Label: TORISEL- temsirolimus kit



- NDC Code(s): 0008-1125-01, 0008-1179-01, 0008-1279-01
- Packager: Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated April 28, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TORISEL® safely and effectively. See full prescribing information for TORISEL.
TORISEL Kit (temsirolimus) injection, for intravenous use
Initial U.S. Approval: 2007INDICATIONS AND USAGE
TORISEL® is a kinase inhibitor indicated for the treatment of advanced renal cell carcinoma. (1)
DOSAGE AND ADMINISTRATION
- •
- The recommended dose of TORISEL is 25 mg administered as an intravenous infusion over a 30–60 minute period once a week. Treat until disease progression or unacceptable toxicity. (2.1)
- •
- Antihistamine pre-treatment is recommended. (2.2)
- •
- Dose reduction is required in patients with mild hepatic impairment. (2.4)
- •
- TORISEL (temsirolimus) injection vial contents must first be diluted with the enclosed diluent before diluting the resultant solution with 250 mL of 0.9% Sodium Chloride Injection. (2.5)
DOSAGE FORMS AND STRENGTHS
TORISEL injection, 25 mg/mL supplied with DILUENT for TORISEL. (3)
CONTRAINDICATIONS
TORISEL is contraindicated in patients with bilirubin > 1.5×ULN. (4)
WARNINGS AND PRECAUTIONS
- •
- Hypersensitivity/Infusion Reactions (including some life-threatening and rare fatal reactions) can occur early in the first infusion of TORISEL. Patients should be monitored throughout the infusion. (5.1)
- •
- To treat hypersensitivity reactions, stop TORISEL and treat with an antihistamine. TORISEL may be restarted at physician discretion at a slower rate. (5.1)
- •
- Hepatic Impairment: Use caution when treating patients with mild hepatic impairment and reduce dose. (2.4, 5.2)
- •
- Hyperglycemia and hyperlipidemia are likely and may require treatment. Monitor glucose and lipid profiles. (5.3, 5.6)
- •
- Infections may result from immunosuppression. (5.4)
- •
- Monitor for symptoms or radiographic changes of interstitial lung disease (ILD). If ILD is suspected, discontinue TORISEL, and consider use of corticosteroids and/or antibiotics. (5.5)
- •
- Bowel perforation may occur. Evaluate fever, abdominal pain, bloody stools, and/or acute abdomen promptly. (5.7)
- •
- Renal failure, sometimes fatal, has occurred. Monitor renal function at baseline and while on TORISEL. (5.8)
- •
- Due to abnormal wound healing, use TORISEL with caution in the perioperative period. (5.9)
- •
- Proteinuria and nephrotic syndrome may occur. Monitor urine protein prior to the start of TORISEL therapy and periodically thereafter. Discontinue TORISEL in patients with who develop nephrotic syndrome. (5.11)
- •
- Live vaccinations and close contact with those who received live vaccines should be avoided. (5.14)
- •
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of the potential hazard to the fetus and to use effective contraception. (5.15, 8.1, 8.3)
- •
- Elderly patients may be more likely to experience certain adverse reactions, including diarrhea, edema and pneumonia. (5.16)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥30%) are rash, asthenia, mucositis, nausea, edema, and anorexia. The most common laboratory abnormalities (incidence ≥30%) are anemia, hyperglycemia, hyperlipidemia, hypertriglyceridemia, elevated alkaline phosphatase, elevated serum creatinine, lymphopenia, hypophosphatemia, thrombocytopenia, elevated AST, and leukopenia. (6)To report SUSPECTED ADVERSE REACTIONS, contact Wyeth Pharmaceuticals Inc. at 1-800-934-5556 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 4/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Advanced Renal Cell Carcinoma
2.2 Premedication
2.3 Dosage Interruption/Adjustment
2.4 Dose Modification Guidelines
2.5 Instructions for Preparation
2.6 Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity/Infusion Reactions
5.2 Hepatic Impairment
5.3 Hyperglycemia/Glucose Intolerance
5.4 Infections
5.5 Interstitial Lung Disease
5.6 Hyperlipidemia
5.7 Bowel Perforation
5.8 Renal Failure
5.9 Wound Healing Complications
5.10 Intracerebral Hemorrhage
5.11 Proteinuria and Nephrotic syndrome
5.12 Co-administration with Inducers or Inhibitors of CYP3A Metabolism
5.13 Concomitant use of TORISEL with sunitinib
5.14 Vaccinations
5.15 Embryo-Fetal Toxicity
5.16 Elderly Patients
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Post-marketing and Other Clinical Experience
7 DRUG INTERACTIONS
7.1 Agents Inducing CYP3A Metabolism
7.2 Agents Inhibiting CYP3A Metabolism
7.3 Angioedema with ACE inhibitors and Calcium Channel Blockers
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Advanced Renal Cell Carcinoma
The recommended dose of TORISEL for advanced renal cell carcinoma is 25 mg administered as an intravenous infusion over a 30 – 60 minute period once a week.
Treatment should continue until disease progression or unacceptable toxicity occurs.
2.2 Premedication
Patients should receive prophylactic intravenous diphenhydramine 25 to 50 mg (or similar antihistamine) approximately 30 minutes before the start of each dose of TORISEL [see Warnings and Precautions (5.1)].
2.3 Dosage Interruption/Adjustment
TORISEL should be held for absolute neutrophil count (ANC) <1,000/mm3, platelet count <75,000/mm3, or NCI CTCAE grade 3 or greater adverse reactions. Once toxicities have resolved to grade 2 or less, TORISEL may be restarted with the dose reduced by 5 mg/week to a dose no lower than 15 mg/week.
2.4 Dose Modification Guidelines
Hepatic Impairment: Use caution when treating patients with hepatic impairment. If TORISEL must be given in patients with mild hepatic impairment (bilirubin >1 – 1.5×ULN or AST >ULN but bilirubin ≤ULN), reduce the dose of TORISEL to 15 mg/week. TORISEL is contraindicated in patients with bilirubin >1.5×ULN [see Contraindications (4), Warnings and Precautions (5.2) and Use in Specific Populations (8.7)].
Concomitant Strong CYP3A4 Inhibitors: The concomitant use of strong CYP3A4 inhibitors should be avoided (e.g. ketoconazole, itraconazole, clarithromycin, atazanavir, indinavir, nefazodone, nelfinavir, ritonavir, saquinavir, telithromycin, and voriconazole). Grapefruit juice may also increase plasma concentrations of sirolimus (a major metabolite of temsirolimus) and should be avoided. If patients must be co-administered a strong CYP3A4 inhibitor, based on pharmacokinetic studies, a TORISEL dose reduction to 12.5 mg/week should be considered. This dose of TORISEL is predicted to adjust the AUC to the range observed without inhibitors. However, there are no clinical data with this dose adjustment in patients receiving strong CYP3A4 inhibitors. If the strong inhibitor is discontinued, a washout period of approximately 1 week should be allowed before the TORISEL dose is adjusted back to the dose used prior to initiation of the strong CYP3A4 inhibitor [see Warnings and Precautions (5.12) and Drug Interactions (7.2)].
