Label: VALSARTAN solution


- NDC Code(s): 70954-310-10, 70954-310-20
- Packager: ANI Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated September 18, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VALSARTAN ORAL SOLUTION safely and effectively. See full prescribing information for VALSARTAN ORAL SOLUTION.
VALSARTAN oral solution
Initial U.S. Approval: 1996
WARNING: FETAL TOXICITY
See full prescribing information for complete boxed warning.
- When pregnancy is detected, discontinue Valsartan Oral Solution as soon as possible. (5.1)
- Drugs that act directly on the renin-angiotensin system can cause injury and death to the developing fetus. (5.1)
INDICATIONS AND USAGE
Valsartan is an angiotensin II receptor blocker (ARB) indicated for: (1)
- Hypertension in adults and children six years and older, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions (1.1)
- Heart failure (NYHA class II-IV); Valsartan oral solution significantly reduces hospitalization for heart failure in patients who are unable to swallow valsartan tablets (1.2)
- Stable left ventricular failure or left ventricular dysfunction following myocardial infarction; Valsartan oral solution reduces cardiovascular mortality in patients who are unable to swallow valsartan tablets (1.3)
DOSAGE AND ADMINISTRATION
Indication
Starting Dose
DoseRange
TargetMaintenance Dose*
Hypertension- adults (2.1)
40 or 80 mg twice daily
40 -160 mg twice daily
---
Hypertension - age 6 to16
years (2.2)
0.65 mg/kg twice daily (up to 40 mg total)
0.65-1.35 mg/kg twice daily (up to 40 mg-160 mg total)
---
Heart Failure (2.3)
40 mg twice daily
40 mg-160 mg twice daily
160 mg twice daily
Post-Myocardial Infarction (2.4)
20 mg twice daily
20 mg to 160 mg twice daily
160 mg twice daily
*as tolerated by patient (2)
DOSAGE FORMS AND STRENGTHS
Oral Solution, 4 mg/mL (3) (3)
WARNINGS AND PRECAUTIONS
ADVERSE REACTIONS
Hypertension: Most common adverse reactions are headache, dizziness, fatigue and abdominal pain (6.1)
Heart Failure: Most common adverse reactions are dizziness, hypotension, diarrhea, arthralgia, back pain, fatigue and hyperkalemia (6.1)
Post-Myocardial Infarction: Most common adverse reactions which caused patients to discontinue therapy are hypotension, cough and increased blood creatinine (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact ANI Pharmaceuticals, Inc. at 1-855-204-1431 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Potassium-sparing diuretics, potassium supplements or salt substitutes may lead to increases in serum potassium, and in heart failure patients, increases in serum creatinine (7)
- NSAIDs increase risk of renal impairment and loss of antihypertensive effect (7)
- Dual inhibition of the renin-angiotensin system: Increased risk of renal impairment, hypotension, and hyperkalemia (7)
- Lithium: Increases in serum lithium concentrations and lithium toxicity (7)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 9/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
BOXED WARNING
1 INDICATIONS & USAGE
1.1 Hypertension
1.2 Heart Failure
1.3 Post-Myocardial Infarction
2 DOSAGE & ADMINISTRATION
2.1 General Considerations
2.2 Adult Hypertension
2.3 Pediatric Hypertension 6 to 16 Years of Age
2.4 Heart Failure
2.5 Post-Myocardial Infarction
3 DOSAGE FORMS & STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
5.2 Hypotension
5.3 Impaired Renal Function
5.4 Hyperkalemia
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Agents Increasing Serum Potassium
7.2 Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
7.3 Dual Blockade of the Renin-Angiotensin System (RAS)
7.4 Lithium
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
13.2 Animal Pharmacology & OR Toxicology
14 CLINICAL STUDIES
14.1 Hypertension
14.2 Heart Failure
14.3 Post-Myocardial Infarction
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- BOXED WARNING (What is this?)
-
1 INDICATIONS & USAGE
1.1 Hypertension
Valsartan is indicated for the treatment of hypertension in adults and children six years and older, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes including the class to which valsartan principally belongs. There are no controlled trials in hypertensive patients demonstrating risk reduction with valsartan.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than one drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program’s Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality also have been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (e.g., patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
Valsartan may be used alone or in combination with other antihypertensive agents.
1.2 Heart Failure
Valsartan Oral Solution is indicated for the treatment of heart failure (NYHA class II-IV) to reduce the risk of hospitalization for heart failure in patients who are unable to swallow valsartan tablets. There is no evidence that valsartan provides added benefits when it is used with an adequate dose of an ACE inhibitor [see Warnings and Precautions (5.2), Clinical Pharmacology (12.3) and Clinical Studies (14.2)].
1.3 Post-Myocardial Infarction
Valsartan Oral Solution is indicated to reduce the risk of cardiovascular death in clinically stable patients with left ventricular failure or left ventricular dysfunction following myocardial infarction who are unable to swallow valsartan tablets [see Warnings and Precautions (5.2), Clinical Pharmacology (12.3) and Clinical Studies (14.3)].
-
2 DOSAGE & ADMINISTRATION
2.1 General Considerations
Valsartan Oral Solution is not therapeutically equivalent to the tablet formulation of Diovan. The peak concentration of valsartan with Valsartan Oral Solution is higher than with Diovan [see Warnings and Precautions (5.2), Clinical Pharmacology (12.3)]. Follow dosing instructions given here.
2.2 Adult Hypertension
The recommended starting dose of Valsartan Oral Solution is 40 mg or 80 mg twice daily when used as monotherapy in patients who are not volume-depleted. Patients requiring greater reductions in blood pressure may be started at 80 mg administered twice a day. Valsartan Oral Solution may be used over a total daily dose range of 80 mg to 320 mg.
The antihypertensive effect is substantially present within 2 weeks and maximal reduction is generally attained after 4 weeks. If additional antihypertensive effect is required over the starting dose range, the total daily dose may be increased to a maximum of 320 mg or a diuretic may be added. Addition of a diuretic has a greater effect than dose increases beyond 80 mg.
No initial dosage adjustment is required for elderly patients, for patients with mild or moderate renal impairment, or for patients with mild or moderate liver insufficiency. Monitor closely patients with severe hepatic or renal impairment.
Valsartan Oral Solution may be administered with other antihypertensive agents.
2.3 Pediatric Hypertension 6 to 16 Years of Age
The recommended starting dose is 0.65 mg/kg twice daily (up to 40 mg total daily dose). The dosage should be adjusted according to blood pressure response. Doses higher than 1.35 mg/kg twice daily (or >160 mg total daily dose) have not been studied in pediatric patients 6 to 16 years old.
