Label: VYLEESI- bremelanotide injection



-
Contains inactivated NDC Code(s)
NDC Code(s): 64011-701-01, 64011-701-04, 64011-701-12 - Packager: AMAG Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated January 12, 2021
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VYLEESI™ safely and effectively. See full prescribing information for VYLEESI™.
VYLEESI (bremelanotide injection), for subcutaneous use
Initial U.S. Approval: 2019
INDICATIONS AND USAGE
VYLEESI is a melanocortin receptor agonist indicated for the treatment of premenopausal women with acquired, generalized hypoactive sexual desire disorder (HSDD) as characterized by low sexual desire that causes marked distress or interpersonal difficulty and is NOT due to:
- A co-existing medical or psychiatric condition,
- Problems with the relationship, or
- The effects of a medication or drug substance (1)
Limitations of Use (1):
- Not indicated for treatment of HSDD in postmenopausal women or in men.
- Not indicated to enhance sexual performance.
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Subcutaneous injection: 1.75 mg/0.3 mL solution. (3)
CONTRAINDICATIONS
Uncontrolled hypertension or known cardiovascular disease. (4)
WARNINGS AND PRECAUTIONS
- Transient increase in blood pressure and decrease in heart rate: Occurs after each dose and usually resolves within 12 hours. Consider the patient’s cardiovascular risk before initiating VYLEESI and periodically during treatment and ensure blood pressure is well-controlled. VYLEESI is not recommended in patients at high risk for cardiovascular disease. (5.1)
- Focal hyperpigmentation: Reported by 1% of patients who received up to 8 doses per month, including involvement of the face, gingiva and breasts. Higher risk in patients with darker skin and with daily dosing. Resolution was not confirmed in some patients. Consider discontinuing VYLEESI if hyperpigmentation develops. (5.2)
- Nausea: Reported by 40% of patients who received up to 8 monthly doses, requiring anti-emetic therapy in 13% of patients and leading to premature discontinuation for 8% of patients. Improved for most patients with the second dose. Consider discontinuing VYLEESI or initiating anti-emetic therapy for persistent or severe nausea. (5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence > 4%) are nausea, flushing, injection site reactions, headache, and vomiting. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AMAG Pharmaceuticals at 1-877-411-2510 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- VYLEESI may slow gastric emptying and impact absorption of concomitantly administered oral medications. (7.1)
- VYLEESI may significantly decrease the systemic exposure of orally-administered naltrexone; avoid use with orally administered naltrexone-containing products intended to treat alcohol or opioid addiction. (7.2)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 6/2019
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Discontinuation of VYLEESI
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Transient Increase in Blood Pressure and Reduction in Heart Rate
5.2 Focal Hyperpigmentation
5.3 Nausea
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effect of VYLEESI on Other Drugs
7.2 Naltrexone
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
VYLEESI is indicated for the treatment of premenopausal women with acquired, generalized hypoactive sexual desire disorder (HSDD), as characterized by low sexual desire that causes marked distress or interpersonal difficulty and is NOT due to:
- A co-existing medical or psychiatric condition,
- Problems with the relationship, or
- The effects of a medication or drug substance.
Acquired HSDD refers to HSDD that develops in a patient who previously had no problems with sexual desire. Generalized HSDD refers to HSDD that occurs regardless of the type of stimulation, situation or partner.
Limitations of Use
- VYLEESI is not indicated for the treatment of HSDD in postmenopausal women or in men.
- VYLEESI is not indicated to enhance sexual performance.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dosage of VYLEESI is 1.75 mg administered subcutaneously in the abdomen or thigh, as needed, at least 45 minutes before anticipated sexual activity. The duration of efficacy after each dose is unknown and the optimal window for VYLEESI administration has not been fully characterized. Patients may decide the optimal time for VYLEESI administration based on how they experience the duration of effect on desire and any adverse reactions such as nausea [see Warnings and Precautions (5.3)].
Patients should not administer more than one dose within 24 hours. The efficacy of consecutive doses within 24 hours has not been established and administering doses close together may increase the risk of additive effects on blood pressure [see Warnings and Precautions (5.1)].
Administering more than 8 doses per month is not recommended. Few patients in the phase 3 program received more than 8 doses per month. Also, more frequent dosing increases the risk for focal hyperpigmentation and the length of time per month when blood pressure is increased [see Warnings and Precautions (5.1, 5.2)].
VYLEESI is self-administered via a prefilled autoinjector pen. Visually inspect the drug product for particulate matter and discoloration prior to administration, whenever solution and container permit. Discard if the solution is cloudy, discolored, or visible particles are observed.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
VYLEESI is contraindicated in patients who have uncontrolled hypertension or known cardiovascular disease [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Transient Increase in Blood Pressure and Reduction in Heart Rate
VYLEESI transiently increases blood pressure and reduces heart rate after each dose. In clinical studies, VYLEESI induced maximal increases of 6 mmHg in systolic blood pressure (SBP) and 3 mmHg in diastolic blood pressure (DBP) that peaked between 2 to 4 hours post dose. There was a corresponding reduction in heart rate up to 5 beats per minute. Blood pressure and heart rate returned to baseline usually within 12 hours post-dose. No additive effects were seen for blood pressure or heart rate following repeat daily dosing 24-hours apart for up to 16 days [see Clinical Pharmacology (12.2)].
Before initiating VYLEESI, and periodically during treatment, consider the patient’s cardiovascular risk and ensure blood pressure is well-controlled. VYLEESI is not recommended for patients at high risk for cardiovascular disease and is contraindicated in patients with uncontrolled hypertension or known cardiovascular disease [see Contraindications (4)].
To minimize the risk of more pronounced blood pressure effects, advise patients to not take more than one VYLEESI dose within 24 hours [see Dosage and Administration (2.1)].
5.2 Focal Hyperpigmentation
In the phase 3 placebo-controlled trials, focal hyperpigmentation, including involvement of the face, gingiva, and breasts, was reported in 1% of patients who received up to 8 doses per month of VYLEESI compared to no placebo-treated patients. In another clinical study, 38% of patients developed focal hyperpigmentation after receiving VYLEESI daily for 8 days; among patients who continued VYLEESI for 8 more consecutive days, an additional 14% developed new focal pigmentary changes. Patients with dark skin were more likely to develop focal hyperpigmentation. Resolution of the focal hyperpigmentation was not confirmed in all patients after discontinuation of VYLEESI. More than 8 monthly doses of VYLEESI is not recommended. Consider discontinuing VYLEESI if hyperpigmentation develops.
