Label: VYVGART- efgartigimod alfa injection

- NDC Code(s): 73475-3041-5
- Packager: argenx US
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated January 23, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use VYVGART safely and effectively. See full prescribing information for VYVGART.
VYVGART® (efgartigimod alfa-fcab) injection, for intravenous use
Initial U.S. Approval: 2021INDICATIONS AND USAGE
VYVGART is a neonatal Fc receptor blocker indicated for the treatment of generalized myasthenia gravis (gMG) in adult patients who are anti-acetylcholine receptor (AChR) antibody positive. (1)
DOSAGE AND ADMINISTRATION
- Evaluate the need to administer age-appropriate vaccines according to immunization guidelines before initiation of a new treatment cycle with VYVGART. (2.1)
- The recommended dosage is 10 mg/kg administered as an intravenous infusion over one hour once weekly for 4 weeks. In patients weighing 120 kg or more, the recommended dose is 1200 mg per infusion. (2.2)
- Administer subsequent treatment cycles based on clinical evaluation; safety of initiating subsequent cycles sooner than 50 days from the start of the previous treatment cycle has not been established. (2.2)
- Must be diluted with 0.9% Sodium Chloride Injection, USP prior to administration. (2.3)
- Administer as an intravenous infusion over one hour via a 0.2 micron in-line filter. (2.3)
DOSAGE FORMS AND STRENGTHS
Injection: 400 mg in 20 mL (20 mg/mL) single-dose vial. (3)
CONTRAINDICATIONS
VYVGART is contraindicated in patients with serious hypersensitivity to efgartigimod alfa products or to any of the excipients of VYVGART. (4)
WARNINGS AND PRECAUTIONS
- Infections: Delay administration of VYVGART to patients with an active infection. Monitor for signs and symptoms of infection in patients treated with VYVGART. If serious infection occurs, administer appropriate treatment and consider withholding VYVGART until the infection has resolved. (5.1)
- Hypersensitivity Reactions: Anaphylaxis, hypotension leading to syncope, angioedema, dyspnea, and rash have occurred. If a hypersensitivity reaction occurs, the healthcare professional should institute appropriate measures if needed or the patient should seek medical attention.(4, 5.2)
- Infusion-Related Reactions: If a severe infusion-related reaction occurs, discontinue the infusion and initiate appropriate therapy; consider risks and benefits of readministering. If a mild to moderate infusion-related reaction occurs, may rechallenge with close clinical observation, slower infusion rates, and pre-medications. (5.3)
ADVERSE REACTIONS
Most common adverse reactions ( ≥10%) in patients treated with gMG are respiratory tract infections, headache, and urinary tract infection. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact argenx at 1-833-argx411 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
Closely monitor for reduced effectiveness of medications that bind to the human neonatal Fc receptor. When concomitant long-term use of such medications is essential for patient care, consider discontinuing VYVGART and using alternative therapies. (7)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 1/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination
2.2 Recommended Dose and Dose Schedules
2.3 Preparation and Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Infections
5.2 Hypersensitivity Reactions
5.3 Infusion-Related Reactions
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
6.3 Immunogenicity
7 DRUG INTERACTIONS
7.1 Effect of VYVGART on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Vaccination
Because VYVGART causes transient reduction in IgG levels, immunization with live-attenuated or live vaccines is not recommended during treatment with VYVGART. Evaluate the need to administer age-appropriate immunizations according to immunization guidelines before initiation of a new treatment cycle with VYVGART [see Dosage and Administration (2.2) and Warnings and Precautions (5.1)].
2.2 Recommended Dose and Dose Schedules
Dilute VYVGART prior to administration. Administer via intravenous infusion only [see Dosage and Administration (2.3)].
The recommended dosage of VYVGART is 10 mg/kg administered as an intravenous infusion over one hour once weekly for 4 weeks. In patients weighing 120 kg or more, the recommended dose of VYVGART is 1200 mg (3 vials) per infusion.
