Label: FROVA- frovatriptan succinate tablet, film coated

-
Contains inactivated NDC Code(s)
NDC Code(s): 16590-396-09 - Packager: STAT RX USA LLC
- This is a repackaged label.
- Source NDC Code(s): 63481-025
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated June 18, 2010
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
FROVA (frovatriptan succinate) tablets contain frovatriptan succinate, a selective 5-hydroxy-tryptamine1 (5-HT1B/1D) receptor subtype agonist, as the active ingredient. Frovatriptan succinate is chemically designated as R-(+) 3-methylamino-6-carboxamido-1,2,3,4-tetrahydrocarbazole monosuccinate monohydrate and it has the following structure:
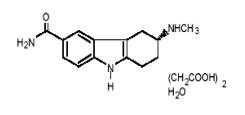
The empirical formula is C14H17N3O.C4H6O4.H2O, representing a molecular weight of 379.4. Frovatriptan succinate is a white to off-white powder that is soluble in water. Each FROVA tablet for oral administration contains 3.91 mg frovatriptan succinate, equivalent to 2.5 mg of frovatriptan base. Each tablet also contains the inactive ingredients lactose NF, microcrystalline cellulose NF, colloidal silicon dioxide NF, sodium starch glycolate NF, magnesium stearate NF, hydroxypropylmethylcellulose USP, polyethylene glycol 3000 USP, triacetin USP, and titanium dioxide USP.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Frovatriptan is a 5-HT receptor agonist that binds with high affinity for 5-HT1B and 5-HT1D receptors. Frovatriptan has no significant effects on GABAA mediated channel activity and has no significant affinity for benzodiazepine binding sites.
Frovatriptan is believed to act on extracerebral, intracranial arteries and to inhibit excessive dilation of these vessels in migraine. In anesthetized dogs and cats, intravenous administration of frovatriptan produced selective constriction of the carotid vascular bed and had no effect on blood pressure (both species) or coronary resistance (in dogs).
Pharmacokinetics
Mean maximum blood concentrations (Cmax) in patients are achieved approximately 2 - 4 hours after administration of a single oral dose of frovatriptan 2.5 mg. The absolute bioavailability of an oral dose of frovatriptan 2.5 mg in healthy subjects is about 20% in males and 30% in females. Food has no significant effect on the bioavailability of frovatriptan, but delays tmax by one hour.
Binding of frovatriptan to serum proteins is low (approximately 15%). Reversible binding to blood cells at equilibrium is approximately 60%, resulting in a blood:plasma ratio of about 2:1 in both males and females. The mean steady state volume of distribution of frovatriptan following intravenous administration of 0.8 mg is 4.2 L/kg in males and 3.0 L/kg in females.
In vitro, cytochrome P450 1A2 appears to be the principal enzyme involved in the metabolism of frovatriptan. Following administration of a single oral dose of radiolabeled frovatriptan 2.5 mg to healthy male and female subjects, 32% of the dose was recovered in urine and 62% in feces. Radiolabeled compounds excreted in urine were unchanged frovatriptan, hydroxylated frovatriptan, N-acetyl desmethyl frovatriptan, hydroxylated N-acetyl desmethyl frovatriptan and desmethyl frovatriptan, together with several other minor metabolites. Desmethyl frovatriptan has lower affinity for 5-HT1B/1D receptors compared to the parent compound. The N-acetyl desmethyl metabolite has no significant affinity for 5-HT receptors. The activity of the other metabolites is unknown.
After an intravenous dose, mean clearance of frovatriptan was 220 and 130 mL/min in males and females, respectively. Renal clearance accounted for about 40% (82 mL/min) and 45% (60 mL/min) of total clearance in males and females, respectively. The mean terminal elimination half-life of frovatriptan in both males and females is approximately 26 hours.
The pharmacokinetics of frovatriptan are similar in migraine patients and healthy subjects.
Special Populations
Age:
Mean AUC of frovatriptan was 1.5- to 2-fold higher in healthy elderly subjects (age 65 – 77 years) compared to those in healthy younger subjects (age 21 - 37 years). There was no difference in tmax or t1/2 between the two populations.
Gender:
There was no difference in the mean terminal elimination half-life of frovatriptan in males and females. Bioavailability was higher, and systemic exposure to frovatriptan was approximately 2-fold greater, in females than males, irrespective of age.
Renal Impairment:
Since less than 10% of FROVA is excreted in urine after an oral dose, it is unlikely that the exposure to frovatriptan will be affected by renal impairment. The pharmacokinetics of frovatriptan following a single oral dose of 2.5 mg was not different in patients with renal impairment (5 males and 6 females, creatinine clearance 16 - 73 mL/min) and in subjects with normal renal function.
Hepatic Impairment:
There is no clinical or pharmacokinetic experience with FROVA in patients with severe hepatic impairment. The AUC in subjects with mild (Child-Pugh 5 - 6) to moderate (Child-Pugh 7 - 9) hepatic impairment is about twice as high as the AUC in young, healthy subjects, but within the range found among normal elderly subjects.
