Label: TEKTURNA- aliskiren hemifumarate tablet, film coated

- NDC Code(s): 50090-2985-0
- Packager: A-S Medication Solutions
- This is a repackaged label.
- Source NDC Code(s): 70839-300
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated February 13, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TEKTURNA safely and effectively. See full prescribing information for TEKTURNA.
TEKTURNA ® (aliskiren) tablets, for oral use
Initial U.S. Approval: 2007INDICATIONS AND USAGE
Tekturna is a renin inhibitor (RI) indicated for the treatment of hypertension in adults and in pediatric patients weighing 50 kg or greater who are at least 6 years of age, to lower blood pressure. Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. ( 1.1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 150 mg, 300 mg ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Avoid concomitant use with ARBs or ACEIs particularly in patients with renal impairment [creatinine clearance (CrCl) <60 mL/min]. ( 5.2, 5.4)
- Anaphylactic Reactions and Head and Neck Angioedema. ( 5.3)
- Hypotension: Correct imbalances in volume and/or salt depleted patients. ( 5.4)
- Impaired Renal Function: Monitor serum creatinine periodically. ( 5.5)
- Hyperkalemia: Monitor potassium levels periodically. ( 5.6)
DRUG INTERACTIONS
ADVERSE REACTIONS
Most common adverse reaction: diarrhea (incidence 2.3%) ( 6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact LXO US Inc. at 1-844-800-8007 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: FETAL TOXICITY
1 INDICATIONS AND USAGE
1.1 Hypertension
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Relationship to Meals
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
5.2 Renal Impairment/Hyperkalemia/Hypotension when Tekturna is Given in Combination with ARBs or ACEIs
5.3 Anaphylactic Reactions and Head and Neck Angioedema
5.4 Hypotension
5.5 Impaired Renal Function
5.6 Hyperkalemia
5.7 Cyclosporine or Itraconazole
7 DRUG INTERACTIONS
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Aliskiren Monotherapy
14.2 Aliskiren in Combination with Other Antihypertensives
14.3 Aliskiren in Patients with Diabetes Treated with ARB or ACEI (ALTITUDE study)
14.4 Pediatric Hypertension
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- BOXED WARNING (What is this?)
-
1 INDICATIONS AND USAGE
1.1 Hypertension
Tekturna is indicated for the treatment of hypertension in adults and in pediatric patients weighing 50 kg or greater who are at least 6 years of age and older to lower blood pressure.
Lowering blood pressure reduces the risk of fatal and nonfatal cardiovascular events, primarily strokes and myocardial infarctions. These benefits have been seen in controlled trials of antihypertensive drugs from a wide variety of pharmacologic classes. There are no controlled trials demonstrating risk reduction with Tekturna.
Control of high blood pressure should be part of comprehensive cardiovascular risk management, including, as appropriate, lipid control, diabetes management, antithrombotic therapy, smoking cessation, exercise, and limited sodium intake. Many patients will require more than 1 drug to achieve blood pressure goals. For specific advice on goals and management, see published guidelines, such as those of the National High Blood Pressure Education Program's Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure (JNC).
Numerous antihypertensive drugs, from a variety of pharmacologic classes and with different mechanisms of action, have been shown in randomized controlled trials to reduce cardiovascular morbidity and mortality, and it can be concluded that it is blood pressure reduction, and not some other pharmacologic property of the drugs, that is largely responsible for those benefits. The largest and most consistent cardiovascular outcome benefit has been a reduction in the risk of stroke, but reductions in myocardial infarction and cardiovascular mortality have also been seen regularly.
Elevated systolic or diastolic pressure causes increased cardiovascular risk, and the absolute risk increase per mmHg is greater at higher blood pressures, so that even modest reductions of severe hypertension can provide substantial benefit. Relative risk reduction from blood pressure reduction is similar across populations with varying absolute risk, so the absolute benefit is greater in patients who are at higher risk independent of their hypertension (e.g., patients with diabetes or hyperlipidemia), and such patients would be expected to benefit from more aggressive treatment to a lower blood pressure goal.
Some antihypertensive drugs have smaller blood pressure effects (as monotherapy) in black patients, and many antihypertensive drugs have additional approved indications and effects (e.g., on angina, heart failure, or diabetic kidney disease). These considerations may guide selection of therapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
In adult patients and in pediatric patients weighing 50 kg or greater who are at least 6 years of age, the recommended starting dose of Tekturna is 150 mg once daily. In patients whose blood pressure is not adequately controlled, the daily dose may be increased to 300 mg once daily. Doses above 300 mg did not give an increased blood pressure response but resulted in an increased rate of diarrhea. The antihypertensive effect of a given dosage is substantially attained (85% to 90%) by 2 weeks.
2.2 Relationship to Meals
Patients should establish a routine pattern for taking Tekturna with regard to meals. High-fat meals decrease absorption substantially [see Clinical Pharmacology ( 12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
Do not use Tekturna with ARBs or ACEIs in patients with diabetes [see Warnings and Precautions ( 5.2) and Clinical Studies ( 14.3)].
Tekturna is contraindicated in patients with known hypersensitivity to any of the components [see Warnings and Precautions ( 5.3)].
Tekturna is contraindicated in pediatric patients less than 2 years of age because of the risk of high aliskiren exposures identified in juvenile animals due to immaturity of transporters and metabolic enzymes [see Use in Specific Populations ( 8.4)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Fetal Toxicity
Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death. Resulting oligohydramnios can be associated with fetal lung hypoplasia and skeletal deformations. Potential neonatal adverse effects include skull hypoplasia, anuria, hypotension, renal failure, and death. When pregnancy is detected, discontinue Tekturna as soon as possible [see Use in Specific Populations ( 8.1)].
5.2 Renal Impairment/Hyperkalemia/Hypotension when Tekturna is Given in Combination with ARBs or ACEIs
Tekturna is contraindicated in patients with diabetes who are receiving ARBs or ACEIs because of the increased risk of renal impairment, hyperkalemia, and hypotension. In general, avoid combined use of Tekturna with ACE inhibitors or ARBs, particularly in patients with creatinine clearance (CrCl) less than 60 mL/min [see Contraindications ( 4), Drug Interactions ( 7) and Clinical Studies ( 14.3)] .