5.3 Nausea
In the phase 3 placebo-controlled trials, nausea was the most commonly reported adverse reaction, reported in 40% of VYLEESI-treated patients, requiring anti-emetic therapy in 13% of VYLEESI-treated patients and leading to premature discontinuation from the trials for 8% of VYLEESI-treated patients. Nausea improves for most patients with the second dose [see Adverse Reactions (6.1)]. Consider discontinuing VYLEESI for persistent or severe nausea or initiating anti-emetic therapy for those patients who are bothered by nausea but wish to continue with VYLEESI treatment.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in greater detail elsewhere in labeling:
- Transient increases in blood pressure and reductions in heart rate [see Warnings and Precautions (5.1) and Clinical Pharmacology (12.2)]
- Focal hyperpigmentation [see Warnings and Precautions (5.2)]
- Nausea [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to the rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The efficacy and safety of VYLEESI was studied in two identical, 24-week, randomized, double-blind, placebo-controlled trials in 1247 premenopausal women with acquired, generalized HSDD. The age range was 19-56 years old with a mean age of 39 years old; 86% were White and 12% were Black. Both trials also included a 52-week open-label, uncontrolled extension phase during which 684 patients received VYLEESI [see Clinical Studies (14)]. Most patients used VYLEESI two to three times per month and no more than once a week.
Serious adverse reactions were reported in 1.1% of VYLEESI-treated patients and 0.5% of placebo-treated patients.
Adverse Reactions Leading to Study Discontinuation
The discontinuation rate due to adverse reactions was 18% among patients treated with VYLEESI and 2% among patients treated with placebo. The most common adverse reactions leading to study drug discontinuation in the VYLEESI group were nausea (8%), headache (2%), vomiting (1%), flushing (1%), injection site reactions (1%), flu-like symptoms (<1%) and increased blood pressure (<1%).
Common Adverse Reactions
Table 1 provides the incidence of common adverse reactions (those reported in at least 2% of patients in the VYLEESI treatment group and at an incidence that was greater than in the placebo group). The most common adverse reactions included nausea, flushing, injection site reactions and headache. The majority of events were reported to be mild (31%) to moderate (40%) in intensity and transient.
Table 1: Adverse Reactions Occurring in ≥ 2% of Patients in Randomized, Double Blind Controlled Trials with VYLEESI in Premenopausal Women with HSDD aIncludes injection site pain, unspecified injection site reactions, erythema, hematoma, pruritus, hemorrhage, bruising, paresthesia and hypoesthesia
VYLEESI
(n = 627)
%Placebo
(n= 620)
%Nausea 40.0 1.3 Flushing 20.3 0.3 Injection site reactionsa 13.2 8.4 Headache 11.3 1.9 Vomiting 4.8 0.2 Cough 3.3 1.3 Fatigue 3.2 0.5 Hot flush 2.7 0.2 Paraesthesia 2.6 0.0 Dizziness 2.2 0.5 Nasal congestion 2.1 0.5 Nausea
In the pooled phase 3 placebo-controlled trials, nausea was the most common adverse reaction, reported in 40% of VYLEESI-treated patients compared to 1% of placebo-treated patients. The median onset of nausea was within one-hour post-dose and lasted about two hours in duration. The incidence of nausea was highest after the first VYLEESI dose (reported in 21% of patients) then declined to about 3% after subsequent doses. Thirteen percent of VYLEESI-treated patients received an anti-emetic medication. Overall, 8% of VYLEESI-treated patients and no placebo-treated patients prematurely discontinued the trials due to nausea. [see Warnings and Precautions (5.3)]
Headache
In the pooled phase 3 placebo-controlled trials, headache occurred at a higher incidence in VYLEESI-treated patients (11%) than placebo-treated patients (2%). One patient experienced a headache event that was serious (intractable pain leading to hospitalization) and 1% of patients who received VYLEESI discontinued the study due to headache.
Flushing
In the pooled phase 3 placebo-controlled trials, flushing occurred more frequently in VYLEESI-treated patients (20%) than placebo-treated patients (<1%). None of the flushing events were serious and few were severe (<1%), and 1% of patients who received VYLEESI discontinued the study due to flushing.
Less Common Adverse Reactions
Less common adverse reactions occurring in <2% of VYLEESI-treated patients and at an incidence greater than in the placebo group were upper abdominal pain, diarrhea, myalgia, arthralgia, pain, restless leg syndrome, rhinorrhea, increased creatine phosphokinase, blood pressure increased, pain in extremity and focal skin hyperpigmentation.
Acute hepatitis
In the open-label, uncontrolled extension phase of one study, a single case of acute hepatitis was reported in a patient who had received 10 doses of VYLEESI over one year. She presented with serum transaminases exceeding 40 times the upper limit of normal (ULN), total bilirubin 6 times the ULN, and alkaline phosphatase less than 2 times ULN. Liver tests returned to normal 4 months after study drug discontinuation. Because another etiology was not identified, the role of VYLEESI could not definitively be excluded. There was no imbalance between treatment groups in serum transaminase outliers or other signals for hepatotoxicity in the clinical development program.
-
7 DRUG INTERACTIONS
7.1 Effect of VYLEESI on Other Drugs
VYLEESI may slow gastric emptying and thus has the potential to reduce the rate and extent of absorption of concomitantly administered oral medications. Instruct patients to avoid the use of VYLEESI when taking concomitant oral drugs that are dependent on threshold concentrations for efficacy (e.g., antibiotics). In addition, patients should consider discontinuing VYLEESI if there is a delayed drug effect of concomitant oral medications when a quick onset of drug effect is desired (e.g. drugs for pain relief such as indomethacin).