Drug Interactions (see also PRECAUTIONS, Drug Interactions)
Frovatriptan is not an inhibitor of human monoamine oxidase (MAO) enzymes or cytochrome P450 (isozymes 1A2, 2C9, 2C19, 2D6, 2E1, 3A4) in vitro at concentrations up to 250 to 500- fold higher than the highest blood concentrations observed in man at a dose of 2.5 mg. No induction of drug metabolizing enzymes was observed following multiple dosing of frovatriptan to rats or on addition to human hepatocytes in vitro. Although no clinical studies have been performed, it is unlikely that frovatriptan will affect the metabolism of co-administered drugs metabolized by these mechanisms.
Oral contraceptives:
Retrospective analysis of pharmacokinetic data from females across trials indicated that the mean Cmax and AUC of frovatriptan are 30% higher in those subjects taking oral contraceptives compared to those not taking oral contraceptives.
Ergotamine:
The AUC and Cmax of frovatriptan (2 x 2.5 mg dose) were reduced by approximately 25% when co-administered with ergotamine tartrate.
Propranolol:
Propranolol increased the AUC of frovatriptan 2.5 mg in males by 60% and in females by 29%. The Cmax of frovatriptan was increased 23% in males and 16% in females in the presence of propranolol. The tmax as well as half-life of frovatriptan, though slightly longer in the females, were not affected by concomitant administration of propranolol.
Clinical Trials
The efficacy of FROVA in the acute treatment of migraine headaches was demonstrated in five randomized, double-blind, placebo-controlled, outpatient trials. Two of these were dose-finding studies in which patients were randomized to receive doses of frovatriptan ranging from 0.5 - 40 mg. The three studies evaluating only one dose studied 2.5 mg. In these controlled short-term studies combined, patients were predominately female (88%) and Caucasian (94%) with a mean age of 42 years (range 18 - 69). Patients were instructed to treat a moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed for up to 24 hours after dosing. The associated symptoms nausea, vomiting, photophobia and phonophobia were also assessed. Maintenance of response was assessed for up to 24 hours post dose. In two of the trials a second dose of FROVA was provided after the initial treatment, to treat recurrence of the headache within 24 hours. Other medication, excluding other 5-HT1 agonists and ergotamine containing compounds, was permitted from 2 hours after the first dose of FROVA. The frequency and time to use of additional medications were also recorded.
In all five placebo-controlled trials, the percentage of patients achieving a headache response 2 hours after treatment was significantly greater for those taking FROVA compared to those taking placebo (Table 1).
Lower doses of frovatriptan (1 mg or 0.5 mg) were not effective at 2 hours. Higher doses (5 mg to 40 mg) of frovatriptan showed no added benefit over 2.5 mg but did cause a greater incidence of adverse events.
Table 1 Percentage of Patients with Headache Response (Mild or No Headache) 2 Hours Following Treatmenta aITT observed data, excludes patients who had missing data or were asleep;
*p<0.05,
**p<0.001 in comparison with placebo
Trial FROVA
(frovatriptan 2.5 mg)Placebo 1 42%* (n=90) 22% (n=91) 2 38%* (n=121) 25% (n=115) 3 39%* (n=187) 21% (n=99) 4 46%** (n=672) 27% (n=347) 5 37%** (n=438) 23% (n=225) Comparisons of drug performance based upon results obtained in different clinical trials are never reliable. Because trials are conducted at different times, with different samples of patients, by different investigators, employing different criteria and/or different interpretations of the same criteria, under different conditions (dose, dosing regimen, etc.), quantitative estimates of treatment response and the timing of response may be expected to vary considerably from study to study.
The estimated probability of achieving an initial headache response by 2 hours following treatment is depicted in Figure 1.
Figure 1
Estimated Probability of Achieving Initial Headache Response Within 2 Hours
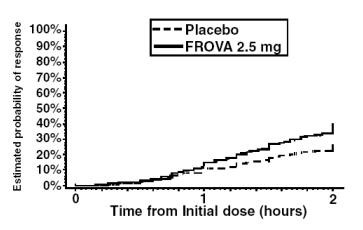
Figure 1 shows a Kaplan-Meier plot of the probability over time of obtaining headache response (no or mild pain) following treatment with frovatriptan 2.5 mg or placebo. The probabilities displayed are based on pooled data from four placebo-controlled trials described in Table 1(Trials 1, 3, 4 and 5). Patients who did not achieve a response were censored at 24 hours.