No data are available in pediatric patients either undergoing dialysis or with a glomerular filtration rate <30 mL/min/1.73 m2[see Use in Specific Populations (8.4)].
Valsartan Oral Solution is not recommended for patients under 6 years of age [see Adverse Reactions (6.1), Use in Specific Populations (8.4), Clinical Studies (14.1)].
2.4 Heart Failure
The recommended starting dose of Valsartan Oral Solution is 40 mg twice daily. Titrate to 80 mg and 160 mg twice daily, as tolerated by the patient. Consider reducing the dose of concomitant diuretics. The maximum daily dose administered in clinical trials is 320 mg in divided doses.
2.5 Post-Myocardial Infarction
Valsartan Oral Solution may be initiated as early as 12 hours after a myocardial infarction. The recommended starting dose of Valsartan Oral Solution is 20 mg twice daily. Patients may be up titrated within 7 days to 40 mg twice daily, with subsequent titrations to a target maintenance dose of 160 mg twice daily, as tolerated by the patient. If symptomatic hypotension or renal dysfunction occurs, consider dosage reduction. Valsartan Oral Solution may be given with other standard post-myocardial infarction treatment, including thrombolytics, aspirin, beta-blockers, and statins.
- 3 DOSAGE FORMS & STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue Valsartan Oral Solution as soon as possible [see Use in Specific Populations (8.1)].
5.2 Hypotension
In patients with an activated renin-angiotensin system, such as volume- and/or salt-depleted patients receiving high doses of diuretics, symptomatic hypotension may occur. This condition should be corrected prior to administration of valsartan, or the treatment should start under close medical supervision.
Peak plasma concentrations of valsartan are higher following administration of Valsartan Oral Solution and may result in increased risk of hypotension as compared to administration of valsartan tablets [see Clinical Pharmacology (12.3)]. Patients with heart failure or post-myocardial infarction patients given valsartan tablets in clinical trials commonly had some reduction in blood pressure. Only use Valsartan Oral Solution in heart failure or post-myocardial infarction patients who are unable to swallow valsartan tablets. In clinical trials of valsartan tablets, discontinuation of therapy because of continuing symptomatic hypotension usually was not necessary. In controlled trials in heart failure patients, the incidence of hypotension in valsartan-treated patients was 5.5% compared to 1.8% in placebo-treated patients. In the Valsartan in Acute Myocardial Infarction Trial (VALIANT), hypotension in post- myocardial infarction patients led to permanent discontinuation of therapy in 1.4% of valsartan-treated patients and 0.8% of captopril-treated patients.
If symptomatic hypotension occurs, place the patient in the supine position and, if necessary, give an intravenous infusion of normal saline. A transient hypotensive response is not a contraindication to further treatment, which usually can be continued without difficulty once the blood pressure has stabilized.
5.3 Impaired Renal Function
Changes in renal function including acute renal failure can be caused by drugs that inhibit the renin-angiotensin system and by diuretics. Patients whose renal function may depend in part on the activity of the renin-angiotensin system (e.g., patients with renal artery stenosis, chronic kidney disease, severe congestive heart failure, or volume depletion) may be at particular risk of developing acute renal failure on valsartan. Monitor renal function periodically in these patients. Consider withholding or discontinuing therapy in patients who develop a clinically significant decrease in renal function on valsartan [see Drug Interactions (7)].
5.4 Hyperkalemia
Some patients with heart failure have developed increases in potassium. These effects are usually minor and transient, and they are more likely to occur in patients with pre-existing renal impairment. Dosage reduction and/or discontinuation of Valsartan Oral Solution may be required [see Adverse Reactions (6.1)].
-
6 ADVERSE REACTIONS
6.1 Clinical Studies Experience
Because clinical studies are conducted under widely varying conditions, adverse reaction rates observed in the clinical studies of a drug cannot be directly compared to rates in the clinical studies of another drug and may not reflect the rates observed in practice.
Adult Hypertension
Valsartan has been evaluated for safety in more than 4,000 patients, including over 400 treated for over 6 months, and more than 160 for over 1 year. Adverse reactions have generally been mild and transient in nature and have only infrequently required discontinuation of therapy. The overall incidence of adverse reactions with valsartan was similar to placebo.
The overall frequency of adverse reactions was neither dose-related nor related to gender, age, race, or regimen. Discontinuation of therapy due to side effects was required in 2.3% of valsartan patients and 2.0% of placebo patients. The most common reasons for discontinuation of therapy with valsartan were headache and dizziness.
The adverse reactions that occurred in placebo-controlled clinical trials in at least 1% of patients treated with valsartan and at a higher incidence in valsartan (n=2,316) than placebo (n=888) patients included fatigue (2% vs. 1%) and abdominal pain (2% vs. 1%).
Headache, dizziness, upper respiratory infection, cough, diarrhea, rhinitis, sinusitis, nausea, pharyngitis, edema, and arthralgia occurred at a more than 1% rate, but at about the same incidence in placebo and valsartan patients.
In trials in which valsartan was compared to an ACE inhibitor with or without placebo, the incidence of dry cough was significantly greater in the ACE-inhibitor group (7.9%) than in the groups who received valsartan (2.6%) or placebo (1.5%). In a 129-patient trial limited to patients who had dry cough when they had previously received ACE inhibitors, the incidences of cough in patients who received valsartan, HCTZ, or lisinopril were 20%, 19%, and 69% respectively (p <0.001).
Dose-related orthostatic effects were seen in less than 1% of patients. An increase in the incidence of dizziness was observed in patients treated with valsartan 320 mg (8%) compared to 10 to 160 mg (2% to 4%).
Valsartan has been used concomitantly with hydrochlorothiazide without evidence of clinically important adverse interactions.
Other adverse reactions that occurred in controlled clinical trials of patients treated with valsartan (>0.2% of valsartan patients) are listed below. It cannot be determined whether these events were causally related to valsartan.
Body as a Whole: Allergic reaction and asthenia
Cardiovascular: Palpitations
Dermatologic: Pruritus and rash
Digestive: Constipation, dry mouth, dyspepsia, and flatulence
Musculoskeletal: Back pain, muscle cramps, and myalgia
Neurologic and Psychiatric: Anxiety, insomnia, paresthesia, and somnolence
Respiratory: Dyspnea
Special Senses: Vertigo
Urogenital: Impotence
Other reported events seen less frequently in clinical trials included chest pain, syncope, anorexia, vomiting, and angioedema.