7.2 Naltrexone
As VYLEESI may significantly decrease the systemic exposure of orally-administered naltrexone, patients should avoid using VYLEESI with an orally administered naltrexone-containing product that is intended to treat alcohol and opioid addiction due to the severe consequence of naltrexone treatment failure [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There will be a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VYLEESI during pregnancy. Pregnant women exposed to VYLEESI and healthcare providers are encouraged to call the VYLEESI Pregnancy Exposure Registry at (877) 411- 2510.
Risk Summary
The few pregnancies in women exposed to VYLEESI in clinical trials are insufficient for determining whether there is a drug-associated risk for major birth defects, miscarriage or adverse maternal or fetal outcomes.
Based on findings in animal studies, the use of VYLEESI in pregnant women may be associated with the potential for fetal harm. In animal reproduction and development studies, daily subcutaneous administration of bremelanotide to pregnant dogs during the period of organogenesis at exposures greater than or equal to 16 times the maximum recommended dose (based on area under the concentration-time curve or AUC) produced fetal harm. In mice subcutaneously dosed with bremelanotide during pregnancy and lactation, developmental effects were observed in the offspring at greater than or equal to 125-times the maximum recommended dose (based on AUC) [see Data]. However, the lowest bremelanotide dose associated with fetal harm has not been identified for either species. For this reason, women should use effective contraception while taking VYLEESI and discontinue VYLEESI if pregnancy is suspected.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Human Data
There were 7 pregnancies reported in the clinical trials of more than 1057 patients treated with VYLEESI for up to 12 months. Among these 7 pregnancies, no major congenital anomalies were reported. There was one spontaneous abortion (miscarriage), five full-term live births, and one outcome was unknown due to loss to follow-up.
Animal Data
An embryofetal development study was conducted in the dog and a pre- and postnatal development study was conducted in the mouse to inform developmental risk. These two species are not routinely used for reproductive toxicity assessment but were the only two species that could be successfully dosed by the subcutaneous route during gestation.
Bremelanotide was administered subcutaneously to pregnant dogs (8/dose) at 2, 8, or 20 mg/kg from gestation day (GD) 18-35, corresponding to the period from implantation to late embryogenesis in the dog. Embryofetal toxicity, as measured by post-implantation loss, was elevated approximately 3 to 8-fold compared to controls across all treated groups but was not dose-dependent. A developmental no-observed-effect level (NOEL) was not set. At the low dose of 2 mg/kg/day in the dog, exposure was approximately 16 times the human exposure based on AUC.
In a pre- and postnatal development study, female mice (30/dose) were dosed subcutaneously at 0, 30, 75, and 150 mg/kg/day from GD 6 through lactation day (LD) 28, and two generations of offspring were assessed (F1 and F2). There were no effects on reproductive parameters in parental (F0) or F1 generation animals at doses up to 150 mg/kg/day (approximately 760 times the human AUC). However, developmental delays were observed in the F1 generation mice at ≥ 30 mg/kg/day (approximately 125 times the human AUC). For that reason, a developmental NOEL was not set. There were no significant effects on the growth and development of F2 generation pups.
8.2 Lactation
Risk Summary
There is no information on the presence of bremelanotide or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for VYLEESI and any potential adverse effects on the breastfed child from VYLEESI or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Use of VYLEESI during pregnancy is not recommended [see Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception while taking VYLEESI, and to discontinue VYLEESI if pregnancy is suspected.
8.4 Pediatric Use
The safety and effectiveness of VYLEESI have not been established in pediatric patients.
8.5 Geriatric Use
The safety and effectiveness of VYLEESI have not been established in geriatric patients.
8.6 Renal Impairment
No dosing adjustments are recommended for patients with mild to moderate (eGFR 30-89 mL/min/1.73 m2) renal impairment. Use with caution in patients with severe (eGFR <30 mL/min/1.73 m2) renal impairment, because these patients may have an increase in the incidence and severity of adverse reactions (e.g., nausea and vomiting) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosing adjustments are recommended for patients with mild to moderate (Child-Pugh A and B; score 5-9) hepatic impairment. VYLEESI has not been evaluated in patients with severe hepatic impairment. Use with caution in patients with severe (Child-Pugh C; score 10-15) hepatic impairment, because these patients may have an increase in the incidence and severity of adverse reactions (e.g., nausea and vomiting) [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
VYLEESI (bremelanotide injection) contains bremelanotide, a melanocortin receptor agonist for subcutaneous administration via an autoinjector. Bremelanotide acetate is a synthetic, cyclic heptapeptide with a free acid at the carboxyl terminus and an acetylated amino group at the amino terminus of the peptide with the following structure:
Ac-Nle-cyclo-(Asp-His-D-Phe-Arg-Trp-Lys-OH) • xCH3COOH
The molecular formula of bremelanotide acetate is C50H68N14O10 • xCH3COOH (1≤ x ≤ 2) and the molecular weight is 1025.2 (free base).
VYLEESI (bremelanotide injection) is supplied as a sterile, clear solution in a pre-filled syringe contained in a single-dose autoinjector for subcutaneous administration. Each pre-filled syringe contains 1.75 mg of bremelanotide (equivalent to 1.89 mg bremelanotide acetate) in 0.3 mL solution. Inactive ingredients consist of 2.5% glycerin, sterile water for injection, and hydrochloric acid or sodium hydroxide added to adjust the pH.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Bremelanotide is a melanocortin receptor (MCR) agonist that nonselectively activates several receptor subtypes with the following order of potency: MC1R, MC4R, MC3R, MC5R, MC2R. At therapeutic dose levels, binding to MC1R and MC4R is most relevant. Neurons expressing MC4R are present in many areas of the central nervous system (CNS). The mechanism by which VYLEESI improves HSDD in women is unknown. The MC1R is expressed on melanocytes; binding at this receptor leads to melanin expression and increased pigmentation.
12.2 Pharmacodynamics
Transient Increases in Blood Pressure
In an open-label ambulatory blood pressure monitoring study of 127 premenopausal women receiving VYLEESI once daily, there was a mean increase of 1.9 mmHg (95% CI: 1.0 to 2.7) in daytime systolic blood pressure (SBP) and a mean increase of 1.7 mmHg (95% CI: 0.9 to 2.4) in daytime diastolic blood pressure (DBP) after 8 days of dosing. The increase in SBP and DBP was transient with a mean peak effect in SBP of 2.8 mmHg between 4 to 8 hours post-dose and 2.7 mmHg for DBP at 0 to 4 hours post-dose. The increase in BP after 8 days of dosing was accompanied by a simultaneous and transient mean decrease in heart rate of 0.5 beats per minute (95% CI: -1.6 to -0.7). The SBP and DBP values 12 to 24-hours post-dose were similar to the pre-dose values [see Warnings and Precautions (5.1)].
