Label: STIVARGA- regorafenib tablet, film coated

-
NDC Code(s):
50419-171-00,
50419-171-01,
50419-171-03,
50419-171-04, view more50419-171-05, 50419-171-06
- Packager: Bayer HealthCare Pharmaceuticals Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated December 9, 2020
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use STIVARGA safely and effectively. See full prescribing information for STIVARGA.
STIVARGA® (regorafenib) tablets, for oral use
Initial U.S. Approval: 2012WARNING: HEPATOTOXICITY
See full prescribing information for complete boxed warning.
- •
- Severe and sometimes fatal hepatotoxicity has occurred in clinical trials. (5.1)
- •
- Monitor hepatic function prior to and during treatment. (5.1)
- •
- Interrupt and then reduce or discontinue STIVARGA for hepatotoxicity as manifested by elevated liver function tests or hepatocellular necrosis, depending upon severity and persistence. (2.2)
INDICATIONS AND USAGE
STIVARGA is a kinase inhibitor indicated for the treatment of patients with:
- •
- Metastatic colorectal cancer (CRC) who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF therapy, and, if RAS wild-type, an anti-EGFR therapy. (1.1)
- •
- Locally advanced, unresectable or metastatic gastrointestinal stromal tumor (GIST) who have been previously treated with imatinib mesylate and sunitinib malate. (1.2)
- •
- Hepatocellular carcinoma (HCC) who have been previously treated with sorafenib (1.3)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 40 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- •
- Hepatotoxicity: Monitor liver function tests. Withhold and then reduce or discontinue STIVARGA based on severity and duration. (5.1)
- •
- Infections: Withhold STIVARGA in patients with worsening or severe infections. (5.2)
- •
- Hemorrhage: Permanently discontinue STIVARGA for severe or life-threatening hemorrhage. (5.3)
- •
- Gastrointestinal perforation or fistula: Discontinue STIVARGA. (5.4)
- •
- Dermatologic toxicity: Withhold and then reduce or discontinue STIVARGA depending on severity and persistence of dermatologic toxicity. (5.5)
- •
- Hypertension: Temporarily or permanently withhold STIVARGA for severe or uncontrolled hypertension. (5.6)
- •
- Cardiac ischemia and infarction: Withhold STIVARGA for new or acute cardiac ischemia/infarction and resume only after resolution of acute ischemic events. (5.7)
- •
- Reversible posterior leukoencephalopathy syndrome (RPLS): Discontinue STIVARGA. (5.8)
- •
- Risk of impaired wound healing: Withhold for at least 2 weeks prior to elective surgery. Do not administer for at least 2 weeks after major surgery and until adequate wound healing. The safety of resumption of STIVARGA after resolution of wound healing complications has not been established. (5.9)
- •
- Embryo-fetal toxicity: Can cause fetal harm. Advise women of potential risk to a fetus and to use effective contraception during treatment and for 2 months after the final dose. Advise males to use effective contraception for 2 months after the final dose. (5.10,8.1, 8.3)
ADVERSE REACTIONS
The most common adverse reactions (≥20%) are pain (including gastrointestinal and abdominal pain), HFSR, asthenia/fatigue, diarrhea, decreased appetite/food intake, hypertension, infection, dysphonia, hyperbilirubinemia, fever, mucositis, weight loss, rash, and nausea. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Bayer HealthCare Pharmaceuticals Inc. at 1-888-842-2937 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
Nursing Mothers: Discontinue drug or nursing, taking into consideration the importance of the drug to the mother. (8.3)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 12/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: HEPATOTOXICITY
1 INDICATIONS AND USAGE
1.1 Colorectal Cancer
1.2 Gastrointestinal Stromal Tumors
1.3 Hepatocellular Carcinoma
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
2.2 Dose Modifications
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
5.2 Infections
5.3 Hemorrhage
5.4 Gastrointestinal Perforation or Fistula
5.5 Dermatologic Toxicity
5.6 Hypertension
5.7 Cardiac Ischemia and Infarction
5.8 Reversible Posterior Leukoencephalopathy Syndrome
5.9 Risk of Impaired Wound Healing
5.10 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Effect of Strong CYP3A4 Inducers on Regorafenib
7.2 Effect of Strong CYP3A4 Inhibitors on Regorafenib
7.3 Effect of Regorafenib on Breast Cancer Resistance Protein (BCRP) Substrates
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
8.8 Race
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Colorectal Cancer
14.2 Gastrointestinal Stromal Tumors
14.3 Hepatocellular Carcinoma (HCC)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: HEPATOTOXICITY
- •
- Severe and sometimes fatal hepatotoxicity has occurred in clinical trials [see Warnings and Precautions (5.1)].
- •
- Monitor hepatic function prior to and during treatment [see Warnings and Precautions (5.1)].
- •
- Interrupt and then reduce or discontinue STIVARGA for hepatotoxicity as manifested by elevated liver function tests or hepatocellular necrosis, depending upon severity and persistence [see Dosage and Administration (2.2)].
-
1 INDICATIONS AND USAGE
1.1 Colorectal Cancer
STIVARGA is indicated for the treatment of patients with metastatic colorectal cancer (CRC) who have been previously treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF therapy, and, if RAS wild- type, an anti-EGFR therapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dose
The recommended dose is 160 mg STIVARGA (four 40 mg tablets) taken orally once daily for the first 21 days of each 28-day cycle. Continue treatment until disease progression or unacceptable toxicity.
Take STIVARGA at the same time each day. Swallow tablet whole with water after a low-fat meal that contains less than 600 calories and less than 30% fat [see Clinical Pharmacology (12.3)]. Do not take two doses of STIVARGA on the same day to make up for a missed dose from the previous day.
2.2 Dose Modifications
If dose modifications are required, reduce the dose in 40 mg (one tablet) increments; the lowest recommended daily dose of STIVARGA is 80 mg daily.
Interrupt STIVARGA for the following:
- •
- Grade 2 hand-foot skin reaction (HFSR) [palmar-plantar erythrodysesthesia syndrome (PPES)] that is recurrent or does not improve within 7 days despite dose reduction; interrupt therapy for a minimum of 7 days for Grade 3 HFSR
- •
- Symptomatic Grade 2 hypertension
- •
- Any Grade 3 or 4 adverse reaction
- •
- Worsening infection of any grade
Reduce the dose of STIVARGA to 120 mg:
- •
- For the first occurrence of Grade 2 HFSR of any duration
- •
- After recovery of any Grade 3 or 4 adverse reaction except infection
- •
- For Grade 3 aspartate aminotransferase (AST)/alanine aminotransferase (ALT) elevation, only resume if the potential benefit outweighs the risk of hepatotoxicity
Reduce the dose of STIVARGA to 80 mg:
- •
- For re-occurrence of Grade 2 HFSR at the 120 mg dose
- •
- After recovery of any Grade 3 or 4 adverse reaction at the 120 mg dose (except hepatotoxicity or infection)
Discontinue STIVARGA permanently for the following:
- •
- Failure to tolerate 80 mg dose
- •
- Any occurrence of AST or ALT more than 20 times the upper limit of normal (ULN)
- •
- Any occurrence of AST or ALT more than 3 times ULN with concurrent bilirubin more than 2 times ULN
- •
- Re-occurrence of AST or ALT more than 5 times ULN despite dose reduction to 120 mg
- •
- For any Grade 4 adverse reaction; only resume if the potential benefit outweighs the risks
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatotoxicity
Severe drug-induced liver injury with fatal outcome occurred in STIVARGA-treated patients in clinical trials. In most cases, liver dysfunction occurred within the first 2 months of therapy and was characterized by a hepatocellular pattern of injury.