Administer subsequent treatment cycles based on clinical evaluation. The safety of initiating subsequent cycles sooner than 50 days from the start of the previous treatment cycle has not been established.
If a scheduled infusion is missed, VYVGART may be administered up to 3 days after the scheduled time point. Thereafter, resume the original dosing schedule until the treatment cycle is completed.
2.3 Preparation and Administration Instructions
Prior to administration, VYVGART single-dose vials require dilution in 0.9% Sodium Chloride Injection, USP, to make a total volume to be administered of 125 mL (see Preparation).
Check that the VYVGART solution is clear to slightly opalescent and colorless to slightly yellow. Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit. Do not use if opaque particles, discoloration, or other foreign particles are present.
Use aseptic technique when preparing the VYVGART diluted solution for intravenous infusion. Each vial is for single-dose only.
Discard any unused portion.
Preparation
- Calculate the dose (mg), total drug volume (mL) of VYVGART solution required, and the number of vials needed based on the recommended dose according to the patient's body weight [see Dosage and Administration (2.2)]. Each vial contains a total of 400 mg of VYVGART at a concentration of 20 mg per mL.
- Gently withdraw the calculated dose of VYVGART from the vial(s) with a sterile syringe and needle. Discard any unused portion of the vials.
- Dilute the withdrawn VYVGART with 0.9% Sodium Chloride Injection, USP to make a total volume of 125 mL for intravenous infusion.
- Gently invert the infusion bag containing the diluted VYVGART without shaking to ensure thorough mixing of the product and the diluent.
- The diluted solution can be administered using polyethylene (PE), polyvinyl chloride (PVC), ethylene vinyl acetate (EVA), or ethylene/polypropylene copolymer bags (polyolefins bags), and with PE, PVC, EVA, or polyurethane/polypropylene infusion lines.
Storage Conditions of the Diluted Solution
- VYVGART does not contain preservatives. Administer immediately after dilution and complete the infusion within 4 hours of dilution.
- If immediate use is not possible, the diluted solution may be stored refrigerated at 2°C to 8°C (36°F to 46°F) for up to 8 hours. Do not freeze. Protect from light. Allow the diluted drug to reach room temperature before administration. Complete the infusion within 4 hours of removal from the refrigerator. Do not heat the diluted drug in any manner other than via ambient air.
Administration
- VYVGART should be administered via intravenous infusion by a healthcare professional.
- Visually inspect VYVGART diluted solution for particles or discoloration prior to administration. Do not use if it is discolored, or if opaque or foreign particles are seen.
- Infuse the total 125 mL of diluted solution intravenously over one hour via a 0.2 micron in-line filter.
- After administration of VYVGART, flush the entire line with 0.9% Sodium Chloride Injection, USP.
- Monitor patients during administration and for 1 hour thereafter for clinical signs and symptoms of hypersensitivity reactions. If a hypersensitivity reaction occurs during administration, discontinue administration of VYVGART and institute appropriate supportive measures [see Warnings and Precautions (5.2)].
- Other medications should not be injected into infusion side ports or mixed with VYVGART.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
VYVGART is contraindicated in patients with serious hypersensitivity to efgartigimod alfa products or to any of the excipients of VYVGART. Reactions have included anaphylaxis and hypotension leading to syncope [see Warnings and Precautions (5.2)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Infections
VYVGART may increase the risk of infection. The most common infections observed in Study 1 were urinary tract infection (10% of VYVGART-treated patients compared to 5% of placebo-treated patients) and respiratory tract infections (33% of VYVGART-treated patients compared to 29% of placebo-treated patients) [see Adverse Reactions (6.1) and Clinical Studies (14)]. A higher frequency of patients who received VYVGART compared to placebo were observed to have below normal levels for white blood cell counts (12% versus 5%, respectively), lymphocyte counts (28% versus 19%, respectively), and neutrophil counts (13% versus 6%, respectively). The majority of infections and hematologic abnormalities were mild to moderate in severity. Delay VYVGART administration in patients with an active infection until the infection is resolved. During treatment with VYVGART, monitor for clinical signs and symptoms of infections. If serious infection occurs, administer appropriate treatment and consider withholding VYVGART until the infection has resolved.
