Label: FOSCAVIR- foscarnet sodium injection, solution


- NDC Code(s): 76310-024-01, 76310-024-11
- Packager: Clinigen Limited
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated December 11, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
WARNING
RENAL IMPAIRMENT IS THE MAJOR TOXICITY OF FOSCAVIR. FREQUENT MONITORING OF SERUM CREATININE, WITH DOSE ADJUSTMENT FOR CHANGES IN RENAL FUNCTION, AND ADEQUATE HYDRATION WITH ADMINISTRATION OF FOSCAVIR IS IMPERATIVE. (See ADMINISTRATION section; Hydration.)
SEIZURES, RELATED TO ALTERATIONS IN PLASMA MINERALS AND ELECTROLYTES, HAVE BEEN ASSOCIATED WITH FOSCAVIR TREATMENT. THEREFORE, PATIENTS MUST BE CAREFULLY MONITORED FOR SUCH CHANGES AND THEIR POTENTIAL SEQUELAE. MINERAL AND ELECTROLYTE SUPPLEMENTATION MAY BE REQUIRED.
FOSCAVIR IS INDICATED FOR USE ONLY IN IMMUNOCOMPROMISED PATIENTS WITH CMV RETINITIS AND MUCOCUTANEOUS ACYCLOVIR-RESISTANT HSV INFECTIONS. (See INDICATIONS section).
-
DESCRIPTION
FOSCAVIR is the brand name for foscarnet sodium. The chemical name of foscarnet sodium is phosphonoformic acid, trisodium salt. Foscarnet sodium is a white, crystalline powder containing 6 equivalents of water of hydration with an empirical formula of Na3CO5P•6 H2O and a molecular weight of 300.1. The structural formula is:
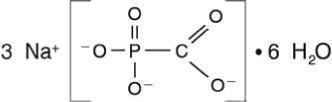
FOSCAVIR has the potential to chelate divalent metal ions, such as calcium and magnesium, to form stable coordination compounds. FOSCAVIR INJECTION is a sterile, isotonic aqueous solution for intravenous administration only. The solution is clear and colorless. Each milliliter of FOSCAVIR contains 24 mg of foscarnet sodium hexahydrate in Water for Injection, USP. Hydrochloric acid may have been added to adjust the pH of the solution to 7.4. FOSCAVIR INJECTION contains no preservatives.
-
VIROLOGY
Mechanism of Action
Foscarnet exerts its antiviral activity by a selective inhibition at the pyrophosphate binding site on virus-specific DNA polymerases at concentrations that do not affect cellular DNA polymerases. Foscarnet does not require activation (phosphorylation) by thymidine kinase or other kinases.
Antiviral Activity in Cell Culture
The quantitative relationship between the cell culture susceptibility of human cytomegalovirus (CMV) or herpes simplex virus 1 and 2 (HSV-1 and HSV-2) to foscarnet and clinical response to therapy has not been established and virus sensitivity testing has not been standardized. Sensitivity test results, expressed as the concentration of drug required to inhibit by 50% the growth of virus in cell culture (EC50), vary greatly depending on the assay method used, cell type employed and the laboratory performing the test. A number of sensitive viruses and their EC50 values are listed below (Table 1). The combination antiviral activity of foscarnet and ganciclovir or acyclovir are not antagonistic in cell culture.
TABLE 1 Foscarnet Inhibition of Virus Replication in Cell Culture *Mean = 269 μM
Virus EC50 value (μM) CMV
Ganciclovir resistant CMV
HSV-1, HSV-2
HSV-TK negative mutant
HSV-DNA polymerase mutants50-800*
190
10-130
67
5-443Antiviral Activity in vivo
Statistically significant decreases in positive CMV cultures from blood and urine have been demonstrated in two studies (FOS-03 and ACTG-015/915) of subjects treated with FOSCAVIR. Although median time to progression of CMV retinitis was increased in subjects treated with FOSCAVIR, reductions in positive blood or urine cultures have not been shown to correlate with clinical efficacy in individual subjects (Table 2).
TABLE 2 Blood and Urine Culture Results from CMV Retinitis Patients* *A total of 77 subjects were treated with FOSCAVIR in two clinical trials (FOS-03 and ACTG-015/915).
Not all subjects had blood or urine cultures done and some subjects had results from both cultures.
†(60 mg/kg FOSCAVIR TID for 2–3 weeks).
Blood +CMV -CMV Baseline 27 34 End of Induction† 1 60 Urine +CMV -CMV Baseline 52 6 End of Induction† 21 37 Resistance
Cell culture: CMV and HSV isolates with reduced susceptibility to foscarnet have been selected in cell culture by passage of wild type virus in the presence of increasing concentrations of the drug. All foscarnet resistant isolates are known to be generated through amino acid substitutions in the viral DNA polymerase pUL54 (CMV) or pUL30 (HSV) (Table 3).
TABLE 3 Summary of Foscarnet Resistance-associated DNA Polymerase Amino Acid Substitutions in Cell Culture CMV pUL54 T419M, T552N, S585A, F595I, Q807A, M844T/V, V946L HSV-1 pUL30 Y577H, E597D, A605V, L702H, V714M, L774F, L788M, D780N, L782I, P797T, L802F, V813M, V817M, Y818C, T821M, R842S, S889A, F891C, V892M, D907V, A910V, SRA914-916LCV, V958L, R959H HSV-2 pUL30 _ In vivo: Limited clinical data are available on the development of clinical resistance to foscarnet and many pathways to resistance likely exist. Substitutions documented in the literature in treated patients as associated with foscarnet resistance, are listed in Table 4.
TABLE 4 Summary of Foscarnet Resistance-associated Amino Acid Substitutions Observed in Treated Patients Note: Many additional pathways to foscarnet resistance likely exist
CMV pUL54 N495K, Q578H/L, D588E/N, T700A, V715M, E756D/K/Q, L773V, L776M, V781I, V787L, L802M, A809V, V812L, T813S, T821I, A834P, T838A, G841A/S, del 981-982 HSV-1 pUL30 S599L, D672N, R700G, V715G, A719T/V, S724N, E798K, G841C/S, A910T, Y941H HSV-2 pUL30 A724T, S725G, S729N, Q732R, L783M, D785N, T844I, L850I, D912V The possibility of viral resistance should be considered in patients who show poor clinical response or experience persistent viral excretion during therapy.
Cross-Resistance: The amino acid substitutions that resulted in reduced susceptibility to foscarnet and either ganciclovir, acyclovir and/or cidofovir are summarized in Tables 5 and 6.