Concomitant Strong CYP3A4 Inducers: The use of concomitant strong CYP3A4 inducers should be avoided (e.g. dexamethasone, phenytoin, carbamazepine, rifampin, rifabutin, rifampacin, phenobarbital). If patients must be co-administered a strong CYP3A4 inducer, based on pharmacokinetic studies, a TORISEL dose increase from 25 mg/week up to 50 mg/week should be considered. This dose of TORISEL is predicted to adjust the AUC to the range observed without inducers. However, there are no clinical data with this dose adjustment in patients receiving strong CYP3A4 inducers. If the strong inducer is discontinued the temsirolimus dose should be returned to the dose used prior to initiation of the strong CYP3A4 inducer [see Warnings and Precautions (5.12) and Drug Interactions (7.1)].
2.5 Instructions for Preparation
TORISEL is a cytotoxic drug. Follow applicable special handling and disposal procedures1.
TORISEL must be stored under refrigeration at 2°–8°C (36°–46°F) and protected from light. During handling and preparation of admixtures, TORISEL should be protected from excessive room light and sunlight. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
In order to minimize the patient exposure to the plasticizer DEHP (di-2-ethylhexyl phthalate), which may be leached from PVC infusion bags or sets, the final TORISEL dilution for infusion should be stored in bottles (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administered through polyethylene-lined administration sets.
TORISEL 25 mg/mL injection must be diluted with the supplied diluent before further dilution in 0.9% Sodium Chloride Injection, USP.
Please note that both the TORISEL injection and diluent vials contain an overfill to ensure the recommended volume can be withdrawn.
Follow this two-step dilution process in an aseptic manner.
Step 1:
DILUTION OF TORISEL INJECTION 25 MG/ML WITH SUPPLIED DILUENT
- •
- Each Vial of TORISEL (temsirolimus) must first be mixed with 1.8 mL of the enclosed diluent. The resultant solution contains 30 mg/3 mL (10 mg/mL).
- •
- Mix well by inversion of the vial. Allow sufficient time for the air bubbles to subside. The solution should be clear to slightly turbid, colorless to light-yellow solution, essentially free from visual particulates.
The concentrate-diluent mixture is stable below 25ºC for up to 24 hours.
Step 2:
DILUTION OF CONCENTRATE-DILUENT MIXTURE WITH 0.9% SODIUM CHLORIDE INJECTION, USP
- •
- Withdraw precisely the required amount of concentrate-diluent mixture containing temsirolimus 10 mg/mL as prepared in Step 1 from the vial (i.e., 2.5 mL for a temsirolimus dose of 25 mg) and further dilute into an infusion bag containing 250 mL of 0.9% Sodium Chloride Injection, USP.
- •
- Mix by inversion of the bag or bottle, avoiding excessive shaking, as this may cause foaming.
The resulting solution should be inspected visually for particulate matter and discoloration prior to administration. The admixture of TORISEL in 0.9% Sodium Chloride Injection, USP should be protected from excessive room light and sunlight.
2.6 Administration
- •
- Administration of the final diluted solution should be completed within six hours from the time that TORISEL is first added to 0.9% Solution Chloride Injection, USP.
- •
- TORISEL is administered as an intravenous infusion over a 30- to 60-minute period once weekly. The use of an infusion pump is the preferred method of administration to ensure accurate delivery of the product.
- •
- Appropriate administration materials should be composed of glass, polyolefin, or polyethylene to avoid excessive loss of product and diethylhexylpthalate (DEHP) extraction. The administration materials should consist of non-DEHP, non-polyvinylchloride (PVC) tubing with appropriate filter. In the case when a PVC administration set has to be used, it should not contain DEHP. An in-line polyethersulfone filter with a pore size of not greater than 5 microns is recommended for administration to avoid the possibility of particles bigger than 5 microns being infused. If the administration set available does not have an in-line filter incorporated, a polyethersulfone filter should be added at the set (i.e., an end-filter) before the admixture reaches the vein of the patient. Different end-filters can be used, ranging in filter pore size from 0.2 microns up to 5 microns. The use of both an in-line and end-filter is not recommended.
- •
- TORISEL, when diluted, contains polysorbate 80, which is known to increase the rate of DEHP extraction from PVC. This should be considered during the preparation and administration of TORISEL, including storage time elapsed when in direct contact with PVC following constitution.
Compatibilities and Incompatibilities
Undiluted TORISEL injection should not be added directly to aqueous infusion solutions. Direct addition of TORISEL injection to aqueous solutions will result in precipitation of drug. Always combine TORISEL injection with DILUENT for TORISEL before adding to infusion solutions. It is recommended that TORISEL be administered in 0.9% Sodium Chloride Injection after combining with diluent. The stability of TORISEL in other infusion solutions has not been evaluated. Addition of other drugs or nutritional agents to admixtures of TORISEL in 0.9% Sodium Chloride Injection has not been evaluated and should be avoided. Temsirolimus is degraded by both acids and bases, and thus combinations of temsirolimus with agents capable of modifying solution pH should be avoided.
-
3 DOSAGE FORMS AND STRENGTHS
TORISEL (temsirolimus) is supplied as a kit consisting of the following:
TORISEL (temsirolimus) injection (25 mg/mL). The TORISEL vial contains temsirolimus at a concentration of 25 mg/mL. The vial contains an overfill of 0.2 mL to ensure the ability to withdraw the recommended dose.
DILUENT for TORISEL. The DILUENT vial includes a deliverable volume of 1.8 mL. This vial contains an overfill in order to ensure that the appropriate volume can be withdrawn.
-
4 CONTRAINDICATIONS
TORISEL is contraindicated in patients with bilirubin >1.5×ULN [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity/Infusion Reactions
Hypersensitivity/infusion reactions, including but not limited to flushing, chest pain, dyspnea, hypotension, apnea, loss of consciousness, hypersensitivity and anaphylaxis, have been associated with the administration of temsirolimus. These reactions can occur very early in the first infusion, but may also occur with subsequent infusions. Patients should be monitored throughout the infusion and appropriate supportive care should be available. Temsirolimus infusion should be interrupted in all patients with severe infusion reactions and appropriate medical therapy administered.
TORISEL should be used with caution in persons with known hypersensitivity to temsirolimus or its metabolites (including sirolimus), polysorbate 80, or to any other component (including the excipients) of TORISEL.
An H1 antihistamine should be administered to patients before the start of the intravenous temsirolimus infusion. TORISEL should be used with caution in patients with known hypersensitivity to an antihistamine, or patients who cannot receive an antihistamine for other medical reasons.
If a patient develops a hypersensitivity reaction during the TORISEL infusion, the infusion should be stopped and the patient should be observed for at least 30 to 60 minutes (depending on the severity of the reaction). At the discretion of the physician, treatment may be resumed with the administration of an H1-receptor antagonist (such as diphenhydramine), if not previously administered [see Dosage and Administration (2.2)], and/or an H2-receptor antagonist (such as intravenous famotidine 20 mg or intravenous ranitidine 50 mg) approximately 30 minutes before restarting the TORISEL infusion. The infusion may then be resumed at a slower rate (up to 60 minutes).
A benefit-risk assessment should be done prior to the continuation of temsirolimus therapy in patients with severe or life-threatening reactions.
5.2 Hepatic Impairment
The safety and pharmacokinetics of TORISEL were evaluated in a dose escalation phase 1 study in 110 patients with normal or varying degrees of hepatic impairment. Patients with baseline bilirubin >1.5×ULN experienced greater toxicity than patients with baseline bilirubin ≤1.5×ULN when treated with TORISEL. The overall frequency of ≥ grade 3 adverse reactions and deaths, including deaths due to progressive disease, were greater in patients with baseline bilirubin >1.5×ULN due to increased risk of death [see Contraindications (4)].