In the CORRECT study, fatal hepatic failure occurred in 1.6% of patients in the regorafenib arm and in 0.4% of patients in the placebo arm. In the GRID study, fatal hepatic failure occurred in 0.8% of patients in the regorafenib arm. In the RESORCE study, there was no increase in the incidence of fatal hepatic failure as compared to placebo [see Adverse Reactions (6.1)].
Obtain liver function tests (ALT, AST, and bilirubin) before initiation of STIVARGA and monitor at least every two weeks during the first 2 months of treatment. Thereafter, monitor monthly or more frequently as clinically indicated. Monitor liver function tests weekly in patients experiencing elevated liver function tests until improvement to less than 3 times the ULN or baseline.
Temporarily hold and then reduce or permanently discontinue STIVARGA depending on the severity and persistence of hepatotoxicity as manifested by elevated liver function tests or hepatocellular necrosis [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
5.2 Infections
STIVARGA caused an increased risk of infections. The overall incidence of infection (Grades 1-5) was higher (32% vs. 17%) in 1142 STIVARGA-treated patients as compared to the control arm in randomized placebo‑controlled trials.The incidence of grade 3 or greater infections in STIVARGA treated patients was 9%. The most common infections were urinary tract infections (5.7%), nasopharyngitis (4.0%), mucocutaneous and systemic fungal infections (3.3%) and pneumonia (2.6%). Fatal outcomes caused by infection occurred more often in patients treated with STIVARGA (1.0%) as compared to patients receiving placebo (0.3%); the most common fatal infections were respiratory (0.6% in STIVARGA-treated patients vs 0.2% in patients receiving placebo).
Withhold STIVARGA for Grade 3 or 4 infections, or worsening infection of any grade. Resume STIVARGA at the same dose following resolution of infection [see Dosage and Administration (2.2)].
5.3 Hemorrhage
STIVARGA caused an increased incidence of hemorrhage. The overall incidence (Grades 1-5) was 18.2% in 1142 patients treated with STIVARGA and 9.5% in patients receiving placebo in randomized, placebo‑controlled trials. The incidence of grade 3 or greater hemorrhage in patients treated with STIVARGA was 3.0%. The incidence of fatal hemorrhagic events was 0.7%, involving the central nervous system or the respiratory, gastrointestinal, or genitourinary tracts.
Permanently discontinue STIVARGA in patients with severe or life-threatening hemorrhage. Monitor INR levels more frequently in patients receiving warfarin [see Clinical Pharmacology (12.3)].
5.4 Gastrointestinal Perforation or Fistula
Gastrointestinal perforation occurred in 0.6% of 4518 patients treated with STIVARGA across all clinical trials of STIVARGA administered as a single agent; this included eight fatal events.
Gastrointestinal fistula occurred in 0.8% of patients treated with STIVARGA and 0.2% of patients in placebo arm across randomized, placebo-controlled trials. Permanently discontinue STIVARGA in patients who develop gastrointestinal perforation or fistula.
5.5 Dermatologic Toxicity
In randomized, placebo-controlled trials, adverse skin reactions occurred in 71.9% of patients in the regorafenib arm and in 25.5% of patients in the placebo arm, including hand-foot skin reaction (HFSR) also known as palmar-plantar erythrodysesthesia syndrome (PPES), and severe rash requiring dose modification.
In the randomized, placebo-controlled trials, the overall incidence of HFSR was higher in 1142 STIVARGA-treated patients (53%) than in the placebo-treated patients (8%). Most cases of HFSR in STIVARGA-treated patients appeared during the first cycle of treatment. The incidences of Grade 3 HFSR (16% versus <1%), Grade 3 rash (3% versus <1%), serious adverse reactions of erythema multiforme (<0.1% vs. 0%) and Stevens-Johnson Syndrome (<0.1% vs. 0%) were also higher in STIVARGA-treated patients [see Adverse Reactions (6.1)]. Across all trials, a higher incidence of HFSR was observed in Asian patients treated with STIVARGA (all grades: 72%; Grade 3: 18%) [see Use in Specific Populations (8.8 )].
Toxic epidermal necrolysis occurred in 0.02% of 4518 STIVARGA-treated patients across all clinical trials of STIVARGA administered as a single agent.
Withhold STIVARGA, reduce the dose, or permanently discontinue STIVARGA depending on the severity and persistence of dermatologic toxicity [see Dosage and Administration (2.2)]. Institute supportive measures for symptomatic relief.
5.6 Hypertension
In randomized, placebo-controlled trials, hypertensive crisis occurred in 0.2% of patients in the regorafenib arms and in none of the patients in the placebo arms. STIVARGA caused an increased incidence of hypertension (30% versus 8% in CORRECT, 59% versus 27% in GRID, and 31% versus 6% in RESORCE) [see Adverse Reactions (6.1)]. The onset of hypertension occurred during the first cycle of treatment in most patients who developed hypertension (67% in randomized, placebo-controlled trials).
Do not initiate STIVARGA unless blood pressure is adequately controlled. Monitor blood pressure weekly for the first 6 weeks of treatment and then every cycle, or more frequently, as clinically indicated. Temporarily or permanently withhold STIVARGA for severe or uncontrolled hypertension [see Dosage and Administration (2.2)].
5.7 Cardiac Ischemia and Infarction
STIVARGA increased the incidence of myocardial ischemia and infarction (0.9% vs 0.2%) in randomized placebo-controlled trials [see Adverse Reactions (6.1)]. Withhold STIVARGA in patients who develop new or acute onset cardiac ischemia or infarction. Resume STIVARGA only after resolution of acute cardiac ischemic events, if the potential benefits outweigh the risks of further cardiac ischemia.
5.8 Reversible Posterior Leukoencephalopathy Syndrome
Reversible posterior leukoencephalopathy syndrome (RPLS), a syndrome of subcortical vasogenic edema diagnosed by characteristic finding on MRI, occurred in one of 4800 STIVARGA-treated patients across all clinical trials. Perform an evaluation for RPLS in any patient presenting with seizures, severe headache, visual disturbances, confusion or altered mental function. Discontinue STIVARGA in patients who develop RPLS.
5.9 Risk of Impaired Wound Healing
Impaired wound healing complications can occur in patients who receive drugs that inhibit the VEGF signaling pathway. Therefore, STIVARGA has the potential to adversely affect wound healing.
Withhold STIVARGA for at least 2 weeks prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of STIVARGA after resolution of wound healing complications has not been established.
5.10 Embryo-Fetal Toxicity
Based on animal studies and its mechanism of action, STIVARGA can cause fetal harm when administered to a pregnant woman. There are no available data on STIVARGA use in pregnant women. Regorafenib was embryolethal and teratogenic in rats and rabbits at exposures lower than human exposures at the recommended dose, with increased incidences of cardiovascular, genitourinary, and skeletal malformations. Advise pregnant women of the potential risk to a fetus.
Advise females of reproductive potential to use effective contraception during treatment with STIVARGA and for 2 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with STIVARGA and for 2 months after the final dose [see Use in Specific Populations (8.1), (8.3)].
-
6 ADVERSE REACTIONS
The following serious adverse reactions are discussed elsewhere in the labeling:
- •
- Hepatotoxicity [see Warnings and Precautions (5.1)]
- •
- Infections [see Warnings and Precautions (5.2)]
- •
- Hemorrhage [see Warnings and Precautions (5.3)]
- •
- Gastrointestinal Perforation or Fistula [see Warnings and Precautions (5.4)]
- •
- Dermatological Toxicity [see Warnings and Precautions (5.5)]
- •
- Hypertension [see Warnings and Precautions (5.6)]
- •
- Cardiac Ischemia and Infarction [see Warnings and Precautions (5.7)]
- •
- Reversible Posterior Leukoencephalopathy Syndrome (RPLS) [see Warnings and Precautions (5.8)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rate observed in practice.