TABLE 5 Summary of CMV DNA polymerase Amino Acid Substitutions Conferring Foscarnet Resistance with Cross-Resistance to Ganciclovir and/or Cidofovir Cross-resistant to ganciclovir CMV pUL54 Q578H, D588N, E756K, L773V, L776M, V781I, V787L, L802M, A809V, V812L, T813S, T821I, A834P, G841A/S, del 981-982 Cross-resistant to cidofovir CMV pUL54 Q578H, D588N, E756K, L773V, V812L, T813S, A834P, G841A, del 981-982 TABLE 6 Summary of HSV DNA polymerase Amino Acid Substitutions Conferring Foscarnet Resistance with Cross-Resistance to Acyclovir and/or Cidofovir Cross-resistant to
acyclovir
HSV-1 pUL30
E597D, S599L, A605V, D672N, R700G, L702H, V714M, V715G, A719T/V, S724N, L774F, L778M, D780N, L782I, P797T, E798K, L802F, V813M, V817M, Y818C, T821M, G841C/S, R842S, S889A, F891C/Y, V892M, D907V, A910V/T, SRA914-916LCV, Y941H, V958L, V959H
HSV-2 pUL30
A724T, S725G, S729N, Q732R, L783M, D785N, T844I, D912V
Marginally
cross-resistant to cidofovir
HSV-1 pUL30
V714M, A719V, S724N, L778M, L802F, Y818C, T821M, G841S
HSV-2 pUL30
L783M
-
CLINICAL PHARMACOLOGY
Pharmacokinetics
The pharmacokinetics of foscarnet has been determined after administration as an intermittent intravenous infusion during induction therapy in AIDS patients with CMV retinitis. Observed plasma foscarnet concentrations in four studies (FOS-01, ACTG-015, FP48PK, FP49PK) are summarized in Table 7:
TABLE 7 Foscarnet Pharmacokinetic Characteristics* *Values expressed as mean S.D. (number of subjects studied) for each parameter
†50 mg/kg Q8h for 28 days, samples taken 3 hrs after end of 1 hr infusion (Astra Report 815-04 AC025-1)
‡90 mg/kg Q12hr for 28 days, samples taken 1 hr after end of 2 hr infusion (Hengge et al., 1993)
Parameter 60 mg/kg Q8h 90 mg/kg Q12h Cmax at steady-state (μM) 589 ± 192 (24) 623 ± 132 (19) Ctrough at steady-state (μM) 114 ± 91 (24) 63 ± 57 (17) Volume of distribution (L/kg) 0.41 ± 0.13 (12) 0.52 ± 0.20 (18) Plasma half-life (hr) 4.0 ± 2.0 (24) 3.3 ± 1.4 (18) Systemic clearance (L/hr) 6.2 ± 2.1 (24) 7.1 ± 2.7 (18) Renal clearance (L/hr) 5.6 ± 1.9 (5) 6.4 ± 2.5 (13) CSF: plasma ratio 0.69 ± 0.19 (9) † 0.66 ± 0.11 (5) ‡ Distribution
In vitro studies have shown that 14 – 17% of foscarnet is protein bound at plasma drug concentrations of 1 – 1000 μM.
The foscarnet terminal half-life determined by urinary excretion was 87.5 ± 41.8 hours, possibly due to release of foscarnet from bone. Postmortem data on several patients in European clinical trials provide evidence that foscarnet does accumulate in bone in humans; however, the extent to which this occurs has not been determined.
Special Populations
Adults with Impaired Renal Function: The pharmacokinetic properties of foscarnet have been determined in a small group of adult subjects with normal and impaired renal function, as summarized in Table 8:
TABLE 8 Pharmacokinetic Parameters (mean ± S.D.) After a Single 60 mg/kg Dose of FOSCAVIR in 4 Groups* of Adults with Varying Degrees of Renal Function *Group 1 patients had normal renal function defined as a creatinine clearance (CrCl) of >80 mL/min, Group 2 CrCl was 50 – 80 mL/min, Group 3 CrCl was 25 – 49 mL/min and Group 4 CrCl was 10 – 24 mL/min.
Parameter Group 1
(N=6)Group 2
(N=6)Group 3
(N=6)Group 4
(N=4)Creatinine clearance (mL/min) 108 ± 16 68 ± 8 34 ± 9 20 ± 4 Foscarnet CL (mL/min/kg) 2.13 ± 0.71 1.33 ± 0.43 0.46 ± 0.14 0.43 ± 0.26 Foscarnet half-life (hr) 1.93 ± 0.12 3.35 ± 0.87 13.0 ± 4.05 25.3 ± 18.7 Total systemic clearance (CL) of foscarnet decreased and half-life increased with diminishing renal function (as expressed by creatinine clearance). Based on these observations, it is necessary to modify the dosage of foscarnet in patients with renal impairment (see DOSAGE AND ADMINISTRATION).
-
CLINICAL TRIALS
CMV Retinitis
A prospective, randomized, controlled clinical trial (FOS-03) was conducted in 24 patients with AIDS and CMV retinitis comparing treatment with FOSCAVIR to no treatment. Patients received induction treatment of FOSCAVIR, 60 mg/kg every 8 hours for 3 weeks, followed by maintenance treatment with 90 mg/kg/day until retinitis progression (appearance of a new lesion or advancement of the border of a posterior lesion greater than 750 microns in diameter). All diagnoses and determinations of retinitis progression were made from masked reading of retinal photographs. The 13 patients randomized to treatment with FOSCAVIR had a significant delay in progression of CMV retinitis compared to untreated controls. Median times to retinitis progression from study entry were 93 days (range 21 – >364) and 22 days (range 7 – 42), respectively.
In another prospective clinical trial of CMV retinitis in patients with AIDS (ACTG-915), 33 patients were treated with two to three weeks of FOSCAVIR induction (60 mg/kg TID) and then randomized to either 90 mg/kg/day or 120 mg/kg/day maintenance therapy. The median times from study entry to retinitis progression were not significantly different between the treatment groups, 96 (range 14 – >176) days and 140 (range 16 – >233) days, respectively.
In study ACTG 129/FGCRT SOCA study 107 patients with newly diagnosed CMV retinitis were randomized to treatment with FOSCAVIR (induction: 60 mg/kg TID for 2 weeks; maintenance: 90 mg/kg QD) and 127 were randomized to treatment with ganciclovir (induction: 5 mg/kg BID; maintenance: 5 mg/kg QD). The median time to progression on the two drugs was similar (Fos=59 and Gcv=56 days).