Alcohol Interaction
A placebo-controlled, randomized, double-blind, three-period, three-way crossover study was conducted to assess the safety of a single intranasal 20 mg dose of bremelanotide co-administered with alcohol in 12 healthy male and 12 healthy female subjects. Intranasal bremelanotide or placebo spray was administered 10 minutes after consumption of placebo drink or 0.6 g/kg ethanol (equivalent of three 12 ounce cans of beer containing 5% alcohol content, three 5 ounce glasses of wine containing 12% alcohol content, or three 1.5 ounce shots of 80-proof spirit in a 70 kg person).
The 20 mg intranasal dose achieves a 2.5-fold higher mean Cmax than that of VYLEESI. Alcohol consumption had no effect on the pharmacokinetic profile of bremelanotide. The incidence of flushing was higher with bremelanotide plus ethanol compared to ethanol alone, but similar to the incidence with bremelanotide alone. The incidence of headache was higher with bremelanotide plus ethanol compared to bremelanotide alone, but similar to the incidence with ethanol alone. The incidence of other adverse reactions was similar across the treatment groups. The incidence of abnormal orthostatic blood pressure reductions was comparable between the bremelanotide plus ethanol group and the ethanol alone group. No participants discontinued due to adverse reactions.
Cardiac Electrophysiology
A 20 mg intranasal dose of bremelanotide does not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
Following subcutaneous administration of VYLEESI, the mean plasma Cmax and AUC of bremelanotide are 72.8 ng/mL and 276 hr*ng/mL, respectively. Mean plasma concentrations of bremelanotide increase in a less than dose proportional manner in the dose range of 0.3 to 10 mg, with mean Cmax levels reaching a plateau at the 7.5 mg subcutaneous dose level (approximately 4.3 times the maximum recommended dose).
Absorption
Bremelanotide median Tmax is approximately 1.0 hour (range: 0.5 - 1.0 hours) in plasma. The absolute bioavailability of bremelanotide following subcutaneous administration of VYLEESI was about 100%. The site of subcutaneous administration (abdomen and thigh) had no significant effect on the systemic exposure to bremelanotide.
Distribution
Twenty-one percent of bremelanotide binds to human serum protein. The mean (SD) volume of distribution after a single subcutaneous administration of VYLEESI is 25.0 ± 5.8 L.
Elimination
Following a single subcutaneous administration of VYLEESI, mean terminal half-life of bremelanotide is approximately 2.7 hours (range: 1.9– 4.0 hours) and the mean (± SD) clearance (CL/F) is 6.5 ±1.0 L/hr.
Metabolism
As a peptide with 7 amino acids, the primary metabolic pathway of bremelanotide involves multiple hydrolyses of the amide bond of the cyclic peptide
Excretion
Following administration of a radiolabeled dose, 64.8% of the total radioactivity was recovered in urine and 22.8% in feces.
Specific Populations
Patients with Renal Impairment
Following a single subcutaneous dose of VYLEESI, bremelanotide exposure (AUC) increased 1.2-fold in patients with mild (eGFR, 60 to 89 mL/min/1.73 m2) renal impairment, 1.5-fold in patients with moderate (eGFR, 30 to 59 mL/min/1.73 m2) renal impairment, and 2-fold in patients with severe (eGFR, <30 mL/min/1.73 m2) renal impairment [see Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
Following a single subcutaneous dose of VYLEESI, bremelanotide exposure (AUC0-inf) increased 1.2-fold in patients with mild (Child-Pugh A; score of 5-6) hepatic impairment and 1.7-fold in patients with moderate (Child-Pugh B; score of 7-9) hepatic impairment [see Use in Specific Populations (8.7)]. The effect of severe hepatic impairment on the pharmacokinetics of bremelanotide was not studied.
Drug Interaction Studies
Potential for VYLEESI to Influence the Pharmacokinetics of Other Drugs
VYLEESI may reduce the rate and extent of absorption of concomitantly administered oral medications, likely due to slowing gastric motility. In clinical pharmacology studies, VYLEESI did not affect the absorption of the tested orally administered concomitant medications to any clinically relevant degree, except for naltrexone and indomethacin [see Drug Interactions (7)].
The effects of bremelanotide on the pharmacokinetics of other drugs are summarized below as change relative to the other drug administered alone (test/reference) (Figure 1).
Figure 1: Effects of Bremelanotide 1.75 mg SC on the Pharmacokinetic Exposures of Orally Administered Medications
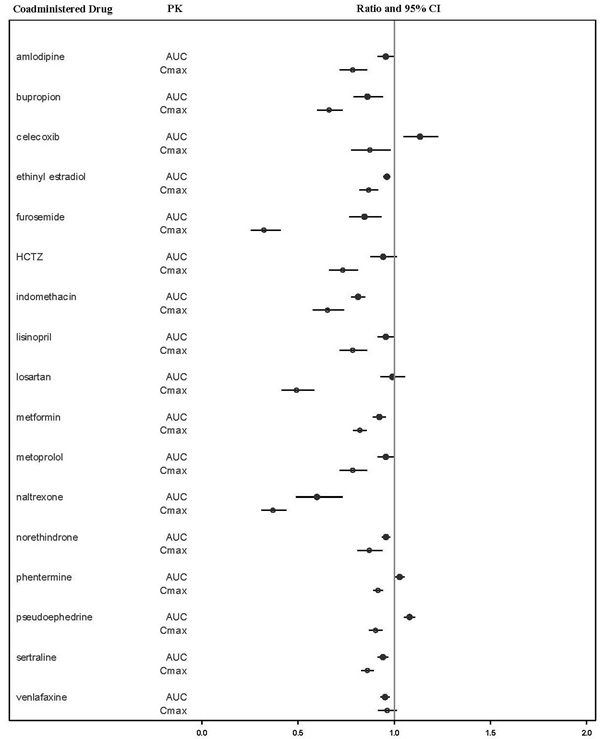
Note: AUC = area under the plasma concentration-time curve; BMT = bremelanotide; CI = confidence interval; Cmax = maximum plasma concentration; SC = subcutaneous.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
There were no significant increases in tumor incidence in two-year carcinogenicity studies with intranasal administration (0.5, 2.5, and 5 mg/animal/day) of bremelanotide to male and female rats, and subcutaneous administration (3, 9, and 15 mg/kg/day) to male and female mice. Multiples of exposure were calculated based on average Cmax at the high dose over the course of the study and were 1.1-fold and 111-fold the human Cmax for rats and mice, respectively.