The data described in the WARNINGS AND PRECAUTIONS section reflect exposure to STIVARGA in more than 4800 patients who were enrolled in four randomized, placebo-controlled trials (n=1142), an expanded access program (CONSIGN, n=2864), or single arm clinical trials (single agent or in combination with other agents). There were 4518 patients who received STIVARGA as a single agent; the distribution of underlying malignancies was 80% CRC, 4% GIST, 10% HCC, 6% other solid tumors; and 74% were White, 11% Asian, and 15% race not known. Among these 4518 patients, 83% received STIVARGA for at least 21 days and 20% received STIVARGA for 6 months or longer.
In randomized placebo-controlled trials (CORRECT, GRID, RESORCE and CONCUR), the most frequently observed adverse drug reactions (≥20%) in patients receiving STIVARGA are pain (including gastrointestinal and abdominal pain), HFSR, asthenia/fatigue, diarrhea, decreased appetite/food intake, hypertension, infection, dysphonia, hyperbilirubinemia, fever, mucositis, weight loss, rash, and nausea.
Colorectal Cancer
The safety data described below, except where noted, are derived from a randomized (2:1), double-blind, placebo-controlled trial (CORRECT) in which 500 patients (median age 61 years; 61% men) with previously-treated metastatic colorectal cancer (CRC) received STIVARGA as a single agent at the dose of 160 mg daily for the first 3 weeks of each 4 week treatment cycle and 253 patients (median age 61 years; 60% men) received placebo. The median duration of therapy was 1.7 months (range 2 days, 10.8 months) for patients receiving STIVARGA. Due to adverse reactions, 61% of the patients receiving STIVARGA required a dose interruption and 38% of the patients had their dose reduced. Adverse reactions that resulted in treatment discontinuation occurred in 8.2% of STIVARGA-treated patients compared to 1.2% of patients who received placebo. Hand-foot skin reaction (HFSR) and rash were the most common reasons for permanent discontinuation of STIVARGA.
Table 1 provides the incidence of adverse reactions (≥10%) in patients in CORRECT.
Table 1: Adverse drug reactions reported in ≥10% of patients treated with STIVARGA in CORRECT and reported more commonly than in patients receiving placeboa
Adverse ReactionsSTIVARGA
(N=500)Placebo
(N=253)Grade Grade All
%≥ 3
%All
%≥ 3
%General disorders and administration site conditions
Asthenia/fatigue
Pain
Fever
64
59
28
15
9
2
46
48
15
9
7
0
Metabolism and nutrition disorders
Decreased appetite and food intake
47
5
28
4
Skin and subcutaneous tissue disorders
HFSR/PPES
Rash b
45
26
17
6
7
4
0
<1
Gastrointestinal disorders
Diarrhea
Mucositis
43
33
8
4
17
5
2
0
Investigations
Weight loss
32
<1
10
0
Infections and infestations
Infection c
31
9
17
6
Vascular disorders
Hypertension
Hemorrhage c
30
21
8
2
8
8
<1
<1
Respiratory, thoracic and mediastinal disorders
Dysphonia
30
0
6
0
Nervous system disorders
Headache
10
<1
7
0
- a Adverse reactions graded according to National Cancer Institute Common Toxicity for Adverse Events version 3.0 (NCI CTCAE v3.0).
- bThe term rash represents reports of events of drug eruption, rash, erythematous rash, generalized rash, macular rash, maculo-papular rash, papular rash, and pruritic rash.
cFatal outcomes observed.
Table 2 provides laboratory abnormalities observed in CORRECT.
Table 2: Laboratory test abnormalities reported in CORRECT Laboratory Parameter STIVARGA
(N=500 a)Placebo
(N=253 a)Grade b Grade b All
%3
%4
%All
%3
%4
%Blood and lymphatic system disorders
Anemia
79
5
1
66
3
0
Thrombocytopenia
41
2
<1
17
<1
0
Neutropenia
3
1
0
0
0
0
- Lymphopenia
54
9
0
35
4
<1
Metabolism and nutrition disorders
Hypocalcemia
59
1
<1
18
1
0
Hypokalemia
26
4
0
8
<1
0
Hyponatremia
30
7
1
22
4
0
Hypophosphatemia
57
31
1
11
4
0
Hepatobiliary disorders
Hyperbilirubinemia
45
10
3
17
5
3
Increased AST
65
5
1
46
4
1
Increased ALT
45
5
1
30
3
<1
Renal and urinary disorders
Proteinuriac
84
2
0
61
1
0
Investigations
Increased INRd
24
4
N/A
17
2
N/A
Increased Lipase
46
9
2
19
3
2
Increased Amylase
26
2
<1
17
2
<1
a % based on number of patients with post-baseline samples which may be less than 500 (regorafenib) or 253 (placebo).
b NCI CTCAE v3.0.
cBased on urine protein-creatinine ratio data.
dInternational normalized ratio: No Grade 4 denoted in NCI CTCAE, v3.0.
Gastrointestinal Stromal Tumors
The safety data described below are derived from a randomized (2:1), double-blind, placebo-controlled trial (GRID) in which 132 patients (median age 60 years; 64% men) with previously-treated GIST received STIVARGA as a single agent at a dose of 160 mg daily for the first 3 weeks of each 4 week treatment cycle and 66 patients (median age 61 years; 64% men) received placebo. The median duration of therapy was 5.7 months (range 1 day, 11.7 months) for patients receiving STIVARGA. Dose interruptions for adverse events were required in 58% of patients receiving STIVARGA and 50% of patients had their dose reduced. Adverse reactions that resulted in treatment discontinuation were reported in 2.3% of STIVARGA-treated patients compared to 1.5% of patients who received placebo.
Table 3 provides the incidence of adverse reactions (≥10%) in patients in GRID.
Table 3: Adverse reactions reported in ≥10% patients treated with STIVARGA in GRID and reported more commonly than in patients receiving placeboa Adverse Reactions STIVARGA
(N=132)Placebo
(N=66)Grade Grade All
%≥ 3
%All
%≥ 3
%Skin and subcutaneous tissue disorders
HFSR/PPE
Rash b
Alopecia
67
30
24
22
7
2
12
3
2
2
0
0
General disorders and administration site conditions
Asthenia/Fatigue
Fever
52
21
4
0
39
11
2
2
Vascular disorders
Hypertension
Hemorrhage
59
11
28
4
27
3
5
0
Gastrointestinal disorders
Pain
Diarrhea
Mucositis
Nausea
Vomiting
60
47
40
20
17
8
8
2
2
<1
55
9
8
12
8
14
0
2
2
0
Respiratory, thoracic and mediastinal disorders
Dysphonia
39
0
9
0
Infections and infestations
Infection c
32
5
5
0
Metabolism and nutrition disorders
Decreased appetite and food intake
Hypothyroidism d
31
18
<1
0
21
6
3
0
Nervous system disorders
Headache
16
0
9
0
Investigations
Weight loss
14
0
8
0
Musculoskeletal and connective tissue disorders
Muscle spasms
14
0
3
0
a Adverse reactions graded according to NCI CTCAE v4.0.
- bThe term rash represents reports of events of rash, erythematous rash, macular rash, maculo-papular rash, papular rash and pruritic rash.
- cFatal outcomes observed.
dHypothyroidism incidence based on subset of patients with normal TSH and no thyroid supplementation at baseline.
Table 4 provides laboratory abnormalities observed in GRID.
Table 4: Laboratory test abnormalities reported in GRID Laboratory Parameter STIVARGA
(N=132a)Placebo
(N=66 a)Grade b Grade b All
%3
%4
%All
%3
%4
%Blood and lymphatic system disorders
Thrombocytopenia
Neutropenia
Lymphopenia
13
16
30
1
2
8
0
1
0
2
12
24
0
3
3
2
0
0
Metabolism and nutrition disorders
Hypocalcemia
Hypokalemia
Hypophosphatemia
17
21
55
2
3
20
0
0
2
5
3
3
0
0
2
0
0
0
Hepatobiliary disorders
Hyperbilirubinemia
Increased AST
Increased ALT
33
58
39
3
3
4
1
1
1
12
47
39
2
3
2
0
0
0
Renal and urinary disorders
Proteinuria c
59
3
- d
53
3
- d
Investigations
Increased Lipase
14
0
1
5
0
0
- a Percent based on number of patients with post-baseline samples which may be less than 132 (regorafenib) or 66 (placebo).
b NCI CTCAE v4.0.
c Based on urine protein-creatinine ratio data.
d No Grade 4 denoted in NCI CTCAE v4.0.