Pediatric Hypertension
Valsartan has been evaluated for safety in over 400 pediatric patients aged 6 to 17 years and more than 160 pediatric patients aged 6 months to 5 years. No relevant differences were identified between the adverse experience profile for pediatric patients aged 6 to 16 years and that previously reported for adult patients. Headache and hyperkalemia were the most common adverse events suspected to be study drug-related in older children (6 to 17 years old) and younger children (6 months to 5 years old), respectively. Hyperkalemia was mainly observed in children with underlying renal disease.
Neurocognitive and developmental assessment of pediatric patients aged 6 to 16 years revealed no overall clinically relevant adverse impact after treatment with valsartan for up to 1 year.
Valsartan is not recommended for pediatric patients under 6 years of age. In a study (n=90) of pediatric patients (1 to 5 years), two deaths and three cases of on-treatment transaminase elevations were seen in the one-year open-label extension phase. These 5 events occurred in a study population in which patients frequently had significant co-morbidities. A causal relationship to valsartan has not been established. In a second study of 6-months duration in 75 children aged 1 to 5 years, there were no deaths; one case of marked liver transaminase elevations occurred following 6 months of treatment.
Heart Failure
The adverse experience profile of valsartan in heart failure patients was consistent with the pharmacology of the drug and the health status of the patients. In the Valsartan Heart Failure Trial, comparing valsartan in total daily doses up to 320 mg (n=2,506) to placebo (n=2,494), 10% of valsartan patients discontinued for adverse reactions vs. 7% of placebo patients.
The table shows adverse reactions in double-blind short-term heart failure trials, including the first 4 months of the Valsartan Heart Failure Trial, with an incidence of at least 2% that were more frequent in valsartan-treated patients than in placebo-treated patients. All patients received standard drug therapy for heart failure, frequently as multiple medications, which could include diuretics, digitalis, beta-blockers. About 93% of patients received concomitant ACE inhibitors.
Valsartan (n=3,282)
Placebo (n=2,740)
Dizziness
17%
9%
Hypotension
7%
2%
Diarrhea
5%
4%
Arthralgia
3%
2%
Fatigue
3%
2%
Back Pain
3%
2%
Dizziness, postural
2%
1%
Hyperkalemia
2%
1%
Hypotension, postural
2%
1%
Discontinuations occurred in 0.5% of valsartan-treated patients and 0.1% of placebo patients for each of the following: elevations in creatinine and elevations in potassium.
Other adverse reactions with an incidence greater than 1% and greater than placebo included headache, nausea, renal impairment, syncope, blurred vision, upper abdominal pain and vertigo.
From the long-term data in the Valsartan Heart Failure Trial, there did not appear to be any significant adverse reactions not previously identified.
Post-Myocardial Infarction
The safety profile of valsartan was consistent with the pharmacology of the drug and the background diseases, cardiovascular risk factors, and clinical course of patients treated in the post-myocardial infarction setting. The table shows the percentage of patients discontinued in the valsartan and captopril-treated groups in the Valsartan in Acute Myocardial Infarction Trial (VALIANT) with a rate of at least 0.5% in either of the treatment groups.
Discontinuations due to renal dysfunction occurred in 1.1% of valsartan-treated patients and 0.8% of captopril-treated patients.
Valsartan (n=4,885)
Captopril (n=4,879)
Discontinuation for adverse reaction
5.8%
7.7%
Adverse reactions
Hypotension NOS
1.4%
0.8%
Cough
0.6%
2.5%
Blood creatinine increased
0.6%
0.4%
Rash NOS
0.2%
0.6%
In controlled clinical trials, clinically important changes in standard laboratory parameters were rarely associated with administration of valsartan.
Creatinine: Minor elevations in creatinine occurred in 0.8% of patients taking valsartan and 0.6% given placebo in controlled clinical trials of hypertensive patients. In heart failure trials, greater than 50% increases in creatinine were observed in 3.9% of valsartan-treated patients compared to 0.9% of placebo-treated patients. In post-myocardial infarction patients, doubling of serum creatinine was observed in 4.2% of valsartan-treated patients and 3.4% of captopril-treated patients.
Hemoglobin and Hematocrit: Greater than 20% decreases in hemoglobin and hematocrit were observed in 0.4% and 0.8%, respectively, of valsartan patients, compared with 0.1% and 0.1% in placebo-treated patients.
Liver Function Tests: Occasional elevations (greater than 150%) of liver chemistries occurred in valsartan-treated patients. Three patients (<0.1%) treated with valsartan discontinued treatment for elevated liver chemistries.
Neutropenia: Neutropenia was observed in 1.9% of patients treated with valsartan and 0.8% of patients treated with placebo.
Serum Potassium: In hypertensive patients, greater than 20% increases in serum potassium were observed in 4.4% of valsartan-treated patients compared to 2.9% of placebo-treated patients. In heart failure patients, greater than 20% increases in serum potassium were observed in 10.0% of valsartan-treated patients compared to 5.1% of placebo-treated patients.
Blood Urea Nitrogen (BUN): In heart failure trials, greater than 50% increases in BUN were observed in 16.6% of valsartan-treated patients compared to 6.3% of placebo-treated patients.
6.2 Postmarketing Experience
The following additional adverse reactions have been reported in postmarketing experience:
Hypersensitivity: There are rare reports of angioedema. Some of these patients previously experienced angioedema with other drugs including ACE inhibitors. Valsartan should not be re-administered to patients who have had angioedema.
Digestive: Elevated liver enzymes and very rare reports of hepatitis
Renal: Impaired renal function, renal failure
Clinical Laboratory Tests: Hyperkalemia
Dermatologic: Alopecia, bullous dermatitis
Blood and Lymphatic: There are very rare reports of thrombocytopenia
Vascular: Vasculitis
Rare cases of rhabdomyolysis have been reported in patients receiving angiotensin II receptor blockers.
Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to estimate their frequency reliably or establish a causal relationship to drug exposure.
-
7 DRUG INTERACTIONS
7.1 Agents Increasing Serum Potassium
Concomitant use of valsartan with other agents that block the renin-angiotensin system, potassium-sparing diuretics (e.g., spironolactone, triamterene, amiloride), potassium supplements, salt substitutes containing potassium or other drugs that may increase potassium levels (e.g., heparin) may lead to increases in serum potassium and in heart failure patients to increases in serum creatinine. If co-medication is considered necessary, monitor serum potassium.
7.2 Non-Steroidal Anti-Inflammatory Agents including Selective Cyclooxygenase-2 Inhibitors (COX-2 Inhibitors)
In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, coadministration of NSAIDs, including selective COX-2 inhibitors, with angiotensin II receptor antagonists, including valsartan, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving valsartan and NSAID therapy.
The antihypertensive effect of angiotensin II receptor antagonists, including valsartan, may be attenuated by NSAIDs including selective COX-2 inhibitors.