Immunization
Immunization with vaccines during VYVGART treatment has not been studied. The safety of immunization with live or live-attenuated vaccines and the response to immunization with any vaccine are unknown. Because VYVGART causes a reduction in IgG levels, vaccination with live-attenuated or live vaccines is not recommended during treatment with VYVGART. Evaluate the need to administer age-appropriate vaccines according to immunization guidelines before initiation of a new treatment cycle with VYVGART.
5.2 Hypersensitivity Reactions
In clinical trials, hypersensitivity reactions, including rash, angioedema, and dyspnea were observed in VYVGART-treated patients. Hypersensitivity reactions were mild or moderate, occurred within one hour to three weeks of administration, and did not lead to treatment discontinuation.
Anaphylaxis and hypotension leading to syncope have been reported in postmarketing experience with VYVGART. Anaphylaxis and hypotension occurred during or within an hour of administration and led to infusion discontinuation and in some cases to permanent treatment discontinuation.
Monitor patients during administration and for 1 hour thereafter for clinical signs and symptoms of hypersensitivity reactions. If a hypersensitivity reaction occurs, the healthcare professional should institute appropriate measures if needed or the patient should seek medical attention. VYVGART is contraindicated in patients with a history of serious hypersensitivity to efgartigimod alfa products or to any of the excipients of VYVGART [see Contraindications (4)].
5.3 Infusion-Related Reactions
Infusion-related reactions have been reported with VYVGART in postmarketing experience. The most frequent symptoms and signs were hypertension, chills, shivering, and thoracic, abdominal, and back pain. Infusion-related reactions occurred during or within an hour of administration and led to infusion discontinuation. If a severe infusion-related reaction occurs during administration, discontinue VYVGART infusion and initiate appropriate therapy. Consider the risks and benefits of readministering VYVGART following a severe infusion-related reaction. If a mild to moderate infusion-related reaction occurs, patients may be rechallenged with close clinical observation, slower infusion rates, and pre-medications.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Infections [see Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Infusion-Related Reactions [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In clinical studies, the safety of VYVGART has been evaluated in 246 patients who received at least one dose of VYVGART, including 57 patients exposed to at least 7 treatment cycles and 8 patients exposed to at least 10 treatment cycles.
In a placebo-controlled study (Study 1) in patients with gMG, 84 patients received VYVGART 10 mg/kg [see Clinical Studies (14)]. Of these 84 patients, approximately 75% were female, 82% were White, 11% were Asian, and 8% were of Hispanic or Latino ethnicity. The mean age at study entry was 46 years (range 19 to 78).
The minimum time between treatment cycles, specified by study protocol, was 50 days. On average, VYVGART-treated patients received 2 cycles in Study 1. The mean and median times to the second treatment cycle were 94 days and 72 days from the initial infusion of the first treatment cycle, respectively, for VYVGART-treated patients.
Adverse reactions reported in at least 5% of patients treated with VYVGART and more frequently than placebo are summarized in Table 1. The most common adverse reactions (reported in at least 10% of VYVGART-treated patients) were respiratory tract infection, headache, and urinary tract infection.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of VYVGART. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Hypersensitivity reactions including anaphylaxis and hypotension, and infusion-related reactions [see Warnings and Precautions (5.2, 5.3)].
6.3 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to VYVGART in the studies described below with the incidence of antibodies in other studies or to other products may be misleading.
In up to 26 weeks of treatment in Study 1, 20% (17/83) of patients developed antibodies to VYVGART. Seven percent (6/83) of patients developed neutralizing antibodies.
Because few patients tested positive for anti-efgartigimod alfa-fcab antibodies and neutralizing antibodies, the available data are too limited to make definitive conclusions regarding immunogenicity and the effect on pharmacokinetics, safety, or efficacy of VYVGART.