Use caution when treating patients with mild hepatic impairment. Concentrations of temsirolimus and its metabolite sirolimus were increased in patients with elevated AST or bilirubin levels. If TORISEL must be given in patients with mild hepatic impairment (bilirubin >1 – 1.5×ULN or AST >ULN but bilirubin ≤ULN), reduce the dose of TORISEL to 15 mg/week [see Dosage and Administration (2.4)].
5.3 Hyperglycemia/Glucose Intolerance
The use of TORISEL is likely to result in increases in serum glucose. In the phase 3 trial, 89% of patients receiving TORISEL had at least one elevated serum glucose while on treatment, and 26% of patients reported hyperglycemia as an adverse event. This may result in the need for an increase in the dose of, or initiation of, insulin and/or oral hypoglycemic agent therapy. Serum glucose should be tested before and during treatment with TORISEL. Patients should be advised to report excessive thirst or any increase in the volume or frequency of urination.
5.4 Infections
The use of TORISEL may result in immunosuppression. Patients should be carefully observed for the occurrence of infections, including opportunistic infections [see Adverse Reactions (6.1)].
Pneumocystis jiroveci pneumonia (PJP), including fatalities, has been reported in patients who received temsirolimus. This may be associated with concomitant use of corticosteroids or other immunosuppressive agents. Prophylaxis of PJP should be considered when concomitant use of corticosteroids or other immunosuppressive agents are required.
5.5 Interstitial Lung Disease
Cases of interstitial lung disease, some resulting in death, occurred in patients who received TORISEL. Some patients were asymptomatic, or had minimal symptoms, with infiltrates detected on computed tomography scan or chest radiograph. Others presented with symptoms such as dyspnea, cough, hypoxia, and fever. Some patients required discontinuation of TORISEL and/or treatment with corticosteroids and/or antibiotics, while some patients continued treatment without additional intervention. Patients should be advised to report promptly any new or worsening respiratory symptoms.
It is recommended that patients undergo baseline radiographic assessment by lung computed tomography scan or chest radiograph prior to the initiation of TORISEL therapy. Follow such assessments periodically, even in the absence of clinical respiratory symptoms.
It is recommended that patients be followed closely for occurrence of clinical respiratory symptoms. If clinically significant respiratory symptoms develop, consider withholding TORISEL administration until after recovery of symptoms and improvement of radiographic findings related to pneumonitis. Empiric treatment with corticosteroids and/or antibiotics may be considered. Opportunistic infections such as PJP should be considered in the differential diagnosis. For patients who require use of corticosteroids, prophylaxis of PJP may be considered.
5.6 Hyperlipidemia
The use of TORISEL is likely to result in increases in serum triglycerides and cholesterol. In the phase 3 trial, 87% of patients receiving TORISEL had at least one elevated serum cholesterol value and 83% had at least one elevated serum triglyceride value. This may require initiation, or increase in the dose, of lipid-lowering agents. Serum cholesterol and triglycerides should be tested before and during treatment with TORISEL.
5.7 Bowel Perforation
Cases of fatal bowel perforation occurred in patients who received TORISEL. These patients presented with fever, abdominal pain, metabolic acidosis, bloody stools, diarrhea, and/or acute abdomen. Patients should be advised to report promptly any new or worsening abdominal pain or blood in their stools.
5.8 Renal Failure
Cases of rapidly progressive and sometimes fatal acute renal failure not clearly related to disease progression occurred in patients who received TORISEL. Some of these cases were not responsive to dialysis.
5.9 Wound Healing Complications
Use of TORISEL has been associated with abnormal wound healing. Therefore, caution should be exercised with the use of TORISEL in the perioperative period.
5.10 Intracerebral Hemorrhage
Patients with central nervous system tumors (primary CNS tumor or metastases) and/or receiving anticoagulation therapy may be at an increased risk of developing intracerebral bleeding (including fatal outcomes) while receiving TORISEL.
5.11 Proteinuria and Nephrotic syndrome
Proteinuria (including cases of nephrotic syndrome) has occurred in patients treated with TORISEL. Monitor urine protein prior to the start of TORISEL therapy and periodically thereafter. Discontinue TORISEL in patients who develop nephrotic syndrome.
5.12 Co-administration with Inducers or Inhibitors of CYP3A Metabolism
Agents Inducing CYP3A Metabolism:
Strong inducers of CYP3A4/5 such as dexamethasone, carbamazepine, phenytoin, phenobarbital, rifampin, rifabutin, and rifampacin may decrease exposure of the active metabolite, sirolimus. If alternative treatment cannot be administered, a dose adjustment should be considered. St. John's Wort may decrease TORISEL plasma concentrations unpredictably. Patients receiving TORISEL should not take St. John's Wort concomitantly [see Dosage and Administration (2.4) and Drug Interactions (7.1)].
Agents Inhibiting CYP3A Metabolism:
Strong CYP3A4 inhibitors such as atazanavir, clarithromycin, indinavir, itraconazole, ketoconazole, nefazodone, nelfinavir, ritonavir, saquinavir, and telithromycin may increase blood concentrations of the active metabolite sirolimus. If alternative treatments cannot be administered, a dose adjustment should be considered [see Dosage and Administration (2.4) and Drug Interactions (7.2)].
5.13 Concomitant use of TORISEL with sunitinib
The combination of TORISEL and sunitinib resulted in dose-limiting toxicity. Dose-limiting toxicities (Grade 3/4 erythematous maculopapular rash, and gout/cellulitis requiring hospitalization) were observed in two out of three patients treated in the first cohort of a phase 1 study at doses of TORISEL 15 mg IV per week and sunitinib 25 mg oral per day (Days 1–28 followed by a 2-week rest).
5.14 Vaccinations
The use of live vaccines and close contact with those who have received live vaccines should be avoided during treatment with TORISEL. Examples of live vaccines are: intranasal influenza, measles, mumps, rubella, oral polio, BCG, yellow fever, varicella, and TY21a typhoid vaccines.
5.15 Embryo-Fetal Toxicity
Based on findings in animal studies and its mechanism of action, TORISEL can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, daily oral administration of temsirolimus to pregnant animals during organogenesis caused adverse embryo-fetal effects in rats and rabbits at approximately 0.04 and 0.12 times the AUC in patients at the recommended human dose, respectively. Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TORISEL and for 3 months after the last dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with TORISEL and for 3 months after the last dose [see Clinical Pharmacology (12.1) and Use in Specific Populations (8.1, 8.3)].
5.16 Elderly Patients
Based on the results of a phase 3 study, elderly patients may be more likely to experience certain adverse reactions including diarrhea, edema, and pneumonia [see Use in Specific Populations (8.5)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions have been associated with TORISEL in clinical trials and are discussed in greater detail in other sections of the label [see Warnings and Precautions (5)].