Relapsed CMV Retinitis
The CMV Retinitis Retreatment Trial (ACTG 228/SOCA CRRT) was a randomized, open-label comparison of FOSCAVIR or ganciclovir monotherapy to the combination of both drugs for the treatment of persistently active or relapsed CMV retinitis in patients with AIDS. Subjects were randomized to one of the three treatments: FOSCAVIR 90 mg/kg BID induction followed by 120 mg/kg QD maintenance (Fos); ganciclovir 5 mg/kg BID induction followed by 10 mg/kg QD maintenance (Gcv); or the combination of the two drugs, consisting of continuation of the subject's current therapy and induction dosing of the other drug (as above), followed by maintenance with FOSCAVIR 90 mg/kg QD plus ganciclovir 5 mg/kg QD (Cmb). Assessment of retinitis progression was performed by masked evaluation of retinal photographs. The median times to retinitis progression or death were 39 days for the FOSCAVIR group, 61 days for the ganciclovir group and 105 days for the combination group. For the alternative endpoint of retinitis progression (censoring on death), the median times were 39 days for the FOSCAVIR group, 61 days for the ganciclovir group and 132 days for the combination group. Due to censoring on death, the latter analysis may overestimate the treatment effect. Treatment modifications due to toxicity were more common in the combination group than in the FOSCAVIR or ganciclovir monotherapy groups (see ADVERSE REACTIONS section).
Mucocutaneous Acyclovir Resistant HSV Infections
In a controlled trial, patients with AIDS and mucocutaneous, acyclovir-resistant HSV infection were randomized to either FOSCAVIR (N=8) at a dose of 40 mg/kg TID or vidarabine (N=6) at a dose of 15 mg/kg per day. Eleven patients were nonrandomly assigned to receive treatment with FOSCAVIR because of prior intolerance to vidarabine. Lesions in the eight patients randomized to FOSCAVIR healed after 11 to 25 days; seven of the 11 patients nonrandomly treated with FOSCAVIR healed their lesions in 10 to 30 days. Vidarabine was discontinued because of intolerance (N=4) or poor therapeutic response (N=2). In a second trial, forty AIDS patients and three bone marrow transplant recipients with mucocutaneous, acyclovir-resistant HSV infections were randomized to receive FOSCAVIR at a dose of either 40 mg/kg BID or 40 mg/kg TID. Fifteen of the 43 patients had healing of their lesions in 11 to 72 days with no difference in response between the two treatment groups.
-
INDICATIONS
CMV Retinitis
FOSCAVIR is indicated for the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS). Combination therapy with FOSCAVIR and ganciclovir is indicated for patients who have relapsed after monotherapy with either drug. SAFETY AND EFFICACY OF FOSCAVIR HAVE NOT BEEN ESTABLISHED FOR TREATMENT OF OTHER CMV INFECTIONS (e.g., PNEUMONITIS, GASTROENTERITIS); CONGENITAL OR NEONATAL CMV DISEASE; OR NONIMMUNOCOMPROMISED INDIVIDUALS.
Mucocutaneous Acyclovir Resistant HSV Infections
FOSCAVIR is indicated for the treatment of acyclovir-resistant mucocutaneous HSV infections in immunocompromised patients. SAFETY AND EFFICACY OF FOSCAVIR HAVE NOT BEEN ESTABLISHED FOR TREATMENT OF OTHER HSV INFECTIONS (e.g., RETINITIS, ENCEPHALITIS); CONGENITAL OR NEONATAL HSV DISEASE; OR HSV IN NONIMMUNOCOMPROMISED INDIVIDUALS.
- CONTRAINDICATIONS
-
WARNINGS
Renal Impairment
THE MAJOR TOXICITY OF FOSCAVIR IS RENAL IMPAIRMENT (see ADVERSE REACTIONS section). Renal impairment is most likely to become clinically evident during the second week of induction therapy, but may occur at any time during FOSCAVIR treatment. Renal function should be monitored carefully during both induction and maintenance therapy (see PATIENT MONITORING section). Elevations in serum creatinine are usually, but not always, reversible following discontinuation or dose adjustment of FOSCAVIR. Safety and efficacy data for patients with baseline serum creatinine levels greater than 2.8 mg/dL or measured 24-hour creatinine clearances <50 mL/min are limited.
SINCE FOSCAVIR HAS THE POTENTIAL TO CAUSE RENAL IMPAIRMENT, DOSE ADJUSTMENT BASED ON SERUM CREATININE IS NECESSARY. Hydration may reduce the risk of nephrotoxicity. It is recommended that 750–1000 mL of normal saline or 5% dextrose solution should be given prior to the first infusion of FOSCAVIR to establish diuresis. With subsequent infusions, 750–1000 mL of hydration fluid should be given with 90-120 mg/kg of FOSCAVIR, and 500 mL with 40–60 mg/kg of FOSCAVIR. Hydration fluid may need to be decreased if clinically warranted.
After the first dose, the hydration fluid should be administered concurrently with each infusion of FOSCAVIR.
Mineral and Electrolyte Abnormalities
FOSCAVIR has been associated with changes in serum electrolytes including hypocalcemia, hypophosphatemia, hyperphosphatemia, hypomagnesemia, and hypokalemia (see ADVERSE REACTIONS section). FOSCAVIR may also be associated with a dose-related decrease in ionized serum calcium which may not be reflected in total serum calcium. This effect is likely to be related to chelation of divalent metal ions such as calcium by foscarnet. Patients should be advised to report symptoms of low ionized calcium such as perioral tingling, numbness in the extremities and paresthesias. Particular caution and careful management of serum electrolytes is advised in patients with altered calcium or other electrolyte levels before treatment and especially in those with neurologic or cardiac abnormalities and those receiving other drugs known to influence minerals and electrolytes (see PATIENT MONITORING and Drug Interactions sections). Physicians should be prepared to treat these abnormalities and their sequelae such as tetany, seizures or cardiac disturbances. The rate of FOSCAVIR infusion may also affect the decrease in ionized calcium. Therefore, an infusion pump must be used for administration to prevent rapid intravenous infusion (see DOSAGE AND ADMINISTRATION section). Slowing the infusion rate may decrease or prevent symptoms.
Seizures
Seizures related to mineral and electrolyte abnormalities have been associated with FOSCAVIR treatment (see WARNING section; Mineral and Electrolyte Abnormalities). Several cases of seizures were associated with death. Cases of status epilepticus have been reported. Risk factors associated with seizures included impaired baseline renal function, low total serum calcium, and underlying CNS conditions.