7.3 Dual Blockade of the Renin-Angiotensin System (RAS)
Dual blockade of the RAS with angiotensin receptor blockers, ACE inhibitors, or aliskiren is associated with increased risks of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Most patients receiving the combination of two RAS inhibitors do not obtain any additional benefit compared to monotherapy. In general, avoid combined use of RAS inhibitors. Closely monitor blood pressure, renal function and electrolytes in patients on Valsartan Oral Solution and other agents that affect the RAS.
Do not coadminister aliskiren with Valsartan Oral Solution in patients with diabetes. Avoid use of aliskiren with Valsartan Oral Solution in patients with renal impairment (GFR < 60 mL/min).
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Valsartan Oral Solution can cause fetal harm when administered to a pregnant woman. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. Published reports include cases of anhydramnios and oligohydramnios in pregnant women treated with valsartan (see Clinical Considerations). Studies in rats and rabbits with valsartan showed fetotoxicity only at maternally toxic doses (see Data). When pregnancy is detected, discontinue Valsartan Oral Solution as soon as possible.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major malformations and miscarriage in clinically recognized pregnancies is 2-4%, and 15-20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage). Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death. Pregnant women with hypertension should be carefully monitored and managed accordingly.
Fetal/Neonatal adverse reactions
Oligohydramnios in pregnant women who use drugs affecting the renin-angiotensin system in the second and third trimesters of pregnancy can result in the following: reduced fetal renal function leading to anuria and renal failure, fetal lung hypoplasia and skeletal deformations, including skull hypoplasia, hypotension, and death. In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus.
In patients taking Valsartan Oral Solution during pregnancy, perform serial ultrasound examinations to assess the intra-amniotic environment. Fetal testing may be appropriate, based on the week of gestation. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to Valsartan Oral Solution for hypotension, oliguria, and hyperkalemia. If oliguria or hypotension occur in neonates with a history of in utero exposure to Valsartan Oral Solution, support blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and substituting for disordered renal function.
Data
Animal Data
No teratogenic effects were observed when valsartan was administered to pregnant mice and rats at oral doses up to 600 mg/kg/day and to pregnant rabbits at oral doses up to 10 mg/kg/day. However, significant decreases in fetal weight, pup birth weight, pup survival rate, and slight delays in developmental milestones were observed in studies in which parental rats were treated with valsartan at oral, maternally toxic (reduction in body weight gain and food consumption) doses of 600 mg/kg/day during organogenesis or late gestation and lactation. In rabbits, fetotoxicity (i.e., resorptions, litter loss, abortions, and low body weight) associated with maternal toxicity (mortality) was observed at doses of 5 and 10 mg/kg/day. The no observed adverse effect doses of 600, 200 and 2 mg/kg/day in mice, rats and rabbits represent 9, 6, and 0.1 times, respectively, the maximum recommended human dose on a mg/m2 basis. Calculations assume an oral dose of 320 mg/day and a 60-kg patient.
8.2 Lactation
Risk Summary
There are no data on the presence of Valsartan Oral Solution in human milk, the effects on the breastfed infant, or the effects on milk production. Valsartan is present in rat milk (see Data). Because of the potential for valsartan to affect postnatal renal development in nursing infants, advise a nursing woman not to breastfeed during treatment with Valsartan Oral Solution.
Data
Valsartan was detected in the milk of lactating rats 15 minutes after administration of a 3 mg/kg dose.
8.4 Pediatric Use
Valsartan is not recommended for pediatric patients under 6 years of age due to safety findings for which a relationship to treatment could not be excluded [see Adverse Reactions (6.1)]. Furthermore, it is unknown whether post-natal use of valsartan before maturation of renal function is complete has long- term deleterious effects on the kidney. In humans, nephrogenesis is thought to be complete around birth; however, maturation of other aspects of kidney function (such as glomerular filtration and tubular function) may continue until approximately 2 years of age.
The antihypertensive effects of valsartan have been evaluated in two randomized, double-blind clinical studies in pediatric patients from 1-5 and 6-16 years of age [see Clinical Studies (14.1)]. The pharmacokinetics of valsartan have been evaluated in pediatric patients 1 to 16 years of age [see Clinical Pharmacology (12.3)]. Valsartan was generally well tolerated in children 6-16 years and the adverse experience profile was similar to that described for adults.
In children and adolescents with hypertension where underlying renal abnormalities may be more common, renal function and serum potassium should be closely monitored as clinically indicated.
No data are available in pediatric patients either undergoing dialysis or with a glomerular filtration rate <30 mL/min/1.73 m2.
There is limited clinical experience with valsartan in pediatric patients with mild to moderate hepatic impairment [see Warnings and Precautions (5.3)].
8.5 Geriatric Use
In the controlled clinical trials of valsartan, 1,214 (36.2%) hypertensive patients treated with valsartan were ≥65 years and 265 (7.9%) were ≥75 years. No overall difference in the efficacy or safety of valsartan was observed in this patient population, but greater sensitivity of some older individuals cannot be ruled out.
Of the 2,511 patients with heart failure randomized to valsartan in the Valsartan Heart Failure Trial, 45% (1,141) were 65 years of age or older. In the Valsartan in Acute Myocardial Infarction Trial (VALIANT), 53% (2,596) of the 4,909 patients treated with valsartan and 51% (2,515) of the 4,885 patients treated with valsartan + captopril were 65 years of age or older. There were no notable differences in efficacy or safety between older and younger patients in either trial.
-
10 OVERDOSAGE
Limited data are available related to overdosage in humans. The most likely manifestations of overdosage would be hypotension and tachycardia; bradycardia could occur from parasympathetic (vagal) stimulation. Depressed level of consciousness, circulatory collapse and shock have been reported. If symptomatic hypotension should occur, supportive treatment should be instituted.
Valsartan is not removed from the plasma by hemodialysis.
Valsartan was without grossly observable adverse effects at single oral doses up to 2000 mg/kg in rats and up to 1000 mg/kg in marmosets, except for salivation and diarrhea in the rat and vomiting in the marmoset at the highest dose (60 and 31 times, respectively, the maximum recommended human dose on a mg/m2 basis). (Calculations assume an oral dose of 320 mg/day and a 60-kg patient.)
-
11 DESCRIPTION
Valsartan is a nonpeptide, orally active, and specific angiotensin II receptor blocker acting on the AT1 receptor subtype.
