Label: VISTIDE- cidofovir injection

-
Contains inactivated NDC Code(s)
NDC Code(s): 61958-0101-1 - Packager: Gilead Sciences, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated September 24, 2013
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
WARNING
RENAL IMPAIRMENT IS THE MAJOR TOXICITY OF VISTIDE. CASES OF ACUTE RENAL FAILURE RESULTING IN DIALYSIS AND/OR CONTRIBUTING TO DEATH HAVE OCCURRED WITH AS FEW AS ONE OR TWO DOSES OF VISTIDE. TO REDUCE POSSIBLE NEPHROTOXICITY, INTRAVENOUS PREHYDRATION WITH NORMAL SALINE AND ADMINISTRATION OF PROBENECID MUST BE USED WITH EACH VISTIDE INFUSION. RENAL FUNCTION (SERUM CREATININE AND URINE PROTEIN) MUST BE MONITORED WITHIN 48 HOURS PRIOR TO EACH DOSE OF VISTIDE AND THE DOSE OF VISTIDE MODIFIED FOR CHANGES IN RENAL FUNCTION AS APPROPRIATE (SEE DOSAGE AND ADMINISTRATION). VISTIDE IS CONTRAINDICATED IN PATIENTS WHO ARE RECEIVING OTHER NEPHROTOXIC AGENTS.
NEUTROPENIA HAS BEEN OBSERVED IN ASSOCIATION WITH VISTIDE TREATMENT. THEREFORE, NEUTROPHIL COUNTS SHOULD BE MONITORED DURING VISTIDE THERAPY.
VISTIDE IS INDICATED ONLY FOR THE TREATMENT OF CMV RETINITIS IN PATIENTS WITH ACQUIRED IMMUNODEFICIENCY SYNDROME.
IN ANIMAL STUDIES CIDOFOVIR WAS CARCINOGENIC, TERATOGENIC AND CAUSED HYPOSPERMIA (SEE CARCINOGENESIS, MUTAGENESIS, & IMPAIRMENT OF FERTILITY).
-
DESCRIPTION
VISTIDE® is the brand name for cidofovir injection. The chemical name of cidofovir is 1-[(S)-3-hydroxy-2-(phosphonomethoxy)propyl]cytosine dihydrate (HPMPC), with the molecular formula of C8H14N3O6P•2H2O and a molecular weight of 315.22 (279.19 for anhydrous). The chemical structure is:
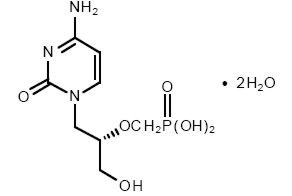
Cidofovir is a white crystalline powder with an aqueous solubility of ≥ 170 mg/mL at pH 6–8 and a log P (octanol/aqueous buffer, pH 7.1) value of -3.3.
VISTIDE is a sterile, hypertonic aqueous solution for intravenous infusion only. The solution is clear and colorless. It is supplied in clear glass vials, each containing 375 mg of anhydrous cidofovir in 5 mL aqueous solution at a concentration of 75 mg/mL. The formulation is pH-adjusted to 7.4 with sodium hydroxide and/or hydrochloric acid and contains no preservatives. The appropriate volume of VISTIDE must be removed from the single-use vial and diluted prior to administration (see DOSAGE AND ADMINISTRATION).
-
MICROBIOLOGY
Mechanism of Action
Cidofovir suppresses cytomegalovirus (CMV) replication by selective inhibition of viral DNA synthesis. Biochemical data support selective inhibition of CMV DNA polymerase by cidofovir diphosphate, the active intracellular metabolite of cidofovir. Cidofovir diphosphate inhibits herpesvirus polymerases at concentrations that are 8- to 600-fold lower than those needed to inhibit human cellular DNA polymerases alpha, beta, and gamma1, 2, 3. Incorporation of cidofovir into the growing viral DNA chain results in reductions in the rate of viral DNA synthesis.
In Vitro Susceptibility
Cidofovir is active in vitro against a variety of laboratory and clinical isolates of CMV and other herpesviruses (Table 1). Controlled clinical studies of efficacy have been limited to patients with AIDS and CMV retinitis.
Table 1. Cidofovir Inhibition of Virus Multiplication in Cell Culture Virus IC50 (µM) Wild-type CMV Isolates 0.5 – 2.8 HSV-1, HSV-2 12.7 – 31.7 Resistance
CMV isolates with reduced susceptibility to cidofovir have been selected in vitro in the presence of high concentrations of cidofovir4. IC50 values for selected resistant isolates ranged from 7–15 µM.
There are insufficient data at this time to assess the frequency or the clinical significance of the development of resistant isolates following VISTIDE administration to patients.
The possibility of viral resistance should be considered for patients who show a poor clinical response or experience recurrent retinitis progression during therapy.
Cross Resistance
Cidofovir-resistant isolates selected in vitro following exposure to increasing concentrations of cidofovir were assessed for susceptibility to ganciclovir and foscarnet4. All were cross resistant to ganciclovir, but remained susceptible to foscarnet. Ganciclovir- or ganciclovir/foscarnet-resistant isolates that are cross resistant to cidofovir have been obtained from drug naive patients and from patients following ganciclovir or ganciclovir/ foscarnet therapy. To date, the majority of ganciclovir-resistant isolates are UL97 gene product (phosphokinase) mutants and remain susceptible to cidofovir5. Reduced susceptibility to cidofovir, however, has been reported for DNA polymerase mutants of CMV which are resistant to ganciclovir6–9. To date, all clinical isolates which exhibit high level resistance to ganciclovir, due to mutations in both the DNA polymerase and UL97 genes, have been shown to be cross resistant to cidofovir. Cidofovir is active against some, but not all, CMV isolates which are resistant to foscarnet10–12. The incidence of foscarnet-resistant isolates that are resistant to cidofovir is not known.
A few triple-drug resistant isolates have been described. Genotypic analysis of two of these triple-resistant isolates revealed several point mutations in the CMV DNA polymerase gene. The clinical significance of the development of these cross-resistant isolates is not known.