Hypersensitivity
Serious acute hypersensitivity reactions (e.g., anaphylactic shock, urticaria, angioedema) have been reported postmarketing in patients receiving FOSCAVIR (see ADVERSE REACTIONS section). If such an acute reaction occurs, therapy should be discontinued and appropriate medical therapy immediately instituted.
QT prolongation and torsade de pointes
FOSCAVIR has been associated with prolongation of the QT interval, an ECG abnormality that has been associated with torsades de pointes, which has been reported during postmarketing surveillance for FOSCAVIR (see ADVERSE REACTIONS section). Some of these patients had confounding risk factors such as underlying cardiac disease, electrolyte abnormalities and other concomitant medications.
Use with caution in patients who have a history of QT prolongation, in patients who are taking medications known to prolong the QT interval (see PRECAUTIONS section), in patients with electrolyte disturbances, or in patients who have other risk factors for QT prolongation. Electrocardiograms (ECGs) and measurement of electrolytes should be obtained prior to treatment initiation and periodically during treatment with FOSCAVIR.
-
PRECAUTIONS
General
Care must be taken to infuse solutions containing FOSCAVIR only into veins with adequate blood flow to permit rapid dilution and distribution to avoid local irritation (see DOSAGE AND ADMINISTRATION). Local irritation and ulcerations of penile epithelium have been reported in male patients receiving FOSCAVIR, possibly related to the presence of drug in the urine. Cases of male and female genital irritation/ulceration have been reported in patients receiving FOSCAVIR. Adequate hydration with close attention to personal hygiene may minimize the occurrence of such events.
Due to the sodium content of FOSCAVIR (240 micromoles (5.5 mg) of sodium per mL), avoid FOSCAVIR use when intravenous infusion of a large amount of sodium or water may not be tolerated (e.g. in patients with cardiomyopathy). FOSCAVIR should also be avoided in patients on a controlled sodium diet.
Hematopoietic System
Anemia has been reported in 33% of patients receiving FOSCAVIR in controlled studies. Granulocytopenia has been reported in 17% of patients receiving FOSCAVIR in controlled studies; however, only 1% (2/189) were terminated from these studies because of neutropenia.
Information for Patients
CMV Retinitis: Patients should be advised that FOSCAVIR is not a cure for CMV retinitis, and that they may continue to experience progression of retinitis during or following treatment. They should be advised to have regular ophthalmologic examinations.
Mucocutaneous Acyclovir-Resistant HSV Infections: Patients should be advised that FOSCAVIR is not a cure for HSV infections. While complete healing is possible, relapse occurs in most patients. Because relapse may be due to acyclovir-sensitive HSV, sensitivity testing of the viral isolate is advised. In addition, repeated treatment with FOSCAVIR has led to the development of resistance associated with poorer response. In the case of poor therapeutic response, sensitivity testing of the viral isolate also is advised.
Effects on Ability to Drive and Use Machines: Adverse effects such as dizziness and convulsions may occur during FOSCAVIR therapy. Patients who experience seizures, dizziness, somnolence or other adverse reactions that could result in impairment, should be advised to avoid driving or operating machinery.
General: Patients should be informed that the major toxicities of foscarnet are renal impairment, electrolyte disturbances, and seizures, and that dose modifications and possibly discontinuation may be required. The importance of close monitoring while on therapy must be emphasized. Patients should be advised of the importance of reporting to their physicians symptoms of perioral tingling, numbness in the extremities or paresthesias during or after infusion as possible symptoms of electrolyte abnormalities. Patients should also be advised to promptly report any cardiac symptoms. Should such symptoms occur, the infusion of FOSCAVIR should be stopped, appropriate laboratory samples for assessment of electrolyte concentrations obtained, and a physician consulted before resuming treatment. The rate of infusion must be no more than 1 mg/kg/minute. The potential for renal impairment may be minimized by accompanying FOSCAVIR administration with hydration adequate to establish and maintain a diuresis during dosing.
Drug Interactions
A possible drug interaction of FOSCAVIR and intravenous pentamidine has been described. Concomitant treatment of four patients in the United Kingdom with FOSCAVIR and intravenous pentamidine may have caused hypocalcemia; one patient died with severe hypocalcemia. Toxicity associated with concomitant use of aerosolized pentamidine has not been reported. Because FOSCAVIR can reduce serum levels of ionized calcium, extreme caution is advised when used concurrently with other drugs known to influence serum calcium levels (e.g., intravenous pentamidine). Renal impairment and symptomatic hypocalcemia have been observed during concurrent treatment with FOSCAVIR and intravenous pentamidine.
Because of foscarnet's tendency to cause renal impairment, the use of FOSCAVIR should be avoided in combination with potentially nephrotoxic drugs such as aminoglycosides, amphotericin B, cyclosporine, acyclovir, methotrexate, tacrolimus and intravenous pentamidine (see above) unless the potential benefits outweigh the risks to the patient.
When diuretics are indicated, thiazides are recommended over loop diuretics because the latter inhibit renal tubular secretion, and may impair elimination of FOSCAVIR, potentially leading to toxicity.
Abnormal renal function has been observed in clinical practice during the use of FOSCAVIR and ritonavir, or FOSCAVIR, ritonavir, and saquinavir. (See DOSAGE and ADMINISTRATION.)
Because of the risk of QT prolongation and the potential for torsades de pointes, the use of FOSCAVIR should be avoided in combination with agents known to prolong the QT interval including Class IA (e.g., quinidine or procainamide) or Class III (e.g., dofetilide, amiodarone, sotalol) antiarrhythmic agents, phenothiazines, tricyclic antidepressants, and certain macrolides and fluoroquinolones.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were conducted in rats and mice at oral doses of 500 mg/kg/day and 250 mg/kg/day. Oral bioavailability in unfasted rodents is < 20%. No evidence of oncogenicity was reported at plasma drug levels equal to 1/3 and 1/5, respectively, of those in humans (at the maximum recommended human daily dose) as measured by the area-under-the-time/concentration curve (AUC).
FOSCAVIR showed genotoxic effects in the BALB/3T3 in vitro transformation assay at concentrations greater than 0.5 mcg/mL and an increased frequency of chromosome aberrations in the sister chromatid exchange assay at 1000 mcg/mL. A high dose of foscarnet (350 mg/kg) caused an increase in micronucleated polychromatic erythrocytes in vivo in mice at doses that produced exposures (area under curve) comparable to that anticipated clinically.
Pregnancy
There are no adequate and well-controlled studies of FOSCAVIR in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Animal Data: FOSCAVIR did not adversely affect fertility and general reproductive performance in rats. The results of peri- and post-natal studies in rats were also negative. However, these studies used exposures that are inadequate to define the potential for impairment of fertility at human drug exposure levels.