- •
- Hypersensitivity/Infusion Reactions [see Warnings and Precautions (5.1)]
- •
- Hepatic Impairment [see Warnings and Precautions (5.2)]
- •
- Hyperglycemia/Glucose Intolerance [see Warnings and Precautions (5.3)]
- •
- Infections [see Warnings and Precautions (5.4)]
- •
- Interstitial Lung Disease [see Warnings and Precautions (5.5)]
- •
- Hyperlipidemia [see Warnings and Precautions (5.6)]
- •
- Bowel Perforation [see Warnings and Precautions (5.7)]
- •
- Renal Failure [see Warnings and Precautions (5.8)]
- •
- Wound Healing Complications [see Warnings and Precautions (5.9)]
- •
- Intracerebral Hemorrhage [see Warnings and Precautions (5.10)]
The most common (≥30%) adverse reactions observed with TORISEL are rash, asthenia, mucositis, nausea, edema, and anorexia. The most common (≥30%) laboratory abnormalities observed with TORISEL are anemia, hyperglycemia, hyperlipidemia, hypertriglyceridemia, lymphopenia, elevated alkaline phosphatase, elevated serum creatinine, hypophosphatemia, thrombocytopenia, elevated AST, and leukopenia.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other trials and may not reflect the rates observed in clinical practice.
In the phase 3 randomized, open-label study of interferon alfa (IFN-α) alone, TORISEL alone, and TORISEL and IFN-α, a total of 616 patients were treated. Two hundred patients received IFN-α weekly, 208 received TORISEL 25 mg weekly, and 208 patients received a combination of TORISEL and IFN-α weekly [see Clinical Studies (14)].
Treatment with the combination of TORISEL 15 mg and IFN-α was associated with an increased incidence of multiple adverse reactions and did not result in a significant increase in overall survival when compared with IFN-α alone.
Table 1 shows the percentage of patients experiencing treatment emergent adverse reactions. Reactions reported in at least 10% of patients who received TORISEL 25 mg alone or IFN-α alone are listed. Table 2 shows the percentage of patients experiencing selected laboratory abnormalities. Data for the same adverse reactions and laboratory abnormalities in the IFN-α alone arm are shown for comparison:
Table 1 – Adverse Reactions Reported in at Least 10% of Patients Who Received 25 mg IV TORISEL or IFN-α in the Randomized Trial Adverse Reaction TORISEL
25 mg
n = 208IFN-α
n = 200All Grades*
n (%)Grades 3&4*
n (%)All Grades*
n (%)Grades 3&4*
n (%)- *
- Common Toxicity Criteria for Adverse Events (CTCAE), Version 3.0.
- †
- Includes edema, facial edema, and peripheral edema
- ‡
- Includes aphthous stomatitis, glossitis, mouth ulceration, mucositis, and stomatitis
- §
- Includes infections not otherwise specified (NOS) and the following infections that occurred infrequently as distinct entities: abscess, bronchitis, cellulitis, herpes simplex, and herpes zoster
- ¶
- Includes cystitis, dysuria, hematuria, urinary frequency, and urinary tract infection
- #
- Includes eczema, exfoliative dermatitis, maculopapular rash, pruritic rash, pustular rash, rash (NOS), and vesiculobullous rash
- Þ
- Includes taste loss and taste perversion
General disorders
Asthenia
106 (51)
23 (11)
127 (64)
52 (26)
Edema†
73 (35)
7 (3)
21 (11)
1 (1)
Pain
59 (28)
10 (5)
31 (16)
4 (2)
Pyrexia
50 (24)
1 (1)
99 (50)
7 (4)
Weight Loss
39 (19)
3 (1)
50 (25)
4 (2)
Headache
31 (15)
1 (1)
30 (15)
0 (0)
Chest Pain
34 (16)
2 (1)
18 (9)
2 (1)
Chills
17 (8)
1 (1)
59 (30)
3 (2)
Gastrointestinal disorders
Mucositis‡
86 (41)
6 (3)
19 (10)
0 (0)
Anorexia
66 (32)
6 (3)
87 (44)
8 (4)
Nausea
77 (37)
5 (2)
82 (41)
9 (5)
Diarrhea
56 (27)
3 (1)
40 (20)
4 (2)
Abdominal Pain
44 (21)
9 (4)
34 (17)
3 (2)
Constipation
42 (20)
0 (0)
36 (18)
1 (1)
Vomiting
40 (19)
4 (2)
57 (29)
5 (3)
Infections
Infections§
42 (20)
6 (3)
19 (10)
4 (2)
Urinary tract infection¶
31 (15)
3 (1)
24 (12)
3 (2)
Pharyngitis
25 (12)
0 (0)
3 (2)
0 (0)
Rhinitis
20 (10)
0 (0)
4 (2)
0 (0)
Musculoskeletal and connective tissue disorders
Back Pain
41 (20)
6 (3)
28 (14)
7 (4)
Arthralgia
37 (18)
2 (1)
29 (15)
2 (1)
Myalgia
16 (8)
1 (1)
29 (15)
2 (1)
Respiratory, thoracic and mediastinal disorders
Dyspnea
58 (28)
18 (9)
48 (24)
11 (6)
Cough
53 (26)
2 (1)
29 (15)
0 (0)
Epistaxis
25 (12)
0 (0)
7 (4)
0 (0)
Skin and subcutaneous tissue disorders
Rash#
97 (47)
10 (5)
14 (7)
0 (0)
Pruritus
40 (19)
1 (1)
16 (8)
0 (0)
Nail Disorder
28 (14)
0 (0)
1 (1)
0 (0)
Dry Skin
22 (11)
1 (1)
14 (7)
0 (0)
Acne
21 (10)
0 (0)
2 (1)
0 (0)
Nervous system disorders
DysgeusiaÞ
41 (20)
0 (0)
17 (9)
0 (0)
Insomnia
24 (12)
1 (1)
30 (15)
0 (0)
Depression
9 (4)
0 (0)
27 (14)
4 (2)
The following selected adverse reactions were reported less frequently (<10%).
Gastrointestinal Disorders – Gastrointestinal hemorrhage (1%), rectal hemorrhage (1%).
Eye Disorders – Conjunctivitis (including lacrimation disorder) (8%).
Immune System – Angioneurotic edema-type reactions (including delayed reactions occurring two months following initiation of therapy) have been observed in some patients who received TORISEL and ACE inhibitors concomitantly.
Infections – Pneumonia (8%), upper respiratory tract infection (7%), wound infection/post-operative wound infection (1%), sepsis (1%).
General Disorders and Administration Site Conditions - Diabetes mellitus (5%).
Respiratory, Thoracic and Mediastinal Disorders – Pleural effusion (4%).
Vascular – Hypertension (7%), venous thromboembolism (including deep vein thrombosis and pulmonary embolus [including fatal outcomes]) (2%), thrombophlebitis (1%), pericardial effusion (1%).
Nervous System Disorders – Convulsion (1%).