-
CLINICAL PHARMACOLOGY
PHARMACOKINETICS
VISTIDE must be administered with probenecid. The pharmacokinetics of cidofovir, administered both without and with probenecid, are described below.
The pharmacokinetics of cidofovir without probenecid were evaluated in 27 HIV-infected patients with or without asymptomatic CMV infection. Dose-independent pharmacokinetics were demonstrated after one hr infusions of 1.0 (n = 5), 3.0 (n = 10), 5.0 (n = 2) and 10.0 (n = 8) mg/kg (See Table 2 for pharmacokinetic parameters). There was no evidence of cidofovir accumulation after 4 weeks of repeated administration of 3 mg/kg/week (n = 5) without probenecid. In patients with normal renal function, approximately 80 to 100% of the VISTIDE dose was recovered unchanged in urine within 24 hr (n = 27). The renal clearance of cidofovir was greater than creatinine clearance, indicating renal tubular secretion contributes to the elimination of cidofovir.
The pharmacokinetics of cidofovir administered with probenecid were evaluated in 12 HIV-infected patients with or without asymptomatic CMV infection and 10 patients with relapsing CMV retinitis. Dose-independent pharmacokinetics were observed for cidofovir, administered with probenecid, after one hr infusions of 3.0 (n = 12), 5.0 (n = 6), and 7.5 (n = 4) mg/kg (See Table 2). Approximately 70 to 85% of the VISTIDE dose administered with concomitant probenecid was excreted as unchanged drug within 24 hr. When VISTIDE was administered with probenecid, the renal clearance of cidofovir was reduced to a level consistent with creatinine clearance, suggesting that probenecid blocks active renal tubular secretion of cidofovir.
Table 2. Cidofovir Pharmacokinetic Parameters Following 3.0 and 5.0 mg/kg Infusions, Without and With Probenecid* PARAMETERS VISTIDE ADMINISTERED
WITHOUT PROBENECIDVISTIDE ADMINISTERED
WITH PROBENECID3 mg/kg (n = 10) 5 mg/kg (n = 2) 3 mg/kg (n = 12) 5 mg/kg (n = 6) AUC (µg∙hr/mL) 20.0 ± 2.3 28.3 25.7 ± 8.5 40.8 ± 9.0 Cmax (end of infusion)
(µg/mL)7.3 ± 1.4 11.5 9.8 ± 3.7 19.6 ± 7.2 Vdss (mL/kg) 537 ± 126
(n = 12)410 ± 102
(n = 18)Clearance
(mL/min/1.73 m2)179 ± 23.1
(n = 12)148 ± 38.8
(n = 18)Renal Clearance
(mL/min/1.73 m2)150 ± 26.9
(n = 12)98.6 ± 27.9
(n = 11)In vitro, cidofovir was less than 6% bound to plasma or serum proteins over the cidofovir concentration range 0.25 to 25 µg/mL. CSF concentrations of cidofovir following intravenous infusion of VISTIDE 5 mg/kg with concomitant probenecid and intravenous hydration were undetectable (< 0.1 µg/mL, assay detection threshold) at 15 minutes after the end of a 1 hr infusion in one patient whose corresponding serum concentration was 8.7 µg/mL.
SPECIAL POPULATIONS
Renal Insufficiency
Pharmacokinetic data collected from subjects with creatinine clearance values as low as 11 mL/min indicate that cidofovir clearance decreases proportionally with creatinine clearance.
High-flux hemodialysis has been shown to reduce the serum levels of cidofovir by approximately 75%.
Initiation of therapy with VISTIDE is contraindicated in patients with serum creatinine > 1.5 mg/dL, a calculated creatinine clearance ≤ 55 mL/min, or a urine protein ≥ 100 mg/dL (equivalent to ≥2+ proteinuria) (See CONTRAINDICATIONS).
-
INDICATION AND USAGE
VISTIDE is indicated for the treatment of CMV retinitis in patients with acquired immunodeficiency syndrome (AIDS). THE SAFETY AND EFFICACY OF VISTIDE HAVE NOT BEEN ESTABLISHED FOR TREATMENT OF OTHER CMV INFECTIONS (SUCH AS PNEUMONITIS OR GASTROENTERITIS), CONGENITAL OR NEONATAL CMV DISEASE, OR CMV DISEASE IN NON-HIV-INFECTED INDIVIDUALS.
-
DESCRIPTION OF CLINICAL TRIALS
Three phase II/III controlled trials of VISTIDE have been conducted in HIV-infected patients with CMV retinitis.
Delayed Versus Immediate Therapy (Study 105)
In stage 1 of this open-label trial, conducted by the Studies of the Ocular Complications of AIDS (SOCA) Clinical Research Group, 29 previously untreated patients with peripheral CMV retinitis were randomized to either immediate treatment with VISTIDE (5 mg/kg once a week for 2 weeks, then 3 mg/kg every other week) or to have VISTIDE delayed until progression of CMV retinitis13. In stage 2 of this trial, an additional 35 previously untreated patients with peripheral CMV retinitis were randomized to either immediate treatment with VISTIDE (5 mg/kg once a week for 2 weeks, then 5 mg/kg every other week), immediate treatment with VISTIDE (5 mg/kg once a week for 2 weeks, then 3 mg/kg every other week), or to have VISTIDE delayed until progression of CMV retinitis. Of the 64 patients in this study, 12 were randomized to 5 mg/kg maintenance therapy, 26 to 3 mg/kg maintenance therapy, and 26 to delayed therapy. Of the 12 patients enrolled in the 5 mg/kg maintenance group, 5 patients progressed, 5 patients discontinued therapy and 2 patients had no progression at study completion. Based on masked readings of retinal photographs, the median [95% confidence interval (CI)] time to retinitis progression was not reached (25, not reached) for the 5 mg/kg maintenance group. Median (95% CI) time to the alternative endpoint of retinitis progression or study drug discontinuation was 44 days (24, 207) for the 5 mg/kg maintenance group. Patients receiving 5 mg/kg maintenance had delayed time to retinitis progression compared to patients receiving 3 mg/kg maintenance or deferred therapy.