Daily subcutaneous doses up to 75 mg/kg administered to female rats prior to and during mating, during gestation, and 21 days post-partum caused a slight increase (< 5%) in the number of skeletal anomalies compared with the control group. Daily subcutaneous doses up to 75 mg/kg administered to rabbits and 150 mg/kg administered to rats during gestation caused an increase in the frequency of skeletal anomalies/variations. On the basis of estimated drug exposure (as measured by AUC), the 150 mg/kg dose in rats and 75 mg/kg dose in rabbits were approximately one-eighth (rat) and one-third (rabbit) the estimated maximal daily human exposure. These studies are inadequate to define the potential teratogenicity at levels to which women will be exposed.
Nursing Mothers
It is not known whether FOSCAVIR is excreted in human milk; however, in lactating rats administered 75 mg/kg, FOSCAVIR was excreted in maternal milk at concentrations three times higher than peak maternal blood concentrations. Because of the potential for serious adverse events in nursing infants, a decision should be made whether to discontinue nursing or discontinue drug, taking into consideration the importance of the drug to the mother. The Centers for Disease Control and Prevention recommend that HIV-infected mothers not breast-feed their infants to avoid risking postnatal transmission of HIV.
Pediatric Use
The safety and effectiveness of FOSCAVIR in pediatric patients have not been established. FOSCAVIR is deposited in teeth and bone and deposition is greater in young and growing animals. FOSCAVIR has been demonstrated to adversely affect development of tooth enamel in mice and rats. The effects of this deposition on skeletal development have not been studied.
Since deposition in human bone has also been shown to occur, it is likely that it does so to a greater degree in developing bone in pediatric patients. Administration to pediatric patients should be undertaken only after careful evaluation and only if the potential benefits for treatment outweigh the risks.
Geriatric Use
No studies of the efficacy or safety of FOSCAVIR in persons 65 years of age or older have been conducted. However, FOSCAVIR has been used in patients age 65 years of age and older. The pattern of adverse events seen in these patients is consistent across all age groups. This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and renal function should be monitored. (See DOSAGE AND ADMINISTRATION).
-
ADVERSE REACTIONS
THE MAJOR TOXICITY OF FOSCAVIR IS RENAL IMPAIRMENT (see WARNINGS section). Approximately 33% of 189 patients with AIDS and CMV retinitis who received FOSCAVIR (60 mg/kg TID), without adequate hydration, developed significant impairment of renal function (serum creatinine ≥ 2.0 mg/dL). The incidence of renal impairment in subsequent clinical trials in which 1000 mL of normal saline or 5% dextrose solution was given with each infusion of FOSCAVIR was 12% (34/280).
FOSCAVIR has been associated with changes in serum electrolytes including hypocalcemia (15–30%), hypophosphatemia (8–26%) and hyperphosphatemia (6%), hypomagnesemia (15–30%), and hypokalemia (16–48%) (see WARNINGS section). The higher percentages were derived from those patients receiving hydration.
FOSCAVIR treatment was associated with seizures in 18/189 (10%) AIDS patients in the initial five controlled studies (see WARNINGS section). Risk factors associated with seizures included impaired baseline renal function, low total serum calcium, and underlying CNS conditions predisposing the patient to seizures. The rate of seizures did not increase with duration of treatment. Three cases were associated with overdoses of FOSCAVIR (see OVERDOSAGE section).
In five controlled U.S. clinical trials the most frequently reported adverse events in patients with AIDS and CMV retinitis are shown in Table 9. These figures were calculated without reference to drug relationship or severity.
TABLE 9 Adverse Events Reported in Five Controlled US Clinical Trials n = 189 n = 189 Fever 65% Abnormal Renal Function 27% Nausea 47% Vomiting 26% Anemia 33% Headache 26% Diarrhea 30% Seizures 10% From the same controlled studies, adverse events categorized by investigator as “severe” are shown in Table 10. Although death was specifically attributed to FOSCAVIR in only one case, other complications of FOSCAVIR (i.e., renal impairment, electrolyte abnormalities, and seizures) may have contributed to patient deaths (see WARNINGS section).
TABLE 10 Severe Adverse Events n = 189 Death 14% Abnormal Renal Function 14% Marrow Suppression 10% Anemia 9% Seizures 7% From the five initial U.S. controlled trials of FOSCAVIR, the following list of adverse events has been compiled regardless of causal relationship to FOSCAVIR. Evaluation of these reports was difficult because of the diverse manifestations of the underlying disease and because most patients received numerous concomitant medications.
Incidence of 5% or Greater
Body as a Whole: fever, fatigue, rigors, asthenia, malaise, pain, infection, sepsis, death
Central and Peripheral Nervous System: headache, paresthesia, dizziness, involuntary muscle contractions, hypoesthesia, neuropathy, seizures including grand mal seizures (see WARNINGS)
Gastrointestinal System: anorexia, nausea, diarrhea, vomiting, abdominal pain
Hematologic: anemia, granulocytopenia, leukopenia, neutropenia (see PRECAUTIONS)
Metabolic and Nutritional: mineral and electrolyte imbalances (see WARNINGS) including hypokalemia, hypocalcemia, hypomagnesemia, hypophosphatemia, hyperphosphatemia
Psychiatric: depression, confusion, anxiety
Respiratory System: coughing, dyspnea
Skin and Appendages: rash, increased sweating
Urinary System: alterations in renal function including increased serum creatinine, decreased creatinine clearance, and abnormal renal function (see WARNINGS)
Special Senses: vision abnormalities
Incidence between 1% and 5%
Application Site: injection site pain, injection site inflammation
Body as a Whole: back pain, chest pain (including reports of transient chest pain as part of infusion reactions), edema, influenza-like symptoms, bacterial infections, moniliasis, fungal infections, abscess
Cardiovascular: hypertension, palpitations, ECG abnormalities including sinus tachycardia, first degree AV block and non-specific ST-T segment changes, hypotension, flushing, cerebrovascular disorder (see WARNINGS)
Central and Peripheral Nervous System: tremor, ataxia, dementia, stupor, generalized spasms, sensory disturbances, meningitis, aphasia, abnormal coordination, leg cramps, EEG abnormalities (see WARNINGS)
Gastrointestinal: constipation, dysphagia, dyspepsia, rectal hemorrhage, dry mouth, melena, flatulence, ulcerative stomatitis, pancreatitis
Hematologic: thrombocytopenia, platelet abnormalities, thrombosis, white blood cell abnormalities, lymphadenopathy
Liver and Biliary: abnormal A-G ratio, abnormal hepatic function, increased SGPT, increased SGOT
Metabolic and Nutritional: hyponatremia, decreased weight, increased alkaline phosphatase, increased LDH, increased BUN, acidosis, cachexia, thirst
Musculo-Skeletal: arthralgia, myalgia
Neoplasms: lymphoma-like disorder, sarcoma
Psychiatric: insomnia, somnolence, nervousness, amnesia, agitation, aggressive reaction, hallucination
Respiratory System: pneumonia, sinusitis, pharyngitis, rhinitis, respiratory disorders, respiratory insufficiency, pulmonary infiltration, stridor, pneumothorax, hemoptysis, bronchospasm
Skin and Appendages: pruritus, skin ulceration, seborrhea, erythematous rash, maculo-papular rash, skin discoloration
Special Senses: taste perversions, eye abnormalities, eye pain, conjunctivitis
Urinary System: albuminuria, dysuria, polyuria, urethral disorder, urinary retention, urinary tract infections, acute renal failure, nocturia, facial edema
Selected adverse events occurring at a rate of less than 1% in the five initial U.S. controlled clinical trials of FOSCAVIR include: syndrome of inappropriate antidiuretic hormone secretion, pancytopenia, hematuria, dehydration, hypoproteinemia, increases in amylase and creatinine phosphokinase, cardiac arrest, coma, and other cardiovascular and neurologic complications.