Valsartan is chemically described as N-(1-oxopentyl)-N-[[2′-(1H-tetrazol-5-yl) [1,1′-biphenyl]-4-yl]methyl]-L-valine. Its empirical formula is C24H29N5O3, its molecular weight is 435.5, and its structural formula is:
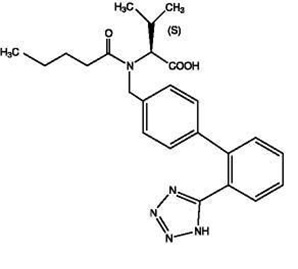
Valsartan is an off white to white powder. It is soluble in methanol.
Valsartan Oral Solution is formulated at a concentration of 4 mg/mL valsartan, USP as a clear colorless, grape flavored solution, free from visible particulate matter for oral administration. The inactive ingredients are: grape flavor, methylparaben, poloxamer 188, potassium sorbate, propylene glycol, purified water, sodium citrate dihydrate, sodium hydroxide and sucralose.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Angiotensin II is formed from angiotensin I in a reaction catalyzed by angiotensin-converting enzyme (ACE, kininase II). Angiotensin II is the principal pressor agent of the renin-angiotensin system, with effects that include vasoconstriction, stimulation of synthesis and release of aldosterone, cardiac stimulation, and renal reabsorption of sodium. Valsartan blocks the vasoconstrictor and aldosterone-secreting effects of angiotensin II by selectively blocking the binding of angiotensin II to the AT1 receptor in many tissues, such as vascular smooth muscle and the adrenal gland. Its action is therefore independent of the pathways for angiotensin II synthesis.
There is also an AT2 receptor found in many tissues, but AT2 is not known to be associated with cardiovascular homeostasis. Valsartan has much greater affinity (about 20,000-fold) for the AT1 receptor than for the AT2 receptor. The increased plasma levels of angiotensin II following AT1 receptor blockade with valsartan may stimulate the unblocked AT2 receptor. The primary metabolite of valsartan is essentially inactive with an affinity for the AT1 receptor about one-200th that of valsartan itself.
Blockade of the renin-angiotensin system with ACE inhibitors, which inhibit the biosynthesis of angiotensin II from angiotensin I, is widely used in the treatment of hypertension. ACE inhibitors also inhibit the degradation of bradykinin, a reaction also catalyzed by ACE. Because valsartan does not inhibit ACE (kininase II), it does not affect the response to bradykinin. Whether this difference has clinical relevance is not yet known. Valsartan does not bind to or block other hormone receptors or ion channels known to be important in cardiovascular regulation.
Blockade of the angiotensin II receptor inhibits the negative regulatory feedback of angiotensin II on renin secretion, but the resulting increased plasma renin activity and angiotensin II circulating levels do not overcome the effect of valsartan on blood pressure.
12.2 Pharmacodynamics
Valsartan inhibits the pressor effect of angiotensin II infusions. An oral dose of 80 mg inhibits the pressor effect by about 80% at peak with approximately 30% inhibition persisting for 24 hours. No information on the effect of larger doses is available.
Removal of the negative feedback of angiotensin II causes a 2- to 3-fold rise in plasma renin and consequent rise in angiotensin II plasma concentration in hypertensive patients. Minimal decreases in plasma aldosterone were observed after administration of valsartan; very little effect on serum potassium was observed.
In multiple-dose studies in hypertensive patients with stable renal insufficiency and patients with renovascular hypertension, valsartan had no clinically significant effects on glomerular filtration rate, filtration fraction, creatinine clearance, or renal plasma flow.
In multiple-dose studies in hypertensive patients, valsartan had no notable effects on total cholesterol, fasting triglycerides, fasting serum glucose, or uric acid.
12.3 Pharmacokinetics
For an equivalent dose, Valsartan Oral Solution has 86% higher peak concentration (Cmax) and 25% higher area under the plasma concentration over time curve (AUC) for valsartan compared to Diovan. AUC and Cmax of valsartan increase approximately linearly with increasing dose over the clinical dosing range. Valsartan does not accumulate appreciably in plasma following repeated administration.
AbsorptionValsartan Oral Solution Cmax is achieved 0.7 to 3.7 hours after dosing.
Effect of Food
High-fat, high-calorie meal decreased the AUC of Valsartan Oral Solution by about 8% and Cmax by about 44%.
Distribution
The steady state volume of distribution of valsartan after intravenous administration is small (17 L), indicating that valsartan does not distribute into tissues extensively. Valsartan is highly bound to serum proteins (95%), mainly serum albumin.
Elimination
Following intravenous administration, plasma clearance of valsartan is about 2 L/h. Renal clearance of valsartan is 0.62 L/h (about 30% of total body clearance). Valsartan shows bi-exponential decay kinetics following intravenous administration, with an average elimination half-life of about 6 hours.
Metabolism
The primary metabolite, accounting for about 9% of dose, is valeryl 4-hydroxy valsartan. In vitro metabolism studies involving recombinant CYP 450 enzymes indicated that the CYP 2C9 isoenzyme is responsible for the formation of valeryl-4-hydroxy valsartan. Valsartan does not inhibit CYP 450 isozymes at clinically relevant concentrations. CYP 450 mediated drug interaction between valsartan and co-administered drugs are unlikely because of the low extent of metabolism.
Excretion
When administered as an oral solution, 83% of the dose is recovered in feces and about 13% is recovered in urine. The recovery is mainly as unchanged drug, with only about 20% of dose recovered as metabolites.
Special Populations:
Geriatric Patients: Exposure (measured by AUC) to valsartan is higher by 70% and the half-life is longer by 35% in the elderly than in the young.
Pediatric Patients: In a study of pediatric hypertensive patients (n=26, 1 to 16 years of age) given single doses of a suspension of valsartan (mean: 0.9 to 2 mg/kg), the clearance (L/h/kg) of valsartan for children was similar to that of adults receiving the same formulation.
Male and Female Patients: Pharmacokinetics of valsartan does not differ significantly between males and females.
Heart Failure Patients: The average time to peak concentration and elimination half-life of valsartan in heart failure patients are similar to those observed in healthy volunteers. AUC and Cmax values of valsartan increase linearly and are almost proportional with increasing dose over the clinical dosing range (40 to 160 mg twice a day). The average accumulation factor is about 1.7. The apparent clearance of valsartan following oral administration is approximately 4.5 L/h. Age does not affect the apparent clearance in heart failure patients.
Patients with Renal Impairment: There is no apparent correlation between renal function (measured by creatinine clearance) and exposure (measured by AUC) to valsartan in patients with different degrees of renal impairment. Consequently, dose adjustment is not required in patients with mild-to-moderate renal dysfunction. No studies have been performed in patients with severe impairment of renal function (creatinine clearance <10 mL/min). Valsartan is not removed from the plasma by hemodialysis. In the case of severe renal disease, monitor closely [see Dosage and Administration (2.1)].