5.3 Anaphylactic Reactions and Head and Neck Angioedema
Hypersensitivity reactions such as anaphylactic reactions and angioedema of the face, extremities, lips, tongue, glottis and/or larynx have been reported in patients treated with Tekturna and has necessitated hospitalization and intubation. This may occur at any time during treatment and has occurred in patients with and without a history of angioedema with ACEIs or angiotensin receptor antagonists. Anaphylactic reactions have been reported from postmarketing experience with unknown frequency. If angioedema involves the throat, tongue, glottis or larynx, or if the patient has a history of upper respiratory surgery, airway obstruction may occur and be fatal. Patients who experience these effects, even without respiratory distress, require prolonged observation and appropriate monitoring measures since treatment with antihistamines and corticosteroids may not be sufficient to prevent respiratory involvement. Prompt administration of subcutaneous epinephrine solution 1:1000 (0.3 mL to 0.5 mL) and measures to ensure a patent airway may be necessary.
Discontinue Tekturna immediately in patients who develop anaphylactic reactions or angioedema, and do not readminister [see Dosage and Administration ( 2.1) and Contraindications ( 4)].
5.4 Hypotension
Symptomatic hypotension may occur after initiation of treatment with Tekturna in patients with marked volume depletion, patients with salt depletion, or with combined use of Tekturna and other agents acting on the renin-angiotensin-aldosterone system (RAAS). The volume or salt depletion should be corrected prior to administration of Tekturna, or the treatment should start under close medical supervision.
A transient hypotensive response is not a contraindication to further treatment, which usually can be continued without difficulty once the blood pressure has stabilized.
5.5 Impaired Renal Function
Monitor renal function periodically in patients treated with Tekturna. Changes in renal function, including acute renal failure, can be caused by drugs that affect the RAAS. Patients whose renal function may depend in part on the activity of the RAAS (e.g., patients with renal artery stenosis, severe heart failure, post-myocardial infarction or volume depletion) or patients receiving ARB, ACEI or nonsteroidal anti-inflammatory drug (NSAID), including selective Cyclooxygenase-2 inhibitors (COX-2 inhibitors), therapy may be at particular risk for developing acute renal failure on Tekturna [see Warnings and Precautions ( 5.2), Drug Interactions ( 7), Use in Specific Populations ( 8.6), and Clinical Studies ( 14.3)] .
Consider withholding or discontinuing therapy in patients who develop a clinically significant decrease in renal function.
5.6 Hyperkalemia
Monitor serum potassium periodically in patients receiving Tekturna. Drugs that affect the RAAS can cause hyperkalemia. Risk factors for the development of hyperkalemia include renal insufficiency, diabetes, combination use with ARBs or ACEIs [see Contraindications ( 4), Warnings and Precautions ( 5.2), and Clinical Studies ( 14.3)] , NSAIDs, or potassium supplements or potassium sparing diuretics.
5.7 Cyclosporine or Itraconazole
When Tekturna was given with cyclosporine or itraconazole, the blood concentrations of aliskiren were significantly increased. Avoid concomitant use of Tekturna with cyclosporine or itraconazole [see Drug Interactions ( 7)] .
-
7 DRUG INTERACTIONS
Cyclosporine: Avoid coadministration of cyclosporine with aliskiren [see Warnings and Precautions ( 5.7) and Clinical Pharmacology ( 12.3)] .
Itraconazole: Avoid coadministration of itraconazole with aliskiren [see Warnings and Precautions ( 5.7) and Clinical Pharmacology ( 12.3)].
Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) including selective Cyclooxygenase-2 inhibitors (COX-2 inhibitors): In patients who are elderly, volume-depleted (including those on diuretic therapy), or with compromised renal function, coadministration of NSAIDs, including selective COX-2 inhibitors with agents that affect the RAAS, including aliskiren, may result in deterioration of renal function, including possible acute renal failure. These effects are usually reversible. Monitor renal function periodically in patients receiving aliskiren and NSAID therapy.
The antihypertensive effect of aliskiren may be attenuated by NSAIDs.
Dual Blockade of the Renin-Angiotensin-Aldosterone System (RAAS): The concomitant use of aliskiren with other agents acting on the RAAS such as ACEIs or ARBs is associated with an increased risk of hypotension, hyperkalemia, and changes in renal function (including acute renal failure) compared to monotherapy. Most patients receiving the combination of two drugs that inhibit the renin-angiotensin system do not obtain any additional benefit compared to monotherapy. In general, avoid combined use of aliskiren with ACE inhibitors or ARBs, particularly in patients with CrCl less than 60 mL/min. Monitor blood pressure, renal function, and electrolytes in patients taking aliskiren and other agents that affect the RAAS [see Warnings and Precautions ( 5.4, 5.5, 5.6)].
The concomitant use of aliskiren with an ARB or an ACEI in diabetic patients is contraindicated [see Contraindications ( 4)].
Furosemide: Oral coadministration of aliskiren and furosemide reduced exposure to furosemide. Monitor diuretic effects when furosemide is coadministered with aliskiren.
-
6 ADVERSE REACTIONS
Most common adverse reaction: diarrhea (incidence 2.3%) ( 6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact LXO US Inc. at 1-844-800-8007 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
The following serious adverse reactions are discussed in greater detail in other sections of the label:
- Fetal Toxicity [see Warnings and Precautions ( 5.1)]
- Anaphylactic Reactions and Head and Neck Angioedema [see Warnings and Precautions ( 5.3)]
- Hypotension [see Warnings and Precautions ( 5.4)]
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
Adult Hypertension
Data described below reflect the evaluation of the safety of Tekturna in more than 6,460 patients, including over 1,740 treated for longer than 6 months, and more than 1,250 patients for longer than 1 year. In placebo-controlled clinical trials, discontinuation of therapy due to a clinical adverse event, including uncontrolled hypertension, occurred in 2.2% of patients treated with Tekturna versus 3.5% of patients given placebo. These data do not include information from the ALTITUDE study which evaluated the use of aliskiren in combination with ARBs or ACEIs [see Contraindications ( 4), Warnings and Precautions ( 5.2), and Clinical Studies ( 14.3)].
Angioedema: Two cases of angioedema with respiratory symptoms were reported with Tekturna use in the clinical studies. Two other cases of periorbital edema without respiratory symptoms were reported as possible angioedema and resulted in discontinuation. The rate of these angioedema cases in the completed studies was 0.06%.
In addition, 26 other cases of edema involving the face, hands, or whole body were reported with Tekturna use including 4 leading to discontinuation.
In the placebo-controlled studies, however, the incidence of edema involving the face, hands, or whole body was 0.4% with Tekturna compared with 0.5% with placebo. In a long-term active-control study with Tekturna and hydrochlorothiazide (HCTZ) arms, the incidence of edema involving the face, hand or whole body was 0.4% in both treatment arms [see Warnings and Precautions ( 5.2)].
