/0.5 mg
(n=37)Placebo
(n=40)
Lopreeza
1 mg/0.5 mg
(n=38)Lumbar spine
-2.1 ± 2.9
2.3 ± 2.8 *
3.8 ± 3.0 *
-0.9 ± 4.0
5.4 ± 4.8 *
Femoral neck
-2.3 ± 3.4
0.3 ± 2.9 **
1.8 ± 4.1 *
-1.0 ± 4.6
0.7 ± 6.1
Femoral trochanter
-2.0 ± 4.3
1.7 ± 4.1***
3.7 ± 4.3 *
0.8 ± 6.9
6.3 ± 7.6 *
US= United States, EU = European
† While Lopreeza 0.5 mg/0.1 mg was not directly studied in these trials, the US trial showed that addition of NETA to estradiol enhances the effect on BMD; therefore the BMD changes expected from treatment with Lopreeza 0.5 mg/0.1 mg should be at least as great as observed with estradiol 0.5 mg.
* Significantly (p<0.001) different from placebo** Significantly (p<0.007) different from placebo
***Significantly (p<0.002) different from placebo
The overall difference in mean percentage change in BMD at the lumbar spine in the US trial (1000 mg per day calcium) between Lopreeza 1 mg/0.5 mg and placebo was 5.9 percent and between estradiol 0.5 mg and placebo was 4.4 percent. In the European trial (500 mg per day calcium), the overall difference in mean percentage change in BMD at the lumbar spine was 6.3 percent. Lopreeza 1 mg/0.5 mg and estradiol 0.5 mg also increased BMD at the femoral neck and femoral trochanter compared to placebo. The increase in lumbar spine BMD in the US and European clinical trials for Lopreeza 1 mg/0.5 mg and estradiol 0.5 mg is displayed in Figure 6.
Figure 6
Percentage Change in Bone Mineral Density (BMD) ± SEM of the Lumbar Spine (L1-L4) for
Lopreeza 1 mg/0.5 mg and Estradiol 0.5 mg
(Intent to Treat Analysis with Last Observation Carried Forward)
14.5 Women’s Health Initiative Studies
The WHI enrolled approximately 27,000 predominantly healthy postmenopausal women in two substudies to assess the risks and benefits of daily oral CE (0.625 mg)-alone or in combination with MPA (2.5 mg) compared to placebo in the prevention of certain chronic diseases. The primary endpoint was the incidence of CHD (defined as nonfatal MI, silent MI and CHD death), with invasive breast cancer as the primary adverse outcome. A “global index” included the earliest occurrence of CHD, invasive breast cancer, stroke, PE, endometrial cancer (only in the CE plus MPA substudy), colorectal cancer, hip fracture, or death due to other cause. These substudies did not evaluate the effects of CE plus MPA or CE-alone on menopausal symptoms.
WHI Estrogen Plus Progestin Substudy
The WHI estrogen plus progestin substudy was stopped early. According to the predefined stopping rule, after an average follow-up of 5.6 years of treatment, the increased risk of invasive breast cancer and cardiovascular events exceeded the specified benefits included in the “global index.” The absolute excess risk of events included in the “global index” was 19 per 10,000 women-years.
For those outcomes included in the WHI “global index,” that reached statistical significance after 5.6 years of follow-up, the absolute excess risks per 10,000 women-years in the group treated with CE plus MPA were 7 more CHD events, 8 more strokes, 10 more PEs, and 8 more invasive breast cancers, while the absolute risk reductions per 10,000 women-years were 6 fewer colorectal cancers and 5 fewer hip fractures.
Results of the CE plus MPA substudy, which included 16,608 women (average 63 years of age, range 50 to 79; 83.9 percent White, 6.8 percent Black, 5.4 percent Hispanic, 3.9 percent Other) are presented in Table 6. These results reflect centrally adjudicated data after an average follow-up of 5.6 years.