Delayed Versus Immediate Therapy (Study 106)
In an open-label trial, 48 previously untreated patients with peripheral CMV retinitis were randomized to either immediate treatment with VISTIDE (5 mg/kg once a week for 2 weeks, then 5 mg/kg every other week), or to have VISTIDE delayed until progression of CMV retinitis14. Patient baseline characteristics and disposition are shown in Table 3. Of 25 and 23 patients in the immediate and delayed groups respectively, 23 and 21 were evaluable for retinitis progression as determined by retinal photography. Based on masked readings of retinal photographs, the median [95% confidence interval (CI)] times to retinitis progression were 120 days (40, 134) and 22 days (10, 27) for the immediate and delayed therapy groups, respectively. This difference was statistically significant. However, because of the limited number of patients remaining on treatment over time (3 of 25 patients received VISTIDE for 120 days or longer), the median time to progression for the immediate therapy group was difficult to precisely estimate. Median (95% CI) times to the alternative endpoint of retinitis progression or study drug discontinuation (including adverse events, withdrawn consent, and systemic CMV disease) were 52 days (37, 85) and 22 days (13, 27) for the immediate and delayed therapy groups, respectively. This difference was statistically significant. Time to progression estimates from this study may not be directly comparable to estimates reported for other therapies.
Table 3. Patient Characteristics and Disposition (Study 106) Immediate Therapy Delayed Therapy (n = 25) (n = 23) Baseline Characteristics Age (years) 38 38 Sex (M/F) 24/1 22/1 Median CD4 Cell Count 6 9 Endpoints CMV Retinitis Progression 10 18 Discontinued Due to Adverse Event 6 0 Withdrew Consent 3* 1 Discontinued Due to Intercurrent Illness 2† 1† Discontinued Based
on Ophthalmological Examination1‡ 1‡ No Progression at Study Completion 1 0 Not Evaluable at Baseline 2 2 Dose-response study of VISTIDE (Study 107)
In an open-label trial, 100 patients with relapsing CMV retinitis were randomized to receive 5 mg/kg once a week for 2 weeks and then either 5 mg/kg (n = 49) or 3 mg/kg (n = 51) every other week. Enrolled patients had been diagnosed with CMV retinitis an average of 390 days prior to randomization and had received a median of 3.8 prior courses of systemic CMV therapy. Eighty-four of the 100 patients were considered evaluable for progression by serial retinal photographs (43 randomized to 5 mg/kg and 41 randomized to 3 mg/kg). Twenty-six and 21 patients discontinued therapy due to either an adverse event, intercurrent illness, excluded medication, or withdrawn consent in the 5 mg/kg and 3 mg/kg groups, respectively. Thirty-eight of the 100 randomized patients had progressed according to masked assessment of serial retinal photographs (13 randomized to 5 mg/kg and 25 randomized to 3 mg/kg). Using retinal photographs, the median (95% CI) times to retinitis progression for the 5 mg/kg and 3 mg/kg groups were 115 days (70, not reached) and 49 days (35, 52), respectively. This difference was statistically significant. Similar to Study 106, the median time to retinitis progression for the 5 mg/kg group was difficult to precisely estimate due to the limited number of patients remaining on treatment over time (4 of the 49 patients in the 5 mg/kg group were treated for 115 days or longer). Median (95% CI) times to the alternative endpoint of retinitis progression or study drug discontinuation were 49 days (38, 63) and 35 days (27, 39) for the 5 mg/kg and 3 mg/kg groups, respectively. This difference was statistically significant.
-
CONTRAINDICATIONS
Initiation of therapy with VISTIDE is contraindicated in patients with a serum creatinine > 1.5 mg/dL, a calculated creatinine clearance ≤ 55 mL/min, or a urine protein ≥ 100 mg/dL (equivalent to ≥ 2+ proteinuria).
VISTIDE is contraindicated in patients receiving agents with nephrotoxic potential. Such agents must be discontinued at least seven days prior to starting therapy with VISTIDE.
VISTIDE is contraindicated in patients with hypersensitivity to cidofovir.
VISTIDE is contraindicated in patients with a history of clinically severe hypersensitivity to probenecid or other sulfa-containing medications.
Direct intraocular injection of VISTIDE is contraindicated; direct injection of cidofovir has been associated with iritis, ocular hypotony, and permanent impairment of vision.
-
WARNINGS
Nephrotoxicity
Dose-dependent nephrotoxicity is the major dose-limiting toxicity related to VISTIDE administration. Cases of acute renal failure resulting in dialysis and/or contributing to death have occurred with as few as one or two doses of VISTIDE. Renal function (serum creatinine and urine protein) must be monitored within 48 hours prior to each dose of VISTIDE. Dose adjustment or discontinuation is required for changes in renal function (serum creatinine and/or urine protein) while on therapy. Proteinuria, as measured by urinalysis in a clinical laboratory, may be an early indicator of VISTIDE-related nephrotoxicity. Continued administration of VISTIDE may lead to additional proximal tubular cell injury, which may result in glycosuria, decreases in serum phosphate, uric acid, and bicarbonate, elevations in serum creatinine, and/or acute renal failure, in some cases, resulting in the need for dialysis. Patients with these adverse events occurring concurrently and meeting a criteria of Fanconi's syndrome have been reported. Renal function that did not return to baseline after drug discontinuation has been observed in clinical studies of VISTIDE.
Intravenous normal saline hydration and oral probenecid must accompany each VISTIDE infusion. Probenecid is known to interact with the metabolism or renal tubular excretion of many drugs (see PRECAUTIONS). The safety of VISTIDE has not been evaluated in patients receiving other known potentially nephrotoxic agents, such as intravenous aminoglycosides (e.g., tobramycin, gentamicin, and amikacin), amphotericin B, foscarnet, intravenous pentamidine, vancomycin, and non-steroidal anti-inflammatory agents (see DOSAGE AND ADMINISTRATION).