Hepatocellular Carcinoma
The safety data described below are derived from a randomized (2:1), double-blind, placebo-controlled trial (RESORCE) in which patients with previously-treated HCC received either STIVARGA (n=374) 160 mg orally on days 1-21 of each 4 week treatment cycle or placebo (n=193). The median age was 63 years, 88% were men, 98% had Child-Pugh A cirrhosis, 66% had an ECOG performance status (PS) of 0 and 34% had PS of 1. The median duration of therapy was 3.5 months (range 1 day to 29.4 months) for patients receiving STIVARGA. Of the patients receiving STIVARGA, 33% were exposed to STIVARGA for greater than or equal to 6 months and 14% were exposed to STIVARGA for greater than or equal to 12 months. Dose interruptions for adverse events were required in 58.3% of patients receiving STIVARGA and 48% of patients had their dose reduced. The most common adverse reactions requiring dose modification (interruption or dose reduction) were HFSR/PPES (20.6%), blood bilirubin increase (5.9%), fatigue (5.1%) and diarrhea (5.3%). Adverse reactions that resulted in treatment discontinuation were reported in 10.4% of STIVARGA-treated patients compared to 3.6% of patients who received placebo; the most common adverse reactions requiring discontinuation of STIVARGA were HFSR/PPES (1.9%) and AST increased (1.6%).
Table 5 provides the incidence of adverse reactions (≥10%) in patients in RESORCE.
Table 5: Adverse reactions reported in ≥10% of patients treated with STIVARGA in RESORCE and reported more commonly than in patients receiving placeboa Adverse Reactions STIVARGA
(N=374)Placebo
(N=193)Grade Grade All
%≥ 3
%All
%≥ 3
%Skin and subcutaneous tissue disorders
HFSR/PPE
51
12
7
<1
General disorders and administration site conditions
Pain
Asthenia/Fatigue
Fever
55
42
20
9
10
0
44
33
7
8
5
0
Vascular disorders
Hypertension
Hemorrhage b
31
18
15
5
6
16
5
8
Gastrointestinal disorders
Diarrhea
Nausea
Vomiting
Mucositis
41
17
13
13
3
<1
<1
1
15
13
7
2
0
0
<1
≤1
Respiratory, thoracic and mediastinal disorders
Dysphonia
18
0
2
0
Infections and infestations
Infection b
31
8
18
6
Metabolism and nutrition disorders
Decreased appetite and food intake
31
3
15
2
Investigations
Weight loss
13
2
4
0
Musculoskeletal and connective tissue disordersMuscle spasms
10
0
2
0
a Adverse reactions graded according to NCI CTCAE v4.0.
- bFatal outcomes observed.
Other clinically significant adverse reactions observed in less than 10% of STIVARGA-treated patients were: alopecia (7%), hypothyroidism (6.4%), pancreatitis (1.6%), exfoliative rash (1.3%), tremor (1.3%), erythema multiforme (0.8%), myocardial ischemia (0.8%), gastrointestinal fistula (0.3%), and myocardial infarction (0.3%).
Table 6 provides laboratory abnormalities observed in RESORCE.
Table 6: Laboratory test abnormalities reported in RESORCE Laboratory Parameter STIVARGA
(N=374a)Placebo
(N=193 a)Grade b Grade b All
%3
%4
%All
%3
%4
%Blood and lymphatic system disorders
Thrombocytopenia
Neutropenia
Lymphopenia
63
14
68
5
3
16
<1
0
2
50
15
59
0
<1
11
0
<1
<1
Metabolism and nutrition disorders
Hypocalcemia
Hypokalemia
Hypophosphatemia
23
31
70
<1
4
32
0
<1
2
10
9
31
0
2
7
0
0
0
Hepatobiliary disorders
Hyperbilirubinemia
Increased AST
Increased ALT
78
93
70
13
16
6
3
2
<1
55
84
59
11
17
5
5
3
0
Renal and urinary disorders
Proteinuria c
51
17
- d
37
3
- d
Investigations
Increased INR
Increased Lipase
Increased Amylase
44
41
23
<1
11
3
- d
3
<1
35
27
19
2
8
2
- d
1
<1
- a Percent based on number of patients with post-baseline samples which may be less than 374 (regorafenib) or 193 (placebo).
b NCI CTCAE v4.0.
c Based on dipstick data.
d No Grade 4 denoted in NCI CTCAE v4.0.
6.2 Postmarketing Experience
The following adverse reaction has been identified during postapproval use of STIVARGA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure:
- •
- hypersensitivity reaction
- •
- nephrotic syndrome
- •
- cardiac failure
- •
- arterial (including aortic) aneurysms, dissections, and rupture
-
7 DRUG INTERACTIONS
7.1 Effect of Strong CYP3A4 Inducers on Regorafenib
Co-administration of a strong CYP3A4 inducer with STIVARGA decreased the plasma concentrations of regorafenib, increased the plasma concentrations of the active metabolite M-5, and resulted in no change in the plasma concentrations of the active metabolite M-2 [see Clinical Pharmacology (12.3)], and may lead to decreased efficacy. Avoid concomitant use of STIVARGA with strong CYP3A4 inducers (e.g. rifampin, phenytoin, carbamazepine, phenobarbital, and St. John’s Wort).
7.2 Effect of Strong CYP3A4 Inhibitors on Regorafenib
Co-administration of a strong CYP3A4 inhibitor with STIVARGA increased the plasma concentrations of regorafenib and decreased the plasma concentrations of the active metabolites M-2 and M-5 [see Clinical Pharmacology (12.3)], and may lead to increased toxicity. Avoid concomitant use of STIVARGA with strong CYP3A4 inhibitors (e.g. clarithromycin, grapefruit juice, itraconazole, ketoconazole, nefazodone, posaconazole, telithromycin, and voriconazole).
7.3 Effect of Regorafenib on Breast Cancer Resistance Protein (BCRP) Substrates
Co-administration of STIVARGA with a BCRP substrate increased the plasma concentrations of the BCRP substrate [see Clinical Pharmacology (12.3)]. Monitor patients closely for signs and symptoms of exposure related toxicity to the BCRP substrate (e.g. methotrexate, fluvastatin, atorvastatin). Consult the concomitant BCRP substrate product information when considering administration of such products together with STIVARGA.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on animal studies and its mechanism of action, STIVARGA can cause fetal harm when administered to a pregnant woman. There are no available data on STIVARGA use in pregnant women. Administration of regorafenib was embryolethal and teratogenic in rats and rabbits at exposures lower than human exposures at the recommended dose, with increased incidences of cardiovascular, genitourinary, and skeletal malformations [see Data]. Advise pregnant women of the potential hazard to a fetus.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4 % and 15 to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies, a total loss of pregnancy (100% resorption of litter) was observed in rats at doses as low as 1 mg/kg (approximately 6% of the recommended human dose, based on body surface area) and in rabbits at doses as low as 1.6 mg/kg (approximately 25% of the human exposure at the clinically recommended dose measured by AUC).