Gastrointestinal: Tekturna produces dose-related gastrointestinal (GI) adverse reactions. Diarrhea was reported by 2.3% of patients at 300 mg, compared to 1.2% in placebo patients. In women and the elderly (age 65 years and older) increases in diarrhea rates were evident starting at a dose of 150 mg daily, with rates for these subgroups at 150 mg comparable to those seen at 300 mg for men or younger patients (all rates about 2.0% to 2.3%). Other GI symptoms included abdominal pain, dyspepsia, and gastroesophageal reflux, although increased rates for abdominal pain and dyspepsia were distinguished from placebo only at 600 mg daily. Diarrhea and other GI symptoms were typically mild and rarely led to discontinuation.
Cough: Tekturna was associated with a slight increase in cough in the placebo-controlled studies (1.1% for any Tekturna use versus 0.6% for placebo). In active-controlled trials with ACE inhibitor (ramipril, lisinopril) arms, the rates of cough for the Tekturna arms were about one-third to one-half the rates in the ACE inhibitor arms.
Seizures: Single episodes of tonic-clonic seizures with loss of consciousness were reported in 2 patients treated with Tekturna in the clinical trials. One of these patients did have predisposing causes for seizures and had a negative electroencephalogram (EEG) and cerebral imaging following the seizures (for the other patient EEG and imaging results were not reported). Tekturna was discontinued and there was no rechallenge.
Other adverse effects with increased rates for Tekturna compared to placebo included rash (1% versus 0.3%), elevated uric acid (0.4% versus 0.1%), gout (0.2% versus 0.1%) and renal stones (0.2% versus 0%).
Aliskiren's effect on ECG intervals was studied in a randomized, double-blind, placebo and active-controlled (moxifloxacin), 7-day repeat dosing study with Holter-monitoring and 12 lead ECGs throughout the interdosing interval. No effect of aliskiren on QT interval was seen.
Pediatric Hypertension
Aliskiren has been evaluated for safety in 267 pediatric hypertensive patients 6 to 17 years of age; including 208 patients treated for 52 weeks [see Clinical Studies ( 14.4)] . These studies did not reveal any unanticipated adverse reactions. Adverse reactions in pediatric patients 6 years of age and older are expected to be similar to those seen in adults.
Clinical Laboratory Findings
In controlled clinical trials, clinically relevant changes in standard laboratory parameters were rarely associated with the administration of Tekturna in patients with hypertension not concomitantly treated with an ARB or ACEI. In multiple- dose studies in hypertensive patients, Tekturna had no clinically important effects on total cholesterol, HDL, fasting triglycerides, or fasting glucose.
Blood Urea Nitrogen, Creatinine: In patients with hypertension not concomitantly treated with an ARB or ACEI, minor increases in blood urea nitrogen (BUN) or serum creatinine were observed in less than 7% of patients treated with Tekturna alone versus 6% on placebo [see Warnings and Precautions ( 5.2)] .
Hemoglobin and Hematocrit: Small decreases in hemoglobin and hematocrit (mean decreases of approximately 0.08 g/dL and 0.16 volume percent, respectively, for all aliskiren monotherapy) were observed. The decreases were dose-related and were 0.24 g/dL and 0.79 volume percent for 600 mg daily. This effect is also seen with other agents acting on the renin angiotensin system, such as angiotensin inhibitors and ARBs, and may be mediated by reduction of angiotensin II which stimulates erythropoietin production via the AT1 receptor. These decreases led to slight increases in rates of anemia with aliskiren compared to placebo were observed (0.1% for any aliskiren use, 0.3% for aliskiren 600 mg daily, versus 0% for placebo). No patients discontinued therapy due to anemia.
Serum Potassium: In patients with hypertension not concomitantly treated with an ARB or ACEI, increases in serum potassium greater than 5.5 mEq/L were infrequent (0.9% compared to 0.6% with placebo) [see Contraindications ( 4) and Warnings and Precautions ( 5.6)] .
Serum Uric Acid: Aliskiren monotherapy produced small median increases in serum uric acid levels (about 6 micromol/L) while HCTZ produced larger increases (about 30 micromol/L). The combination of aliskiren with HCTZ appears to be additive (about 40 micromol/L increase). The increases in uric acid appear to lead to slight increases in uric acid-related AEs: elevated uric acid (0.4% versus 0.1%), gout (0.2% versus. 0.1%), and renal stones (0.2% versus 0%).
Creatine Kinase: Increases in creatine kinase of greater than 300% were recorded in about 1% of aliskiren monotherapy patients versus 0.5% of placebo patients. Five cases of creatine kinase rises, 3 leading to discontinuation and 1 diagnosed as subclinical rhabdomyolysis, and another as myositis, were reported as adverse events with aliskiren use in the clinical trials. No cases were associated with renal dysfunction.
6.2 Postmarketing Experience
The following adverse reactions have been reported in aliskiren postmarketing experience. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to estimate their frequency or establish a causal relationship to drug exposure.
Hypersensitivity: anaphylactic reactions and angioedema requiring airway management and hospitalization
Urticaria
Peripheral edema
Hepatic enzyme increase with clinical symptoms of hepatic dysfunction
Severe cutaneous adverse reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis
Pruritus
Erythema
Hyponatremia
Nausea, Vomiting
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Tekturna can cause fetal harm when administered to a pregnant woman. Use of drugs that act on the renin-angiotensin system during the second and third trimesters of pregnancy reduces fetal renal function and increases fetal and neonatal morbidity and death [see Clinical Considerations] . Most epidemiologic studies examining fetal abnormalities after exposure to antihypertensive use in the first trimester have not distinguished drugs affecting the renin-angiotensin system from other antihypertensive agents. When pregnancy is detected, discontinue Tekturna as soon as possible.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major malformations and miscarriage in clinically recognized pregnancies is 2-4%, and 15-20%, respectively.
Clinical Considerations
Disease-Associated Maternal and/or Embryo/Fetal Risk
Hypertension in pregnancy increases the maternal risk for pre-eclampsia, gestational diabetes, premature delivery, and delivery complications (e.g., need for cesarean section, and post-partum hemorrhage).
Hypertension increases the fetal risk for intrauterine growth restriction and intrauterine death. Pregnant women with hypertension should be carefully monitored and managed accordingly.
Fetal/Neonatal Adverse Reactions
Use of drugs that act on the renin-angiotensin system in the second and third trimesters of pregnancy can result in the following: reduced fetal renal function leading to anuria and renal failure, oligohydramnios, fetal lung hypoplasia and skeletal deformations, including skull hypoplasia, hypotension, and death. In the unusual case that there is no appropriate alternative to therapy with drugs affecting the renin-angiotensin system for a particular patient, apprise the mother of the potential risk to the fetus.