Preexisting Renal Impairment
Initiation of therapy with VISTIDE is contraindicated in patients with a baseline serum creatinine > 1.5 mg/dL, a creatinine clearance ≤ 55 mL/min, or a urine protein ≥ 100 mg/dL (equivalent to ≥ 2+ proteinuria).
Hematological Toxicity
Neutropenia may occur during VISTIDE therapy. Neutrophil count should be monitored while receiving VISTIDE therapy.
Decreased Intraocular Pressure/Ocular Hypotony
Decreased intraocular pressure may occur during VISTIDE therapy, and in some instances has been associated with decreased visual acuity. Intraocular pressure should be monitored during VISTIDE therapy.
Metabolic Acidosis
Decreased serum bicarbonate associated with proximal tubule injury and renal wasting syndrome (including Fanconi's syndrome) have been reported in patients receiving VISTIDE (see ADVERSE REACTIONS). Cases of metabolic acidosis in association with liver dysfunction and pancreatitis resulting in death have been reported in patients receiving VISTIDE.
-
PRECAUTIONS
General
Due to the potential for increased nephrotoxicity, doses greater than the recommended dose must not be administered and the frequency or rate of administration must not be exceeded (see DOSAGE AND ADMINISTRATION).
VISTIDE is formulated for intravenous infusion only and must not be administered by intraocular injection. Administration of VISTIDE by infusion must be accompanied by oral probenecid and intravenous saline prehydration (see DOSAGE AND ADMINISTRATION).
Uveitis/Iritis
Uveitis or iritis was reported in clinical trials and during postmarketing in patients receiving VISTIDE therapy. Treatment with topical corticosteroids with or without topical cycloplegic agents should be considered. Patients should be monitored for signs and symptoms of uveitis/iritis during VISTIDE therapy.
Information for Patients
Patients should be advised that VISTIDE is not a cure for CMV retinitis, and that they may continue to experience progression of retinitis during and following treatment. Patients receiving VISTIDE should be advised to have regular follow-up ophthalmologic examinations. Patients may also experience other manifestations of CMV disease despite VISTIDE therapy.
HIV-infected patients may continue taking antiretroviral therapy, but those taking zidovudine should be advised to temporarily discontinue zidovudine administration or decrease their zidovudine dose by 50%, on days of VISTIDE administration only, because probenecid reduces metabolic clearance of zidovudine.
Patients should be informed of the major toxicity of VISTIDE, namely renal impairment, and that dose modification, including reduction, interruption, and possibly discontinuation, may be required. Close monitoring of renal function (routine urinalysis and serum creatinine) while on therapy should be emphasized.
The importance of completing a full course of probenecid with each VISTIDE dose should be emphasized. Patients should be warned of potential adverse events caused by probenecid (e.g., headache, nausea, vomiting, and hypersensitivity reactions). Hypersensitivity/allergic reactions may include rash, fever, chills and anaphylaxis. Administration of probenecid after a meal or use of antiemetics may decrease the nausea. Prophylactic or therapeutic antihistamines and/or acetaminophen can be used to ameliorate hypersensitivity reactions.
Patients should be advised that cidofovir causes tumors, primarily mammary adenocarcinomas, in rats. VISTIDE should be considered a potential carcinogen in humans (See Carcinogenesis, Mutagenesis, & Impairment of Fertility). Women should be advised of the limited enrollment of women in clinical trials of VISTIDE.
Patients should be advised that VISTIDE caused reduced testes weight and hypospermia in animals. Such changes may occur in humans and cause infertility. Women of childbearing potential should be advised that cidofovir is embryotoxic in animals and should not be used during pregnancy. Women of childbearing potential should be advised to use effective contraception during and for 1 month following treatment with VISTIDE. Men should be advised to practice barrier contraceptive methods during and for 3 months after treatment with VISTIDE.
Drug Interactions
Probenecid
Probenecid is known to interact with the metabolism or renal tubular excretion of many drugs (e.g., acetaminophen, acyclovir, angiotensin-converting enzyme inhibitors, aminosalicylic acid, barbiturates, benzodiazepines, bumetanide, clofibrate, methotrexate, famotidine, furosemide, nonsteroidal anti-inflammatory agents, theophylline, and zidovudine). Concomitant medications should be carefully assessed. Zidovudine should either be temporarily discontinued or decreased by 50% when coadministered with probenecid on the day of VISTIDE infusion.
Nephrotoxic agents
Concomitant administration of VISTIDE and agents with nephrotoxic potential [e.g., intravenous aminoglycosides (e.g., tobramycin, gentamicin, and amikacin), amphotericin B, foscarnet, intravenous pentamidine, vancomycin, and nonsteroidal anti-inflammatory agents] is contraindicated. Such agents must be discontinued at least seven days prior to starting therapy with VISTIDE.
Carcinogenesis, Mutagenesis, & Impairment of Fertility
Chronic, two-year carcinogenicity studies in rats and mice have not been carried out to evaluate the carcinogenic potential of cidofovir. However, a 26-week toxicology study evaluating once weekly subscapular subcutaneous injections of cidofovir in rats was terminated at 19 weeks because of the induction, in females, of palpable masses, the first of which was detected after six doses. The masses were diagnosed as mammary adenocarcinomas which developed at doses as low as 0.6 mg/kg/week, equivalent to 0.04 times the human systemic exposure at the recommended intravenous VISTIDE dose based on AUC comparisons.
In a 26-week intravenous toxicology study in which rats received 0.6, 3, or 15 mg/kg cidofovir once weekly, a significant increase in mammary adenocarcinomas in female rats as well as a significant incidence of Zymbal's gland carcinomas in male and female rats were seen at the high dose but not at the lower two doses. The high dose was equivalent to 1.1 times the human systemic exposure at the recommended dose of VISTIDE, based on comparisons of AUC measurements. In light of the results of these studies, cidofovir should be considered to be a carcinogen in rats as well as a potential carcinogen in humans.