Table 6: Relative and Absolute Risk Seen in the Estrogen Plus Progestin Substudy of WHI at an Average of 5.6 Yearsa,b
Event
Relative Risk
CE/MPA versus Placebo
(95% nCIc)CE/MPA
n = 8,506Placebo
n = 8,102Absolute Risk per 10,000 Women-Years
CHD events
1.23 (0.99-1.53)
41
34
Non-fatal MI
1.28 (1.00-1.63)
31
25
CHD death
1.10 (0.70-1.75)
8
8
All strokes
1.31 (1.03–1.68)
33
25
Ischemic stroke
1.44 (1.09–1.90)
26
18
Deep vein thrombosisd
1.95 (1.43–2.67)
26
13
Pulmonary embolism
2.13 (1.45–3.11)
18
8
Invasive breast cancere
1.24 (1.01–1.54)
41
33
Colorectal cancer
0.61 (0.42–0.87)
10
16
Endometrial cancerd
0.81 (0.48–1.36)
6
7
Cervical cancerd
1.44 (0.47–4.42)
2
1
Hip fracture
0.67 (0.47–0.96)
11
16
Vertebral fracturesd
0.65 (0.46–0.92)
11
17
Lower arm/wrist fracturesd
0.71 (0.59–0.85)
44
62
Total fracturesd
0.76 (0.69–0.83)
152
199
Overall Mortalityf
1.00 (0.83-1.19)
52
52
Global Indexg
1.13 (1.02-1.25)
184
165
a Adapted from numerous WHI publications. WHI publications can be viewed at www.nhlbi.nih.gov/whi.
b Results are based on centrally adjudicated data.
c Nominal confidence intervals unadjusted for multiple looks and multiple comparisons.
d Not included in “global index”.
eIncludes metastatic and non-metastatic breast cancer, with the exception of in situ breast cancer.
f All deaths, except from breast or colorectal cancer, definite or probable CHD, PE or cerebrovascular disease.
g A subset of the events was combined in a “global index” defined as the earliest occurrence of CHD events, invasive breast cancer, stroke, pulmonary embolism, colorectal cancer, hip fracture, or death due to other causes.
Timing of the initiation of estrogen plus progestin therapy relative to the start of menopause may affect the overall risk benefit profile. The WHI estrogen plus progestin substudy, stratified by age, showed in women 50 to 59 years of age a non-significant trend toward reduced risk for overall mortality [hazard ratio (HR) 0.69 (95 percent CI, 0.44-1.07)].
WHI Estrogen-Alone Substudy
The WHI estrogen-alone substudy was stopped early because an increased risk of stroke was observed, and it was deemed that no further information would be obtained regarding the risks and benefits of estrogen-alone in predetermined primary endpoints.
Results of the estrogen-alone substudy, which included 10,739 women (average 63 years of age, range 50 to 79; 75.3 percent White, 15.1 percent Black, 6.1 percent Hispanic, 3.6 percent Other), after an average follow-up of 7.1 years, are presented in Table 7.
Table 7: Relative and Absolute Risk Seen in the Estrogen-Alone Substudy of WHIa
Event
Relative Risk
CE versus Placebo
(95% nCIb)CE
n = 5,310Placebo
n = 5,429Absolute Risk per 10,000 Women-Years
CHD eventsc
0.95 (0.78–1.16)
54
57
Non-fatal MIc
0.91 (0.73–1.14)
40
43
CHD deathc
1.01(0.71–1.43)
16
16
All strokesc
1.33 (1.05-1.68)
45
33
Ischemic strokeb
1.55 (1.19 – 2.01)
38
25
Deep vein thrombosisc,d
1.47 (1.06–2.06)
23
15
Pulmonary embolismc
1.37 (0.90–2.07)
14
10
Invasive breast cancerc
0.80 (0.62–1.04)
28
34
Colorectal cancere
1.08 (0.75–1.55)
17
16
Hip fracturec
0.65 (0.45–0.94)
12
19
Vertebral fracturesc,d
0.64 (0.44-0.93)
11
18
Lower arm/wrist fracturesc,d
0.58 (0.47-0.72)
35
59
Total fracturesc,d
0.71 (0.64-0.80)
144
197
Death due to other causese,f
1.08 (0.88–1.32)
53
50
Overall mortalityc,d
1.04 (0.88–1.22)
79
75
Global Indexg
1.02 (0.92–1.13)
206
201
a Adapted from numerous WHI publications. WHI publications can be viewed at www.nhlbi.nih.gov/whi.
b Nominal confidence intervals unadjusted for multiple looks and multiple comparisons.
c Results are based on centrally adjudicated data for an average follow-up of 7.1 years.
d Not included in “global index”.
e Results are based on an average follow-up of 6.8 years.
f All deaths, except from breast or colorectal cancer, definite or probable CHD, PE or cerebrovascular disease.
g A subset of the events was combined in a “global index,” defined as the earliest occurrence of CHD events, invasive breast cancer, stroke, pulmonary embolism, colorectal cancer, hip fracture, or death due to other causes.