In patients taking Tekturna during pregnancy, perform serial ultrasound examinations to assess the intra- amniotic environment. Fetal testing may be appropriate, based on the week of gestation. Patients and physicians should be aware, however, that oligohydramnios may not appear until after the fetus has sustained irreversible injury. Closely observe infants with histories of in utero exposure to Tekturna for hypotension, oliguria, and hyperkalemia. If oliguria or hypotension occur in neonates with a history of in utero exposure to aliskiren, support blood pressure and renal perfusion. Exchange transfusions or dialysis may be required as a means of reversing hypotension and substituting for disordered renal function.
Data
Animal Data
In developmental toxicity studies, pregnant rats and rabbits received oral aliskiren hemifumarate during organogenesis at doses up to 20 and 7 times the maximum recommended human dose (MRHD) based on body surface area (mg/m 2), respectively, in rats and rabbits. (Actual animal doses were up to 600 mg/kg/day in rats and up to 100 mg/kg/day in rabbits.) No teratogenicity was observed; however, fetal birth weight was decreased in rabbits at doses 3.2 times the MRHD based on body surface area (mg/m 2). Aliskiren was present in placentas, amniotic fluid and fetuses of pregnant rabbits.
8.2 Lactation
Risk Summary
There is no information regarding the presence of aliskiren in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for serious adverse reactions, including hypotension, hyperkalemia and renal impairment in nursing infants, advise a nursing woman that breastfeeding is not recommended during treatment with Tekturna.
8.4 Pediatric Use
Tekturna is contraindicated in patients less than 2 years of age [see Contraindications ( 4)].
Tekturna is indicated for treatment of hypertension in pediatric patients 6 years of age and older weighing 50 kg or more. The safety and effectiveness of aliskiren have been established in pediatric patients 6 years of age and older weighing 20 kg or more, but Tekturna is not approved in patients 6 years of age and older weighing 20 kg to less than 50 kg because of the lack of an appropriate dosage form. Use of Tekturna in pediatric patients 6 years and older is supported by evidence from a pharmacokinetic trial and two randomized, double-blind clinical trials in pediatric patients with hypertension 6 years to 17 years of age weighing 20 kg or more [see Clinical Pharmacology ( 12.3), Clinical Studies ( 14.4) ].
The safety and effectiveness of Tekturna have not been established in pediatric patients younger than 6 years of age and patients less than 20 kg. Avoid use in patients 2 years to less than 6 years and patients weighing less than 20 kg due to the limited information about aliskiren metabolism and exposures in this age group. No data are available in pediatric patients weighing less than 20 kg or in pediatric patients with a glomerular filtration rate <30 mL/min/1.73 m 2.
Juvenile Animal Toxicity Data
Toxicology studies in juvenile animals the approximate human age equivalent of children less than 2 years of age identified 85- to 385-fold increased systemic exposure to aliskiren compared to adult rats. The increased aliskiren exposure in juvenile rats was attributed to immaturity in aliskiren drug transporters and metabolizing enzymes. Increased aliskiren exposures were associated with premature deaths. Although a definitive pathology-based cause of death could not be ascertained, the premature deaths were attributed to the immaturity in aliskiren metabolism. The nonclinical findings suggest a distinct age-dependent relationship between dose and exposure.
8.5 Geriatric Use
Of the total number of patients receiving aliskiren in clinical studies, 1,275 (19%) were 65 years or older and 231 (3.4%) were 75 years or older. No overall differences in safety or effectiveness were observed between these subjects and younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
Safety and effectiveness of Tekturna in patients with severe renal impairment [creatinine clearance (CrCl) less than 30 mL/min] have not been established as these patients were excluded in efficacy trials [see Clinical Studies ( 14)].
-
10 OVERDOSAGE
Limited data are available related to overdosage in humans. The most likely manifestation of overdosage would be hypotension. If symptomatic hypotension occurs, supportive treatment should be initiated.
Aliskiren is poorly dialyzed. Therefore, hemodialysis is not adequate to treat aliskiren overexposure [see Clinical Pharmacology ( 12.3)] .
-
11 DESCRIPTION
Tekturna contains aliskiren hemifumarate, adirect renin inhibitor. Aliskiren hemifumarate is chemically described as (2S,4S,5S,7S)-N-(2-carbamoyl-2-methylpropyl)-5-amino-4-hydroxy- 2,7-diisopropyl-8-[4-methoxy-3-(3-methoxypropoxy)phenyl]-octanamide hemifumarate and its structural formula is:
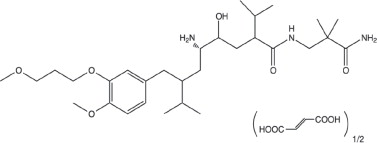
Molecular formula: C 30H 53N 3O 6 • 0.5 C 4H 4O 4
Aliskiren hemifumarate is a white to slightly yellowish crystalline powder with a molecular weight of 609.8 (free base- 551.8). It is soluble in phosphate buffer, n-octanol, and highly soluble in water.
Tekturna is available as film-coated tablets, which contains 165.75 mg or 331.5 mg aliskiren hemifumerate (equivalent to 150 mg or 300 mg aliskiren) and the following excipients: crospovidone; magnesium stearate; microcrystalline cellulose; povidone; silica, colloidal anhydrous; hypromellose; macrogol; talc; iron oxide, black (E 172); iron oxide, red (E 172); titanium dioxide (E 171).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Renin is secreted by the kidney in response to decreases in blood volume and renal perfusion. Renin cleaves angiotensinogen to form the inactive decapeptide angiotensin I (Ang I). Ang I is converted to the active octapeptide angiotensin II (Ang II) by ACE and non-ACE pathways. Ang II is a powerful vasoconstrictor and leads to the release of catecholamines from the adrenal medulla and prejunctional nerve endings. It also promotes aldosterone secretion and sodium reabsorption. Together, these effects increase blood pressure. Ang II also inhibits renin release, thus providing a negative feedback to the system. This cycle, from renin through angiotensin to aldosterone and its associated negative feedback loop, is known as the renin-angiotensin-aldosterone system (RAAS). Aliskiren is a direct renin inhibitor, decreasing plasma renin activity (PRA) and inhibiting the conversion of angiotensinogen to Ang I. Whether aliskiren affects other RAAS components, e.g., ACE or non-ACE pathways, is not known.