Cynomolgus monkeys received intravenous cidofovir, alone and in conjunction with concomitant oral probenecid, intravenously once weekly for 52 weeks at doses resulting in exposures of approximately 0.7 times the human systemic exposure at the recommended dose of VISTIDE. No tumors were detected. However, the study was not designed as a carcinogenicity study due to the small number of animals at each dose and the short duration of treatment.
No mutagenic response was observed in microbial mutagenicity assays involving Salmonella typhimurium (Ames) and Escherichia coli in the presence and absence of metabolic activation. An increase in micronucleated polychromatic erythrocytes in vivo was seen in mice receiving ≥ 2000 mg/kg, a dosage approximately 65-fold higher than the maximum recommended clinical intravenous VISTIDE dose based on body surface area estimations. Cidofovir induced chromosomal aberrations in human peripheral blood lymphocytes in vitro without metabolic activation. At the 4 cidofovir levels tested, the percentage of damaged metaphases and number of aberrations per cell increased in a concentration-dependent manner.
Studies showed that cidofovir caused inhibition of spermatogenesis in rats and monkeys. However, no adverse effects on fertility or reproduction were seen following once weekly intravenous injections of cidofovir in male rats for 13 consecutive weeks at doses up to 15 mg/kg/week (equivalent to 1.1 times the recommended human dose based on AUC comparisons). Female rats dosed intravenously once weekly at 1.2 mg/kg/week (equivalent to 0.09 times the recommended human dose based on AUC) or higher, for up to 6 weeks prior to mating and for 2 weeks post mating had decreased litter sizes and live births per litter and increased early resorptions per litter. Peri- and post-natal development studies in which female rats received subcutaneous injections of cidofovir once daily at doses up to 1.0 mg/kg/day from day 7 of gestation through day 21 postpartum (approximately 5 weeks) resulted in no adverse effects on viability, growth, behavior, sexual maturation or reproductive capacity in the offspring.
Pregnancy
Category C
Cidofovir was embryotoxic (reduced fetal body weights) in rats at 1.5 mg/kg/day and in rabbits at 1.0 mg/kg/day, doses which were also maternally toxic, following daily intravenous dosing during the period of organogenesis. The no-observable-effect levels for embryotoxicity in rats (0.5 mg/kg/day) and in rabbits (0.25 mg/kg/day) were approximately 0.04 and 0.05 times the clinical dose (5 mg/kg every other week) based on AUC, respectively. An increased incidence of fetal external, soft tissue and skeletal anomalies (meningocele, short snout, and short maxillary bones) occurred in rabbits at the high dose (1.0 mg/kg/day) which was also maternally toxic. There are no adequate and well-controlled studies in pregnant women. VISTIDE should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether cidofovir is excreted in human milk. Since many drugs are excreted in human milk and because of the potential for adverse reactions as well as the potential for tumorigenicity shown for cidofovir in animal studies, VISTIDE should not be administered to nursing mothers. The U.S. Public Health Service Centers for Disease Control and Prevention advises HIV-infected women not to breast-feed to avoid postnatal transmission of HIV to a child who may not yet be infected.
Pediatric Use
Safety and effectiveness in children have not been studied. The use of VISTIDE in children with AIDS warrants extreme caution due to the risk of long-term carcinogenicity and reproductive toxicity. Administration of VISTIDE to children should be undertaken only after careful evaluation and only if the potential benefits of treatment outweigh the risks.
Geriatric Use
No studies of the safety or efficacy of VISTIDE in patients over the age of 60 have been conducted. Since elderly individuals frequently have reduced glomerular filtration, particular attention should be paid to assessing renal function before and during VISTIDE administration (see DOSAGE AND ADMINISTRATION).
-
ADVERSE REACTIONS
- 1.
- Nephrotoxicity: Renal toxicity, as manifested by ≥ 2+ proteinuria, serum creatinine elevations of ≥ 0.4 mg/dL, or decreased creatinine clearance ≤ 55 mL/min, occurred in 79 of 135 (59%) patients receiving VISTIDE at a maintenance dose of 5 mg/kg every other week. Maintenance dose reductions from 5 mg/kg to 3 mg/kg due to proteinuria or serum creatinine elevations were made in 12 of 41 (29%) patients who had not received prior therapy for CMV retinitis (Study 106) and in 19 of 74 (26%) patients who had received prior therapy for CMV retinitis (Study 107). Prior foscarnet use has been associated with an increased risk of nephrotoxicity; therefore, such patients must be monitored closely (see CONTRAINDICATIONS, WARNINGS, DOSAGE AND ADMINISTRATION).
- 2.
- Neutropenia: In clinical trials, at the 5 mg/kg maintenance dose, a decrease in absolute neutrophil count to ≤ 500 cells/mm3 occurred in 24% of patients. Granulocyte colony stimulating factor (GCSF) was used in 39% of patients.
- 3.
- Decreased Intraocular Pressure/Ocular Hypotony: Among the subset of patients monitored for intraocular pressure changes, a ≥ 50% decrease from baseline intraocular pressure was reported in 17 of 70 (24%) patients at the 5 mg/kg maintenance dose. Severe hypotony (intraocular pressure of 0–1 mm Hg) has been reported in 3 patients. Risk of ocular hypotony may be increased in patients with preexisting diabetes mellitus.
- 4.
- Anterior Uveitis/Iritis: Uveitis or iritis has been reported in clinical trials and during postmarketing in patients receiving VISTIDE therapy. Uveitis or iritis was reported in 15 of 135 (11%) patients receiving 5 mg/kg maintenance dosing. Treatment with topical corticosteroids with or without topical cycloplegic agents may be considered. Patients should be monitored for signs and symptoms of uveitis/iritis during VISTIDE therapy.
- 5.