In a single dose distribution study in pregnant rats, there was increased penetration of regorafenib across the blood-brain barrier in fetuses compared to dams. Daily administration of regorafenib to pregnant rats during organogenesis resulted in fetal findings of delayed ossification at doses > 0.8 mg/kg (approximately 5% of the recommended human dose based on body surface area) and dose-dependent increases in skeletal malformations including cleft palate and enlarged fontanelle at doses ≥ 1 mg/kg (approximately 10% of the clinical exposure based on AUC). At doses ≥ 1.6 mg/kg (approximately 11% of the recommended human dose based on body surface area), there were dose-dependent increases in the incidence of cardiovascular malformations, external abnormalities, diaphragmatic hernia, and dilation of the renal pelvis.
In pregnant rabbits administered regorafenib daily during organogenesis, there were findings of ventricular septal defects evident at the lowest tested dose of 0.4 mg/kg (approximately 7% of the AUC in patients at the recommended dose). At doses of ≥ 0.8 mg/kg (approximately 15% of the human exposure at the recommended human dose based on AUC), administration of regorafenib resulted in dose-dependent increases in the incidence of additional cardiovascular malformations and skeletal anomalies, as well as significant adverse effects on the urinary system including missing kidney/ureter; small, deformed and malpositioned kidney; and hydronephrosis. The proportion of viable fetuses that were male decreased with increasing dose in two rabbit embryo-fetal toxicity studies.
8.2 Lactation
Risk Summary
There are no data on the presence of regorafenib or its metabolites in human milk, the effects of regorafenib on the breastfed infant, or on milk production. In rats, regorafenib and its metabolites are excreted in milk. Because of the potential for serious adverse reactions in breastfed infants from STIVARGA, do not breastfeed during treatment with STIVARGA and for 2 weeks after the final dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Use effective contraception during treatment and for 2 months after completion of therapy.
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 2 months following the final dose of STIVARGA [see Nonclinical Toxicology (13.1)].
Infertility
There are no data on the effect of STIVARGA on human fertility. Results from animal studies indicate that regorafenib can impair male and female fertility [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and efficacy of STIVARGA in pediatric patients less than 18 years of age have not been established.
Animal Data
In 28-day repeat-dose studies in rats there were dose-dependent findings of dentin alteration and angiectasis. These findings occurred at regorafenib doses as low as 4 mg/kg (approximately 25% of the AUC in humans at the recommended dose). In 13-week repeat-dose studies in dogs there were similar findings of dentin alteration at doses as low as 20 mg/kg (approximately 43% of the AUC in humans at the recommended dose). Administration of regorafenib in these animals also led to persistent growth and thickening of the femoral epiphyseal growth plate.
8.5 Geriatric Use
Of the 1142 STIVARGA-treated patients enrolled in randomized, placebo-controlled trials, 40% were 65 years of age and over, while 10% were 75 and over. No overall differences in efficacy were observed between these patients and younger patients. There was an increased incidence of Grade 3 hypertension (18% versus 9%) in the placebo-controlled trials among STIVARGA-treated patients 65 years of age and older as compared to younger patients. In addition, one Grade 4 hypertension event has been reported in the 65 years and older age group and none in the younger age group.
8.6 Hepatic Impairment
No dose adjustment is recommended in patients with mild (total bilirubin ≤ULN and AST >ULN, or total bilirubin >ULN to ≤1.5 times ULN) or moderate (total bilirubin >1.5 to ≤3 times ULN and any AST) hepatic impairment, [see Clinical Pharmacology (12.3)]. Closely monitor patients with hepatic impairment for adverse reactions [see Warnings and Precautions (5.1)].
STIVARGA is not recommended for use in patients with severe hepatic impairment (total bilirubin >3x ULN) as STIVARGA has not been studied in this population.
8.7 Renal Impairment
No dose adjustment is recommended for patients with renal impairment. The pharmacokinetics of regorafenib have not been studied in patients who are on dialysis and there is no recommended dose for this patient population [see Clinical Pharmacology (12.3)].
8.8 Race
Based on pooled data from three placebo-controlled trials (CORRECT, GRID and CONCUR), a higher incidence of HFSR and liver function test abnormalities occurred in Asian patients treated with STIVARGA as compared with Whites [see Warnings and Precautions (5.1, 5.5)]. No starting dose adjustment is necessary based on race.
-
10 OVERDOSAGE
The highest dose of STIVARGA studied clinically is 220 mg per day. The most frequently observed adverse drug reactions at this dose were dermatological events, dysphonia, diarrhea, mucosal inflammation, dry mouth, decreased appetite, hypertension, and fatigue. There is no known antidote for STIVARGA overdose. In the event of suspected overdose, interrupt STIVARGA, institute supportive care, and observe until clinical stabilization.
-
11 DESCRIPTION
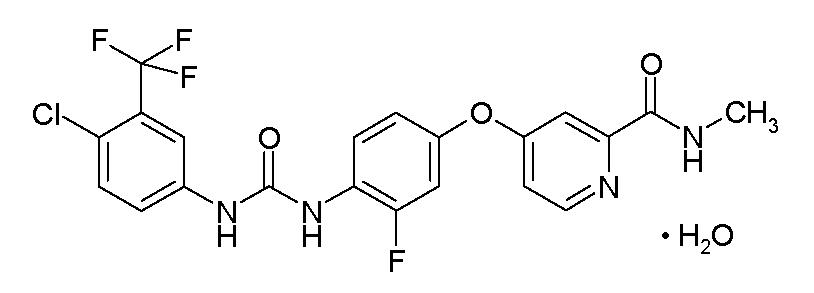
STIVARGA (regorafenib) is a multikinase inhibitor with the chemical name 4-[4-({[4-chloro-3-(trifluoromethyl) phenyl] carbamoyl} amino)-3-fluorophenoxy]-N-methylpyridine-2-carboxamide monohydrate. Regorafenib has the following structural formula:
Regorafenib is a monohydrate and it has a molecular formula C21H15ClF4N4O3 • H2O and a molecular weight of 500.83. Regorafenib is practically insoluble in water, slightly soluble in acetonitrile, methanol, ethanol, and ethyl acetate and sparingly soluble in acetone.
STIVARGA tablets for oral administration are formulated as light pink, oval-shaped tablets debossed with "BAYER" on one side and "40" on the other. Each tablet contains 40 mg of regorafenib in the anhydrous state, which corresponds to 41.49 mg of regorafenib monohydrate, and the following inactive ingredients: cellulose microcrystalline, croscarmellose sodium, magnesium stearate, povidone, and colloidal silicon dioxide. The film-coating contains the following inactive ingredients: ferric oxide red, ferric oxide yellow, lecithin (soy), polyethylene glycol 3350, polyvinyl alcohol, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Regorafenib is a small molecule inhibitor of multiple membrane-bound and intracellular kinases involved in normal cellular functions and in pathologic processes such as oncogenesis, tumor angiogenesis, metastasis and tumor immunity. In in vitro biochemical or cellular assays, regorafenib or its major human active metabolites M-2 and M-5 inhibited the activity of RET, VEGFR1, VEGFR2, VEGFR3, KIT, PDGFR-alpha, PDGFR-beta, FGFR1, FGFR2, TIE2, DDR2, TrkA, Eph2A, RAF-1, BRAF, BRAF V600E, SAPK2, PTK5, Abl and CSF1R at concentrations of regorafenib that have been achieved clinically. In in vivo models, regorafenib demonstrated anti-angiogenic activity in a rat tumor model and inhibition of tumor growth in several mouse xenograft models including some for human colorectal carcinoma, gastrointestinal stromal and hepatocellular carcinoma. Regorafenib also demonstrated anti-metastatic activity in a mouse xenograft model and two mouse orthotopic models of human colorectal carcinoma.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of multiple doses of STIVARGA (160 mg once daily for 21 days) on the QTc interval was evaluated in an open-label, single-arm study in 25 patients with advanced solid tumors. No large changes in the mean QTc interval (i.e., > 20 msec) were detected in the study.