-
7 DRUG INTERACTIONS
7.1 Effect of VYVGART on Other Drugs
Concomitant use of VYVGART with medications that bind to the human neonatal Fc receptor (FcRn) (e.g., immunoglobulin products, monoclonal antibodies, or antibody derivates containing the human Fc domain of the IgG subclass) may lower systemic exposures and reduce effectiveness of such medications. Closely monitor for reduced effectiveness of medications that bind to the human neonatal Fc receptor. When concomitant long-term use of such medications is essential for patient care, consider discontinuing VYVGART and using alternative therapies.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VYVGART during pregnancy. Healthcare providers and patients may call 1-855-272-6524 or go to https://www.Vyvgartpregnancy.com to enroll in or to obtain information about the registry.
Risk Summary
There are no available data on the use of VYVGART during pregnancy. There is no evidence of adverse developmental outcomes following the administration of VYVGART at up to 100 mg/kg/day in rats and rabbits (see Data).
The background rate of major birth defects and miscarriage in the indicated population is unknown. In the U.S. general population, the estimated background rate of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Monoclonal antibodies are increasingly transported across the placenta as pregnancy progresses, with the largest amount transferred during the third trimester. Therefore, efgartigimod alfa-fcab may be transmitted from the mother to the developing fetus.
As VYVGART is expected to reduce maternal IgG antibody levels, reduction in passive protection to the newborn is anticipated. Risk and benefits should be considered prior to administering live or live-attenuated vaccines to infants exposed to VYVGART in utero [see Warnings and Precautions (5.1)].
Data
Animal Data
Intravenous administration of efgartigimod alfa-fcab (0, 30, or 100 mg/kg/day) to pregnant rats and rabbits throughout organogenesis resulted in no adverse effects on embryofetal development in either species. The doses tested are 3 and 10 times the recommended human dose (RHD) of 10 mg/kg, on a body weight (mg/kg) basis.
Intravenous administration of efgartigimod alfa-fcab (0, 30, or 100 mg/kg/day) to rats throughout gestation and lactation resulted in no adverse effects on pre- or postnatal development. The doses tested are 3 and 10 times the recommended human dose (RHD) of 10 mg/kg, on a body weight (mg/kg) basis.
8.2 Lactation
Risk Summary
There is no information regarding the presence of efgartigimod alfa-fcab in human milk, the effects on the breastfed infant, or the effects on milk production. Maternal IgG is known to be present in human milk.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VYVGART and any potential adverse effects on the breastfed infant from VYVGART or from the underlying maternal condition.
8.5 Geriatric Use
Clinical studies of VYVGART did not include sufficient numbers of patients aged 65 and older to determine whether they respond differently from younger adult patients.
8.6 Renal Impairment
No dose adjustment of VYVGART is needed for patients with mild renal impairment. There are insufficient data to evaluate the impact of moderate renal impairment (eGFR 30-59 mL/min/1.73 m2) and severe renal impairment (eGFR <30 mL/min/1.73 m2) on pharmacokinetic parameters of efgartigimod alfa-fcab [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
Efgartigimod alfa-fcab is a human immunoglobulin G1 (IgG1)-derived Fc fragment (fragment, crystallized) of the za allotype. The efgartigimod alfa-fcab Fc fragment is a homodimer consisting of two identical peptide chains each consisting of 227 amino acids linked together by two interchain disulfide bonds with affinity for FcRn.The molecular weight of efgartigimod alfa-fcab is approximately 54 kDa.
VYVGART (efgartigimod alfa-fcab) injection is a sterile, preservative free, clear to slightly opalescent, colorless to slightly yellow solution supplied in a single-dose vial for infusion after dilution.
Each 20 mL single-dose vial contains 400 mg of efgartigimod alfa-fcab at a concentration of 20 mg/mL. In addition, each mL of solution contains L-arginine hydrochloride (31.6 mg), polysorbate 80 (0.2 mg), sodium chloride (5.8 mg), sodium phosphate dibasic anhydrous (2.4 mg), sodium phosphate monobasic monohydrate (1.1 mg) and water for injection, USP, at a pH of 6.7.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Efgartigimod alfa-fcab is a human IgG1 antibody fragment that binds to the neonatal Fc receptor (FcRn), resulting in the reduction of circulating IgG.