All agents that inhibit the RAAS, including renin inhibitors, suppress the negative feedback loop, leading to a compensatory rise in plasma renin concentration. When this rise occurs during treatment with ACEIs and ARBs, the result is increased levels of PRA. During treatment with aliskiren, however, the effect of increased renin levels is blocked so that PRA, Ang I and Ang II are all reduced, whether aliskiren is used as monotherapy or in combination with other antihypertensive agents.
12.2 Pharmacodynamics
In placebo-controlled clinical trials, PRA was decreased in a range of 50% to 80%. This reduction in PRA was not dose- related and did not correlate with blood pressure reductions. The clinical implications of the differences in effect on PRA are not known.
12.3 Pharmacokinetics
Aliskiren is poorly absorbed (bioavailability about 2.5%) with an approximate accumulation half-life of 24 hours. Steady state blood levels are reached in about 7 to 8 days.
Absorption and Distribution
Following oral administration, peak plasma concentrations of aliskiren are reached within 1 to 3 hours. When taken with a high-fat meal, mean AUC and C max of aliskiren are decreased by 71% and 85% respectively. In the clinical trials of aliskiren, it was administered without requiring a fixed relation of administration to meals.
Metabolism and Elimination
About one-fourth of the absorbed dose appears in the urine as parent drug. How much of the absorbed dose is metabolized is unknown. Based on the in vitro studies, the major enzyme responsible for aliskiren metabolism appears to be CYP3A4. Aliskiren does not inhibit the CYP450 isoenzymes (CYP 1A2, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A) or induce CYP3A4.
Transporters: Pgp (MDR1/Mdr1a/1b) was found to be the major efflux system involved in intestinal absorption and elimination via biliary excretion of aliskiren in preclinical studies. The potential for drug interactions at the Pgp site will likely depend on the degree of inhibition of this transporter.
Drug Interactions
The effect of coadministered drugs on the pharmacokinetics of aliskiren and vice versa, were studied in several single- and multiple-dose studies. Pharmacokinetic measures indicating the magnitude of these interactions are presented in Figure 1(impact of coadministered drugs on aliskiren) and Figure 2(impact of aliskiren on coadministered drugs).
Figure 1: The Impact of Coadministered Drugs on the Pharmacokinetics of Aliskiren
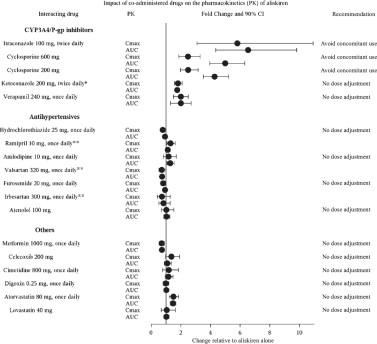
*Ketoconazole: A 400 mg once daily dose was not studied, but would be expected to increase aliskiren blood levels further.
**Ramipril, valsartan, irbesartan: In general, avoid combined use of aliskiren with ACE inhibitors or ARBs, particularly in patients with CrCl less than 60 mL/min [see Drug Interactions ( 7)].
Warfarin: There was no clinically significant effect of a single dose of warfarin 25 mg on the pharmacokinetics of aliskiren.
Figure 2: The Impact of Aliskiren on the Pharmacokinetics of Coadministered Drugs
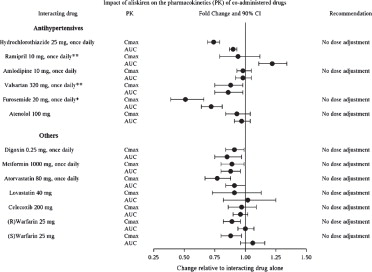
*Furosemide: Patients receiving furosemide could find its effects diminished after starting aliskiren. In patients with heart failure, coadministration of aliskiren (300 mg/day) reduced plasma AUC and C maxof oral furosemide (60 mg/day) by 17% and 27%, respectively, and reduced 24-hour urinary furosemide excretion by 29%. This change in exposure did not result in statistically significant difference in total urine volume and urinary sodium excretion over 24 hours. However, a transient decrease in urinary sodium excretion and urine volume effects up to 12 hours were observed when furosemide was coadministered with aliskiren 300 mg/day.
**Ramipril, valsartan: In general, avoid combined use of aliskiren with ACE inhibitors or ARBs, particularly in patients with CrCl less than 60 mL/min [see Drug Interactions ( 7)].
Specific Populations
Renally Impaired Patients: Aliskiren was evaluated in adult patients with varying degrees of renal insufficiency. The rate and extent of exposure (AUC and C max) of aliskiren in subjects with renal impairment did not show a consistent correlation with the severity of renal impairment. Adjustment of the starting dose is not required in these patients [see Warnings and Precautions ( 5.2)] .
The pharmacokinetics of aliskiren following administration of a single oral dose of 300 mg was evaluated in adult patients with End Stage Renal Disease (ESRD) undergoing hemodialysis. When compared to matched healthy subjects, changes in the rate and extent of aliskiren exposure (C max and AUC) in ESRD patients undergoing hemodialysis were not clinically significant.
Timing of hemodialysis did not significantly alter the pharmacokinetics of aliskiren in ESRD patients. Therefore, no dose adjustment is warranted in ESRD patients receiving hemodialysis.
Hepatically Impaired Patients: The pharmacokinetics of aliskiren were not significantly affected in patients with mild to severe liver disease. Consequently, adjustment of the starting dose is not required in these patients.
Pediatric Patients:
The pharmacokinetics of aliskiren were evaluated in an 8-day pharmacokinetic study in 39 pediatric patients with hypertension 6 years to 17 years of age. Aliskiren was given as daily doses of 2 mg/kg (0.67 times the lowest approved recommended dosage in a 50 kg pediatric patient) or 6 mg/kg (the highest recommended approved dosage in a 50 kg pediatric patient), administered as mini-tablets (3.125 mg oral pellets). The pharmacokinetic parameters of aliskiren were similar to those in adults, and the results of this study do not suggest that age or gender have any significant effect on aliskiren systemic exposure in patients 6 years to 17 years of age. Exposure decreased with increase in body weight.