Mutagenesis
Bremelanotide was not genotoxic or mutagenic in a battery of tests, including the in vitro bacterial reverse mutation assay, the in vitro chromosomal aberration test in Chinese Hamster Ovary cells, and the in vivo mouse micronucleus assay.
Impairment of Fertility
There were no effects on fertility in male (75 mg/kg/day, approximately 375 times the human AUC) or female (150 mg/kg/day, approximately 760 times the human AUC) mice following subcutaneous administration.
-
14 CLINICAL STUDIES
The efficacy of VYLEESI for the treatment of HSDD in premenopausal women was evaluated in two identical, Phase 3, randomized, double-blind, placebo-controlled trials: NCT02333071 and NCT02338960 (Study 1 and Study 2). Both trials included premenopausal women with acquired, generalized HSDD of at least 6 months’ duration. All patients in heterosexual relationships were required to use an effective form of contraception. A majority of patients (74% in Study 1 and 67% in Study 2) reported HSDD with concomitant decreased arousal. The trials consisted of two phases: a Core Study Phase (24 week placebo-controlled, double-blind treatment period) and an uncontrolled, 52-week Open-label Extension Study Phase.
Study participants were randomized to subcutaneous injections of VYLEESI 1.75 mg (n= 635) or placebo (n= 632), self-administered by an autoinjector on an as-needed basis. Patients were instructed to administer the drug approximately 45 minutes prior to anticipated sexual activity. Patients were not to administer more than one dose within a 24-hour period and no more than twelve doses per month. Trial participants were mostly Caucasian (86%) or Black (12%). The mean age of study participants was 39 years old (range 19 to 56 years old); the mean duration in a monogamous relationship was 12 years, and the mean duration of HSDD was approximately 4 years. Across the two trials, the median number of VYLEESI injections was 10 in the 24-week double-blind treatment period and 12 during the uncontrolled open-label extension. Most patients used VYLEESI two to three times per month and no more than once a week.
Study 1 and Study 2 had the following co-primary efficacy endpoints:
- Change from baseline to end of study (EOS) in the Desire domain from the Female Sexual Function Index (FSFI) (Questions 1 and 2). Question 1 asks patients “Over the past 4 weeks, how often did you feel sexual desire or interest?”, with responses ranging from 1 (almost never or never) to 5 (almost always or always). Question 2 asks patients “Over the past 4 weeks, how would you rate your level (degree) of sexual desire or interest?”, with responses ranging from 1 (very low or none at all) to 5 (very high). The FSFI Desire domain score was calculated by adding the patient’s responses to these two questions then multiplying that sum by 0.6. The FSFI Desire Domain score ranged from 1.2 to 6. An increase in the FSFI Desire domain score over time denotes improvement in sexual desire.
- Change from baseline to EOS in the score for feeling bothered by low sexual desire as measured by the Female Sexual Distress Scale – Desire/Arousal/Orgasm Question 13 (FSDS-DAO Q13). This question asks patients, “How often did you feel: Bothered by low sexual desire?” Patients assessed their sexual distress over a 30-day recall period and responded on a scale of 0 (never) to 4 (always). A decrease in the FSDS-DAO Q13 score over time denotes improvement in the level of distress associated with low sexual desire.
EOS is defined as the patient’s last study visit during the double-blind treatment period. For patients who completed the double-blind treatment period, the EOS visit occurred at Week 24.
Efficacy results for these co-primary endpoints from Study 1 and Study 2 are summarized in Table 2 and Table 3. In both studies, VYLEESI showed a statistically significant increase in the FSFI Desire Domain score and a statistically significant decrease in the FSDS-DAO Q13 score from baseline to the EOS visit compared to placebo. The magnitude of the treatment differences was similar in both studies.
Table 2: Efficacy Results for the FSFI-Desire Domain Score in Premenopausal HSDD Patients in Study 1 and Study 2 (MITT* Population) 1 FSFI Desire score range: 1.2 to 6.0, with higher scores indicating greater desire.
2 p-value from unadjusted Wilcoxon rank-sum test.
* MITT: modified intent to treat defined as all patients who were randomized, used at least one dose of double-blind study drug, and had at least one double-blind follow-up visit. However, one VYLEESI patient and one placebo patient in Study 1 and two placebo patients in Study 2 did not have either a baseline or EOS efficacy measurement and change from baseline could not be calculated. Therefore, N = the number of patients in the MITT population with an evaluable change measurement.
Study 1 Study 2 VYLEESI
1.75 mg
(N= 313)Placebo
(N= 315)VYLEESI
1.75 mg
(N= 282)Placebo
(N= 288)Mean Baseline (SD)1 2.1 (0.9) 2.0 (0.8) 2.0 (0.8) 2.1 (0.8) Mean Change from Baseline (SD) 0.5 (1.1) 0.2 (1.0) 0.6 (1.0) 0.2 (0.9) Median Change from Baseline 0.6 0 0.6 0 p-value2 0.0002 < 0.0001 Table 3: Efficacy Results for the FSDS-DAO Q13 Score in Premenopausal HSDD Patients in Study 1 and Study 2 (MITT* Population) 1 FSDS-DAO Q13 score range: 0 to 4, with higher scores indicating greater bother.
2 p-value from unadjusted Wilcoxon rank-sum test.
*MITT: modified intent to treat defined as all patients who were randomized, used at least one dose of the double-blind drug and had at least one double-blind follow-up visit. However, one VYLEESI patient and two placebo patients in Study 1 and five placebo patients in Study 2 did not have either a baseline or EOS efficacy measurement and change from baseline could not be calculated. Therefore, N = the number of patients in the MITT population with an evaluable change measurement.