12.2 Pharmacodynamics
In Study 1 [see Clinical Studies (14)], the pharmacological effect of efgartigimod alfa-fcab was assessed by measuring the decrease in serum IgG levels and AChR autoantibody levels. In patients testing positive for AChR antibodies and who were treated with VYVGART, there was a reduction in total IgG levels relative to baseline. Decrease in AChR autoantibody levels followed a similar pattern.
12.3 Pharmacokinetics
Efgartigimod alfa-fcab exhibits linear pharmacokinetics, and following single doses of efgartigimod alfa-fcab, exposures increase proportionally up to 50 mg/kg (5 times the recommended dosage).
Metabolism and Elimination
Efgartigimod alfa-fcab is expected to be degraded by proteolytic enzymes into small peptides and amino acids.
The terminal half-life is 80 to 120 hours (3 to 5 days).
After a single intravenous dose of 10 mg/kg efgartigimod alfa-fcab in healthy subjects, less than 0.1% of the administered dose was recovered in urine.
Specific Populations
Age, Sex, and Race
A population pharmacokinetics analysis assessing the effects of age, sex, and race did not suggest any clinically significant impact of these covariates on efgartigimod alfa-fcab exposures.
Patients with Renal Impairment
No dedicated pharmacokinetic study has been performed in patients with renal impairment.
A population PK analysis of data from the VYVGART clinical studies indicated that patients with mild renal impairment (eGFR 60-89 mL/min/1.72m²) had 22% increase in exposure relative to the exposure in patients with normal renal function [see Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical drug interactions studies have not been performed with efgartigimod alfa-fcab.
P450 Enzymes
Efgartigimod alfa-fcab is not metabolized by cytochrome P450 enzymes; therefore, interactions with concomitant medications that are substrates, inducers, or inhibitors of cytochrome P450 enzymes are unlikely.
Drug Interactions with Other Drugs or Biological Products
Efgartigimod alfa-fcab may decrease concentrations of compounds that bind to the human FcRn [see Drug Interactions (7.1)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
No studies have been conducted to assess the carcinogenic potential of efgartigimod alfa-fcab.
No studies have been conducted to assess the genotoxic potential of efgartigimod alfa-fcab.
Impairment of Fertility
Intravenous administration of efgartigimod alfa-fcab (0, 30, or 100 mg/kg/day) to male and female rats prior to and during mating and continuing in females through gestation day 7 resulted in no adverse effects on fertility. The doses tested are 3 and 10 times the recommended human dose (RHD) of 10 mg/kg, on a body weight (mg/kg) basis.
-
14 CLINICAL STUDIES
The efficacy of VYVGART for the treatment of generalized myasthenia gravis (gMG) in adults who are AChR antibody positive was established in a 26-week, multicenter, randomized, double-blind, placebo-controlled trial (Study 1; NCT03669588).
Study 1 enrolled patients who met the following criteria at screening:
- -
- Myasthenia Gravis Foundation of America (MGFA) clinical classification class II to IV
- -
- MG-Activities of Daily Living (MG-ADL) total score of ≥ 5
- -
- On stable dose of MG therapy prior to screening, that included acetylcholinesterase (AChE) inhibitors, steroids, or non-steroidal immunosuppressive therapies (NSISTs), either in combination or alone
- -
- IgG levels of at least 6 g/L
A total of 167 patients were enrolled in Study 1 and were randomized to receive either VYVGART 10 mg/kg (1200 mg for those weighing 120 kg or more) (n=84) or placebo (n=83). Baseline characteristics were similar between treatment groups. Patients had a median age of 46 years at screening (range: 19 to 81 years) and a median time since diagnosis of 7 years. Seventy-one percent were female, and 84% were White. Median MG-ADL total score was 9, and median Quantitative Myasthenia Gravis (QMG) total score was 16. The majority of patients (n=65 for VYVGART; n=64 for placebo) were positive for AChR antibodies.