In patients with migraine-associated nausea, photophobia and phonophobia at baseline there was a decreased incidence of these symptoms in FROVA treated patients compared to placebo. The estimated probability of patients taking a second dose or other medication for their migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
Figure 2
Estimated Probability of Patients Taking a Second Dose or Other Medication for Migraine Over the 24 Hours Following the Initial Dose of Study Treatment
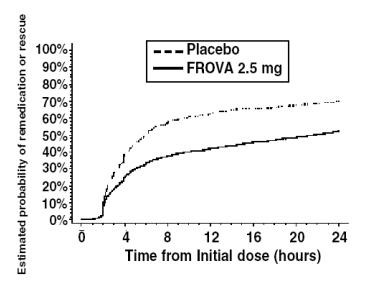
Figure 2 is a Kaplan-Meier plot showing the probability of patients taking a second dose or other medication for migraine over the 24 hours following the initial dose of study medication based on the data from four placebo-controlled trials described in Table 1 (Trials 1, 3, 4 and 5). The plot includes those patients who had a response to the initial dose and those who did not. The protocols did not permit remedication within 2 hours of the initial dose.
Efficacy was unaffected by a history of aura; gender; age, or concomitant medications commonly used by migraine patients FROVA.
-
INDICATIONS AND USAGE
FROVA is indicated for the acute treatment of migraine attacks with or without aura in adults.
FROVA is not intended for the prophylactic therapy of migraine or for use in the management of hemiplegic or basilar migraine (see CONTRAINDICATIONS). The safety and effectiveness of FROVA have not been established for cluster headache, which is present in an older, predominately male, population.
-
CONTRAINDICATIONS
FROVA should not be given to patients with ischemic heart disease (e.g. angina pectoris, history of myocardial infarction, or documented silent ischemia), or to patients who have symptoms or findings consistent with ischemic heart disease, coronary artery vasospasm, including Prinzmetal’s variant angina or other significant underlying cardiovascular disease (see WARNINGS).
FROVA should not be given to patients with cerebrovascular syndromes including (but not limited to) strokes of any type as well as transient ischemic attacks.
FROVA should not be given to patients with peripheral vascular disease including (but is not limited to) ischemic bowel disease (see WARNINGS)
FROVA should not be given to patients with uncontrolled hypertension (see WARNINGS).
FROVA should not be administered to patients with hemiplegic or basilar migraine.
FROVA should not be used within 24 hours of treatment with another 5-HT1 agonist, an ergotamine containing or ergot-type medication such as dihydroergotamine (DHE) or methysergide.
FROVA is contraindicated in patients who are hypersensitive to frovatriptan or any of the inactive ingredients in the tablets.
-
WARNINGS
FROVA should only be used where a clear diagnosis of migraine has been established.
Risk of Myocardial Ischemia and/or Infarction and Other Adverse Cardiac Events:
Because of the potential of this class of compound (5-HT1 agonists) to cause coronary vasospasm, frovatriptan should not be given to patients with documented ischemic or vasospastic coronary artery disease (CAD) (see CONTRAINDICATIONS). It is strongly recommended that frovatriptan not be given to patients in whom unrecognized CAD is predicted by the presence of risk factors (e.g., hypertension, hypercholesterolemia, smoker, obesity, diabetes, strong family history of CAD, female with surgical or physiological menopause, or male over 40 years of age) unless a cardiovascular evaluation provides satisfactory clinical evidence that the patient is reasonably free of coronary artery and ischemic myocardial disease or other significant underlying cardiovascular disease. The sensitivity of cardiac diagnostic procedures to detect cardiovascular disease or predisposition to coronary artery vasospasm is modest, at best. If, during the cardiovascular evaluation, the patient’s medical history, electrocardiographic, or other investigations reveal findings indicative of, or consistent with, coronary artery vasospasm or myocardial ischemia, frovatriptan should not be administered (see CONTRAINDICATIONS).
For patients with risk factors predictive of CAD, who are determined to have a satisfactory cardiovascular evaluation, it is strongly recommended that administration of the first dose of frovatriptan take place in the setting of a physician’s office or similar medically staffed and equipped facility unless the patient has previously received frovatriptan. Because cardiac ischemia can occur in the absence of clinical symptoms, consideration should be given to obtaining on the first occasion of use an electrocardiogram (ECG) during the interval immediately following administration of FROVA in these patients with risk factors.
It is recommended that patients who are intermittent long-term users of 5-HT1 agonists, including FROVA and who have or acquire risk factors predictive of CAD, as described above, undergo periodic cardiovascular evaluation as they continue to use FROVA.
The systematic approach described above is intended to reduce the likelihood that patients with unrecognized cardiovascular disease would be inadvertently exposed to frovatriptan.
Cardiac Events and Fatalities with 5-HT1 Agonists:
Serious adverse cardiac events, including acute myocardial infarction, life-threatening disturbances of cardiac rhythm and death have been reported within a few hours of administration of 5-HT1 agonists. Considering the extent of use of 5-HT1 agonists in patients with migraine, the incidence of these events is extremely low.
Premarketing experience with frovatriptan:
Among more than 3000 patients with migraine who participated in premarketing clinical trials of FROVA no deaths or serious cardiac events were reported which were related to the use of FROVA.