Selected adverse event data from the Foscarnet vs. Ganciclovir CMV Retinitis Trial (FGCRT), performed by the Studies of the Ocular Complications of AIDS (SOCA) Research Group, are shown in Table 11 (see CLINICAL TRIALS section).
TABLE 11 FGCRT: Selected Adverse Events* * Values for the treatment groups refer only to patients who completed at least one follow-up visit – i.e., 133 to 119 patients in the ganciclovir group and 93 to 100 in the foscarnet group. “Events” denotes all events observed and “patients” the number of patients with one or more of the indicated events.
†Per person-year at risk
‡Final frozen SOCA I database dated October 1991
EVENT GANCICLOVIR FOSCARNET No. of
EventsNo. of Patients Rates† No. of
EventsNo. of Patients Rates† Absolute neutrophil count decreasing to <0.50 x 109 per liter 63 41 1.30 31 17 0.72 Serum creatinine increasing to >260 μmol per liter (>2.9 mg/dL) 6 4 0.12 13 9 0.30 Seizure ‡ 21 13 0.37 19 13 0.37 Catheterization-related infection 49 27 1.26 51 28 1.46 Hospitalization 209 91 4.74 202 75 5.03 Selected adverse events from ACTG Study 228 (CRRT) comparing combination therapy with FOSCAVIR or ganciclovir monotherapy are shown in Table 12. The most common reason for a treatment change in patients assigned to either FOSCAVIR or ganciclovir was retinitis progression. The most frequent reason for a treatment change in the combination treatment group was toxicity.
TABLE 12 CRRT: Selected Adverse Events * Pts. = patients with event; †Rate = events/person/year; ‡ANC = absolute neutrophil count
Foscavir
N=88Ganciclovir
N=93Combination
N=93No. Events No. Pts.* Rate† No. Events No. Pts.* Rate† No. Events No. Pts.* Rate† Anemia (Hgb <70g/L) 11 7 0.20 9 7 0.14 19 15 0.33 Neutropenia‡
ANC <0.75 x 109 cells/L
ANC <0.50 x 109 cells/L
86
50
32
25
1.53
0.91
95
49
41
28
1.51
0.80
107
50
51
28
1.91
0.85Thrombocytopenia
Platelets <50 x 109/L
Platelets <20 x 109/L
28
1
14
1
0.50
0.01
19
6
8
2
0.43
0.05
40
7
15
6
0.56
0.18Nephrotoxicity
Creatinine >260 μmol/L
(>2.9 mg/dL)
9
7
0.15
10
7
0.17
11
10
0.20Seizures 6 6 0.17 7 6 0.15 10 5 0.18 Hospitalizations 86 53 1.86 111 59 2.36 118 64 2.36 Adverse events that have been reported in post-marketing surveillance include: administration site extravasation, localized edema, hypersensitivity reactions (including anaphylactic shock, urticaria and angioedema) (see WARNINGS section), gastrointestinal hemorrhage, increased lipase, glomerulonephritis, nephrotic syndrome, proteinuria, status epilepticus, ventricular arrhythmia, prolongation of QT interval, torsade de pointes (see WARNINGS section), gamma GT increased, diabetes insipidus (usually nephrogenic), renal calculus, Fanconi syndrome acquired, renal tubular acidosis, renal tubular necrosis, crystal-induced nephropathy, hypercalcemia, hypernatremia, esophageal ulceration and muscle disorders including myopathy, myositis, muscle weakness and rare cases of rhabdomyolysis. Cases of vesiculobullous eruptions including erythema multiforme, toxic epidermal necrolysis, and Stevens-Johnson syndrome have been reported. In most cases, patients were taking other medications that have been associated with toxic epidermal necrolysis or Stevens-Johnson syndrome.
-
OVERDOSAGE
In controlled clinical trials performed in the United States, overdosage with FOSCAVIR was reported in 10 out of 189 patients. All 10 patients experienced adverse events and all except one made a complete recovery. One patient died after receiving a total daily dose of 12.5 g for three days instead of the intended 10.9 g. The patient suffered a grand mal seizure and became comatose. Three days later the patient expired with the cause of death listed as respiratory/cardiac arrest. The other nine patients received doses ranging from 1.14 times to 8 times their recommended doses with an average of 4 times their recommended doses. Overall, three patients had seizures, three patients had renal function impairment, four patients had paresthesias either in limbs or periorally, and five patients had documented electrolyte disturbances primarily involving calcium and phosphate.
Overdose (up to 20 times the recommended dose) has been reported in post-marketing use of FOSCAVIR. Some of these post-marketing reports were relative overdoses in that the dose of FOSCAVIR had not been adjusted in patients with a reduced renal function. The pattern of adverse events associated with a FOSCAVIR overdose is consistent with the known adverse event profile of the drug.
There is no specific antidote for FOSCAVIR overdose. Hemodialysis and hydration may be of benefit in reducing drug plasma levels in patients who receive an overdosage of FOSCAVIR, but the effectiveness of these interventions has not been evaluated. The patient should be observed for signs and symptoms of renal impairment and electrolyte imbalance. Medical treatment should be instituted if clinically warranted.