For those outcomes included in the WHI “global index” that reached statistical significance, the absolute excess risk per 10,000 women-years in the group treated with CE-alone was 12 more strokes, while the absolute risk reduction per 10,000 women-years was 7 fewer hip fractures.9 The absolute excess risk of events included in the “global index” was a non-significant 5 events per 10,000 women-years. There was no difference between the groups in terms of all-cause mortality.
No overall difference for primary CHD events (nonfatal MI, silent MI and CHD death) and invasive breast cancer incidence in women receiving CE-alone compared with placebo was reported in final centrally adjudicated results from the estrogen-alone substudy, after an average follow up of 7.1 years.
Centrally adjudicated results for stroke events from the estrogen-alone substudy, after an average follow-up of 7.1 years, reported no significant difference in distribution of stroke subtype or severity, including fatal strokes, in women receiving CE-alone compared to placebo. Estrogen-alone increased the risk for ischemic stroke, and this excess risk was present in all subgroups of women examined.10
Timing of the initiation of estrogen-alone therapy relative to the start of menopause may affect the overall risk benefit profile. The WHI estrogen-alone substudy, stratified by age, showed in women 50 to 59 years of age a non-significant trend toward reduced risk for CHD [HR 0.63 (95 percent CI, 0.36-1.09)] and overall mortality [HR 0.71 (95 percent CI, 0.46-1.11)].
14.6 Women’s Health Initiative Memory Study
The WHIMS estrogen plus progestin ancillary study of WHI enrolled 4,532 predominantly healthy postmenopausal women 65 years of age and older (47 percent were 65 to 69 years of age, 35 percent were 70 to 74 years of age, 18 percent were 75 years of age and older) to evaluate the effects of daily CE (0.625 mg) plus MPA (2.5 mg) on the incidence of probable dementia (primary outcome) compared to placebo.
After an average follow-up of 4 years, the relative risk of probable dementia for CE plus MPA versus placebo was 2.05 (95 percent CI, 1.21-3.48). The absolute risk of probable dementia for CE plus MPA versus placebo was 45 versus 22 cases per 10,000 women-years. Probable dementia as defined in this study included Alzheimer’s disease (AD), vascular dementia (VaD) and mixed types (having features of both AD and VaD). The most common classification of probable dementia in the treatment group and the placebo group was AD. Since the ancillary study was conducted in women 65 to 79 years of age, it is unknown whether these findings apply to younger postmenopausal women [see Warnings and Precautions (5.3), and Use in Specific Populations (8.5)].
The WHIMS estrogen-alone ancillary study of WHI study enrolled 2,947 predominantly healthy hysterectomized postmenopausal women 65 to 79 years of age (45 percent were 65 to 69 years of age, 36 percent were 70 to 74 years of age, 19 percent were 75 years of age and older) to evaluate the effects of daily CE (0.625 mg)-alone on the incidence of probable dementia (primary outcome) compared to placebo.
After an average follow-up of 5.2 years, the relative risk of probable dementia for CE-alone versus placebo was 1.49 (95 percent CI, 0.83-2.66). The absolute risk of probable dementia for CE-alone versus placebo was 37 versus 25 cases per 10,000 women-years. Probable dementia as defined in this study included AD, VaD and mixed types (having features of both AD and VaD). The most common classification of probable dementia in the treatment group and the placebo group was AD. Since the ancillary study was conducted in women 65 to 79 years of age, it is unknown whether these findings apply to younger postmenopausal women [see Warnings and Precautions (5.3), and Use in Specific Populations (8.5)].
When data from the two populations were pooled as planned in the WHIMS protocol, the reported overall relative risk for probable dementia was 1.76 (95 percent CI, 1.19-2.60). Differences between groups became apparent in the first year of treatment. It is unknown whether these findings apply to younger postmenopausal women [see Warnings and Precautions (5.3), and Use in Specific Populations (8.5)].
-
15 REFERENCES
1. Rossouw JE, et al. Postmenopausal Hormone Therapy and Risk of Cardiovascular Disease by Age and Years Since Menopause. JAMA. 2007;297:1465-1477.