At baseline, over 80% of patients in each group received AChE inhibitors, over 70% in each treatment group received steroids, and approximately 60% in each treatment group received NSISTs, at stable doses.
Patients were treated with VYVGART at the recommended dosage regimen [see Dosage and Administration (2.2)].
The efficacy of VYVGART was measured using the Myasthenia Gravis-Specific Activities of Daily Living scale (MG-ADL) which assesses the impact of gMG on daily functions of 8 signs or symptoms that are typically affected in gMG. Each item is assessed on a 4-point scale where a score of 0 represents normal function and a score of 3 represents loss of ability to perform that function. A total score ranges from 0 to 24, with the higher scores indicating more impairment. In this study, an MG-ADL responder was defined as a patient with a 2-point or greater reduction in the total MG-ADL score compared to the treatment cycle baseline for at least 4 consecutive weeks, with the first reduction occurring no later than 1 week after the last infusion of the cycle.
The primary efficacy endpoint was the comparison of the percentage of MG-ADL responders during the first treatment cycle between treatment groups in the AChR-Ab positive population. A statistically significant difference favoring VYVGART was observed in the MG-ADL responder rate during the first treatment cycle [67.7% in the VYVGART-treated group vs 29.7% in the placebo-treated group (p <0.0001)].
The efficacy of VYVGART was also measured using the Quantitative Myasthenia Gravis (QMG) total score which is a 13-item categorical grading system that assesses muscle weakness. Each item is assessed on a 4-point scale where a score of 0 represents no weakness and a score of 3 represents severe weakness. A total possible score ranges from 0 to 39, where higher scores indicate more severe impairment. In this study, a QMG responder was defined as a patient who had a 3-point or greater reduction in the total QMG score compared to the treatment cycle baseline for at least 4 consecutive weeks, with the first reduction occurring no later than 1 week after last infusion of the cycle.
The secondary endpoint was the comparison of the percentage of QMG responders during the first treatment cycle between both treatment groups in the AChR-Ab positive patients. A statistically significant difference favoring VYVGART was observed in the QMG responder rate during the first treatment cycle [63.1% in the VYVGART-treated group vs 14.1% in the placebo-treated group (p <0.0001)].
The results are presented in Table 2.
Table 2: MG-ADL and QMG Responders During Cycle 1 in AChR-Ab Positive Patients (mITT Analysis Set) VYVGART
n=65
%Placebo
n=64
%P-value Odds Ratio (95% CI) MG-ADL=Myasthenia Gravis Activities of Daily Living; QMG =Quantitative Myasthenia Gravis; mITT=modified intent-to-treat; n=number of patients for whom the observation was reported; CI = confidence interval;
Logistic regression stratified for AChR-Ab status (if applicable), Japanese/Non-Japanese and standard of care, with baseline MG-ADL as covariate / QMG as covariates
Two-sided exact p-valueMG-ADL Responders 67.7 29.7 < 0.0001 4.951 (2.213, 11.528) QMG Responders 63.1 14.1 < 0.0001 10.842 (4.179, 31.200) Figure 1 shows the mean change from baseline on the MG-ADL during cycle 1.
Figure 1: Mean Change in Total MG-ADL From Cycle 1 Baseline Over Time in AChR-Ab Positive Patients (mITT Analysis Set) 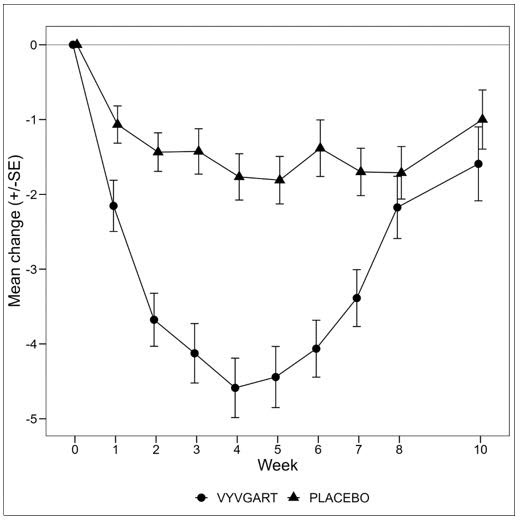
Figure 2 shows the distribution of response on the MG-ADL and QMG during cycle 1, four weeks after the first infusion with VYVGART.