In an 8-week randomized double blind study with aliskiren monotherapy in 267 pediatric patients with hypertension 6 years to 17 years of age [see Clinical Studies ( 14.4)] , fasting trough aliskiren concentrations at Day 28 demonstrated similar drug trough exposure levels to those observed in other trials using similar aliskiren doses in both adults and pediatric patients.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenic potential was assessed in a 2-year rat study and a 6-month transgenic (rasH2) mouse study with aliskiren hemifumarate at oral doses of up to 1500 mg aliskiren/kg/day. Although there were no statistically significant increases in tumor incidence associated with exposure to aliskiren, mucosal epithelial hyperplasia (with or without erosion/ulceration) was observed in the lower gastrointestinal tract at doses of greater than or equal to 750 mg/kg/day in both species, with a colonic adenoma identified in 1 rat and a cecal adenocarcinoma identified in another, rare tumors in the strain of rat studied. On a systemic exposure (AUC 0-24hr) basis, 1500 mg/kg/day in the rat is about 4 times and in the mouse about 1.5 times the maximum recommended human dose (MRHD) (300 mg aliskiren/day). Mucosal hyperplasia in the cecum or colon of rats was also observed at doses of 250 mg/kg/day (the lowest tested dose) as well as at higher doses in 4- and 13- week studies.
Aliskiren hemifumarate was devoid of genotoxic potential in the Ames reverse mutation assay with S. typhimurium and
E. coli, the in vitro Chinese hamster ovary cell chromosomal aberration assay, the in vitro Chinese hamster V79 cell gene mutation test and the in vivo mouse bone marrow micronucleus assay.
Fertility of male and female rats was unaffected at doses of up to 250 mg aliskiren/kg/day (8 times the MRHD of 300 mg Tekturna/60 kg on a mg/m 2 basis.)
13.2 Animal Toxicology and/or Pharmacology
Reproductive Toxicology Studies: Reproductive toxicity studies of aliskiren hemifumarate did not reveal any evidence of teratogenicity at oral doses up to 600 mg aliskiren/kg/day (20 times the MRHD of 300 mg/day on a mg/m 2 basis) in pregnant rats or up to 100 mg aliskiren/kg/day (7 times the MRHD on a mg/m 2 basis) in pregnant rabbits. Fetal birth weight was adversely affected in rabbits at 50 mg/kg/day (3.2 times the MRHD on a mg/m 2 basis). Aliskiren was present in placenta, amniotic fluid and fetuses of pregnant rabbits.
-
14 CLINICAL STUDIES
14.1 Aliskiren Monotherapy
The antihypertensive effects of Tekturna have been demonstrated in 6 randomized, double-blind, placebo-controlled 8- week clinical trials in patients with mild-to-moderate hypertension. The placebo response and placebo-subtracted changes from baseline in seated trough cuff blood pressure are shown in Table 2.
Table 2: Reductions in Seated Trough Cuff Blood Pressure (mmHg systolic/diastolic) in the Placebo-Controlled Studies *p value less than 0.05 versus placebo by ANCOVA with Dunnett's procedure for multiple comparisons
†p value less than 0.05 versus placebo by ANCOVA for the pairwise comparison.
Aliskiren daily dose, mg Study Placebo mean change 75 150 300 600 Placebo-subtracted Placebo-subtracted Placebo-subtracted Placebo-subtracted 1 2.9/3.3 5.7/4 * 5.9/4.5 * 11.2/7.5 * -- 2 5.3/6.3 -- 6.1/2.9* 10.5/5.4 * 10.4/5.2 * 3 10/8.6 2.2/1.7 2.1/1.7 5.1/3.7 * -- 4 7.5/6.9 1.9/1.8 4.8/2 * 8.3/3.3 * -- 5 3.8/4.9 -- 9.3/5.4 * 10.9/6.2 * 12.1/7.6 * 6 4.6/4.1 -- -- 8.4/4.9 † -- The studies included approximately 2,730 patients given doses of 75 mg (0.5 times the lowest recommended dosage) to 600 mg (twice the highest recommended dosage) of aliskiren and 1,231 patients given placebo. The recommended dosage of Tekturna is either 150 or 300 mg once daily [see Dosage and Administration ( 2.1)]. As shown in Table 1, there is some increase in response with administered dose in all studies, with reasonable effects seen at 150mg to 300 mg, and no clear further increases at 600 mg. A substantial proportion (85% to 90%) of the blood pressure-lowering effect was observed within 2 weeks of treatment. Studies with ambulatory blood pressure monitoring showed reasonable control throughout the interdosing interval; the ratios of mean daytime to mean nighttime ambulatory BP range from 0.6 to 0.9.
Patients in the placebo-controlled trials continued open-label aliskiren for up to 1 year. A persistent blood pressure- lowering effect was demonstrated by a randomized withdrawal study (patients randomized to continue drug or placebo), which showed a statistically significant difference between patients kept on aliskiren and those randomized to placebo. With cessation of treatment, blood pressure gradually returned toward baseline levels over a period of several weeks.
There was no evidence of rebound hypertension after abrupt cessation of therapy.
Aliskiren lowered blood pressure in all demographic subgroups, although black patients tended to have smaller reduction than Caucasians and Asians, as has been seen with ACEIs and ARBs.
There are no studies of Tekturna or members of the direct renin inhibitors demonstrating reductions in cardiovascular risk in patients with hypertension.
14.2 Aliskiren in Combination with Other Antihypertensives
Hydrochlorothiazide (HCTZ)
Aliskiren 75, 150, and 300 mg (0.5 times the recommended lowest dosage, the lowest recommended dosage, and the maximum recommended dosage, respectively) and HCTZ 6.25, 12.5, and 25 mg were studied alone and in combination in an 8-week, 2,776-patient, randomized, double-blind, placebo-controlled, parallel-group, 15-arm factorial study. Blood pressure reductions with the combinations were greater than the reductions with the monotherapies as shown in Table 3.
Table 3: Placebo-Subtracted Reductions in Seated Trough Cuff Blood Pressure (mmHg systolic/diastolic) in Combination with Hydrochlorothiazide Hydrochlorothiazide, mg Aliskiren, mg Placebo mean change 0 6.25 12.5 25 Placebo- subtracted Placebo- subtracted Placebo- subtracted Placebo- subtracted 0 7.5/6.9 -- 3.5/2.1 6.4/3.2 6.8/2.4 75 -- 1.9/1.8 6.8/3.8 8.2/4.2 9.8/4.5 150 -- 4.8/2 7.8/3.4 10.1/5 12/5.7 300 -- 8.3/3.3 -- 12.3/7 13.7/7.3 Valsartan
Aliskiren 150 mg and 300 mg and valsartan 160 mg and 320 mg were studied alone and in combination in an 8-week, 1,797-patient, randomized, double-blind, placebo-controlled, parallel-group, 4-arm, dose-escalation study. The dosages of aliskiren and valsartan were started at 150 mg and 160 mg, respectively, and increased at 4 weeks to 300 mg and 320 mg, respectively. Seated trough cuff blood pressure was measured at baseline, 4, and 8 weeks. Blood pressure reductions with the combinations were greater than the reductions with the monotherapies as shown in Table 4. In general, the combination of aliskiren and angiotensin receptor blocker should be avoided [see Contraindications ( 4), Warnings and Precautions ( 5), and Drug Interactions ( 7)].