2. Hsia J, et al. Conjugated Equine Estrogens and Coronary Heart Disease. Arch Int Med. 2006;166:357-365.
3. Cushman M, et al. Estrogen Plus Progestin and Risk of Venous Thrombosis. JAMA. 2004;292:1573-1580.
4. Curb JD, et al. Venous Thrombosis and Conjugated Equine Estrogen in Women Without a Uterus.
Arch Int Med. 2006;166:772-780.5. Chlebowski RT, et al. Influence of Estrogen Plus Progestin on Breast Cancer and Mammography in Healthy Postmenopausal Women. JAMA. 2003;289:3234-3253.
6. Stefanick ML, et al. Effects of Conjugated Equine Estrogens on Breast Cancer and Mammography Screening in Postmenopausal Women With Hysterectomy. JAMA. 2006;295:1647-1657.
7. Anderson GL, et al. Effects of Estrogen Plus Progestin on Gynecologic Cancers and Associated Diagnostic Procedures. JAMA. 2003;290:1739-1748.
8. Shumaker SA, et al. Conjugated Equine Estrogens and Incidence of Probable Dementia and Mild Cognitive Impairment in Postmenopausal Women. JAMA. 2004;291:2947-2958.
9. Jackson RD, et al. Effects of Conjugated Equine Estrogen on Risk of Fractures and BMD in Postmenopausal Women With Hysterectomy: Results From the Women’s Health Initiative Randomized Trial. J Bone Miner Res. 2006;21:817-828.
10. Hendrix SL, et al. Effects of Conjugated Equine Estrogen on Stroke in the Women’s Health Initiative. Circulation. 2006;113:2425-2434.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
Lopreeza (estradiol/norethindrone acetate) tablets, 1 mg/0.5 mg are available as white, round, biconvex, film-coated tablets, debossed with “ALH” on the one side and plain on the other side. (NDC 69238-1610-6). It is supplied as 28 tablets in a blister pack, one blister pack per carton.
Lopreeza (estradiol/norethindrone acetate) tablets, 0.5 mg/0.1 mg are available as white, round, biconvex, film-coated tablets, debossed with “ALL” on the one side and plain on the other side.
(NDC 69238-1609-6). It is supplied as 28 tablets in a blister pack, one blister pack per carton.
-
17 PATIENT COUNSELING INFORMATION
See FDA-approved patient labeling (Patient Information)
17.1 Abnormal Vaginal Bleeding
Inform postmenopausal women of the importance of reporting abnormal vaginal bleeding to their healthcare provider as soon as possible [see Warnings and Precautions (5.2)].
17.2 Possible Serious Adverse Reactions with Estrogen Plus Progestin Therapy
Inform postmenopausal women of possible serious adverse reactions of estrogen plus progestin therapy including Cardiovascular Disorders, Malignant Neoplasms, and Probable Dementia [see Warnings and Precautions (5.1, 5.2, 5.3)].
17.3 Possible Less Serious but Common Adverse Reactions with Estrogen Plus Progestin Therapy
Inform postmenopausal women of possible less serious but common adverse reactions of estrogen plus progestin therapy such as headache, breast pain and tenderness, nausea and vomiting.
Rx Only
Lopreeza® is a registered trademark of Amneal Pharmaceuticals LLC
© 2018 Amneal Pharmaceuticals LLC
For information contact:
Amneal Pharmaceuticals
1-877-835-5472
www.amneal.com
Distributed by:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 07-2018-00
-
PATIENT INFORMATION
Lopreeza™ (lo-PREE-zuh)
(estradiol/norethindrone acetate)
Tablets
Read this Patient Information before you start taking Lopreeza and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your menopausal symptoms or your treatment.
What is the most important information I should know about Lopreeza
(a combination of estrogen and progestin)?
- Do not use estrogens with progestins to prevent heart disease, heart attacks, strokes, or dementia (decline of brain function).
- Taking estrogens with progestins may increase your chances of getting heart attacks, strokes, breast cancer, or blood clots.
- Taking estrogens with progestins may increase your chance of getting dementia, based on a study of women 65 years of age or older.
- Do not use estrogen-alone to prevent heart disease, heart attacks, strokes or dementia.
- Taking estrogen-alone may increase your chance of getting cancer of the uterus (womb).
- Taking estrogen-alone may increase your chances of getting strokes or blood clots.
- Taking estrogen-alone may increase your chance of getting dementia, based on a study of women 65 years of age or older.
- You and your healthcare provider should talk regularly about whether you still need treatment with Lopreeza.
What is Lopreeza?
Lopreeza is a prescription medicine that contains two kinds of hormones, an estrogen and a progestin.