Cerebrovascular Events and Fatalities with 5-HT1 Agonists:
Cerebral hemorrhage, subarachnoid hemorrhage, stroke and other cerebrovascular events have been reported in patients treated with 5-HT1 agonists; and some have resulted in fatalities. In a number of cases, it appears possible that the cerebrovascular events were primary, the agonist having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine, when they were not. It should be noted that patients with migraine may be at increased risk of certain cerebrovascular events (e.g. stroke, hemorrhage, transient ischemic attack).
Other Vasospasm-Related Events:
5-HT1 agonists may cause vasospastic reactions other than coronary artery spasm. Both peripheral vascular ischemia and colonic ischemia with abdominal pain and bloody diarrhea have been reported with 5-HT1 agonists.
Effects on Blood Pressure:
In young healthy subjects, there were statistically significant increases in systolic and diastolic blood pressure after single doses of 80 mg frovatriptan (32 times the clinical dose) and above. These increases were transient, resolved spontaneously and were not clinically significant. At the recommended dose of 2.5 mg, transient changes in systolic blood pressure were recorded in some elderly subjects (65 - 77 years). Any increases were generally small, resolved spontaneously, and blood pressure remained within the normal range. Frovatriptan is contraindicated in patients with uncontrolled hypertension (see CONTRAINDICATIONS).
An 18% increase in mean pulmonary artery pressure was seen following dosing with another 5-HT1 agonist in a study evaluating subjects undergoing cardiac catheterization.
Serotonin Syndrome:
The development of a potentially life-threatening serotonin syndrome may occur with triptans, including FROVA treatment, particularly during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs). If concomitant treatment with FROVA and an SSRI (e.g., fluoxetine, paroxetine, sertraline, fluvoxamine, citalopram, escitalopram) or SNRI (e.g., venlafaxine, duloxetine) is clinically warranted, careful observation of the patient is advised, particularly during treatment initiation and dose increases. Serotonin syndrome symptoms may include mental status changes (e.g., agitation hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination) and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). (See PRECAUTIONS – Drug Interactions).
-
PRECAUTIONS
General:
As with other 5-HT1 agonists, sensations of pain, tightness, pressure and heaviness have been reported in the chest, throat, neck and jaw after treatment with FROVA. These events have not been associated with arrhythmias or ischemic ECG changes in clinical trials with FROVA. Because 5-HT1 agonists may cause coronary vasospasm, patients who experience signs or symptoms suggestive of angina following dosing should be evaluated for the presence of CAD. Patients shown to have CAD and those with Prinzmetal’s variant angina should not receive 5-HT1 agonists (see CONTRAINDICATIONS). Patients who experience other symptoms or signs suggestive of decreased arterial flow, such as ischemic bowel syndrome or Raynaud’s syndrome following the use of any 5-HT1 agonist are candidates for further evaluation. If a patient has no response for the first migraine attack treated with FROVA, the diagnosis of migraine should be reconsidered before frovatriptan is administered to treat any subsequent attacks.
Hepatically Impaired Patients:
There is no clinical or pharmacokinetic experience with FROVA in patients with severe hepatic impairment. The AUC of frovatriptan in patients with mild (Child-Pugh 5-6) to moderate (Child-Pugh 7-9) hepatic impairment was about twice that of young, healthy subjects, but within the range observed in healthy elderly subjects and was considerably lower than the values attained with higher doses of frovatriptan (up to 40 mg), which were not associated with any serious adverse effects. Therefore, no dosage adjustment is necessary when FROVA is given to patients with mild to moderate hepatic impairment (see CLINICAL PHARMACOLOGY, Special Populations).
Binding to Melanin-Containing Tissues:
When pigmented rats were given a single oral dose of 5 mg/kg of radiolabeled frovatriptan, the radioactivity in the eye after 28 days was 87% of the value measured after 8 hours. This suggests that frovatriptan and/or its metabolites may bind to the melanin of the eye. Because there could be accumulation in melanin rich tissues over time, this raises the possibility that frovatriptan could cause toxicity in these tissues after extended use. However, no effects on the retina related to treatment with frovatriptan were noted in the toxicity studies. Although no systematic monitoring of ophthalmologic function was undertaken in clinical trials and no specific recommendations for ophthalmologic monitoring are made, prescribers should be aware of the possibility of long-term ophthalmologic effects.
Information for Patients
Physicians should instruct their patients to read the patient package insert before taking FROVA. See PATIENT INFORMATION at the end of this labeling for the text of the separate leaflet provided for patients.
Patients should be cautioned about the risk of serotonin syndrome with the use of FROVA or other triptans, especially during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs).
Laboratory Tests
No specific laboratory tests are recommended for monitoring patients prior to and/or after treatment with FROVA.
Drug Interactions (see also CLINICAL PHARMACOLOGY, Drug Interactions)
Ergot-containing drugs have been reported to cause prolonged vasospastic reactions. Due to a theoretical risk of a pharmacodynamic interaction, use of ergotamine-containing or ergot-type medications (like dihydroergotamine or methysergide) and FROVA within 24 hours of each other should be avoided (see CONTRAINDICATIONS).