Patients with Hepatic Impairment: On average, patients with mild-to-moderate chronic liver disease have twice the exposure (measured by AUC values) to valsartan of healthy volunteers (matched by age, sex, and weight). In general, no dosage adjustment is needed in patients with mild-to-moderate liver disease. Monitor closely patients with liver disease [see Dosage and Administration (2.1)].
Drug Interactions
No clinically significant pharmacokinetic interactions were observed when valsartan was coadministered with nebivolol, amlodipine, atenolol, cimetidine, digoxin, furosemide, glyburide, hydrochlorothiazide, or indomethacin.
Co-administration of valsartan and warfarin did not change the pharmacokinetics of valsartan or the time- course of the anticoagulant properties of warfarin.
Transporters: The results from an in vitro study with human liver tissue indicate that valsartan is a substrate of the hepatic uptake transporter OATP1B1 and the hepatic efflux transporter MRP2. Coadministration of inhibitors of the uptake transporter (rifampin, cyclosporine) or efflux transporter (ritonavir) may increase the systemic exposure to valsartan.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis & Mutagenesis & Impairment Of Fertility
There was no evidence of carcinogenicity when valsartan was administered in the diet to mice and rats for up to 2 years at doses up to 160 and 200 mg/kg/day, respectively. These doses in mice and rats are about 2.6 and 6 times, respectively, the maximum recommended human dose on a mg/m2 basis. (Calculations assume an oral dose of 320 mg/day and a 60-kg patient.)
Mutagenicity assays did not reveal any valsartan-related effects at either the gene or chromosome level. These assays included bacterial mutagenicity tests with Salmonella (Ames) and E. coli; a gene mutation test with Chinese hamster V79 cells; a cytogenetic test with Chinese hamster ovary cells; and a rat micronucleus test.
Valsartan had no adverse effects on the reproductive performance of male or female rats at oral doses up to 200 mg/kg/day. This dose is 6 times the maximum recommended human dose on a mg/m2 basis.
(Calculations assume an oral dose of 320 mg/day and a 60-kg patient.)
13.2 Animal Pharmacology & OR Toxicology
Daily oral dosing of neonatal/juvenile rats with valsartan at doses as low as 1 mg/kg/day (about 10% of the maximum recommended pediatric dose on a mg/m2 basis) from postnatal day 7 to postnatal day 70 produced persistent, irreversible kidney damage. These kidney effects in neonatal rats represent expected exaggerated pharmacological effects that are observed if rats are treated during the first 13 days of life.
-
14 CLINICAL STUDIES
14.1 Hypertension
Studies evaluating the antihypertensive effects of valsartan were conducted with a formulation that is not therapeutically equivalent to Valsartan Oral Solution [see Clinical Pharmacology (12.3)].
Adult Hypertension
The antihypertensive effects of valsartan were demonstrated principally in 7 placebo-controlled, 4- to 12-week trials (1 in patients over 65 years) of dosages from 10 to 320 mg/day in patients with baseline diastolic blood pressures of 95-115 mmHg. The studies allowed comparison of once-daily and twice-daily regimens of 160 mg/day; comparison of peak and trough effects; comparison (in pooled data) of response by gender, age, and race; and evaluation of incremental effects of hydrochlorothiazide.
Administration of valsartan to patients with essential hypertension results in a significant reduction of sitting, supine, and standing systolic and diastolic blood pressure, usually with little or no orthostatic change.
In most patients, after administration of a single oral dose, onset of antihypertensive activity occurs at approximately 2 hours, and maximum reduction of blood pressure is achieved within 6 hours. The antihypertensive effect persists for 24 hours after dosing, but there is a decrease from peak effect at lower doses (40 mg) presumably reflecting loss of inhibition of angiotensin II. At higher doses, however (160 mg), there is little difference in peak and trough effect. During repeated dosing, the reduction in blood pressure with any dose is substantially present within 2 weeks, and maximal reduction is generally attained after 4 weeks. In long-term follow-up studies (without placebo control), the effect of valsartan appeared to be maintained for up to 2 years. The antihypertensive effect is independent of age, gender or race. The latter finding regarding race is based on pooled data and should be viewed with caution, because antihypertensive drugs that affect the renin-angiotensin system (that is, ACE inhibitors and angiotensin-II blockers) have generally been found to be less effective in low-renin hypertensives (frequently blacks) than in high-renin hypertensives (frequently whites). In pooled, randomized, controlled trials of valsartan that included a total of 140 blacks and 830 whites, valsartan and an ACE-inhibitor control were generally at least as effective in blacks as whites. The explanation for this difference from previous findings is unclear.
Abrupt withdrawal of valsartan has not been associated with a rapid increase in blood pressure.
The blood pressure-lowering effect of valsartan and thiazide-type diuretics are approximately additive.
The 7 studies of valsartan monotherapy included over 2,000 patients randomized to various doses of valsartan and about 800 patients randomized to placebo. Doses below 80 mg were not consistently distinguished from those of placebo at trough, but doses of 80, 160 and 320 mg produced dose-related decreases in systolic and diastolic blood pressure, with the difference from placebo of approximately 6-9/3-5 mmHg at 80 to 160 mg and 9/6 mmHg at 320 mg. In a controlled trial the addition of HCTZ to valsartan 80 mg resulted in additional lowering of systolic and diastolic blood pressure by approximately 6/3 and 12/5 mmHg for 12.5 and 25 mg of HCTZ, respectively, compared to valsartan 80 mg alone.
Patients with an inadequate response to 80 mg once daily were titrated to either 160 mg once daily or 80 mg twice daily, which resulted in a comparable response in both groups.
In controlled trials, the antihypertensive effect of once-daily valsartan 80 mg was similar to that of once-daily enalapril 20 mg or once-daily lisinopril 10 mg.
There are no trials of valsartan demonstrating reductions in cardiovascular risk in patients with hypertension, but at least one pharmacologically similar drug has demonstrated such benefits.
There was essentially no change in heart rate in valsartan-treated patients in controlled trials.
Pediatric Hypertension
The antihypertensive effects of valsartan were evaluated in two randomized, double-blind clinical studies.