What is Lopreeza used for?
Lopreeza is used after menopause to:
-
reduce moderate to severe hot flushes
Estrogens are hormones made by a woman’s ovaries. The ovaries normally stop making estrogens when a woman is between 45 and 55 yrs old. This drop in body estrogen levels causes the “change of life” or menopause, the end of monthly menstrual periods. Sometimes both ovaries are removed during an operation before natural menopause takes place. The sudden drop in estrogen levels causes “surgical menopause.”
When the estrogen levels begin dropping, some women get very uncomfortable symptoms, such as feelings of warmth in the face, neck, and chest, or sudden, intense episodes of heat and sweating (“hot flashes” or “hot flushes”). In some women, the symptoms are mild, and they will not need to take estrogens. In other women, symptoms can be more severe. You and your healthcare provider should talk regularly about whether or not you still need treatment with Lopreeza.
-
treat moderate to severe menopausal changes in and around the vagina
You and your healthcare provider should talk regularly about whether you still need treatment with Lopreeza 1.0 mg/0.5 mg to treat these problems. If you use Lopreeza 1.0 mg/0.5 mg only to treat your menopausal changes in and around your vagina, talk with your healthcare provider about whether a topical vaginal product would be better for you.
- help reduce your chances of getting osteoporosis (thin weak bones)
If you use Lopreeza only to prevent osteoporosis from menopause, talk to your healthcare provider about whether a different treatment or medicine without estrogens might be better for you.
You and your healthcare provider should talk regularly about whether you still need treatment with Lopreeza.
Who should not take Lopreeza?
Do not take Lopreeza if you have had your uterus (womb) removed (hysterectomy).
Lopreeza contains a progestin to decrease the chance of getting cancer of the uterus. If you do not have a uterus, you do not need a progestin and you should not take Lopreeza.
Do not take Lopreeza if you:
-
have unusual vaginal bleeding
Vaginal bleeding after menopause may be a warning sign of cancer of the uterus (womb). Your healthcare provider should check any unusual vaginal bleeding to find out the cause.
- currently have or have had certain cancers
Estrogens may increase the chance of getting certain types of cancers, including cancer of the breast or uterus. If you have or have had cancer, talk with your healthcare provider about whether you should take Lopreeza.
- had a stroke or heart attack
- currently have or have had blood clots
- currently have or have had liver problems
- have been diagnosed with a bleeding disorder
- are allergic to Lopreeza or any of its ingredients
See the list of ingredients in Lopreeza at the end of this leaflet.
-
think you may be pregnant
Lopreeza is not for pregnant women. If you think you may be pregnant, you should have a pregnancy test and know the results. Do not take Lopreeza if the test is positive and talk to your healthcare provider.
What should I tell my healthcare provider before taking Lopreeza?
Before you take Lopreeza, tell your healthcare provider if you:
-
have any unusual vaginal bleeding
Vaginal bleeding after menopause may be a warning sign of cancer of the uterus (womb). Your healthcare provider should check any unusual vaginal bleeding to find out the cause.
-
have any other medical conditions
Your healthcare provider may need to check you more carefully if you have certain conditions, such as asthma (wheezing), epilepsy (seizures), diabetes, migraine, endometriosis, lupus, angioedema (swelling of face and tongue), or problems with your heart, liver, thyroid, kidneys, or have high calcium levels in your blood.
- are going to have surgery or will be on bed rest
Your healthcare provider will let you know if you need to stop taking Lopreeza.
- are breast feeding
The hormones in Lopreeza can pass into your breast milk.
Tell your healthcare provider about all the medicines you take, including prescription and nonprescription medicines, vitamins, and herbal supplements. Some medicines may affect how Lopreeza works. Lopreeza may also affect how your other medicines work. Keep a list of your medicines and show them to your healthcare provider and pharmacist when you get a new medicine.
How should I take Lopreeza?
- Take Lopreeza exactly as your healthcare provider tells you to take it.
- Take 1 Lopreeza at the same time each day.
- You and your healthcare provider should talk regularly (every 3 to 6 months) about the dose you are taking and whether you still need treatment with Lopreeza.
What are the possible side effects of Lopreeza?
Side effects are grouped by how serious they are and how often they happen when you are treated.