-
DOSAGE AND ADMINISTRATION
CAUTION - DO NOT ADMINISTER FOSCAVIR BY RAPID OR BOLUS INTRAVENOUS INJECTION. THE TOXICITY OF FOSCAVIR MAY BE INCREASED AS A RESULT OF EXCESSIVE PLASMA LEVELS. CARE SHOULD BE TAKEN TO AVOID UNINTENTIONAL OVERDOSE BY CAREFULLY CONTROLLING THE RATE OF INFUSION. THEREFORE, AN INFUSION PUMP MUST BE USED. IN SPITE OF THE USE OF AN INFUSION PUMP, OVERDOSES HAVE OCCURRED.
-
ADMINISTRATION
Instructions for Administration and Preparation
FOSCAVIR is administered by controlled intravenous infusion, either by using a central venous line or by using a peripheral vein. The rate of infusion must be no more than 1 mg/kg/minute. An individualized dose of FOSCAVIR should be calculated on the basis of body weight (mg/kg), renal function, indication of use and dosing frequency (refer to DOSAGE subsection). To reduce the risk of nephrotoxicity, creatinine clearance (mL/min/kg) should be calculated even if serum creatinine is within the normal range, and doses should be adjusted accordingly.
An individualized dose at the required concentration (24 mg/mL or 12 mg/mL) for the route of administration (central line or peripheral line) needs to be aseptically prepared prior to dispensing. The standard 24 mg/mL solution may be used with or without dilution when using a central venous catheter for infusion. When a peripheral vein catheter is used, the 24 mg/mL injection must be diluted to a 12 mg/mL concentration with 5% dextrose in water or with a normal saline solution prior to administration to avoid local irritation of peripheral veins. Dilutions and/or removals of excess quantities should be accomplished under aseptic conditions. Solutions thus prepared should be used within 24 hours of first entry into a sealed bottle or infusion bag.
Hydration
Hydration may reduce the risk of nephrotoxicity. Clinically dehydrated patients should have their condition corrected before initiating FOSCAVIR therapy. It is recommended that 750–1000 mL of normal saline or 5% dextrose solution should be given prior to the first infusion of FOSCAVIR to establish diuresis. With subsequent infusions, 750–1000 mL of hydration fluid should be given with 90–120 mg/kg of FOSCAVIR, and 500 mL with 40–60 mg/kg of FOSCAVIR. Hydration fluid may need to be decreased if clinically warranted. Oral rehydration with similar regimens may be considered in certain patients.
After the first dose, the hydration fluid should be administered concurrently with each infusion of FOSCAVIR.
Compatibility With Other Solutions/Drugs
Other drugs and supplements can be administered to a patient receiving FOSCAVIR. However, care must be taken to ensure that FOSCAVIR is only administered with normal saline or 5% dextrose solution and that no other drug or supplement is administered concurrently via the same catheter. Foscarnet has been reported to be chemically incompatible with 30% dextrose, amphotericin B, and solutions containing calcium such as Ringer's lactate and TPN. Physical incompatibility with other IV drugs has also been reported including acyclovir sodium, ganciclovir, trimetrexate glucuronate, pentamidine isethionate, vancomycin, trimethoprim/sulfamethoxazole, diazepam, midazolam, digoxin, phenytoin, leucovorin, and proclorperazine. Because of foscarnet's chelating properties, a precipitate can potentially occur when divalent cations are administered concurrently in the same catheter.
Parenteral drug products must be inspected visually for particulate matter and discoloration prior to administration whenever the solution and container permit. Solutions that are discolored or contain particulate matter should not be used.
-
DOSAGE
THE RECOMMENDED DOSAGE, FREQUENCY, OR INFUSION RATES SHOULD NOT BE EXCEEDED. ALL DOSES MUST BE INDIVIDUALIZED FOR PATIENTS' RENAL FUNCTION.
Induction Treatment
The recommended initial dose of FOSCAVIR for patients with normal renal function is:
- For CMV retinitis patients, either 90 mg/kg (1-1/2 to 2 hour infusion) every twelve hours or 60 mg/kg (minimum one hour infusion) every eight hours over 2-3 weeks depending on clinical response.
- For acyclovir-resistant HSV patients, 40 mg/kg (minimum one hour infusion) either every 8 or 12 hours for 2-3 weeks or until healed.
An infusion pump must be used to control the rate of infusion. Adequate hydration is recommended to establish a diuresis (see Hydration for recommendation), both prior to and during treatment to minimize renal toxicity (see WARNINGS), provided there are no clinical contraindications.
Maintenance Treatment
Following induction treatment the recommended maintenance dose of FOSCAVIR for CMV retinitis is 90 mg/kg/day to 120 mg/kg/day (individualized for renal function) given as an intravenous infusion over 2 hours. Because the superiority of the 120 mg/kg/day has not been established in controlled trials, and given the likely relationship of higher plasma foscarnet levels to toxicity, it is recommended that most patients be started on maintenance treatment with a dose of 90 mg/kg/day. Escalation to 120 mg/kg/day may be considered should early reinduction be required because of retinitis progression. Some patients who show excellent tolerance to FOSCAVIR may benefit from initiation of maintenance treatment at 120 mg/kg/day earlier in their treatment.
An infusion pump must be used to control the rate of infusion with all doses. Again, hydration to establish diuresis both prior to and during treatment is recommended to minimize renal toxicity, provided there are no clinical contraindications (see WARNINGS).
Patients who experience progression of retinitis while receiving FOSCAVIR maintenance therapy may be retreated with the induction and maintenance regimens given above or with a combination of FOSCAVIR and ganciclovir (see CLINICAL TRIALS section). Because of physical incompatibility, FOSCAVIR and ganciclovir must NOT be mixed.
Use in Patients with Abnormal Renal Function
FOSCAVIR should be used with caution in patients with abnormal renal function because reduced plasma clearance of foscarnet will result in elevated plasma levels (see CLINICAL PHARMACOLOGY). In addition, FOSCAVIR has the potential to further impair renal function (see WARNINGS). Safety and efficacy data for patients with baseline serum creatinine levels greater than 2.8 mg/dL or measured 24-hour creatinine clearances < 50 mL/min are limited.
Renal function must be monitored carefully at baseline and during induction and maintenance therapy with appropriate dose adjustments for FOSCAVIR as outlined below (see Dose Adjustment and PATIENT MONITORING). During FOSCAVIR therapy if creatinine clearance falls below the limits of the dosing nomograms (0.4 mL/min/kg), FOSCAVIR should be discontinued, the patient hydrated, and monitored daily until resolution of renal impairment is ensured.