12.3 Pharmacokinetics
Absorption
Following a single 160 mg dose of STIVARGA in patients with advanced solid tumors, regorafenib reaches a geometric mean peak plasma level (Cmax) of 2.5 µg/mL at a median time of 4 hours and a geometric mean area under the plasma concentration vs. time curve (AUC) of 70.4 µg*h/mL. The AUC of regorafenib at steady-state increases less than dose proportionally at doses greater than 60 mg. At steady-state, regorafenib reaches a geometric mean Cmax of 3.9 µg/mL and a geometric mean AUC of 58.3 µg*h/mL. The coefficient of variation of AUC and Cmax is between 35% and 44%.
The mean relative bioavailability of tablets compared to an oral solution is 69% to 83%.
In a food-effect study, 24 healthy men received a single 160 mg dose of STIVARGA on three separate occasions: under a fasted state, with a high-fat meal and with a low-fat meal. A high-fat meal (945 calories and 54.6 g fat) increased the mean AUC of regorafenib by 48% and decreased the mean AUC of the M-2 and M-5 metabolites by 20% and 51%, respectively, as compared to the fasted state. A low-fat meal (319 calories and 8.2 g fat) increased the mean AUC of regorafenib, M-2 and M-5 by 36%, 40% and 23%, respectively as compared to fasted conditions. STIVARGA was administered with a low-fat meal in the CORRECT and GRID studies [see Dosage and Administration (2.1), Clinical Studies (14)].
Distribution
Regorafenib undergoes enterohepatic circulation with multiple plasma concentration peaks observed across the 24-hour dosing interval. Regorafenib is highly bound (99.5%) to human plasma proteins.
Elimination
Following a single 160 mg oral dose of STIVARGA, the geometric mean (minimum to maximum) elimination half-lives for regorafenib and the M-2 metabolite in plasma are 28 hours (14 to 58 hours) and 25 hours (14 to 32 hours), respectively. M-5 has a longer mean (minimum to maximum) elimination half-life of 51 hours (32 to 70 hours).
Metabolism
Regorafenib is metabolized by CYP3A4 and UGT1A9. The main circulating metabolites of regorafenib measured at steady-state in human plasma are M-2 (N-oxide) and M-5 (N-oxide and N-desmethyl). Both metabolites have similar in vitro pharmacological activity and steady-state concentrations as regorafenib. M-2 and M-5 are highly protein bound (99.8% and 99.95%, respectively).
Excretion
Approximately 71% of a radiolabeled dose was excreted in feces (47% as parent compound, 24% as metabolites) and 19% of the dose was excreted in urine (17% as glucuronides) within 12 days after administration of a radiolabeled oral solution at a dose of 120 mg.
Specific Populations
Age, sex, race and weight had no clinically meaningful effect on the pharmacokinetics of regorafenib.
Hepatic Impairment
Based on a population pharmacokinetic analysis, no clinically important differences in the mean total exposure of regorafenib, including M-2 and M-5, were noted amongst patients with normal liver function (total bilirubin and AST ≤ ULN, n=744), mild hepatic impairment (total bilirubin ≤ ULN and AST >ULN or total bilirubin >ULN to ≤1.5x ULN, n=437), and moderate hepatic impairment (total bilirubin >1.5x to ≤3x ULN and any AST, n=36). The pooled analysis included 391 patients with HCC of whom 116, 249, and 26 were categorized as having normal liver function, mild, and moderate hepatic impairment, respectively. The pharmacokinetics of regorafenib were not evaluated in patients with severe hepatic impairment (total bilirubin >3x ULN).
Renal Impairment
The pharmacokinetics of regorafenib, M-2, and M-5 was evaluated in 6 patients with severe renal impairment (CLcr 15-29 mL/min) and 18 patients with normal/mild renal function (CLcr ≥60 mL/min) following the administration of STIVARGA at a dose of 160 mg daily for 21 days. No differences in the mean steady-state exposure of regorafenib, M-2, or M-5 were observed in patients with severe renal impairment compared to patients with normal renal function. The pharmacokinetics of regorafenib has not been studied in patients with end-stage renal disease on dialysis.
Drug Interaction Studies
Effect of Regorafenib on Cytochrome P450 Substrates: In vitro studies suggested that regorafenib is an inhibitor of CYP2C8, CYP2C9, CYP2B6, CYP3A4 and CYP2C19; M-2 is an inhibitor of CYP2C9, CYP2C8, CYP3A4 and CYP2D6, and M-5 is an inhibitor of CYP2C8. In vitro studies suggested that regorafenib is not an inducer of CYP1A2, CYP2B6, CYP2C19, and CYP3A4 enzyme activity.
Patients with advanced solid tumors received single oral doses of CYP substrates, 2 mg of midazolam (CYP3A4), 40 mg of omeprazole (CYP2C19) and 10 mg of warfarin (CYP2C9) or 4 mg of rosiglitazone (CYP2C8) one week before and two weeks after STIVARGA at a dose of 160 mg once daily. No clinically meaningful effect was observed in the mean AUC of rosiglitazone (N=12) or the mean omeprazole (N=11) plasma concentrations measured 6 hours after dosing or the mean AUC of midazolam (N=15). The mean AUC of warfarin (N=8) increased by 25% [see Warnings and Precautions (5.2)].
Effect of CYP3A4 Strong Inducers on Regorafenib: Twenty-two healthy men received a single 160 mg dose of STIVARGA alone and then 7 days after starting rifampin. Rifampin, a strong CYP3A4 inducer, was administered at a dose of 600 mg daily for 9 days. The mean AUC of regorafenib decreased by 50% and mean AUC of M-5 increased by 264%. No change in the mean AUC of M-2 was observed [see Drug Interactions (7.1)].
Effect of CYP3A4 Strong Inhibitors on Regorafenib: Eighteen healthy men received a single 160 mg dose of STIVARGA alone and then 5 days after starting ketoconazole. Ketoconazole, a strong CYP3A4 inhibitor, was administered at a dose of 400 mg daily for 18 days. The mean AUC of regorafenib increased by 33% and the mean AUC of M-2 and M-5 both decreased by 93% [see Drug Interactions (7.2)].
Effect of Neomycin on Regorafenib: Twenty-seven healthy men received a single 160 mg dose of STIVARGA and then 5 days after starting neomycin. Neomycin, a non-absorbable antibiotic, was administered at a dose of 1 gram three times daily for 5 days. No clinically meaningful effect on the mean AUC of regorafenib was observed; however, the mean AUC of M-2 decreased by 76% and the mean AUC of M-5 decreased by 86%. The decreased exposure of M-2 and M-5 may result in a decreased efficacy of STIVARGA. The effects of other antibiotics on the exposure of regorafenib and its active metabolites have not been studied.
Effect of Regorafenib on UGT1A1 Substrates: In vitro studies showed that regorafenib, M-2, and M-5 competitively inhibit UGT1A9 and UGT1A1 at therapeutically relevant concentrations. Eleven patients received irinotecan-containing combination chemotherapy with STIVARGA at a dose of 160 mg. The mean AUC of irinotecan increased by 28% and the mean AUC of SN-38 increased by 44% when irinotecan was administered 5 days after the last of 7 daily doses of STIVARGA.
Effect of Regorafenib on BCRP Substrates: Administration of regorafenib (160 mg for 14 days) prior to administration of a single dose of rosuvastatin (5 mg), a BCRP substrate, resulted in a 3.8-fold increase in mean exposure (AUC) of rosuvastatin and a 4.6-fold increase in Cmax [see Drug Interactions (7.3)].
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Studies examining the carcinogenic potential of regorafenib have not been conducted. Regorafenib itself did not demonstrate genotoxicity in in vitro or in vivo assays; however, a major human active metabolite of regorafenib, (M-2), was positive for clastogenicity, causing chromosome aberration in Chinese hamster V79 cells.