Study 1 Study 2 VYLEESI
1.75 mg
(N= 313)Placebo
(N= 314)VYLEESI
1.75 mg
(N= 282)Placebo
(N= 285)Mean Baseline (SD)1 2.9 (1.0) 2.8 (0.9) 2.9 (0.9) 2.9 (0.9) Mean Change from Baseline (SD) -0.7 (1.2) -0.4 (1.1) -0.7 (1.1) -0.4 (1.1) Median Change from Baseline -1 0 -1 0 p-value2 < 0.00001 0.0053 Supplementary analyses were conducted to help interpret clinical meaningfulness of the observed score change from baseline to EOS in the FSFI-Desire Domain and FSDS-DAO Q13. These analyses defined responders for each coprimary efficacy endpoint by anchoring change from baseline to EOS with multiple anchor measures. Each anchor analysis considered responders to be those who reported experiencing meaningful change at their EOS visit according to the respective anchor measure.
Because a greater percentage of MITT patients in the VYLEESI group prematurely discontinued the 24-week double-blind treatment period compared to placebo patients (40% vs. 13% for Study 1 and 39% vs. 25% for Study 2), an exploratory analysis was performed examining the percentages of patients who were able to complete the treatment period and improved from baseline. Figure 2 displays the percentages of the MITT patients in the two Phase 3 trials who completed the 24-week double-blind treatment period and achieved various levels of increase in the FSFI-Desire Domain Score from baseline (higher scores indicate increased sexual desire). Figure 3 displays the percentages of the MITT patients in the two clinical trials who completed the 24-week double-blind treatment period and achieved various levels of reduction in the FSDS-DAO Q13 score from baseline (higher scores indicate greater reduction in distress).
Figure 2: Percent of Patients (MITT Population) who Completed the 24-Week Double-Blind Treatment Period and Achieved Various Levels of Increases in the FSFI-Desire Domain Score
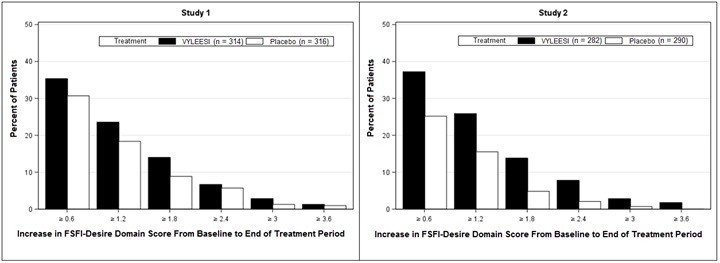
Patients who did not complete the double-blind treatment period or were missing baseline scores are not considered to have experienced an increase in FSFI-Desire Domain score at the end of the double-blind treatment period.
Responder threshold: at least 1.2-point increase from baseline in FSFI-Desire Domain score. The threshold was defined for these studies by anchoring change from baseline to end of treatment with multiple anchor measures.
Figure 3: Percent of Patients (MITT Population) who Completed the 24-Week Double-Blind Treatment Period and Achieved Various Levels of Reductions in the FSDS-DAO Q13 Score
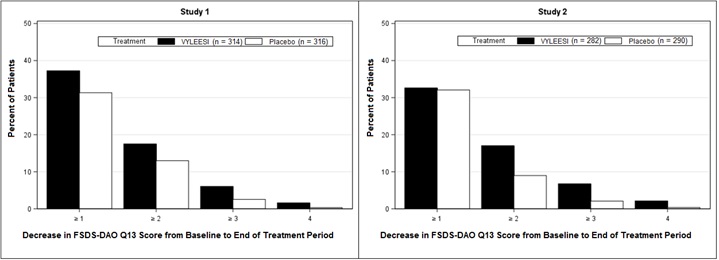
Patients who did not complete the double-blind treatment period or were missing change from baseline scores are not considered to have experienced a decrease in FSDS-DAO Q13 score at the end of the double-blind treatment period.
Responder threshold: at least 1-point decrease from baseline in the FSDS-DAO Q13 score. The threshold was defined for these studies by anchoring change from baseline to end of treatment with multiple anchor measures.
There was no significant difference between treatment groups in the change from baseline to end of study visit in the number of satisfying sexual events (SSEs), a secondary endpoint.
Efficacy results for the number of SSEs are summarized in Table 4.
Table 4: Efficacy Results for the Number of Satisfying Sexual Events in Premenopausal HSDD Patients in Study 1 and Study 2 (MITT* Population) 1 p-value from unadjusted Wilcoxon rank-sum test.
*MITT: modified intent to treat defined as all patients who were randomized, used at least one dose of double-blind drug and had at least 1 double-blind follow-up visit. N = the number of patients in the MITT population.
Study 1 Study 2 VYLEESI
1.75 mg
(N= 314)Placebo
(N= 316)VYLEESI
1.75 mg
(N= 282)Placebo
(N= 285)Mean Baseline (SD) 0.7 (1.0) 0.8 (101) 0.8 (101) 0.7 (1.0) Mean Change from Baseline (SD) 0.0 (1.4) -0.1 (1.4) 0.0 (1.3) 0.0 (1.2) Median Change from Baseline 0 0 0 0 p-value1 0.76 0.70 - 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Transient Increase in Blood Pressure and Decrease in Heart Rate
Advise patients that increases in blood pressure and decreases in heart rate may occur after taking each VYLEESI dose, and that these changes usually resolve within12 hours post-dose [see Warnings and Precautions (5.1)].
Advise patients not to take VYLEESI within 24 hours of a prior dose and that more than 8 doses per month is not recommended. Advise patients that taking VYLEESI more frequently or too close together may lead to more pronounced increases in blood pressure [see Dosage and Administration (2.1)].
Focal Hyperpigmentation
Advise patients that focal hyperpigmentation, including on the face, gingiva and breasts, may occur when VYLEESI is used intermittently, particularly in patients with darker skin. The incidence may increase with daily VYLEESI use. Advise patients that the pigmentary changes may not resolve completely after stopping VYLEESI, and to contact their healthcare provider if they have any concerns about changes to their skin [see Warnings and Precautions (5.2)].