- Metabolic Acidosis: A diagnosis of Fanconi's syndrome, as manifested by multiple abnormalities of proximal renal tubular function, was reported in 1% of patients. Decreases in serum bicarbonate to ≤ 16 mEq/L occurred in 16% of cidofovir-treated patients. Cases of metabolic acidosis in association with liver dysfunction and pancreatitis resulting in death have been reported in patients receiving VISTIDE.
In clinical trials, VISTIDE was withdrawn due to adverse events in 39% of patients treated with 5 mg/kg every other week as maintenance therapy.
The incidence of adverse reactions reported as serious in three controlled clinical studies in patients with CMV retinitis, regardless of presumed relationship to drug, is listed in Table 4.
Table 4. Serious Clinical Adverse Events or Laboratory Abnormalities Occurring in > 5% of Patients N = 135*
# patients (%)- *
- Patients receiving 5 mg/kg maintenance regimen in Studies 105, 106 and 107.
- †
- Defined as decreased intraocular pressure (IOP) to ≤ 50% that at baseline. Based on 70 patients receiving 5 mg/kg maintenance dosing (Studies 105, 106 and 107), for whom baseline and follow-up IOP determinations were recorded.
Proteinuria (≥ 100 mg/dL) 68 (50) Neutropenia (≤ 500 cells/mm3) 33 (24) Decreased Intraocular Pressure† 17 (24) Decreased Serum Bicarbonate (≤ 16 mEq/L) 21 (16) Fever 19 (14) Infection 16 (12) Creatinine Elevation (≥ 2.0 mg/dL) 16 (12) Pneumonia 12 (9) Dyspnea 11 (8) Nausea with Vomiting 10 (7) The most frequently reported adverse events regardless of relationship to study drugs (cidofovir or probenecid) or severity are shown in Table 5.
The following additional list of adverse events/intercurrent illnesses have been observed in clinical studies of VISTIDE and are listed below regardless of causal relationship to VISTIDE. Evaluation of these reports was difficult because of the diverse manifestations of the underlying disease and because most patients received numerous concomitant medicines.
Body as a Whole: abdominal pain, accidental injury, AIDS, allergic reaction, back pain, catheter blocked, cellulitis, chest pain, chills and fever, cryptococcosis, cyst, death, face edema, flu-like syndrome, hypothermia, injection site reaction, malaise, mucous membrane disorder, neck pain, overdose, photosensitivity reaction, sarcoma, sepsis
Cardiovascular System: cardiomyopathy, cardiovascular disorder, congestive heart failure, hypertension, hypotension, migraine, pallor, peripheral vascular disorder, phlebitis, postural hypotension, shock, syncope, tachycardia, vascular disorder, edema
Digestive System: cholangitis, colitis, constipation, esophagitis, dyspepsia, dysphagia, fecal incontinence, flatulence, gastritis, gastrointestinal hemorrhage, gingivitis, hepatitis, hepatomegaly, hepatosplenomegaly, jaundice, abnormal liver function, liver damage, liver necrosis, melena, pancreatitis, proctitis, rectal disorder, stomatitis, aphthous stomatitis, tongue discoloration, mouth ulceration, tooth caries
Endocrine System: adrenal cortex insufficiency
Hemic & Lymphatic System: hypochromic anemia, leukocytosis, leukopenia, lymphadenopathy, lymphoma like reaction, pancytopenia, splenic disorder, splenomegaly, thrombocytopenia, thrombocytopenic purpura
Metabolic & Nutritional System: cachexia, dehydration, edema, hypercalcemia, hyperglycemia, hyperkalemia, hyperlipemia, hypocalcemia, hypoglycemia, hypoglycemic reaction, hypokalemia, hypomagnesemia, hyponatremia, hypophosphatemia, hypoproteinemia, increased alkaline phosphatase, increased BUN, increased lactic dehydrogenase, increased SGOT, increased SGPT, peripheral edema, respiratory alkalosis, thirst, weight loss, weight gain
Musculoskeletal System: arthralgia, arthrosis, bone necrosis, bone pain, joint disorder, leg cramps, myalgia, myasthenia, pathological fracture
Nervous System: abnormal dreams, abnormal gait, acute brain syndrome, agitation, amnesia, anxiety, ataxia, cerebrovascular disorder, confusion, convulsion, delirium, dementia, depression, dizziness, drug dependence, dry mouth, encephalopathy, facial paralysis, hallucinations, hemiplegia, hyperesthesia, hypertonia, hypotony, incoordination, increased libido, insomnia, myoclonus, nervousness, neuropathy, paresthesia, personality disorder, somnolence, speech disorder, tremor, twitching, vasodilatation, vertigo
Respiratory System: asthma, bronchitis, epistaxis, hemoptysis, hiccup, hyperventilation, hypoxia, increased sputum, larynx edema, lung disorder, pharyngitis, pneumothorax, rhinitis, sinusitis
Skin & Appendages: acne, angioedema, dry skin, eczema, exfoliative dermatitis, furunculosis, herpes simplex, nail disorder, pruritus, rash, seborrhea, skin discoloration, skin disorder, skin hypertrophy, skin ulcer, sweating, urticaria
Special Senses: abnormal vision, amblyopia, blindness, cataract, conjunctivitis, corneal lesion, corneal opacity, diplopia, dry eyes, ear disorder, ear pain, eye disorder, eye pain, hyperacusis, iritis, keratitis, miosis, otitis externa, otitis media, refraction disorder, retinal detachment, retinal disorder, taste perversion, tinnitus, uveitis, visual field defect, hearing loss
Urogenital System: decreased creatinine clearance, dysuria, glycosuria, hematuria, kidney stone, mastitis, metorrhagia, nocturia, polyuria, prostatic disorder, toxic nephrophathy, urethritis, urinary casts, urinary incontinence, urinary retention, urinary tract infection
Table 5. All Clinical Adverse Events, Laboratory Abnormalities or Intercurrent Illnesses Regardless of Severity Occurring in > 15% of Patients N = 115*
# patients (%)- *
- Patients receiving 5 mg/kg maintenance regimen in Studies 106 and 107.