Table 4: Placebo-Subtracted Reductions in Seated Trough Cuff Blood Pressure (mmHg systolic/diastolic) in Combination with Valsartan * The placebo change is 5.2/4.8 for week 4 endpoint which was used for the dose groups containing aliskiren 150 mg or valsartan 160 mg.
Aliskiren, mg Placebo mean change Valsartan, mg 0 160 320 0 4.6/4.1 * -- 5.6/3.9 8.2/5.6 150 -- 5.4/2.7 10.0/5.7 -- 300 -- 8.4/4.9 -- 12.6/8.1 Amlodipine
Aliskiren 150 mg and 300 mg and amlodipine besylate 5 mg and 10 mg were studied alone and in combination in an 8- week, 1,685-patient, randomized, double-blind, placebo-controlled, multifactorial study. Treatment with aliskiren and amlodipine resulted overall in significantly greater reductions in diastolic and systolic blood pressure compared to the respective monotherapy components as shown in Table 5.
Table 5: Placebo-Subtracted Reductions in Seated Trough Cuff Blood Pressure (mmHg systolic/diastolic) in Combination with Amlodipine
Aliskiren, mg
Placebo mean changeAmlodipine, mg 0 5 10 0 6.8/5.4 -- 9.0/5.6 14.3/8.5 150 -- 3.9/2.6 13.9/8.6 17.1/10.8 300 -- 8.6/4.9 15.0/9.6 16.4/11.1 14.3 Aliskiren in Patients with Diabetes Treated with ARB or ACEI (ALTITUDE study)
Patients with diabetes with renal disease (defined either by the presence of albuminuria or reduced GFR) were randomized to aliskiren 300 mg daily (n=4296) or placebo (n=4310). All patients were receiving background therapy with an ARB or ACEI. The primary efficacy outcome was the time to the first event of the primary composite endpoint consisting of cardiovascular death, resuscitated sudden death, nonfatal myocardial infarction, nonfatal stroke, unplanned hospitalization for heart failure, onset of end stage renal disease, renal death, and doubling of serum creatinine concentration from baseline sustained for at least 1 month. After a median follow-up of about 32 months, the trial was terminated early for lack of efficacy. Higher risk of renal impairment, hypotension and hyperkalemia was observed in aliskiren compared to placebo-treated patients, as shown in Table 6.
Table 6: Incidence of Selected Adverse Events During the Treatment Phase in ALTITUDE †renal failure, renal failure acute, renal failure chronic, renal impairment
††dizziness, dizziness postural, hypotension, orthostatic hypotension, presyncope, syncope
††† Given the variable baseline potassium levels of patients with renal insufficiency on dual RAAS therapy, the reporting of adverse event of hyperkalemia was at the discretion of the investigator.
* A Serious Adverse Event (SAE) is defined as: an event which is fatal or life-threatening, results in persistent or significant disability/incapacity, constitutes a congenital anomaly/birth defect, requires inpatient hospitalization or prolongation of existing hospitalization, or is medically significant (i.e., defined as an event that jeopardizes the patient or may require medical or surgical intervention to prevent one of the outcomes previously listed).
Aliskiren N=4272 Placebo N=4285 Serious Adverse Events* (%) Adverse Events (%) Serious Adverse Events* (%) Adverse Events (%) Renal impairment † 5.7 14.5 4.3 12.4 Hypotension †† 2.3 19.9 1.9 16.3 Hyperkalemia ††† 1.0 38.9 0.5 28.8 The risk of stroke (3.4% aliskiren versus 2.7% placebo) and death (8.4% aliskiren versus 8.0% placebo) were also numerically higher in aliskiren treated patients.
14.4 Pediatric Hypertension
The efficacy of aliskiren was evaluated in an 8-week randomized, double-blind trial in 267 pediatric patients with hypertension 6 years to 17 years of age (Study CSPP100A2365; NCT01150357). The majority of patients (82%) had primary hypertension, 59% had a BMI ≥95 th percentile, 20% had an estimated GFR between 60 and 90 mL/min/1.73m 2 and < 2% had an estimated GFR < 60 mL/min/1.73m 2. The mean age was 11.8 years and 74% of patients were Caucasian. In the initial 4-week,dose-response phase of the trial patients were randomized to weight-based low, mid and high dosing groups. At the end of this phase, patients entered a 4-week randomized withdrawal phase in which they were re-randomized in each weight category in a 1:1 ratio to continue the same dose of aliskiren or take placebo.
During the initial dose-response phase, aliskiren reduced both systolic and diastolic blood pressure in a weight-based dose-dependent manner. Sitting systolic blood pressure, the trial's primary endpoint, was reduced by 4.8, 5.6 and 8.7 mmHg from baseline in the low, medium and high dose groups, respectively. In the randomized withdrawal phase, the mean difference between the high dose group of aliskiren and placebo in the mean change in sitting systolic blood pressure was -2.7 mmHg.
Following the 8-week trial, 208 patients were enrolled in a 52-week extension trial in which patients were randomized in a 1:1 ratio (irrespective of whether they were on placebo or aliskiren at the end of the 8-week study) to receive either aliskiren or enalapril (CSPP100A2365E1; NCT01151410). The extension study included 3 dose levels based on weight; optional dose up-titrations were allowed during the study to control blood pressure.
At the end of 52 weeks, reductions in blood pressure from baseline were similar in patients receiving aliskiren (7.6/3.9 mmHg) and enalapril (7.9/4.9 mmHg).
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling ( Patient Information) and Instructions for Use.
Pregnancy: Advise female patients of child-bearing age about the consequences of exposure to Tekturna during pregnancy. Discuss treatment options with women planning to become pregnant. Advise patients to report pregnancies to their physicians as soon as possible.
Lactation: Advise nursing women that breastfeeding is not recommended during treatment with Tekturna [see Use in Specific Populations ( 8.2)].
Anaphylactic Reactions and Angioedema: Advise patients to immediately report any signs or symptoms suggesting a severe allergic reaction (difficulty breathing or swallowing, tightness of the chest, hives, general rash, swelling, itching, dizziness, vomiting, or abdominal pain) or angioedema (swelling of face, extremities, eyes, lips, tongue, difficulty in swallowing or breathing) and to take no more drug until they have consulted with the prescribing physicians.
Angioedema, including laryngeal edema, may occur at any time during treatment with Tekturna.