Serious, but less common side effects include:
- heart attack
- stroke
- blood clots
- dementia
- breast cancer
- cancer of the lining of the uterus (womb)
- cancer of the ovary
- high blood pressure
- high blood sugar
- gallbladder disease
- liver problems
- changes in your thyroid hormone levels
- enlargement of benign tumors (“fibroids”)
Call your healthcare provider right away if you get any of the following warning signs or any other unusual symptoms that concern you:
- new breast lumps
- unusual vaginal bleeding
- changes in vision or speech
- sudden new severe headaches
- severe pains in your chest or legs with or without shortness of breath, weakness and fatigue
Less serious, but common side effects include:
- headache
- breast pain
- irregular vaginal bleeding or spotting
- stomach or abdominal cramps, bloating
- nausea and vomiting
- hair loss
- fluid retention
- vaginal yeast infection
These are not all the possible side effects of Lopreeza. For more information, ask your healthcare provider or pharmacist. Tell your healthcare provider if you have any side effect that bothers you or does not go away. You may report side effects to Amneal Pharmaceuticals at 1-877-835-5472 or to FDA at 1-800-FDA-1088.
What can I do to lower my chances of a serious side effect with Lopreeza?
- Talk with your healthcare provider regularly about whether you should continue taking Lopreeza.
- If you have a uterus, talk with your healthcare provider about whether the addition of a progestin is right for you.
- The addition of a progestin is generally recommended for a woman with a uterus to reduce the chance of getting cancer of the uterus (womb).
- See your healthcare provider right away if you get vaginal bleeding while taking Lopreeza.
- Have a pelvic exam, breast exam and mammogram (breast X-ray) every year unless your healthcare provider tells you something else.
- If members of your family have had breast cancer or if you have ever had breast lumps or an abnormal mammogram (breast x-ray), you may need to have breast exams more often.
- If you have high blood pressure, high cholesterol (fat in the blood), diabetes, are overweight, or if you use tobacco, you may have higher chances for getting heart disease.
Ask your healthcare provider for ways to lower your chances for getting heart disease.
How should I store Lopreeza?
- Store Lopreeza at room temperature between 68°F to 77°F (20°C to 25°C).
- Store Lopreeza in a dry place protected from light.
KEEP LOPREEZA and all medicines out of the reach of children.
General information about the safe and effective use of Lopreeza.
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not take Lopreeza for conditions for which it was not prescribed. Do not give Lopreeza to other people, even if they have the same symptoms you have. It may harm them.
This leaflet summarizes the most important information about Lopreeza. If you would like more information, talk with your healthcare provider or pharmacist. You can ask your pharmacist or healthcare provider for information about Lopreeza that is written for health professionals.
For more information go to www.amneal.com or call 1-877-835-5472.
What are the ingredients in Lopreeza?
Active ingredients: estradiol and norethindrone acetate
Inactive Ingredients: lactose monohydrate, starch (corn), talc, magnesium stearate, hypromellose, and triacetin.
The 1 mg/0.5 mg tablet also contains copovidone.
The 0.5 mg/0.1 mg tablet also contains hydroxypropyl cellulose.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Lopreeza® is a trademark of Amneal Pharmaceuticals LLC.
© 2018 Amneal Pharmaceuticals LLC
Distributed by:
Amneal Pharmaceuticals LLC
Bridgewater, NJ 08807
Rev. 07-2018-00
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
LOPREEZA
estradiol/norethindrone acetate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:69238-1610 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ESTRADIOL (UNII: 4TI98Z838E) (ESTRADIOL - UNII:4TI98Z838E) ESTRADIOL 1 mg NORETHINDRONE ACETATE (UNII: 9S44LIC7OJ) (NORETHINDRONE - UNII:T18F433X4S) NORETHINDRONE ACETATE 0.5 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) STARCH, CORN (UNII: O8232NY3SJ) COPOVIDONE K25-31 (UNII: D9C330MD8B) TALC (UNII: 7SEV7J4R1U) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSES (UNII: 3NXW29V3WO) TRIACETIN (UNII: XHX3C3X673) Product Characteristics Color white Score no score Shape ROUND Size 6mm Flavor Imprint Code ALH Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:69238-1610-6 1 in 1 CARTON 01/07/2019 1 28 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020907 01/07/2019 Labeler - Amneal Pharmaceuticals NY LLC (123797875) Establishment Name Address ID/FEI Business Operations Amneal Pharmaceuticals of New York, LLC 123797875 analysis(69238-1610) , label(69238-1610) , manufacture(69238-1610) , pack(69238-1610)