Any Adverse Event 115 (100) Proteinuria (≥ 30 mg/dL) 101 (88) Nausea +/- Vomiting 79 (69) Fever 67 (58) Neutropenia (< 750 cells/mm3) 50 (43) Asthenia 50 (43) Headache 34 (30) Rash 34 (30) Infection 32 (28) Alopecia 31 (27) Diarrhea 30 (26) Pain 29 (25) Creatinine Elevation (> 1.5 mg/dL) 28 (24) Anemia 28 (24) Anorexia 26 (23) Dyspnea 26 (23) Chills 25 (22) Increased Cough 22 (19) Oral Moniliasis 21 (18) Reporting of Adverse Reactions
Malignancies or serious adverse reactions that occur in patients who have received VISTIDE should be reported to Gilead in writing to the Director of Clinical Research, Gilead Sciences, Inc., 333 Lakeside Drive, Foster City, CA 94404 or by calling 1-800-GILEAD-5 (445-3235), or to FDA MedWatch 1-800-FDA-1088/fax 1-800-FDA-0178.
-
OVERDOSAGE
Two cases of cidofovir overdose have been reported. These patients received single doses of VISTIDE at 16.3 mg/kg and 17.4 mg/kg, respectively, with concomitant oral probenecid and intravenous hydration. In both cases, the patients were hospitalized and received oral probenecid (one gram three times daily) and vigorous intravenous hydration with normal saline for 3 to 5 days. Significant changes in renal function were not observed in either patient.
-
DOSAGE AND ADMINISTRATION
VISTIDE MUST NOT BE ADMINISTERED BY INTRAOCULAR INJECTION.
Dosage
THE RECOMMENDED DOSAGE, FREQUENCY, OR INFUSION RATE MUST NOT BE EXCEEDED. VISTIDE MUST BE DILUTED IN 100 MILLILITERS 0.9% (NORMAL) SALINE PRIOR TO ADMINISTRATION. TO MINIMIZE POTENTIAL NEPHROTOXICITY, PROBENECID AND INTRAVENOUS SALINE PREHYDRATION MUST BE ADMINISTERED WITH EACH VISTIDE INFUSION.
Induction Treatment
The recommended induction dose of VISTIDE for patients with a serum creatinine of ≤ 1.5 mg/dL, a calculated creatinine clearance > 55 mL/min, and a urine protein < 100 mg/dL (equivalent to < 2+ proteinuria) is 5 mg/kg body weight (given as an intravenous infusion at a constant rate over 1 hr) administered once weekly for two consecutive weeks. Because serum creatinine in patients with advanced AIDS and CMV retinitis may not provide a complete picture of the patient's underlying renal status, it is important to utilize the Cockcroft-Gault formula to more precisely estimate creatinine clearance (CrCl). As creatinine clearance is dependent on serum creatinine and patient weight, it is necessary to calculate clearance prior to initiation of VISTIDE. CrCl (mL/min) should be calculated according to the following formula:
Creatinine clearance for males = [140-age (years)] × [body wt (kg)]
72 × [serum creatinine (mg/dL)]Creatinine clearance for females = [140-age (years)] × [body wt (kg)] × 0.85
72 × [serum creatinine (mg/dL)]Maintenance Treatment
The recommended maintenance dose of VISTIDE is 5 mg/kg body weight (given as an intravenous infusion at a constant rate over 1 hr), administered once every 2 weeks.
Dose Adjustment
Changes in Renal Function During VISTIDE Therapy
The maintenance dose of VISTIDE must be reduced from 5 mg/kg to 3 mg/kg for an increase in serum creatinine of 0.3 – 0.4 mg/dL above baseline. VISTIDE therapy must be discontinued for an increase in serum creatinine of ≥ 0.5 mg/dL above baseline or development of ≥ 3+ proteinuria.
Probenecid
Probenecid must be administered orally with each VISTIDE dose. Two grams must be administered 3 hr prior to the VISTIDE dose and one gram administered at 2 and again at 8 hr after completion of the 1 hr VISTIDE infusion (for a total of 4 grams).
Ingestion of food prior to each dose of probenecid may reduce drug-related nausea and vomiting. Administration of an antiemetic may reduce the potential for nausea associated with probenecid ingestion. In patients who develop allergic or hypersensitivity symptoms to probenecid, the use of an appropriate prophylactic or therapeutic antihistamine and/or acetaminophen should be considered (see CONTRAINDICATIONS).
Hydration
Patients must receive at least one liter of 0.9% (normal) saline solution intravenously with each infusion of VISTIDE. The saline solution should be infused over a 1–2 hr period immediately before the VISTIDE infusion. Patients who can tolerate the additional fluid load should receive a second liter. If administered, the second liter of saline should be initiated either at the start of the VISTIDE infusion or immediately afterwards, and infused over a 1 to 3 hr period.
Method of Preparation and Administration
Inspect vials visually for particulate matter and discoloration prior to administration. If particulate matter or discoloration is observed, the vial should not be used. With a syringe, extract the appropriate volume of VISTIDE from the vial and transfer the dose to an infusion bag containing 100 mL 0.9% (normal) saline solution. Infuse the entire volume intravenously into the patient at a constant rate over a 1 hr period. Use of a standard infusion pump for administration is recommended.
It is recommended that VISTIDE infusion admixtures be administered within 24 hr of preparation and that refrigerator or freezer storage not be used to extend this 24 hr limit.
If admixtures are not intended for immediate use, they may be stored under refrigeration (2–8°C) for no more than 24 hr. Refrigerated admixtures should be allowed to equilibrate to room temperature prior to use.
The chemical stability of VISTIDE admixtures was demonstrated in polyvinyl chloride composition and ethylene/propylene copolymer composition commercial infusion bags, and in glass bottles. No data are available to support the addition of other drugs or supplements to the cidofovir admixture for concurrent administration.