Symptomatic Hypotension: Advise patients that lightheadedness can occur, especially during the first days of Tekturna therapy, and that it should be reported to the prescribing physician. Advise patients that if syncope occurs, Tekturna should be discontinued until the physician has been consulted.
Caution patients that inadequate fluid intake, excessive perspiration, diarrhea, or vomiting can lead to an excessive fall in blood pressure, with the same consequences of lightheadedness and possible syncope.
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration
PATIENT INFORMATION
Tekturna® (tek-turn-a)
(aliskiren)
tabletsWhat is the most important information I should know about Tekturna?
Tekturna can cause harm or death to your unborn baby.- Talk to your doctor about other ways to lower your blood pressure if you plan to become pregnant.
- If you become pregnant during treatment with Tekturna, stop taking Tekturna and tell your doctor right away.
What is Tekturna?
Tekturna is a prescription medicine used to treat high blood pressure (hypertension) in adults and children weighing 50 kg or greater who are at least 6 years of age to lower blood pressure.
It is not known if Tekturna is safe and effective in children under 6 years of age.Do not take Tekturna if: - you have diabetes and are taking a kind of medicine called an angiotensin receptor blocker (ARB) or angiotensin-converting enzyme inhibitor (ACEI).
- you are allergic to any of the ingredients in Tekturna. See the end of this leaflet for a complete list of the ingredients in Tekturna.
Do not give Tekturna to children less than 2 years of age. Before taking Tekturna, tell your doctor about all of your medical conditions, including if you: - have kidney or heart problems.
- have diabetes.
- have ever had an allergic reaction to another blood pressure medicine.
- are pregnant or plan to become pregnant. See “ What is the most important information I should know about Tekturna?”
- are breastfeeding or plan to breastfeed. It is not known if Tekturna passes into your breast milk. You should not breastfeed during treatment with Tekturna.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Especially tell your doctor if you are taking: - a kind of medicine to control blood pressure called angiotensin receptor blocker (ARB) or angiotensin-converting enzyme inhibitor (ACEI).
- water pills (also called “diuretics”).
- cyclosporine (Gengraf ®, Neoral ®, Sandimmune ®)
- itraconazole (Onmel ®, Sporanox ®).
- potassium-containing medicines, potassium supplements, or salt substitutes containing potassium.
- nonsteroidal anti-inflammatory drugs (NSAIDs).
Ask your doctor if you are not sure if you are taking one of the medicines listed above.
Know the medicines you take. Keep a list of them to show your doctor or pharmacist when you get a new medicine. Your doctor or pharmacist will know what medicines are safe to take together.How should I take Tekturna tablets? - Take Tekturna exactly as your doctor tells you to take it.
- Take Tekturna 1 time a day, about the same time each day.
- Your doctor may change your dose if needed.
What are possible side effects of Tekturna?
Tekturna may cause serious side effects, including:- See “ What is the most important information I should know about Tekturna?”
- Serious allergic reactions that can be life threatening. Stop taking Tekturna and get emergency medical help right away and if you get any of these symptoms of a serious reactions during treatment with Tekturna:
- trouble breathing or swallowing
- chest tightness
- aised bumps (hives)
- rash
- swelling of the face, lips, tongue,
- throat, arms, legs or the whole body
- itching
- dizziness
- vomiting
- stomach (abdomen) pain
- Low blood pressure (hypotension). Your blood pressure may get too low if you also take water pills, are on a low-salt diet, sweat more than normal, receive dialysis treatments, have heart problems, or have vomiting or diarrhea. Lie down if you feel faint or dizzy and call your doctor right away.
- Kidney problems, including kidney failure. Tell your doctor if you urinate less.
- Increased potassium blood levels (hyperkalemia). Your doctor will check your potassium blood level during treatment with Tekturna.
The most common side effect of Tekturna is diarrhea.
These are not all of the possible side effects of Tekturna.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store Tekturna tablets? - Store Tekturna at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep Tekturna in the original container it comes in. Do not remove the desiccant (drying agent) from the bottle.
- Protect Tekturna from moisture.
Keep Tekturna and all medicines out of the reach of children. General information about the safe and effective use of Tekturna.
Medicines are sometimes prescribed for purpose other than those listed in the Patient Information leaflet. Do not take Tekturna for a condition for which it was not prescribed. Do not give Tekturna to other people, even if they have the same symptoms that you have. It may harm them. You can ask your doctor or pharmacist for information that is written for healthcare professionals.What is high blood pressure (hypertension)?
Blood pressure is the force in your blood vessels when your heart beats and when your heart rests. You have high blood pressure when the force is too great. High blood pressure makes the heart work harder to pump blood through the body and causes damage to the blood vessels. Tekturna can help your blood vessels relax so your blood pressure is lower. Medicines that lower your blood pressure lower your chance of having a stroke or heart attack.What are the ingredients in Tekturna?
Tekturna is available as film-coated tablets, which contains 165.75 mg or 331.5 mg aliskiren hemifumerate (equivalent to 150 mg or 300 mg aliskiren).
Active ingredient: aliskiren hemifumarate
Tekturna tablets inactive ingredients: crospovidone, magnesium stearate, microcrystalline cellulose, povidone, silica, colloidal anhydrous, hypromellose, macrogol, talc, iron oxide, black (E172), iron oxide, red (E172), and titanium dioxide (E171).
Distributed by: LXO US Inc., 1690 Sumneytown Pike, Suite 250 Lansdale, PA 19446
For more information about Tekturna, go to www.Tekturna.com, or call 1-844-800-8007. - aliskiren hemifumarate
-
INGREDIENTS AND APPEARANCE
TEKTURNA
aliskiren hemifumarate tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50090-2985(NDC:70839-300) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength ALISKIREN HEMIFUMARATE (UNII: C8A0P8G029) (ALISKIREN - UNII:502FWN4Q32) ALISKIREN 300 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 2S7830E561) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) FERRIC OXIDE RED (UNII: 1K09F3G675) MAGNESIUM STEARATE (UNII: 70097M6I30) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYETHYLENE GLYCOL, UNSPECIFIED (UNII: 3WJQ0SDW1A) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color red (light-red) Score no score Shape OVAL (Ovaliod) Size 18mm Flavor Imprint Code NVR;IU Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50090-2985-0 30 in 1 BOTTLE; Type 0: Not a Combination Product 04/25/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021985 03/05/2007 Labeler - A-S Medication Solutions (830016429) Establishment Name Address ID/FEI Business Operations A-S Medication Solutions 830016429 RELABEL(50090-2985)