Concomitant use of other 5HT1B/1D agonists within 24 hours of FROVA treatment is not recommended (see CONTRAINDICATIONS).
Selective Serotonin Reuptake Inhibitors / Serotonin Norepinephrine Reuptake Inhibitors and Serotonin Syndrome: Cases of life-threatening serotonin syndrome have been reported during combined use of selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs) and triptans. (See WARNINGS).
Drug/Laboratory Test Interactions
FROVA is not known to interfere with commonly employed clinical laboratory tests.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis: The carcinogenic potential of frovatriptan was evaluated in an 84-week study in mice (4, 13, and 40 mg/kg/day), a 104-week study in rats (8.5, 27 and 85 mg/kg/day), and a 26-week study in p53(+/-) transgenic mice (20, 62.5, 200, and 400 mg/kg/day). Although the maximum tolerated dose (MTD) was not achieved in the 84-week mouse study and in female rats, exposures at the highest doses studied were many fold greater than those achieved at the maximum recommended daily human dose (MRHD) of 7.5 mg. There were no increases in tumor incidence in the 84-week mouse study at doses producing 140 times the exposure achieved at the MRHD based on blood AUC comparisons. In the rat study, there was a statistically significant increase in the incidence of pituitary adenomas in males only at 85 mg/kg/day, a dose that produced 250 times the exposure achieved at the MRHD based on AUC comparisons. In the 26-week p53(+/-) transgenic mouse study, there was an increased incidence of subcutaneous sarcomas in females dosed at 200 and 400 mg/kg/day, or 390 and 630 times the human exposure based on AUC comparisons. The incidence of sarcomas was not increased at lower doses that achieved exposures 180 and 60 times the human exposure. These sarcomas were physically associated with subcutaneously implanted animal identification transponders. There were no other increases in tumor incidence of any type in any dose group. These sarcomas are not considered to be relevant to humans.
Mutagenesis: Frovatriptan was clastogenic in human lymphocyte cultures,in the absence of metabolic activation. In the bacterial reverse mutation assay (Ames test), frovatriptan produced an equivocal response in the absence of metabolic activation. No mutagenic or clastogenic activities were seen in an in vitro mouse lymphoma assay, an in vivo mouse bone marrow micronucleus test, or an ex vivo assay for unscheduled DNA synthesis in rat liver.
Impairment of Fertility: Male and female rats were dosed prior to and during mating, and up to implantation, at doses of 100, 500, and 1000 mg/kg/day (equivalent to approximately 130, 650, and 1300 times the MRHD on a mg/m2 basis). At all dose levels there was an increase in the number of females that mated on the first day of pairing compared to control animals. This occurred in conjunction with a prolongation of the estrous cycle. In addition females had a decreased mean number of corpora lutea, and consequently a lower number of live fetuses per litter, which suggested a partial impairment of ovulation. There were no other fertility-related effects.
Pregnancy: Pregnancy Category C
When pregnant rats were administered frovatriptan during the period of organogenesis at oral doses of 100, 500 and 1000 mg/kg/day (equivalent to 130, 650 and 1300 times the maximum recommended human dose [MRHD] on a mg/m2 basis) there were dose related increases in incidences of both litters and total numbers of fetuses with dilated ureters, unilateral and bilateral pelvic cavitation, hydronephrosis, and hydroureters. A no-effect dose for renal effects was not established. This signifies a syndrome of related effects on a specific organ in the developing embryo in all treated groups, which is consistent with a slight delay in fetal maturation. This delay was also indicated by a treatment related increased incidence of incomplete ossification of the sternebrae, skull and nasal bones in all treated groups. Slightly lower fetal weights and an increased incidence of early embryonic deaths in treated rats were observed; although not statistically significant compared to control, the latter effect occurred in both the embryo-fetal developmental study and in the prenatal-postnatal developmental study. There was no evidence of this latter effect at the lowest dose level studied, 100 mg/kg/day (equivalent to 130 times the MRHD on a mg/m2 basis). When pregnant rabbits were dosed throughout organogenesis at doses up to 80 mg/kg/day (equivalent to 210 times the MRHD on a mg/m2 basis) no effects on fetal development were observed.
There are no adequate and well-controlled studies in pregnant women; therefore, frovatriptan should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether frovatriptan is excreted in human milk. Frovatriptan and/or its metabolites are excreted in the milk of lactating rats with the maximum concentration being four-fold higher than that seen in blood. Therefore, caution should be exercised when considering the administration of FROVA to a nursing woman.
Pediatric Use
Safety and effectiveness of FROVA in pediatric patients have not been established; therefore, FROVA is not recommended for use in patients under 18 years of age. Postmarketing experience with other triptans includes a limited number of reports that describe pediatric patients who have experienced clinically serious adverse events that are similar in nature to those reported rarely in adults.