Figure 2: Percentage of Patients with MG-ADL and QMG Total Score Change 4 Weeks Post Initial Infusion of the First Cycle in AChR-Ab Positive Population 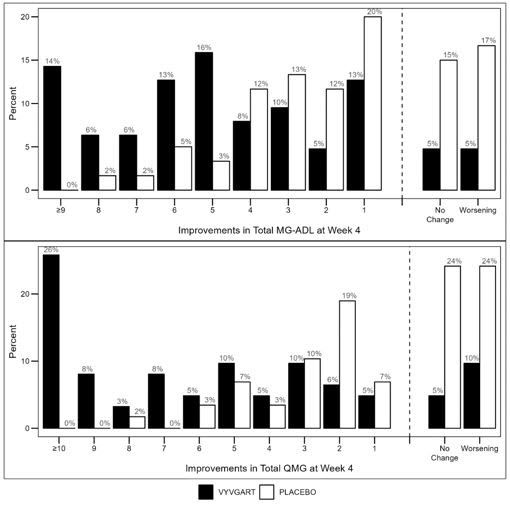
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Pregnancy Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to VYVGART during pregnancy. Encourage participation and advise patients about how they may enroll in the registry [see Use In Specific Populations (8.1)].
Infections
Instruct patients to communicate any history of infections to the healthcare provider and to contact their healthcare provider if they develop any symptoms of an infection. Advise patients to complete age-appropriate vaccines according to immunization guidelines prior to initiation of a new treatment cycle with VYVGART. Administration of live or live-attenuated vaccines is not recommended during treatment with VYVGART [see Warnings and Precautions (5.1)].
Hypersensitivity Reactions
Inform patients that hypersensitivity reactions, including angioedema and anaphylaxis, have occurred in patients who were treated with VYVGART. Inform patients about the signs and symptoms of these reactions, and advise patients to contact their healthcare provider immediately if these occur [see Warnings and Precautions (5.2)].
Infusion-Related Reactions
Advise patients of the potential risk of infusion-related reactions, which can include hypertension, chills, shivering, and chest, abdominal, and back pain [see Warnings and Precautions (5.3)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 20 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
VYVGART
efgartigimod alfa injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:73475-3041 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength EFGARTIGIMOD ALFA (UNII: 961YV2O515) (EFGARTIGIMOD ALFA - UNII:961YV2O515) EFGARTIGIMOD ALFA 20 mg in 1 mL Inactive Ingredients Ingredient Name Strength ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 31.6 mg in 1 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.2 mg in 1 mL SODIUM CHLORIDE (UNII: 451W47IQ8X) 5.8 mg in 1 mL SODIUM PHOSPHATE, DIBASIC, ANHYDROUS (UNII: 22ADO53M6F) 2.4 mg in 1 mL SODIUM PHOSPHATE, MONOBASIC, MONOHYDRATE (UNII: 593YOG76RN) 1.1 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:73475-3041-5 1 in 1 CARTON 12/17/2021 1 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761195 12/17/2021 Labeler - argenx US (116702819) Establishment Name Address ID/FEI Business Operations Vetter (DP) 316126754 MANUFACTURE(73475-3041) Establishment Name Address ID/FEI Business Operations Lonza Biologics, plc. 230794810 API MANUFACTURE(73475-3041) Establishment Name Address ID/FEI Business Operations Lonza Biologics Tuas Pte. Ltd. 936939342 API MANUFACTURE(73475-3041)