In a clinical study involving 261 hypertensive pediatric patients 6 to 16 years of age, patients who weighed < 35 kg received 10, 40 or 80 mg of valsartan daily (low, medium and high doses), and patients who weighed ≥ 35 kg received 20, 80, and 160 mg of valsartan daily (low, medium and high doses). Renal and urinary disorders, and essential hypertension with or without obesity were the most common underlying causes of hypertension in children enrolled in this study. At the end of 2 weeks, valsartan reduced both systolic and diastolic blood pressure in a dose-dependent manner. Overall, the three dose levels of valsartan (low, medium and high) significantly reduced systolic blood pressure by -8, -10, -12 mmHg from the baseline, respectively. Patients were re-randomized to either continue receiving the same dose of valsartan or were switched to placebo. In patients who continued to receive the medium and high doses of valsartan, systolic blood pressure at trough was -4 and -7 mmHg lower than patients who received the placebo treatment. In patients receiving the low dose of valsartan, systolic blood pressure at trough was similar to that of patients who received the placebo treatment. Overall, the dose- dependent antihypertensive effect of valsartan was consistent across all the demographic subgroups.
In a clinical study involving 90 hypertensive pediatric patients 1 to 5 years of age with a similar study design, there was some evidence of effectiveness, but safety findings for which a relationship to treatment could not be excluded mitigate against recommending use in this age group [see Adverse Reactions (6.1)].
14.2 Heart Failure
The Valsartan Heart Failure Trial (Val-HeFT) was a multinational, double-blind study in which 5,010 patients with NYHA class II (62%) to IV (2%) heart failure and LVEF <40%, on baseline therapy chosen by their physicians, were randomized to placebo or valsartan (titrated from 40 mg twice daily to the highest tolerated dose or 160 mg twice daily) and followed for a mean of about 2 years. The VAL-HeFT study was conducted with a formulation of valsartan that is not therapeutically equivalent to Valsartan Oral Solution [see Clinical Pharmacology (12.3)]. Although Val-HeFT’s primary goal was to examine the effect of valsartan when added to an ACE inhibitor, about 7% were not receiving an ACE inhibitor. Other background therapy included diuretics (86%), digoxin (67%), and beta-blockers (36%). The population studied was 80% male, 46% 65 years or older and 89% Caucasian. At the end of the trial, patients in the valsartan group had a blood pressure that was 4 mmHg systolic and 2 mmHg diastolic lower than the placebo group. There were two primary end points, both assessed as time to first event: all-cause mortality and heart failure morbidity, the latter defined as all-cause mortality, sudden death with resuscitation, hospitalization for heart failure, and the need for intravenous inotropic or vasodilatory drugs for at least 4 hours. These results are summarized in the following table.
Placebo
(N=2,499)
Valsartan
(N=2,511)
Hazard Ratio
(95% CI*)
Nominal
p-value
All-cause mortality
484
(19.4%)
495
(19.7%)
1.02
(0.90-1.15)
0.8
HF morbidity
801
(32.1%)
723
(28.8%)
0.87
(0.79-0.97)
0.009
* CI = Confidence Interval
Although the overall morbidity result favored valsartan, this result was largely driven by the 7% of patients not receiving an ACE inhibitor, as shown in the following table.
Without ACE Inhibitor
With ACE Inhibitor
Placebo
(N=181)
Valsartan
(N=185)
Placebo
(N=2,318)
Valsartan
(N=2,326)
Events (%)
77 (42.5%)
46 (24.9%)
724 (31.2%)
677 (29.1%)
Hazard ratio (95% CI)
0.51 (0.35, 0.73)
0.92 (0.82, 1.02)
p-value
0.0002
0.0965
The modest favorable trend in the group receiving an ACE inhibitor was largely driven by the patients receiving less than the recommended dose of ACE inhibitor. Thus, there is little evidence of further clinical benefit when valsartan is added to an adequate dose of ACE inhibitor.
Secondary end points in the subgroup not receiving ACE inhibitors were as follows.
Placebo
(N=181)
Valsartan
(N=185)
Hazard Ratio
(95% CI)
Components of HF morbidity
All-cause mortality
49 (27.1%)
32 (17.3%)
0.59 (0.37, 0.91)
Sudden death with resuscitation
2 (1.1%)
1 (0.5%)
0.47 (0.04, 5.20)
CHF therapy
1 (0.6%)
0 (0.0%)
--
CHF hospitalization
48 (26.5%)
24 (13.0%)
0.43 (0.27, 0.71)
Cardiovascular mortality
40 (22.1%)
29 (15.7%)
0.65 (0.40, 1.05)
Non-fatal morbidity
49 (27.1%)
24 (13.0%)
0.42 (0.26, 0.69)
In patients not receiving an ACE inhibitor, valsartan-treated patients had an increase in ejection fraction and reduction in left ventricular internal diastolic diameter (LVIDD).
Effects were generally consistent across subgroups defined by age and gender for the population of patients not receiving an ACE inhibitor. The number of black patients was small and does not permit a meaningful assessment in this subset of patients.
14.3 Post-Myocardial Infarction
The VALsartan In Acute myocardial iNfarcTion trial (VALIANT) was a randomized, controlled, multinational, double-blind study in 14,703 patients with acute myocardial infarction and either heart failure (signs, symptoms or radiological evidence) or left ventricular systolic dysfunction (ejection fraction ≤40% by radionuclide ventriculography or ≤35% by echocardiography or ventricular contrast angiography). The VALIANT study was conducted with a formulation of valsartan that is not therapeutically equivalent to Valsartan Oral Solution [see Clinical Pharmacology (12.3)]. Patients were randomized within 12 hours to 10 days after the onset of myocardial infarction symptoms to one of three treatment groups: valsartan (titrated from 20 or 40 mg twice daily to the highest tolerated dose up to a maximum of 160 mg twice daily), the ACE inhibitor, captopril (titrated from 6.25 mg three times daily to the highest tolerated dose up to a maximum of 50 mg three times daily), or the combination of valsartan plus captopril. In the combination group, the dose of valsartan was titrated from 20 mg twice daily to the highest tolerated dose up to a maximum of 80 mg twice daily; the dose of captopril was the same as for monotherapy. The population studied was 69% male, 94% Caucasian, and 53% were 65 years of age or older. Baseline therapy included aspirin (91%), beta-blockers (70%), ACE inhibitors (40%), thrombolytics (35%) and statins (34%). The mean treatment duration was 2 years. The mean daily dose of valsartan in the monotherapy group was 217 mg.
The primary endpoint was time to all-cause mortality. Secondary endpoints included (1) time to cardiovascular (CV) mortality, and (2) time to the first event of cardiovascular mortality, reinfarction, or hospitalization for heart failure. The results are summarized in the following table.