Use in the Elderly
Mean blood concentrations of frovatriptan in elderly subjects were 1.5- to 2-times higher than those seen in younger adults (see CLINICAL PHARMACOLOGY, Special Populations). Because migraine occurs infrequently in the elderly, clinical experience with FROVA is limited in such patients.
-
ADVERSE REACTIONS
Serious cardiac events, including some that have been fatal, have occurred following use of 5-HT1 agonists. These events are extremely rare and most have been reported in patients with risk factors predictive of CAD. Events reported have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia and ventricular fibrillation (see CONTRAINDICATIONS, WARNINGS and PRECAUTIONS).
Incidence in Controlled Clinical Trials:
Among 1554 patients treated with FROVA in four placebo-controlled trials (Trials 1, 3, 4 and 5 in Table 1), only 1% (16) patients withdrew because of treatment-emergent adverse events. In a long term, open-label study where patients were allowed to treat multiple migraine attacks with FROVA for up to 1 year, 5% (26/496) patients discontinued due to treatment-emergent adverse events.
The treatment-emergent adverse events that occurred most frequently following administration of frovatriptan 2.5 mg (i.e., in at least 2% of patients), and at an incidence ≥1% greater than with placebo, in the four placebo-controlled trials were dizziness, paresthesia, headache, dry mouth, fatigue, flushing, hot or cold sensation and chest pain.
Table 2 lists treatment-emergent adverse events reported within 48 hours of drug administration that occurred with frovatriptan 2.5 mg at an incidence of ≥ 2% and more often than on placebo, in the first attack in four placebo-controlled trials (Trials 1, 3, 4 and 5 in Table 1). These studies involved 2392 patients (1554 frovatriptan 2.5 mg and 838 placebo). The events cited reflect experience gained under closely monitored conditions of clinical trials in a highly selected patient population. In actual clinical practice or in other clinical trials, these incidence estimates may not apply, as the conditions of use, reporting behavior, and the kinds of patients treated may differ.
Table 2: Treatment-Emergent Adverse Events (Incidence ≥ 2% and Greater Than Placebo) of Patients in Four Placebo-Controlled Migraine Trials Adverse events Frovatriptan 2.5 mg
(n=1554)Placebo
(n=838)Central & peripheral nervous system
Dizziness
Headache
Paresthesia
8%
4%
4%
5%
3%
2%
Gastrointestinal system disorders
Mouth dry
Dyspepsia
3%
2%
1%
1%
Body as a whole – general disorders
Fatigue
Hot or cold sensation
Chest pain
5%
3%
2%
2%
2%
1%
Musculo-skeletal
Skeletal pain
3%
2%
Vascular
Flushing
4%
2%
Other events that occurred at ≥2% on frovatriptan that were equally or more common in the placebo group were somnolence and nausea.
FROVA is generally well tolerated. The incidence of adverse events in clinical trials did not increase when up to 3 doses were used within 24 hours. The majority of adverse events were mild or moderate and transient. The incidence of adverse events in four placebo-controlled clinical trials was not affected by gender, age or concomitant medications commonly used by migraine patients. There were insufficient data to assess the impact of race on the incidence of adverse events.
Other Events Observed in Association with FROVA:
In the paragraphs that follow, the incidence of less commonly reported adverse events in four placebo-controlled trials are presented. Variability associated with adverse event reporting, the terminology used to describe adverse events etc, limit the value of the incidence estimates provided. The incidence of each adverse event is calculated as the number of patients reporting the event at least once divided by the number of patients who used FROVA. All adverse events reported within 48 hours of drug administration in the first attack in four placebo controlled trials involving 2392 patients (1554 frovatriptan 2.5 mg and 838 placebo) are included, except those already listed in Table 2, those too general to be informative, those not reasonably associated with the use of the drug and those which occurred at the same or a greater incidence in the placebo group. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are those occurring in at least 1/100 patients, infrequent adverse events are those occurring in between 1/100 and 1/1000 patients, and rare adverse events are those occurring in fewer than 1/1000 patients.
Central and peripheral nervous system: Frequent: dysesthesia and hypoesthesia. Infrequent: tremor, hyperesthesia, migraine aggravated, involuntary muscle contractions, vertigo, ataxia, abnormal gait and speech disorder. Rare: hypertonia, hypotonia, abnormal reflexes and tongue paralysis.
Gastrointestinal: Frequent: vomiting, abdominal pain and diarrhea. Infrequent: dysphagia, flatulence, constipation, anorexia, esophagospasm and saliva increased. Rare: change in bowel habits, cheilitis, eructation, gastroesophageal reflux, hiccup, peptic ulcer, salivary gland pain, stomatitis and toothache.
Body as a whole: Frequent: pain. Infrequent: asthenia, rigors, fever, hot flushes and malaise. Rare: feeling of relaxation, leg pain and edema mouth.