VISTIDE is supplied in single-use vials. Partially used vials should be discarded (see Handling and Disposal).
Compatibility with Ringer's solution, Lactated Ringer's solution or bacteriostatic infusion fluids has not been evaluated.
Handling and Disposal
Due to the mutagenic properties of cidofovir, adequate precautions including the use of appropriate safety equipment are recommended for the preparation, administration, and disposal of VISTIDE. The National Institutes of Health presently recommends that such agents be prepared in a Class II laminar flow biological safety cabinet and that personnel preparing drugs of this class wear surgical gloves and a closed front surgical-type gown with knit cuffs. If VISTIDE contacts the skin, wash membranes and flush thoroughly with water. Excess VISTIDE and all other materials used in the admixture preparation and administration should be placed in a leak-proof, puncture-proof container. The recommended method of disposal is high temperature incineration.
Patient Monitoring
Serum creatinine and urine protein must be monitored within 48 hours prior to each dose. White blood cell counts with differential should be monitored prior to each dose. In patients with proteinuria, intravenous hydration should be administered and the test repeated. Intraocular pressure, visual acuity and ocular symptoms should be monitored periodically.
- HOW SUPPLIED
- SPL UNCLASSIFIED SECTION
-
REFERENCES
- Ho HT, Woods KL, Bronson JJ, De Boeck H, Martin JC and Hitchcock MJM. Intracellular Metabolism of the Antiherpesvirus Agent (S)-1-[3-hydroxy-2-(phosphonylmethoxy) propyl]cytosine. Mol Pharmacol 41:197–202, 1992.
- Cherrington JM, Allen SJW, McKee BH, and Chen MS. Kinetic Analysis of the Interaction Between the Diphosphate of (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine, zalcitabineTP, zidovudineTP, and FIAUTP with Human DNA Polymerases b and g. Biochem Pharmacol 48:1986–1988, 1994.
- Xiong X, Smith JL, Kim C, Huang E, and Chen MS. Kinetic Analysis of the Interaction of Cidofovir Diphosphate with Human Cytomegalovirus DNA Polymerase. Biochem Pharmacol 51:1563–1567, 1996.
- Cherrington JM, Mulato AS, Fuller MD, Chen MS. In Vitro Selection of a Human Cytomegalovirus (HCMV) that is Resistant to Cidofovir. 35th International Conference on Antimicrobial Agents and Chemotherapy (ICAAC), San Francisco, CA. Abstract H117, 1995.
- Stanat SC, Reardon JE, Erice A, Jordan MC, Drew WL, and Biron KK. Ganciclovir-Resistant Cytomegalovirus Clinical Isolates: Mode of Resistance to Ganciclovir. Antimicrob Agents Chemother 35:2191–2197, 1991.
- Sullivan V, Biron KK, Talarico C, Stanat SC, Davis M, Pozzi M, and Coen DM. A Point Mutation in the Human Cytomegalovirus DNA Polymerase Gene Confers Resistance to Ganciclovir and phosphonylmethoxyalkyl Derivatives. Antimicrob Agents Chemother 37:19–25, 1993.
- Tatarowicz WA, Lurain NS, and Thompson KD. A Ganciclovir-Resistant Clinical Isolate of Human Cytomegalovirus Exhibiting Cross-Resistance to other DNA Polymerase Inhibitors. J Infect Dis 166:904–907, 1992.
- Lurain NS, Thompson KD, Holmes EW, and Read GS. Point Mutations in the DNA Polymerase Gene of Human Cytomegalovirus that Result in Resistance to Antiviral Agents. J Virol 66:7146–7152, 1992.
- Smith IL, Cherrington JM, Jiles RE, Fuller MD, Freeman WR, Spector SA. High-level Resistance of Cytomegalovirus to Ganciclovir is Associated with Alterations in both the UL97 and DNA Polymerase Genes. J Infect Dis 176:69–77, 1997.
- Sullivan V and Coen DM. Isolation of Foscarnet-Resistant Human Cytomegalovirus Patterns of Resistance and Sensitivity to Other Antiviral Drugs. J Infect Dis 164:781–784, 1991.
- Snoeck R, Andrei G, and De Clercq E. Patterns of Resistance and Sensitivity to Antiviral Compounds of Drug-Resistant Strains of Human Cytomegalovirus Selected in Vitro. Eur J Clin Microbiol Infect Dis 15:574–579, 1996.
- Baldanti F, Underwood MR, Stanat SC, Biron KK, Chou S, Sarasini A, Silini E, and Gerna G. Single Amino Acid Changes in the DNA Polymerase Confer Foscarnet Resistance and Slow-Growth Phenotype, While Mutations in the UL97-Encoded Phosphotransferase Confer Ganciclovir Resistance in Three Double-Resistant Human Cytomegalovirus Strains Recovered from Patients with AIDS. J Virol 70:1390–1395, 1996.
- The Studies of Ocular Complications of AIDS Research Group in Collaboration with the AIDS Clinical Trials Group. Cidofovir (HPMPC) for the Treatment of Cytomegalovirus Retinitis in Patients with AIDS: the HPMPC Peripheral Cytomegalovirus Retinitis Trial. Ann Intern Med 126:264–274, 1997.
- Lalezari JP, Stagg RJ, Kupperman BD, et al. Intravenous Cidofovir for Peripheral Cytomegalovirus Retinitis in Patients with AIDS. A Randomized, Controlled Trial. Ann Intern Med 126:257–263, 1997.
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 5 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
VISTIDE
cidofovir injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:61958-0101 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CIDOFOVIR (UNII: JIL713Q00N) (CIDOFOVIR ANHYDROUS - UNII:768M1V522C) CIDOFOVIR ANHYDROUS 75 mg in 1 mL Inactive Ingredients Ingredient Name Strength WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) HYDROCHLORIC ACID (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:61958-0101-1 1 in 1 CARTON 1 5 mL in 1 VIAL, SINGLE-USE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA020638 06/26/1996 Labeler - Gilead Sciences, Inc. (185049848)