Table 2 – Incidence of Selected Laboratory Abnormalities in Patients Who Received 25 mg IV TORISEL or IFN-α in the Randomized Trial Laboratory Abnormality TORISEL
25 mg
n = 208IFN-α
n = 200All Grades*
n (%)Grades 3&4*
n (%)All Grades*
n (%)Grades 3&4*
n (%)Any
208 (100)
162 (78)
195 (98)
144 (72)
Hematology
Hemoglobin Decreased
195 (94)
41 (20)
180 (90)
43 (22)
Lymphocytes Decreased†
110 (53)
33 (16)
106 (53)
48 (24)
Neutrophils Decreased†
39 (19)
10 (5)
58 (29)
19 (10)
Platelets Decreased
84 (40)
3 (1)
51 (26)
0 (0)
Leukocytes Decreased
67 (32)
1 (1)
93 (47)
11 (6)
Chemistry
Alkaline Phosphatase Increased
141 (68)
7 (3)
111 (56)
13 (7)
AST Increased
79 (38)
5 (2)
103 (52)
14 (7)
Creatinine Increased
119 (57)
7 (3)
97 (49)
2 (1)
Glucose Increased
186 (89)
33 (16)
128 (64)
6 (3)
Phosphorus Decreased
102 (49)
38 (18)
61 (31)
17 (9)
Total Bilirubin Increased
16 (8)
2 (1)
25 (13)
4 (2)
Total Cholesterol Increased
181 (87)
5 (2)
95 (48)
2 (1)
Triglycerides Increased
173 (83)
92 (44)
144 (72)
69 (35)
Potassium Decreased
43 (21)
11 (5)
15 (8)
0 (0)
6.2 Post-marketing and Other Clinical Experience
The following adverse reactions have been identified during post approval use of TORISEL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to readily estimate their frequency or establish a causal relationship to drug exposure.
The following adverse reactions have been observed in patients receiving temsirolimus: angioedema, rhabdomyolysis, Stevens-Johnson Syndrome, complex regional pain syndrome (reflex sympathetic dystrophy), pancreatitis, cholecystitis, and cholelithiasis.
There are also post-marketing reports of temsirolimus extravasations resulting in swelling, pain, warmth, and erythema.
-
7 DRUG INTERACTIONS
7.1 Agents Inducing CYP3A Metabolism
Co-administration of TORISEL with rifampin, a potent CYP3A4/5 inducer, had no significant effect on temsirolimus Cmax (maximum concentration) and AUC (area under the concentration versus the time curve) after intravenous administration, but decreased sirolimus Cmax by 65% and AUC by 56% compared to TORISEL treatment alone. If alternative treatment cannot be administered, a dose adjustment should be considered [see Dosage and Administration (2.4)].
7.2 Agents Inhibiting CYP3A Metabolism
Co-administration of TORISEL with ketoconazole, a potent CYP3A4 inhibitor, had no significant effect on temsirolimus Cmax or AUC; however, sirolimus AUC increased 3.1-fold, and Cmax increased 2.2-fold compared to TORISEL alone. If alternative treatment cannot be administered, a dose adjustment should be considered [see Dosage and Administration (2.4)].
7.3 Angioedema with ACE inhibitors and Calcium Channel Blockers
Angioedema has been reported in patients taking mammalian target of rapamycin (mTOR) inhibitors in combination with ramipril and/or amlodipine. Monitor patients for signs and symptoms of angioedema when temsirolimus is given concomitantly with angiotensin converting enzyme (ACE) inhibitors (e.g., ramipril) or calcium channel blockers (CCB) (e.g., amlodipine).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal studies and its mechanism of action, temsirolimus can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. Although there are no data on the use of TORISEL in pregnant women, there are limited data on the use of sirolimus, the active metabolite of temsirolimus, during pregnancy; however, these data are insufficient to inform a drug-associated risk of adverse developmental outcomes. In animal reproductive studies, oral daily administration of temsirolimus to pregnant rats and rabbits during organogenesis caused adverse embryo-fetal effects at approximately 0.04 and 0.12 times the AUC in patients at the recommended dose, respectively (see Data). Advise pregnant women of the potential hazard to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Data
Animal Data
Temsirolimus administered daily as an oral formulation throughout organogenesis caused adverse embryo-fetal effects in rats and rabbits at human sub-therapeutic exposures. Embryo-fetal adverse effects in rats consisted of reduced fetal weight and reduced ossifications, and in rabbits included reduced fetal weight, omphalocele, bifurcated sternabrae, notched ribs, and incomplete ossifications.
In rats, the adverse embryo-fetal effects were observed at the oral dose of 2.7 mg/m2/day (approximately 0.04-fold the AUC in patients with cancer at the human recommended dose). In rabbits, the adverse embryo-fetal effects were observed at the oral dose of ≥7.2 mg/m2/day (approximately 0.12-fold the AUC in patients with cancer at the recommended human dose).
8.2 Lactation
Risk Summary
There is no information regarding the presence of TORISEL or its metabolites in human milk, or their effects on the breastfed child or milk production. Trace amounts of sirolimus, the active metabolite of temsirolimus, were present in milk from lactating rats administered sirolimus. Because of the potential for serious adverse reactions in a breastfed child from TORISEL, advise a lactating woman not to breastfeed during treatment with TORISEL and for 3 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
TORISEL can cause fetal harm when administered to a pregnant woman [see Use in Specific Population (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with TORISEL and for 3 months after the last dose.
Males
Advise males with partners of reproductive potential to use effective contraception during treatment with TORISEL and for 3 months after the last dose [see Nonclinical Toxicology (13.1)].
Infertility
Based on the findings in animal fertility studies, male and female fertility may be compromised by the treatment with Torisel. It is not known if the effects on fertility in animal studies were reversible [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Limited data are available on the use of temsirolimus in pediatric patients. The effectiveness of temsirolimus in pediatric patients with advanced recurrent/refractory solid tumors has not been established.
TORISEL was studied in 71 patients (59 patients ages 1 to 17 years and 12 patients ages 18 to 21 years) with relapsed/refractory solid tumors in a phase 1–2 safety and exploratory pharmacodynamic study.
In phase 1, 19 pediatric patients with advanced recurrent/refractory solid tumors received TORISEL at doses ranging from 10 mg/m2 to 150 mg/m2 as a 60-minute intravenous infusion once weekly in three-week cycles.
In phase 2, 52 pediatric patients with recurrent/relapsed neuroblastoma, rhabdomyosarcoma, or high grade glioma received TORISEL at a weekly dose of 75 mg/m2. One of 19 patients with neuroblastoma achieved a partial response. There were no objective responses in pediatric patients with recurrent/relapsed rhabdomyosarcoma or high grade glioma.
Adverse reactions associated with TORISEL were similar to those observed in adults. The most common adverse reactions (≥20%) in pediatric patients receiving the 75 mg/m2 dose included thrombocytopenia, infections, asthenia/fatigue, fever, pain, leukopenia, rash, anemia, hyperlipidemia, increased cough, stomatitis, anorexia, increased plasma levels of alanine aminotransferase and aspartate aminotransferase, hypercholesterolemia, hyperglycemia, abdominal pain, headache, arthralgia, upper respiratory infection, nausea and vomiting, neutropenia, hypokalemia, and hypophosphatemia.
Pharmacokinetics:
In phase 1 of the above mentioned pediatric trial, the single dose and multiple dose total systemic exposure (AUC) of temsirolimus and sirolimus were less than dose-proportional over the dose range of 10 to 150 mg/m2.
In the phase 2 portion, the multiple dose (Day 1, Cycle 2) pharmacokinetics of TORISEL 75 mg/m2 were characterized in an additional 35 patients ages 28 days to 21 years (median age of 8 years). The geometric mean body surface adjusted clearance of temsirolimus and sirolimus was 9.45 L/h/m2 and 9.26 L/h/m2, respectively. The mean elimination half-life of temsirolimus and sirolimus was 31 hours and 44 hours, respectively.
The exposure (AUCss) to temsirolimus and sirolimus was approximately 6-fold and 2-fold higher, respectively than the exposure in adult patients receiving a 25 mg intravenous infusion.