Psychiatric: Frequent: insomnia and anxiety. Infrequent: confusion, nervousness, agitation, euphoria, impaired concentration, depression, emotional lability, amnesia, thinking abnormal and depersonalization. Rare: depression aggravated, abnormal dreaming and personality disorder.
Musculoskeletal: Infrequent: myalgia, back pain, arthralgia, arthrosis, leg cramps and muscle weakness.
Respiratory: Frequent: sinusitis and rhinitis. Infrequent: pharyngitis, dyspnea, hyperventilation and laryngitis.
Vision disorders: Frequent: vision abnormal. Infrequent: eye pain, conjunctivitis and abnormal lacrimation.
Skin and appendages: Frequent: sweating increased. Infrequent: pruritis, and bullous eruption.
Hearing and vestibular disorders: Frequent: tinnitus. Infrequent: ear ache, and hyperacusis.
Heart rate and rhythm: Frequent: palpitation. Infrequent: tachycardia. Rare: bradycardia.
Metabolic and nutritional disorders: Infrequent: thirst and dehydration. Rare: hypocalcemia and hypoglycemia.
Special senses, other disorders: Infrequent: taste perversion.
Urinary system disorders: Infrequent: micturition frequency and polyuria. Rare: nocturia, renal pain and abnormal urine.
Cardiovascular disorders, general: Infrequent: abnormal ECG.
Platelet, bleeding and clotting disorders: Infrequent: epistaxis. Rare: purpura.
Autonomic nervous system: Rare: syncope.
Postmarketing Experience
Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency. Information is often incomplete so that a definite causal relationship to drug exposure can often not be established.
Central and peripheral nervous system: Seizure.
- DRUG ABUSE AND DEPENDENCE
-
OVERDOSAGE
There is no direct experience of any patient taking an overdose of FROVA. The maximum single dose of frovatriptan given to male and female patients with migraine was 40 mg (16 times the clinical dose) and the maximum single dose given to healthy male subjects was 100 mg (40 times the clinical dose) without significant adverse events.
As with other 5-HT1 receptor agonists, there is no specific antidote for frovatriptan. The elimination half-life of frovatriptan is 26 hours, therefore if overdose occurs, the patient should be monitored closely for at least 48 hours and be given any necessary symptomatic treatment.
The effects of hemo- or peritoneal dialysis on blood concentrations of frovatriptan are unknown.
-
DOSAGE AND ADMINISTRATION
The recommended dose is a single tablet of FROVA (frovatriptan 2.5 mg) taken orally with fluids.
If the headache recurs after initial relief, a second tablet may be taken, providing there is an interval of at least 2 hours between doses. The total daily dose of frovatriptan should not exceed 3 tablets (3 x 2.5 mg per day).
There is no evidence that a second dose of frovatriptan is effective in patients who do not respond to a first dose of the drug for the same headache.
The safety of treating an average of more than 4 migraine attacks in a 30-day period has not been established.
-
HOW SUPPLIED
FROVA tablets, containing 2.5 mg of frovatriptan (base) as the succinate, are available as round, white, film-coated tablets debossed with 2.5 on one side and “E” on the other side. The tablets are available in:
Blister card of 9 tablets, 1 blister card per carton (NDC 63481-025-09)
Store at controlled room temperature, 25°C (77°F) excursions permitted to 15 - 30°C (59°F - 86°F) [see USP Controlled Room Temperature]. Protect from moisture.
U.S. Patent Nos 5,962,501, 5,827,871, 5,637,611 and 5,464,864 and 5,616,603.
Rx Only
Manufactured for:
Endo Pharmaceuticals Inc.
Chadds Ford, PA 19317Manufactured by:
Almac Pharma Services Limited
Craigavon, BT63 5UA, UKFROVA is a registered trademark of Vernalis Development Limited.
© 2007 Endo Pharmaceuticals Inc. PX544-3 / April, 2007
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION: The following wording is contained in a separate leaflet provided for patients.
PATIENT INFORMATION ABOUT
FROVA® (frovatriptan succinate) TabletsRead this information before you start taking FROVA (FRO-va). Also, read the information each time you renew your prescription, in case anything has changed. This leaflet does not contain all of the information about FROVA. For further information or advice ask your doctor or pharmacist. You and your doctor should discuss FROVA before you start taking the medicine and at regular checkups.
What is FROVA?
FROVA is a prescription medicine used to treat migraine attacks in adults. It is in the class of drugs called selective serotonin receptor agonists.
FROVA should only be taken for a migraine headache. Do not use FROVA to treat headaches that might be caused by other conditions. Tell your doctor about your symptoms. Your doctor will decide if you have migraine headaches and if FROVA is for you.
There is more information about migraine at the end of this leaflet.
Who should not take FROVA?