Valsartan vs. Captopril
(N=4,909) (N=4,909)
Valsartan + Captopril vs. Captopril
(N=4,885) (N=4,909)
No. of Deaths
Valsartan/
Captopril
Hazard Ratio
CI
p-value
No. of Deaths
Comb/
Captopril
Hazard Ratio
CI
p-value
All-cause mortality
979 (19.9%) /958 (19.5%)
1.001 (0.902, 1.111)
0.98
941 (19.3%)
/958 (19.5%)
0.984 (0.886, 1.093)
0.73
CV mortality
827 (16.8%)
/830 (16.9%)
0.976 (0.875, 1.090)
CV mortality, hospitalization for HF, and recurrent non-fatal MI
1,529 (31.1%)
/1,567 (31.9%)
0.955 (0.881, 1.035)
There was no difference in overall mortality among the three treatment groups. There was thus no evidence that combining the ACE inhibitor captopril and the angiotensin II blocker valsartan was of value.
The data were assessed to see whether the effectiveness of valsartan could be demonstrated by showing in a non-inferiority analysis that it preserved a fraction of the effect of captopril, a drug with a demonstrated survival effect in this setting. A conservative estimate of the effect of captopril (based on a pooled analysis of 3 post-infarction studies of captopril and 2 other ACE inhibitors) was a 14% to 16% reduction in mortality compared to placebo. Valsartan would be considered effective if it preserved a meaningful fraction of that effect and unequivocally preserved some of that effect. As shown in the table, the upper bound of the CI for the hazard ratio (valsartan/captopril) for overall or CV mortality is 1.09 to 1.11, a difference of about 9% to 11%, thus making it unlikely that valsartan has less than about half of the estimated effect of captopril and clearly demonstrating an effect of valsartan. The other secondary endpoints were consistent with this conclusion.
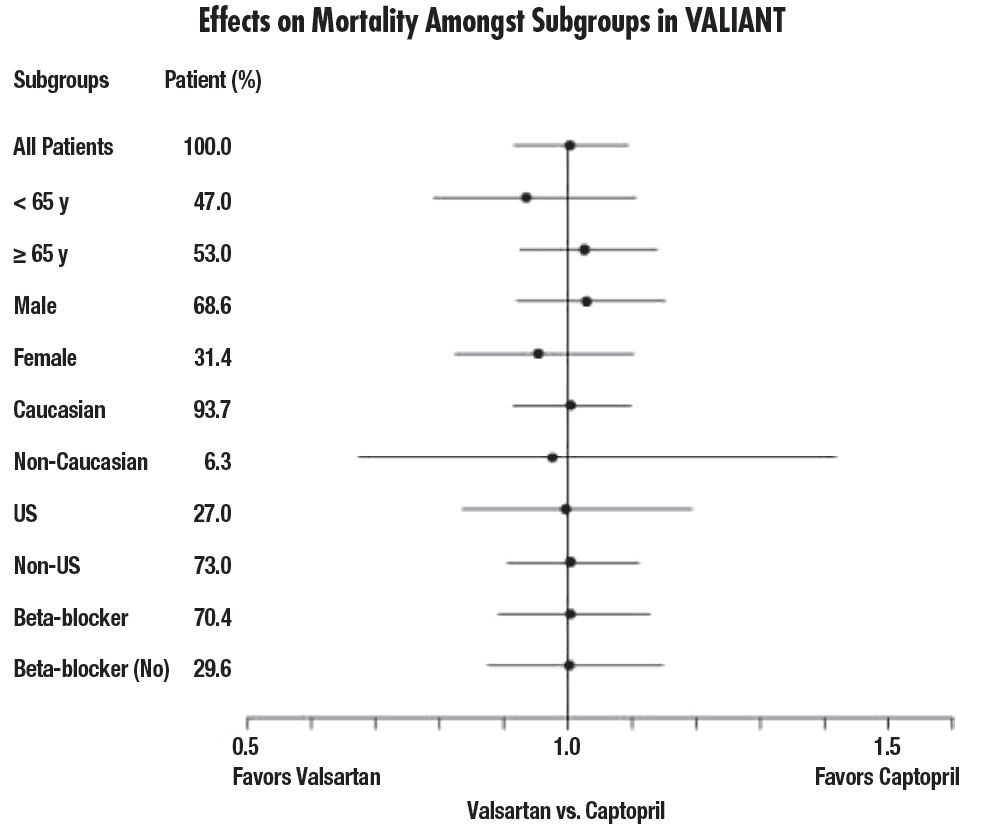
There were no clear differences in all-cause mortality based on age, gender, race, or baseline therapies, as shown in the figure above.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Valsartan Oral Solution containing 4 mg/mL valsartan, USP for oral administration is available as:
White HDPE bottles containing 120 mL: NDC 70954-310-10
White HDPE bottles containing 473 mL: NDC 70954-310-20
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Dispense in tight container (USP).
-
17 PATIENT COUNSELING INFORMATION
Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to notify their healthcare provider with a known or suspected pregnancy [see Warnings and Precautions (5.1) and Use in Specific Populations (8.1)].
Lactation
Advise women not to breastfeed during treatment with Valsartan Oral Solution [see Use in Specific Populations (8.2)].
Symptomatic Hypotension
Advise patients that lightheadedness can occur, especially during the first days of therapy, and that it should be reported to the prescribing physician. Tell patients that if syncope occurs to discontinue Valsartan Oral Solution until the physician has been consulted.
Caution all patients that inadequate fluid intake, excessive perspiration, diarrhea, or vomiting can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope.
Hyperkalemia
Advise patients not to use salt substitutes containing potassium without consulting their physician.
Trademarks are the property of their respective owners.
Distributed by:
ANI Pharmaceuticals, Inc.
Baudette, MN 56623
Issued: 08/2023
LB4631-00
- PACKAGE LABEL.PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
VALSARTAN
valsartan solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70954-310 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength VALSARTAN (UNII: 80M03YXJ7I) (VALSARTAN - UNII:80M03YXJ7I) VALSARTAN 4 mg in 1 mL Inactive Ingredients Ingredient Name Strength PROPYLENE GLYCOL (UNII: 6DC9Q167V3) METHYLPARABEN (UNII: A2I8C7HI9T) POLOXAMER 188 (UNII: LQA7B6G8JG) POTASSIUM SORBATE (UNII: 1VPU26JZZ4) SUCRALOSE (UNII: 96K6UQ3ZD4) TRISODIUM CITRATE DIHYDRATE (UNII: B22547B95K) WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) Product Characteristics Color Score Shape Size Flavor GRAPE Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70954-310-10 120 mL in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 11/02/2021 2 NDC:70954-310-20 473 mL in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 11/02/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA214102 11/02/2021 Labeler - ANI Pharmaceuticals, Inc. (145588013) Establishment Name Address ID/FEI Business Operations Novitium Pharma LLC 080301870 ANALYSIS(70954-310) , LABEL(70954-310) , MANUFACTURE(70954-310) , PACK(70954-310)