8.5 Geriatric Use
Clinical studies of TORISEL did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects. Based on the results of a phase 3 study, elderly patients may be more likely to experience certain adverse reactions including diarrhea, edema, and pneumonia [see Warnings and Precautions (5.16)].
8.6 Renal Impairment
No clinical studies were conducted with TORISEL in patients with decreased renal function. Less than 5% of total radioactivity was excreted in the urine following a 25 mg intravenous dose of [14C]-labeled temsirolimus in healthy subjects. Renal impairment is not expected to markedly influence drug exposure, and no dosage adjustment of TORISEL is recommended in patients with renal impairment.
TORISEL has not been studied in patients undergoing hemodialysis.
8.7 Hepatic Impairment
TORISEL was evaluated in a dose escalation phase 1 study in 110 patients with normal or varying degrees of hepatic impairment as defined by AST and bilirubin levels and patients with liver transplant (Table 3). Patients with moderate and severe hepatic impairment had increased rates of adverse reactions and deaths, including deaths due to progressive disease, during the study (Table 3).
Table 3 – Adverse Reactions in Patients with Advanced Malignancies Plus Normal or Impaired Hepatic Function Hepatic Function* TORISEL Dose Range Adverse Reactions
Grade ≥ 3†
n (%)Death‡
n (%)- *
- Hepatic Function Groups: normal = bilirubin and AST ≤ULN; mild = bilirubin >1 – 1.5×ULN or AST >ULN but bilirubin ≤ULN; moderate = bilirubin >1.5 – 3×ULN; severe = bilirubin >3×ULN; liver transplant = any bilirubin and AST.
- †
- Common Terminology Criteria for Adverse Events, version 3.0, including all causality.
- ‡
- Includes deaths due to progressive disease and adverse reactions.
Normal (n = 25)
25 – 175
20 (80.0)
2 (8.0)
Mild (n = 39)
10 – 25
32 (82.1)
5 (12.8)
Moderate (n = 20)
10 – 25
19 (95.0)
8 (40.0)
Severe (n = 24)
7.5 – 15
23 (95.8)
13 (54.2)
Liver Transplant (n = 2)
10
1 (50.0)
0 (0)
TORISEL is contraindicated in patients with bilirubin >1.5×ULN [see Contraindications (4), and Warnings and Precautions (5.2)]. Use caution when treating patients with mild hepatic impairment. If TORISEL must be given in patients with mild hepatic impairment (bilirubin >1–1.5×ULN or AST >ULN but bilirubin ≤ULN), reduce the dose of TORISEL to 15 mg/week [see Dosage and Administration (2.4)]. Because there is a need for dosage adjustment based upon hepatic function, assessment of AST and bilirubin levels is recommended before initiation of TORISEL and periodically thereafter.
-
10 OVERDOSAGE
There is no specific treatment for TORISEL intravenous overdose. TORISEL has been administered to patients with cancer in phase 1 and 2 trials with repeated intravenous doses as high as 220 mg/m2. The risk of several serious adverse events, including thrombosis, bowel perforation, interstitial lung disease (ILD), seizure, and psychosis, is increased with doses of TORISEL greater than 25 mg.
-
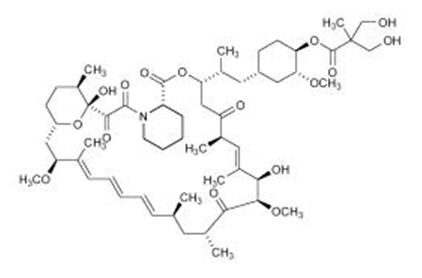
11 DESCRIPTION
Temsirolimus, an inhibitor of mTOR, is an antineoplastic agent.
Temsirolimus is a white to off-white powder with a molecular formula of C56H87NO16 and a molecular weight of 1030.30. It is non-hygroscopic. Temsirolimus is practically insoluble in water and soluble in alcohol. It has no ionizable functional groups, and its solubility is independent of pH.
The chemical name of temsirolimus is (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-Hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-hydroxy-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23, 27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone 4'-[2,2-bis(hydroxymethyl)propionate]; or Rapamycin, 42-[3-hydroxy-2-(hydroxymethyl)-2-methylpropanoate].
TORISEL (temsirolimus) injection, 25 mg/mL, is a clear, colorless to light yellow, non-aqueous, ethanolic, sterile solution. TORISEL (temsirolimus) injection requires two dilutions prior to intravenous infusion. TORISEL (temsirolimus) injection should be diluted only with the supplied DILUENT for TORISEL.
DILUENT for TORISEL is a sterile, non-aqueous solution that is supplied with TORISEL injection, as a kit.
TORISEL (temsirolimus) injection, 25 mg/mL:
Active ingredient: temsirolimus (25 mg/mL)
Inactive ingredients: dehydrated alcohol (39.5% w/v), dl-alpha-tocopherol (0.075% w/v), propylene glycol (50.3% w/v), and anhydrous citric acid (0.0025% w/v).
DILUENT for TORISEL:
Inactive ingredients: polysorbate 80 (40.0% w/v), polyethylene glycol 400 (42.8% w/v) and dehydrated alcohol (19.9% w/v).
After the TORISEL (temsirolimus) injection vial has been diluted with DILUENT for TORISEL, in accordance with the instructions in section 2.5, the solution contains 35.2% alcohol.
TORISEL (temsirolimus) injection and DILUENT for TORISEL are filled in clear glass vials with butyl rubber stoppers.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Temsirolimus is an inhibitor of mTOR (mammalian target of rapamycin). Temsirolimus binds to an intracellular protein (FKBP-12), and the protein-drug complex inhibits the activity of mTOR that controls cell division. Inhibition of mTOR activity resulted in a G1 growth arrest in treated tumor cells. When mTOR was inhibited, its ability to phosphorylate p70S6k and S6 ribosomal protein, which are downstream of mTOR in the PI3 kinase/AKT pathway was blocked. In in vitro studies using renal cell carcinoma cell lines, temsirolimus inhibited the activity of mTOR and resulted in reduced levels of the hypoxia-inducible factors HIF-1 and HIF-2 alpha, and the vascular endothelial growth factor.
12.2 Pharmacodynamics
Effects on Electrocardiogram: There were no clinically relevant QT changes observed at the recommended dose for TORISEL. In a randomized, single-blinded, crossover study, 58 healthy subjects received TORISEL 25 mg, placebo, and a single oral dose of moxifloxacin 400 mg. A supratherapeutic TORISEL dose was not studied in this randomized QT trial. The largest difference between the upper bound 2-sided 90% CI for the mean difference between TORISEL and placebo-corrected QT interval was less than 10 ms. In a different trial in 69 patients with a hematologic malignancy, TORISEL doses up to 175 mg were studied. No patient with a normal QTcF at baseline had an increase in QTcF >60 ms. Additionally, there were no patients with a QTcF interval greater than 500 ms.
12.3 Pharmacokinetics
Absorption
Following administration of a single 25 mg dose of TORISEL in patients with cancer, mean temsirolimus Cmax in whole blood was 585 ng/mL (coefficient of variation, CV = 14%), and mean AUC in blood was 1627 ng∙h/mL (CV = 26%). Typically Cmax occurred at the end of infusion. Over the dose range of 1 mg to 25 mg, temsirolimus exposure increased in a less than dose proportional manner while sirolimus exposure increased proportionally with dose. Following a single 25 mg intravenous dose in patients with cancer, sirolimus AUC was 2.7-fold that of temsirolimus AUC, due principally to the longer half-life of sirolimus.