Dedicated studies to examine the effects of regorafenib on fertility have not been conducted; however, there were histological findings of tubular atrophy and degeneration in the testes, atrophy in the seminal vesicle, and cellular debris and oligospermia in the epididymides in male rats at doses similar to those in human at the clinical recommended dose based on AUC. In female rats, there were increased findings of necrotic corpora lutea in the ovaries at the same exposures. There were similar findings in dogs of both sexes in repeat dose studies at exposures approximately 83% of the human exposure at the recommended human dose based on AUC. These findings suggest that regorafenib may adversely affect fertility in humans.
13.2 Animal Toxicology and/or Pharmacology
In a chronic 26-week repeat dose study in rats there was a dose-dependent increase in the finding of thickening of the atrioventricular valve. At a dose that resulted in an exposure of approximately 12% of the human exposure at the recommended dose, this finding was present in half of the examined animals.
-
14 CLINICAL STUDIES
14.1 Colorectal Cancer
The clinical efficacy and safety of STIVARGA were evaluated in an international, multicenter, randomized (2:1), double-blind, placebo-controlled trial [Study “Patients with metastatic COloRectal cancer treated with REgorafenib or plaCebo after failure of standard Therapy” (CORRECT); NCT 01103323)] in 760 patients with previously-treated metastatic colorectal cancer. The major efficacy outcome measure was overall survival (OS); additional efficacy outcome measures included progression-free survival (PFS) and overall tumor response rate.
Patients were randomized to receive 160 mg regorafenib orally once daily (N=505) plus best supportive care (BSC) or placebo (N=255) plus BSC for the first 21 days of each 28-day cycle. STIVARGA was administered with a low-fat breakfast that contains less than 30% fat [see Dosage and Administration (2.1), Clinical Pharmacology (12.3)]. Treatment continued until disease progression or unacceptable toxicity.
Baseline demographics were: median age 61 years, 61% men, 78% White, and all patients had an ECOG performance status of 0 or 1. The primary sites of disease were colon (65%), rectum (29%), or both (6%). History of KRAS evaluation was reported for 729 (96%) patients; 430 (59%) of these patients were reported to have KRAS mutation. The median number of prior lines of therapy for metastatic disease was 3. All patients received prior treatment with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, and with bevacizumab. All but one patient with KRAS mutation-negative tumors received panitumumab or cetuximab.
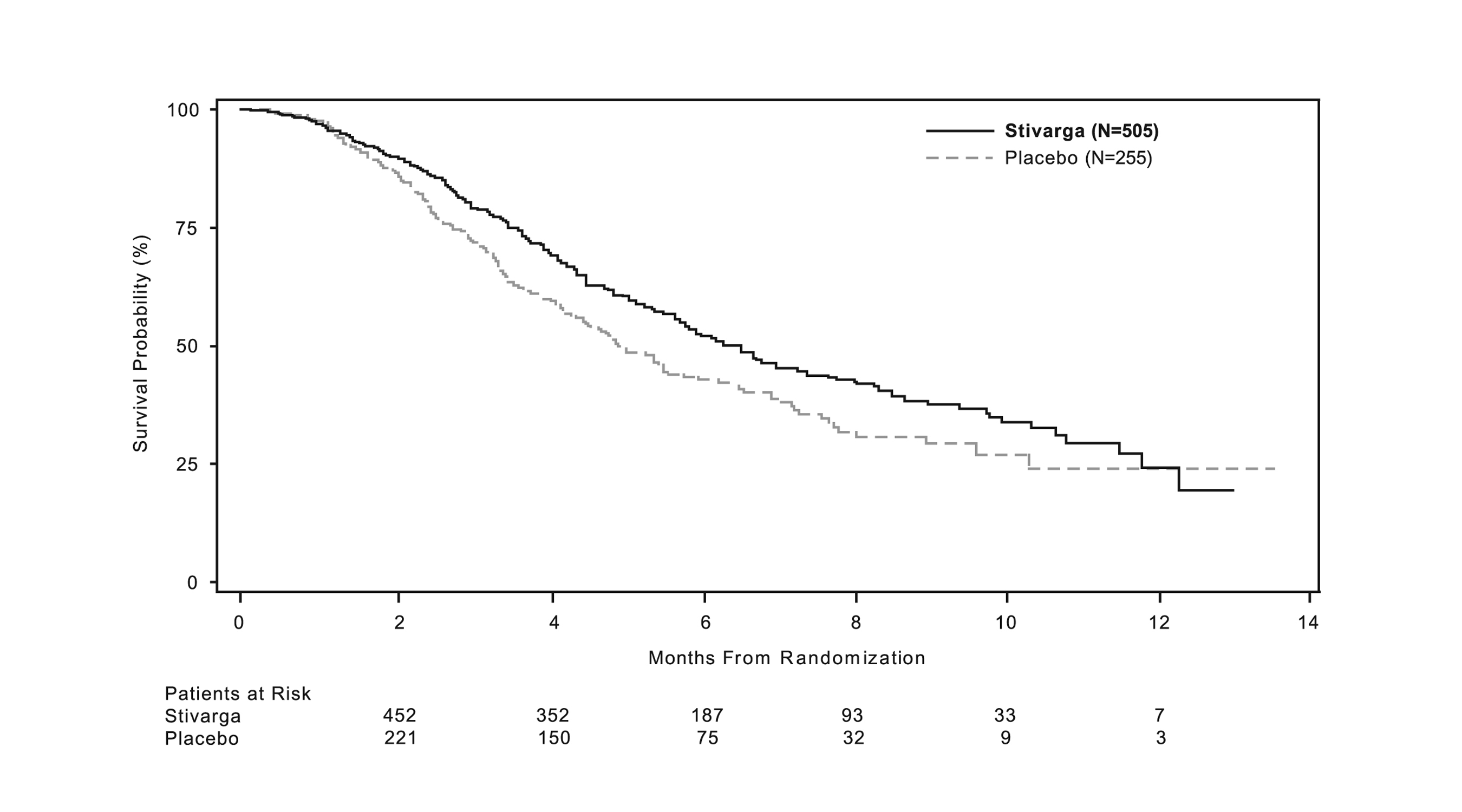
The addition of STIVARGA to BSC resulted in a statistically significant improvement in survival compared to placebo plus BSC (see Table 7 and Figure 1).
Table 7: Efficacy Results from CORRECT STIVARGA
(N=505)
Placebo
(N=255)
Overall Survival
Number of Deaths (%)
275 (55%)
157 (62%)
- Median Overall Survival (months)
6.4
5.0
- 95% CI a
(5.8, 7.3)
(4.4, 5.8)
- Hazard Ratio (HR) (95% CI)
0.77 (0.64, 0.94)
- Stratified log-rank test p-value b, c
0.0102
Progression-Free Survival
Number of Deaths or Progressions (%)
417 (83%)
231 (91%)
- Median Progression-Free Survival (months)
2.0
1.7
- 95% CI
(1.9, 2.3)
(1.7, 1.8)
- HR (95% CI)
0.49 (0.42, 0.58)
- Stratified log-rank test p-value c
<0.0001
Overall Response Rate
- Overall Response, N (%)
5 (1%)
1 (0.4%)
- 95% CI
0.3%, 2.3%
0%, 2.2%
- a CI=confidence interval.
- bStratified by geographic region and time from diagnosis of metastatic disease.
- c Crossed the O’Brien-Fleming boundary (two-sided p-value < 0.018) at second interim analysis.
14.2 Gastrointestinal Stromal Tumors
The efficacy and safety of STIVARGA were evaluated in an international, multicenter, randomized (2:1), double-blind, placebo-controlled trial [Study “GIST Regorafenib In progressive Disease” (GRID); NCT 01271712] in patients with unresectable, locally advanced or metastatic gastrointestinal stromal tumor (GIST), who had been previously treated with imatinib mesylate and sunitinib malate. Randomization was stratified by line of therapy (third vs. four or more) and geographic region (Asia vs. rest of the world).