Do not take FROVA if you:
- have uncontrolled high blood pressure
- have heart disease or a history of heart disease
- have hemiplegic or basilar migraine (if you are not sure about this, ask your doctor)
- have had a stroke
- have circulation (blood flow) problems
- have taken a similar drug (a serotonin receptor agonist) in the last 24 hours. These include sumatriptan (IMITREX®), naratriptan (AMERGE™), zolmitriptan (ZOMIG™), rizatriptan (MAXALT™), eletriptan hydrobromide (RELPAX®),or almotriptan (AXERT™)
- have taken ergotamine type medicines in the last 24 hours. These include BELLERGAL®, CAFERGOT®, ERGOMAR®, WIGRAINE®, DHE 45®, or SANSERT®
- have any allergic reaction to the tablet
What you should tell your doctor before and during treatment with FROVA?
To help your doctor decide if FROVA is right for you, tell your doctor if you:
- are pregnant, or planning to become pregnant
- are breast-feeding or plan to breast-feed
- have any history of chest pain, shortness of breath, or palpitations
- have any risk factors for heart disease, including
- high blood pressure
- diabetes
- high cholesterol
- overweight
- smoking
- a family history of heart disease
- past menopause
- male over 40 years old
- are taking any other medicines, including prescription and non-prescription medicines, and herbal supplements
- have any past or present medical problems
- have previous allergies to any medicine
Tell your doctor if you take
- propranolol
- selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs), two types of drugs for depression or other disorders. Common SSRIs are CELEXA® (citalopram HBr), LEXAPRO® (escitalopram oxalate), PAXIL® (paroxetine), PROZAC®/SARAFEM® (fluoxetine), SYMBYAX® (olanzapine/fluoxetine), ZOLOFT® (sertraline), and fluvoxamine. Common SNRIs are CYMBALTA® (duloxetine) and EFFEXOR® (venlafaxine).
These medicines may affect how FROVA works, or FROVA may affect how these medicines work.
How should you take FROVA?
Take one FROVA tablet anytime after the start of your migraine headache. If your headache comes back after your first dose, you may take a second tablet after two (2) hours. Do not take more than three (3) FROVA tablets in a 24-hour period.
If you take too much medicine, contact your doctor, hospital emergency department, or poison control center right away.
What are the common side effects of FROVA?
The most common side effects associated with use of FROVA are:
- dizziness
- fatigue (tiredness)
- headache (other than a migraine headache)
- paresthesia (feeling of tingling)
- dry mouth
- flushing (hot flashes)
- feeling hot or cold
- chest pain
- dyspepsia (indigestion)
- skeletal pain (pain in joints or bones)
Tell your doctor about any symptoms that you develop while taking FROVA. If you feel dizziness or fatigue, take extra care or avoid driving and operating machinery.
In very rare cases, patients taking this class of medicines experience serious heart problems, stroke, or increased blood pressure. If you develop pain, tightness, heaviness, or pressure in your chest, throat, neck, or jaw, contact your doctor right away.
Also contact your doctor right away if you develop a rash or itching after taking FROVA. You may be allergic to this medicine.
What is a migraine and how does it differ from other headaches?
Migraine is an intense, throbbing headache that often affects one side of the head. It often includes nausea, vomiting, and sensitivity to light and sound. The pain and symptoms from a migraine headache may be worse than the pain and symptoms of a common headache. Migraine headaches usually last for hours or longer.
Some people have problems with vision (an aura) before they get a migraine headache. These include flashing lights, wavy lines, and dark spots.
Only your doctor can determine that your headache is a migraine headache, so it is important that you discuss all of your symptoms with your doctor.
LEXAPRO® / CELEXA® are registered trademarks of Forest Pharmaceuticals, Inc.
PAXIL® is a registered trademark of GlaxoSmithKline.
PROZAC® / SARAFEM® / SYMBYAX® / CYMBALTA® are registered trademarks of Eli Lilly and Company
ZOLOFT® is a registered trademark of Pfizer Pharmaceuticals.
EFFEXOR® is a registered trademark of Wyeth Pharmaceuticals.
FROVA is a registered trademark of Vernalis Development Limited.
© 2007 Endo Pharmaceuticals Inc. PX544-3 / April, 2007
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
FROVA
frovatriptan succinate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:16590-396(NDC:63481-025) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength FROVATRIPTAN SUCCINATE (UNII: D28J6W18HY) (FROVATRIPTAN - UNII:H82Q2D5WA7) FROVATRIPTAN SUCCINATE 2.5 mg Inactive Ingredients Ingredient Name Strength LACTOSE (UNII: J2B2A4N98G) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE (UNII: 3NXW29V3WO) POLYETHYLENE GLYCOL 3000 (UNII: SA1B764746) TRIACETIN (UNII: XHX3C3X673) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white Score no score Shape ROUND Size 8mm Flavor Imprint Code E;2;5 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:16590-396-09 9 in 1 BLISTER PACK Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021006 11/08/2001 Labeler - STAT RX USA LLC (786036330) Establishment Name Address ID/FEI Business Operations STAT RX USA LLC 786036330 repack, relabel
 FROVA 2.5MG LABEL IMAGE
FROVA 2.5MG LABEL IMAGE