Nausea
Advise patients that nausea may occur, most commonly with the first injection of VYLEESI, but could occur intermittently with continued use. Advise patients that nausea most commonly lasts for two hours after taking a dose but could last longer in some patients, and that anti-emetic medications may be necessary. Advise patients to contact their healthcare provider for persistent or severe nausea [see Warnings and Precautions (5.3)].
Females of Reproductive Potential
Advise patients to use effective contraception while taking VYLEESI and to discontinue VYLEESI if pregnancy is suspected. Advise pregnant patients that there is a pregnancy registry that monitors pregnancy outcomes in women exposed to VYLEESI during pregnancy [see Use in Specific Populations (8.1, 8.3)].
Manufactured for:
AMAG Pharmaceuticals, Inc.
1100 Winter Street
Waltham, MA 02451VYLEESI is a trademark of AMAG Pharmaceuticals, Inc.
© 2019 AMAG Pharmaceuticals, Inc. All rights reserved.Version 1.0
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 06/2019
PATIENT INFORMATION
VYLEESI™ (vahy-lee-see)
(bremelanotide injection)
for subcutaneous useWhat is VYLEESI?
VYLEESI is a prescription medicine used to treat hypoactive (low) sexual desire disorder (HSDD) in women who have not gone through menopause, who have not had problems with low sexual desire in the past, and who have low sexual desire no matter the type of sexual activity, the situation, or the sexual partner. Women with HSDD have low sexual desire that is troubling to them. Their low sexual desire is not due to:
- a medical or mental health problem
- problems in the relationship
- medicine or other drug use
VYLEESI is not for the treatment of HSDD in women who have gone through menopause or in men.
VYLEESI is not for use to improve sexual performance.
VYLEESI is not for use in children.
Do not use VYLEESI if you have:
- high blood pressure that is not controlled (uncontrolled hypertension)
- known heart (cardiovascular) disease
Before using VYLEESI, tell your healthcare provider about all of your medical conditions, including if you:
- have high blood pressure.
- have heart problems.
- have kidney problems.
- have liver problems.
- are pregnant or plan to become pregnant. It is not known if VYLEESI will harm your unborn baby.
- Pregnancy Registry: There will be a pregnancy registry for women who use VYLEESI during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry or call the VYLEESI Pregnancy Exposure Registry at 877-411-2510.
- Women who can become pregnant should use effective birth control during treatment with VYLEESI. Talk to your healthcare provider about birth control choices that may be right for you during this time. Stop using VYLEESI and tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with VYLEESI.
- are breastfeeding or plan to breastfeed. It is not known if VYLEESI passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby if you use VYLEESI.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. VYLEESI may affect the way other medicines work, and other medicines may affect the way VYLEESI works.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist each time you get a new medicine.
How should I use VYLEESI?
See the detailed “Instructions for Use” that comes with VYLEESI for information on how to prepare and inject a dose of VYLEESI. Talk to your healthcare provider or pharmacist if you have any questions.
- Use VYLEESI exactly as prescribed by your healthcare provider.
- VYLEESI comes in an autoinjector that you or your caregiver may use at home to give injections.
- VYLEESI is given as an injection under the skin (subcutaneous injection), in your thighs or stomach area (abdomen).
- Inject VYLEESI at least 45 minutes before you think that you will begin sexual activity.
- Do not inject more than 1 dose of VYLEESI within 24 hours of your last dose.
- Do not inject more than 8 doses of VYLEESI within a month.
Tell your healthcare provider if your symptoms of HSDD have not improved after you have used VYLEESI for 8 weeks.
What are the possible side effects of VYLEESI?
VYLEESI can cause serious side effects, including:
- Temporary increase in blood pressure and decrease in heart rate: An increase in blood pressure and decrease in heart rate can happen shortly after you inject VYLEESI. These changes usually go away within 12 hours after your injection. Increases in blood pressure and an increased risk of heart (cardiovascular) problems can happen if you use VYLEESI more often than prescribed by your healthcare provider. See “How should I use VYLEESI?”
- Darkening of the skin on certain parts of the body (focal hyperpigmentation) including the face, gums (gingiva) and breast. The chance of darkening of the skin is increased in people with darker skin color. The chance of darkening of the skin is higher if VYLEESI is used every day. Darkening of the skin may not go away, even after you stop using VYLEESI. Tell your healthcare provider if you have any concerns about changes to your skin
- Nausea. Nausea is common and can also be severe. Nausea most commonly happens after the first VYLEESI injection but can also happen after any dose of VYLEESI. The nausea usually lasts for about 2 hours but can last longer in some people. The nausea usually goes away by itself. Tell your healthcare provider if you have nausea that is severe or does not go away. Your healthcare provider may prescribe an anti-nausea medicine for you.
The most common side effects of VYLEESI include:
- flushing
- vomiting
- hot flush
- nasal congestion
- injection site reactions
- cough
- tingling
- headache
- fatigue
- dizziness
These are not all the possible side effects of VYLEESI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VYLEESI?
- Store at or below 77°F (25°C).
- Do not freeze.
- Protect from light.
Keep VYLEESI and all medicines out of the reach of children.
General information about the safe and effective use of VYLEESI.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VYLEESI for a condition for which it was not prescribed. Do not give VYLEESI to other people, even if they have the same symptoms you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about VYLEESI that is written for health professionals.
What are the ingredients in VYLEESI?
Active ingredient: bremelanotide
Inactive ingredients: 2.5% glycerin, sterile water for injection, and hydrochloric acid or sodium hydroxide is added to adjust the pH
Manufactured for: AMAG Pharmaceuticals, Inc.
VYLEESI is a trademark of AMAG Pharmaceuticals, Inc.
©2019 AMAG Pharmaceuticals, Inc. All rights reserved.
For more information, go to www.VYLEESI.com or call AMAG Pharmaceuticals Customer Service at the toll-free number 1-877-411-2510.
-
INSTRUCTIONS FOR USE
INSTRUCTIONS FOR USE


Important
Information
Storage- Do not use more than 1 dose of VYLEESI in 24 hours.
- Do not use more than 8 doses of VYLEESI within a month.