Distribution
Following a single 25 mg intravenous dose, mean steady-state volume of distribution of temsirolimus in whole blood of patients with cancer was 172 liters. Both temsirolimus and sirolimus are extensively partitioned into formed blood elements.
Metabolism
Cytochrome P450 3A4 is the major isozyme responsible for the formation of five temsirolimus metabolites. Sirolimus, an active metabolite of temsirolimus, is the principal metabolite in humans following intravenous treatment. The remainder of the metabolites account for less than 10% of radioactivity in the plasma. In human liver microsomes temsirolimus was an inhibitor of CYP2D6 and 3A4. However, there was no effect observed in vivo when temsirolimus was administered with desipramine (a CYP2D6 substrate), and no effect is anticipated with substrates of CYP3A4 metabolism.
Elimination
Elimination is primarily via the feces. After a single IV dose of [14C]-temsirolimus approximately 82% of total radioactivity was eliminated within 14 days, with 4.6% and 78% of the administered radioactivity recovered in the urine and feces, respectively. Following a single 25 mg dose of TORISEL in patients with cancer, temsirolimus mean (CV) systemic clearance was 16.2 (22%) L/h. Temsirolimus exhibits a bi-exponential decline in whole blood concentrations and the mean half-lives of temsirolimus and sirolimus were 17.3 hours and 54.6 hours, respectively.
Drug-Transport Systems - P-glycoprotein
Temsirolimus is a substrate of the efflux transporter P-glycoprotein (Pgp) in vitro. If TORISEL is administered with drugs that inhibit Pgp, increased concentrations of temsirolimus are likely and caution should be exercised.
In vitro, temsirolimus inhibited human Pgp (IC50 value of 2 µM). If TORISEL is administered with drugs that are substrates of Pgp, increased concentrations of the substrate drug are likely and caution should be exercised.
Effects of Age and Gender
In population pharmacokinetic-based data analyses, no relationship was apparent between drug exposure and patient age or gender.
Drug Interactions
Effect of Temsirolimus on CYP2D6 or CYP3A
The concentration of desipramine, a CYP2D6 substrate, was unaffected when 25 mg of temsirolimus was co-administered. No clinically significant effect is anticipated when 25 mg of temsirolimus is co-administered with agents that are metabolized by CYP2D6 or CYP3A.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with temsirolimus. However, sirolimus, the major metabolite of temsirolimus in humans, was carcinogenic in mice and rats. The following effects were reported in mice and/or rats in the carcinogenicity studies conducted with sirolimus: lymphoma, hepatocellular adenoma and carcinoma, and testicular adenoma.
Temsirolimus was not genotoxic in a battery of in vitro (bacterial reverse mutation in Salmonella typhimurium and Escherichia coli, forward mutation in mouse lymphoma cells, and chromosome aberrations in Chinese hamster ovary cells) and in vivo (mouse micronucleus) assays.
In a fertility study in male rats, decreased number of pregnancies, decreased sperm concentration and motility, decreased reproductive organ weights, and testicular tubular degeneration were observed. These effects were observed at oral temsirolimus doses ≥3 mg/m2/day (approximately 0.2-fold the human recommended intravenous dose). Fertility was absent at 30 mg/m2/day.
In a fertility study in female rats, an increased incidence of pre- and post-implantation losses occurred at oral doses ≥4.2 mg/m2/day (approximately 0.3-fold the human recommended intravenous dose), resulting in decreased numbers of live fetuses.
-
14 CLINICAL STUDIES
A phase 3, multi-center, three-arm, randomized, open-label study was conducted in previously untreated patients with advanced renal cell carcinoma (clear cell and non-clear cell histologies). The objectives were to compare Overall Survival (OS), Progression-Free Survival (PFS), Objective Response Rate (ORR), and safety in patients receiving IFN-α to those receiving TORISEL or TORISEL plus IFN-α. Patients in this study had 3 or more of 6 pre-selected prognostic risk factors (less than one year from time of initial renal cell carcinoma diagnosis to randomization, Karnofsky performance status of 60 or 70, hemoglobin less than the lower limit of normal, corrected calcium of greater than 10 mg/dL, lactate dehydrogenase >1.5 times the upper limit of normal, more than one metastatic organ site). Patients were stratified for prior nephrectomy status within three geographic regions and were randomly assigned (1:1:1) to receive IFN-α alone (n = 207), TORISEL alone (25 mg weekly; n = 209), or the combination arm (n = 210).
The ITT population for this interim analysis included 626 patients. Demographics were comparable between the three treatment arms with regard to age, gender, and race. The mean age of all groups was 59 years (range 23–86). Sixty-nine percent were male and 31% were female. The racial distribution for all groups was 91% White, 4% Black, 2% Asian, and 3% other. Sixty-seven percent of patients had a history of prior nephrectomy.
The median duration of treatment in the TORISEL arm was 17 weeks (range 1–126 weeks). The median duration of treatment on the IFN arm was 8 weeks (range 1–124 weeks).
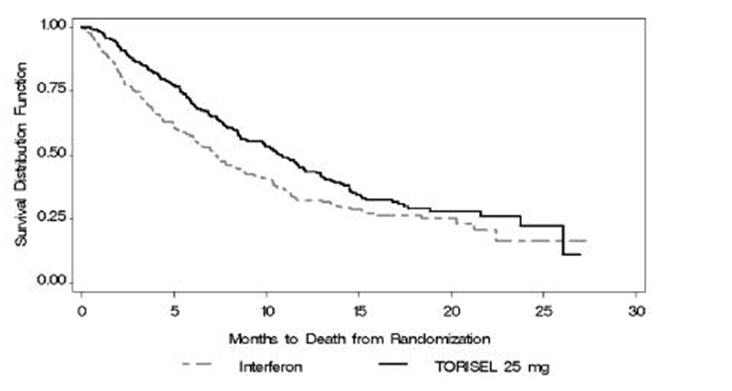
There was a statistically significant improvement in OS (time from randomization to death) in the TORISEL 25 mg arm compared to IFN-α. The combination of TORISEL 15 mg and IFN-α did not result in a significant increase in OS when compared with IFN-α alone. Figure 1 is a Kaplan-Meier plot of OS in this study. The evaluations of PFS (time from randomization to disease progression or death) and ORR, were based on blinded independent radiologic assessment of tumor response. Efficacy results are summarized in Table 4.
Table 4 - Summary of Efficacy Results of TORISEL vs. IFN-α Parameter TORISEL
n = 209IFN-α
n = 207P-value* Hazard Ratio
(95% CI)†CI = confidence interval; NA = not applicable - *
- Based on log-rank test stratified by prior nephrectomy and region.
- †
- Based on Cox proportional hazard model stratified by prior nephrectomy and region.
- ‡
- A comparison is considered statistically significant if the p-value is <0.0159 (O'Brien-Fleming boundary at 446 deaths).
- §
- Not adjusted for multiple comparisons.
- ¶
- Based on Cochran-Mantel-Haenszel test stratified by prior nephrectomy and region.