FOSCAVIR is not recommended in patients undergoing hemodialysis because dosage guidelines have not been established.
Dose Adjustment
FOSCAVIR dosing must be individualized according to the patient's renal function status. Refer to Table 13 below for recommended doses and adjust the dose as indicated. Even patients with serum creatinine in the normal range may require dose adjustment; therefore, the dose should be calculated at baseline and frequently thereafter.
To use this dosing guide, actual 24-hour creatinine clearance (mL/min) must be divided by body weight (kg), or the estimated creatinine clearance in mL/min/kg can be calculated from serum creatinine (mg/dL) using the following formula (modified Cockcroft and Gault equation):
For males: 140 – age (x 0.85 for females) = mL/min/kg
Serum creatinine x 72TABLE 13 FOSCAVIR Dosage Guide Induction HSV: Equivalent to CMV: Equivalent to CrCI
(mL/min/kg)80 mg/kg/day total
(40 mg/kg Q12h)120 mg/kg/day total
(40 mg/kg Q8h)180 mg/kg/day total (60 mg/kg Q8h) (90 mg/kg Q12h) >1.4 40 Q12h 40 Q8h 60 Q8h 90 Q12h >1.0 – 1.4 30 Q12h 30 Q8h 45 Q8h 70 Q12h >0.8 – 1.0 20 Q12h 35 Q12h 50 Q12h 50 Q12h >0.6 – 0.8 35 Q24h 25 Q12h 40 Q12h 80 Q24h >0.5 – 0.6 25 Q24h 40 Q24h 60 Q24h 60 Q24h >0.4 – 0.5 20 Q24h 35 Q24h 50 Q24h 50 Q24h <0.4 Not recommended Not recommended Not recommended Not recommended Maintenance *> means “greater than”, †≥ means “greater than or equal to”, ‡< means “less than”
CMV: Equivalent to CrCI
(mL/min/kg)90 mg/kg/day
(once daily)120 mg/kg/day
(once daily)>*1.4 90 Q24h 120 Q24h >*1.0 – 1.4 70 Q24h 90 Q24h >*0.8 – 1.0 50 Q24h 65 Q24h >*0.6 – 0.8 80 Q48h 105 Q48h >*0.5 – 0.6 60 Q48h 80 Q48h ≥†0.4 – 0.5 50 Q48h 65 Q48h <‡0.4 Not recommended Not recommended -
PATIENT MONITORING
The majority of patients will experience some decrease in renal function due to FOSCAVIR administration. Therefore it is recommended that creatinine clearance, either measured or estimated using the modified Cockcroft and Gault equation based on serum creatinine, be determined at baseline, 2–3 times per week during induction therapy and once weekly during maintenance therapy, with FOSCAVIR dose adjusted accordingly (see Dose Adjustment). More frequent monitoring may be required for some patients. It is also recommended that a 24-hour creatinine clearance be determined at baseline and periodically thereafter to ensure correct dosing (assuming verification of an adequate collection using creatinine index). FOSCAVIR should be discontinued if creatinine clearance drops below 0.4 mL/min/kg.
Due to FOSCAVIR's propensity to chelate divalent metal ions and alter levels of serum electrolytes, patients must be monitored closely for such changes. It is recommended that a schedule similar to that recommended for serum creatinine (see above) be used to monitor serum calcium, magnesium, potassium and phosphorus. Particular caution is advised in patients with decreased total serum calcium or other electrolyte levels before treatment, as well as in patients with neurologic or cardiac abnormalities, and in patients receiving other drugs known to influence serum calcium levels. Any clinically significant metabolic changes should be corrected. Also, patients who experience mild (e.g., perioral numbness or paresthesias) or severe (e.g., seizures) symptoms of electrolyte abnormalities should have serum electrolyte and mineral levels assessed as close in time to the event as possible.
Careful monitoring and appropriate management of electrolytes, calcium, magnesium and creatinine are of particular importance in patients with conditions that may predispose them to seizures (see WARNINGS).
-
HOW SUPPLIED
FOSCAVIR (foscarnet sodium) INJECTION, 24 mg/mL for intravenous infusion, is supplied in 250 mL glass bottles or polypropylene based infusion bags containing 6000 mg foscarnet sodium (24 mg/mL) as follows:
NDC 76310-024-11 250 mL bottle
NDC 76310-024-01 250 mL bottles, cases of 10
NDC 76310-024-31 250 mL infusion bag
NDC 76310-024-35 250 mL infusion bag, cases of 10Single-dose. Discard unused portion.
Store between 20° and 25°C (68° and 77°F) [See USP Controlled Room Temperature]. Protect from excessive heat (above 40°C) and from freezing. If refrigerated or exposed to temperatures below the freezing point, precipitation may occur. By keeping the bottle or infusion bag at room temperature with repeated shaking, the precipitate can be brought into solution again.
FOSCAVIR INJECTION should be used only if the bottle or infusion bag and its seal(s) are intact, a vacuum is present, and the solution is clear and colorless. Do not remove the infusion bag from the overwrap until ready for use.

Manufactured for:
Clinigen Healthcare Ltd., DE14 2WW, UKDistributed by: Hospira, Inc., Lake Forest, IL 60045 USA
FOSCAVIR is a trademark of Clinigen Healthcare Ltd.
Date of revision: October 2020

Novaplus is a registered trademark of Vizient, Inc.
-
BOTTLE LABEL – PRINCIPAL DISPLAY PANEL
NDC 76310-024-11
Foscavir ® (foscarnet sodium) injection
6000 mg/250 mL (24 mg/mL)For Central Intravenous Infusion Only
Must Be Diluted for Peripheral Intravenous InfusionRx only
Single-dose. Discard unused portion.Mfd. for: Clinigen Healthcare Ltd., DE14 2WW, UK.
Distributed by: Hospira Inc., Lake Forest, IL 60045 USA.(Clinigen and Novaplus logos)
Novaplus is a registered trademark of Vizient, Inc.

- INNER CARTON LABEL – PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
FOSCAVIR
foscarnet sodium injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:76310-024 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength foscarnet sodium (UNII: 964YS0OOG1) (foscarnet - UNII:364P9RVW4X) foscarnet sodium 24 mg in 1 mL Inactive Ingredients Ingredient Name Strength hydrochloric acid (UNII: QTT17582CB) water (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:76310-024-01 10 in 1 CARTON 04/01/2014 1 NDC:76310-024-11 1 in 1 CARTON 1 250 mL in 1 BOTTLE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020068 04/01/2014 Labeler - Clinigen Limited (211471076)