- Use 1 autoinjector for your dose of VYLEESI. Throw away (dispose of) the autoinjector after giving your injection. See Step 6: Throw away (dispose of) the VYLEESI autoinjector.
- Inject VYLEESI into the skin of the stomach area (abdomen) or thigh only.
- Do not pull off the clear cap from the VYLEESI autoinjector until you are ready to inject VYLEESI.
Store at or below 77°F (25°C). Do not freeze. Protect from light.
Keep VYLEESI and all medicines out of the reach of children.
Instructions for UseRead this Instructions for Use before you use VYLEESI and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment. Do not inject VYLEESI unless you have been trained by your healthcare provider. Talk to your healthcare provider or pharmacist if you have any questions. Supplies Needed for Your Injection: - 1 alcohol wipe (not included in carton)
- 1 VYLEESI autoinjector
- 1 cotton ball or gauze (not included in carton)
- 1 adhesive bandage (not included in carton)
- 1 sharps disposal container for VYLEESI autoinjector disposal. See Step 6: Throw away (dispose of) the VYLEESI autoinjector.
Read and follow Step 1 to Step 6 to use the VYLEESI autoinjector.
Check the label on the autoinjector for the expiration date (EXP). Do not use the autoinjector if the expiration date has passed.
Check the view window. You should see the gray tip in half of the view window and the medicine in half of the view window. If the view window is purple, the autoinjector will not work. Use a new autoinjector if the view window is purple.
Look at the medicine in the view window. It should be clear and free of particles. Do not use if the medicine is cloudy, discolored, or contains particles.
Check that the clear cap fits tightly on the purple tip of the autoinjector. If the cap does not fit tightly or is damaged, do not use the autoinjector. Call AMAG Pharmaceuticals, Inc. at 1-877-411-2510.
The autoinjector must be used right away after it is activated.
Pull the clear cap from the autoinjector (See Figure A) to activate.
- Do not try to recap the autoinjector.
-
The autoinjector should be used or thrown away right after the cap is removed.
See Step 6: Throw away (dispose of) the VYLEESI autoinjector.
Important: During the injection you will hear two clicks.
Place the purple tip flat against the center of the clean skin at your injection site.
Make sure you can see the view window (See Figure B).
Wash your hands with soap and water.
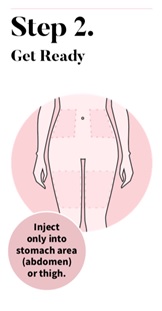
Choose an injection site on your stomach area (abdomen) or the front of your thigh. Avoid the area on your abdomen that is 2 inches around your belly button (navel).
- Do not inject into skin that is irritated, sore, bruised, red, hard, or scarred.
- Do not inject through your clothes.
- Choose a different site each time you give yourself an injection.
Clean the injection site with an alcohol wipe.
Let the injection site air dry. Do not fan or blow on the clean area. Do not touch the injection site again before giving the injection.
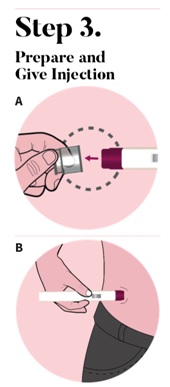
Press down and hold the autoinjector firmly against your skin (See Figure C). You will hear the 1st click right away, which tells you that your injection has started.
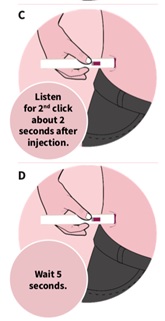
In about 2 seconds, you will hear a 2nd click.
Continue to press and hold the autoinjector firmly against your skin for about 5 seconds after the 2nd click to be sure your injection is complete (See Figure D).

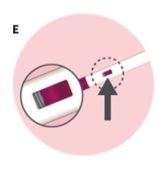
Check that the view window is now purple with a small part of the gray tip still showing. This means that all of the medicine was given (See Figure E).
Remove the autoinjector by lifting it straight off of your skin. After you remove the autoinjector from your skin, the purple tip will lock over the needle.
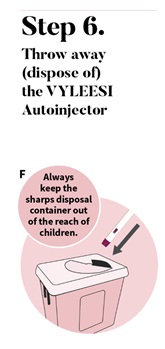
Put your used VYLEESI autoinjector in a FDA-cleared sharps disposal container right after use (See Figure F).
Do not throw away (dispose of) the autoinjector in your household trash.
If you do not have a FDA-cleared sharps disposal container, you may use a household container that is:
- made of heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.

- A cotton ball or gauze can be used initially to apply pressure after the injection if there is bleeding. An adhesive bandage should be applied after this, if needed.
- Apply an adhesive bandage if needed.
- Do not rub the injection site.

When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used needles and syringes.
For more information about safe sharps disposal and for specific information about sharps disposal in the state that you live in, go to the FDA’s website at: http://www.fda.gov/safesharpsdisposal.
Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this. Do not recycle your used sharps disposal container.
This Instructions for Use has been approved by the U.S. Food and Drug Administration.

- PRINCIPAL DISPLAY PANEL - NDC: 64011-701-04 - Autoinjector Carton Label
- PRINCIPAL DISPLAY PANEL - NDC: 64011-701-01 - Autoinjector Label
- PRINCIPAL DISPLAY PANEL - NDC: 64011-701-12 - Professional Sample Autoinjector Carton Label
-
INGREDIENTS AND APPEARANCE
VYLEESI
bremelanotide injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:64011-701 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BREMELANOTIDE ACETATE (UNII: PV2WI7495P) (BREMELANOTIDE - UNII:6Y24O4F92S) BREMELANOTIDE 1.75 mg in 0.3 mL Inactive Ingredients Ingredient Name Strength GLYCERIN (UNII: PDC6A3C0OX) 7.5 mg in 0.3 mL WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:64011-701-04 4 in 1 CARTON 06/21/2019 1 NDC:64011-701-01 0.3 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) 2 NDC:64011-701-12 2 in 1 CARTON 09/09/2019 2 NDC:64011-701-01 0.3 mL in 1 SYRINGE; Type 2: Prefilled Drug Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210557 06/21/2019 Labeler - AMAG Pharmaceuticals, Inc. (017511155) Establishment Name Address ID/FEI Business Operations Catalent Belgium SA 370696762 MANUFACTURE(64011-701)