Median Overall Survival
Months (95% CI)10.9 (8.6, 12.7)
7.3 (6.1, 8.8)
0.0078‡
0.73 (0.58, 0.92)
Median Progression-Free Survival
Months (95% CI)5.5 (3.9, 7.0)
3.1 (2.2, 3.8)
0.0001§
0.66 (0.53, 0.81)
Overall Response Rate
% (95% CI)8.6 (4.8, 12.4)
4.8 (1.9, 7.8)
NA
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
NDC 0008-1179-01 TORISEL (temsirolimus) injection, 25 mg/mL.
Each kit is supplied in a single carton containing one single-use vial of 25 mg/mL of temsirolimus and one DILUENT vial which includes a deliverable volume of 1.8 mL, and must be stored at 2º–8º C (36º–46º F). Protect from light.
TORISEL is a cytotoxic drug. Follow applicable special handling and disposal procedures1.
-
17 PATIENT COUNSELING INFORMATION
Allergic (Hypersensitivity/Infusion) Reactions
Patients should be informed of the possibility of serious allergic reactions, including anaphylaxis (including life threatening and fatal reactions), despite premedication with antihistamines, and to immediately report any facial swelling or difficulty breathing [see Warnings and Precautions (5.1)].
Increased Blood Glucose Levels
Patients are likely to experience increased blood glucose levels while taking TORISEL. This may result in the need for initiation of, or increase in the dose of, insulin and/or hypoglycemic agents. Patients should be directed to report any excessive thirst or frequency of urination to their physician [see Warnings and Precautions (5.3)].
Infections
Patients should be informed that they may be more susceptible to infections while being treated with TORISEL [see Warnings and Precautions (5.4)].
Interstitial Lung Disease
Patients should be warned of the possibility of developing interstitial lung disease, a chronic inflammation of the lungs, which may rarely result in death [see Warnings and Precautions (5.5)]. Patients, including those who are taking or have taken corticosteroids or immunosuppressive agents, should be directed to report promptly any new or worsening respiratory symptoms to their physician.
Increased Blood Triglycerides and/or Cholesterol
Patients are likely to experience elevated triglycerides and/or cholesterol during TORISEL treatment. This may require initiation of, or increase in the dose of, lipid-lowering agents [see Warnings and Precautions (5.6)].
Bowel Perforation
Patients should be warned of the possibility of bowel perforation. Patients should be directed to report promptly any new or worsening abdominal pain or blood in their stools [see Warnings and Precautions (5.7)].
Renal Failure
Patients should be informed of the risk of renal failure [see Warnings and Precautions (5.8)].
Wound Healing Complications
Patients should be advised of the possibility of abnormal wound healing if they have surgery within a few weeks of initiating therapy or during therapy [see Warnings and Precautions (5.9)].
Intracerebral Bleeding
Patients with CNS tumors and/or receiving anticoagulants should be informed of the increased risk of developing intracerebral bleeding (including fatal outcomes) while on TORISEL [see Warnings and Precautions (5.10)].
Proteinuria and Nephrotic Syndrome
Advise patients that treatment with TORISEL may require monitoring for proteinuria and nephrotic syndrome and to contact their health care provider for signs and symptoms of nephrotic syndrome [see Warnings and Precautions (5.11)].
Medications that can interfere with TORISEL
Some medicines can interfere with the breakdown or metabolism of TORISEL. In particular, patients should be directed to inform their physician if they are taking any of the following: Protease inhibitors, anti-epileptic medicines including carbamazepine, phenytoin, and barbiturates, St. John's Wort, rifampicin, rifabutin, nefazodone or selective serotonin re-uptake inhibitors used to treat depression, antibiotics or antifungal medicines used to treat infections [see Warnings and Precautions (5.12)].
Vaccinations
Patients should be advised that vaccinations may be less effective while being treated with TORISEL. In addition, the use of live vaccines, and close contact with those who have received live vaccines, while on TORISEL should be avoided [see Warnings and Precautions (5.14)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential that TORISEL can cause fetal harm. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.15) and Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with TORISEL and for 3 months after receiving the last dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with TORISEL and for 3 months after receiving the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise lactating women not to breastfeed during treatment with TORISEL and for 3 weeks after the last dose [see Use in Specific Populations (8.2)].
Infertility
Advise patients that male and female fertility may be compromised by treatment with TORISEL [see Use in Specific Populations (8.3) and Nonclinical Toxicology (13.1)].
Elderly Patients
Elderly patients should be advised that they may be more likely to experience certain adverse reactions including diarrhea, edema, and pneumonia [see Warnings and Precautions (5.16)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 25 mg/mL Vial Label
- PRINCIPAL DISPLAY PANEL - 2.2 mL Vial Label
-
PRINCIPAL DISPLAY PANEL - Kit Carton
NDC 0008-1179-01
Pfizer
TORISEL®Kit
(temsirolimus) injection25 mg/mL*
*each vial contains 0.2 mL overfill
CONCENTRATED - Requires two dilutions
before administration10 mg/mL after initial dilution
For intravenous infusion onlyEach carton contains:
1 vial TORISEL® (temsirolimus) injection 25 mg/mL*
1 vial DILUENT for TORISEL®Cytotoxic: Handle with caution
Refrigerate
Rx only -
INGREDIENTS AND APPEARANCE
TORISEL
temsirolimus kitProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0008-1179 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0008-1179-01 1 in 1 CARTON; Type 0: Not a Combination Product 07/01/2007 Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-USE 25 mL Part 2 1 VIAL, SINGLE-USE 1.8 mL Part 1 of 2 TORISEL
temsirolimus injection, solution, concentrateProduct Information Item Code (Source) NDC:0008-1279 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TEMSIROLIMUS (UNII: 624KN6GM2T) (TEMSIROLIMUS - UNII:624KN6GM2T) TEMSIROLIMUS 25 mg in 1 mL Inactive Ingredients Ingredient Name Strength ANHYDROUS CITRIC ACID (UNII: XF417D3PSL) 0.025 mg in 1 mL ALCOHOL (UNII: 3K9958V90M) 394.6 mg in 1 mL PROPYLENE GLYCOL (UNII: 6DC9Q167V3) 503.325 mg in 1 mL .ALPHA.-TOCOPHEROL, DL- (UNII: 7QWA1RIO01) 0.75 mg in 1 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0008-1279-01 25 mL in 1 VIAL, SINGLE-USE; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022088 07/01/2007 Part 2 of 2 TORISEL DILUENT
diluent injection, solution, concentrateProduct Information Item Code (Source) NDC:0008-1125 Route of Administration INTRAVENOUS Inactive Ingredients Ingredient Name Strength POLYSORBATE 80 (UNII: 6OZP39ZG8H) 880 mg in 1.8 mL POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) 941 mg in 1.8 mL ALCOHOL (UNII: 3K9958V90M) 438 mg in 1.8 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0008-1125-01 1.8 mL in 1 VIAL, SINGLE-USE; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022088 07/01/2007 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022088 07/01/2007 Labeler - Wyeth Pharmaceuticals LLC, a subsidiary of Pfizer Inc. (113008515) Establishment Name Address ID/FEI Business Operations Pfizer Manufacturing Belgium NV 370156507 ANALYSIS(0008-1179) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 986019327 ANALYSIS(0008-1179) Establishment Name Address ID/FEI Business Operations Wyeth Lederle SRL 542812040 ANALYSIS(0008-1179) , PACK(0008-1179) , LABEL(0008-1179) Establishment Name Address ID/FEI Business Operations Pfizer Ireland Pharmaceuticals 985052076 ANALYSIS(0008-1179) , API MANUFACTURE(0008-1179)