The major efficacy outcome measure of GRID was progression-free survival (PFS) based on disease assessment by independent radiological review using modified RECIST 1.1 criteria, in which lymph nodes and bone lesions were not target lesions and progressively growing new tumor nodule within a pre-existing tumor mass was progression. The key secondary outcome measure was overall survival.
Patients were randomized to receive 160 mg regorafenib orally once daily (N=133) plus best supportive care (BSC) or placebo (N=66) plus BSC for the first 21 days of each 28-day cycle. Treatment continued until disease progression or unacceptable toxicity. In GRID, the median age of patients was 60 years, 64% were men, 68% were White, and all patients had baseline ECOG performance status of 0 (55%) or 1 (45%). At the time of disease progression as assessed by central review, the study blind was broken and all patients were offered the opportunity to take STIVARGA at the investigator’s discretion.
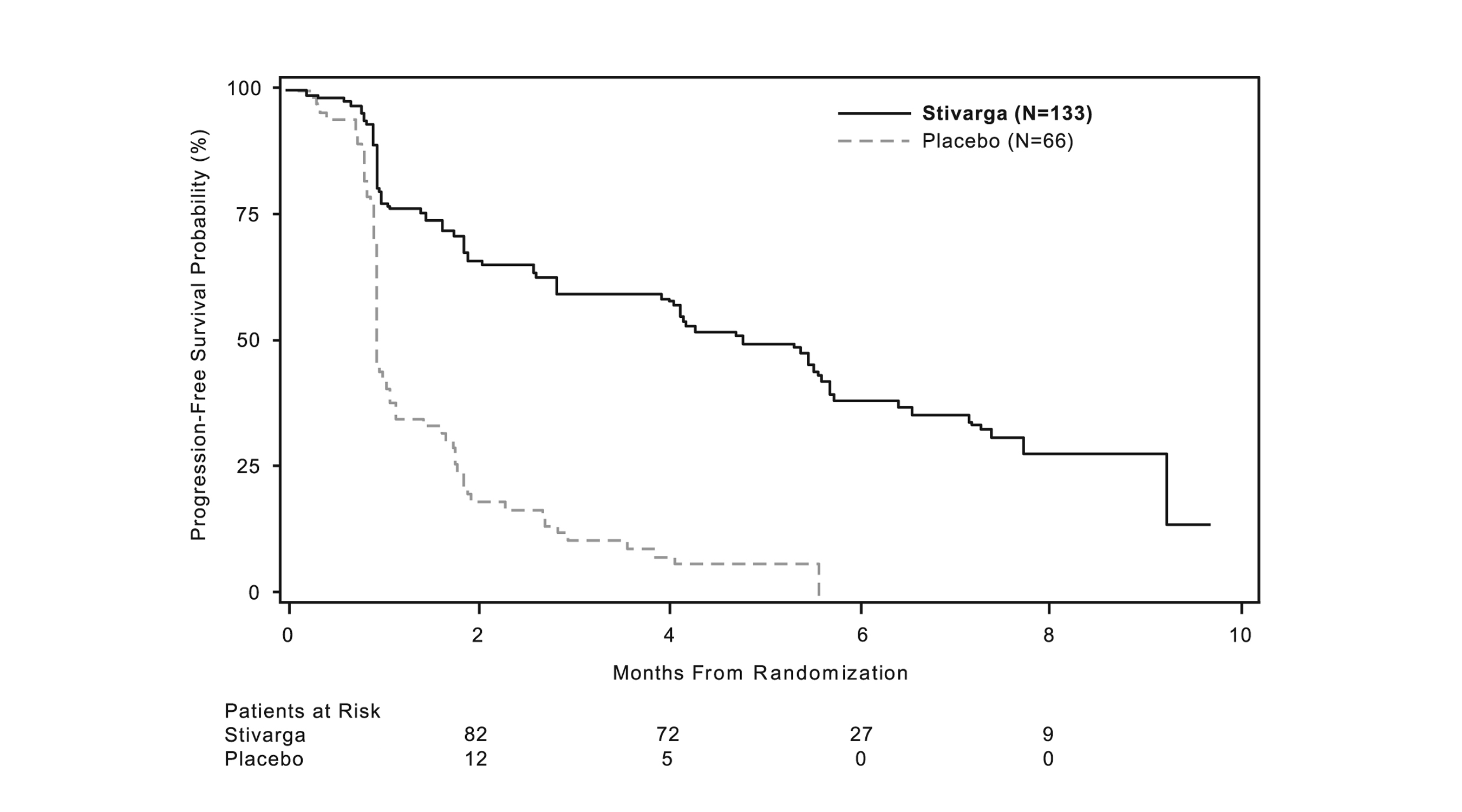
A statistically significant improvement in PFS was demonstrated among patients treated with STIVARGA compared to placebo (see Table 8 and Figure 2).
There was no statistically significant difference in overall survival at the final OS analysis, conducted at 162 OS events (Table 8). Cross-over to open label STIVARGA occurred in 58 (88%) placebo-treated patients after disease progression.
Table 8: Efficacy Results for GRID STIVARGA
(N=133)
Placebo
(N=66)
Progression-Free Survival
Number of Deaths or Progressions (%)
82 (62%)
63 (96%)
Median PFS in months (95% CI)
4.8 (3.9, 5.7)
0.9 (0.9, 1.1)
HR (95% CI)
0.27 (0.19, 0.39)
p-value a
<0.0001
Overall Survival
Number of Deaths (%)
109 (82%)
53 (80.3%)
Median OS in Months (95% CI)
17.4 (14.9, 20.2)
17.4 (12.3, 21.0)
HR (95% CI)
0.91 (0.65, 1.27)
p-value a
0.5716
a 2-sided p-value by log-rank test stratified by line of treatment and geographical region.
14.3 Hepatocellular Carcinoma (HCC)
The clinical efficacy and safety of STIVARGA were evaluated in an international, multicenter, randomized (2:1), double-blind, placebo-controlled trial [Study “REgorafenib after SORafenib in patients with hepatoCEllular carcinoma” (RESORCE); NCT 01774344]. The study enrolled adults with Child-Pugh A and Barcelona Clinic Liver Cancer Stage Category B or C hepatocellular carcinoma, with documented disease progression following sorafenib. The median duration of previous sorafenib treatment was 7.8 months; patients who permanently discontinued sorafenib due to toxicity or were unable to tolerate sorafenib doses of 400 mg once daily were ineligible.
Patients were randomized to receive 160 mg regorafenib orally once daily plus best supportive care (BSC) or matching placebo plus BSC for the first 21 days of each 28-day cycle until disease progression or unacceptable toxicity. Randomization was stratified by geographical region (Asia vs rest of world), ECOG performance status (0 vs 1), alpha-fetoprotein levels (<400 ng/mL vs ≥400 ng/mL), extrahepatic disease (presence vs absence), and macrovascular invasion (presence vs absence). The major efficacy outcome measure was overall survival (OS). Additional outcome measures were progression-free survival (PFS), overall tumor response rate (ORR) and duration of response as assessed by investigators using RECIST 1.1 and using modified RECIST (mRECIST) for HCC. Patients continued therapy with STIVARGA until clinical or radiological disease progression or unacceptable toxicity.
The characteristics of the study population were a median age of 63 years (range 19 to 85 years); 88% male; 41% Asian, 36% White, and 21% not reported; 66% had ECOG performance status (PS) of 0 and 34% had ECOG PS of 1; 98% had Child-Pugh A and 2% had Child-Pugh B. Risk factors for underlying cirrhosis included hepatitis B (38%), alcohol use (25%), hepatitis C (21%), and non-alcoholic steato hepatitis (7%). Macroscopic vascular invasion or extra-hepatic tumor spread was present in 81% of patients. Barcelona Clinic Liver Cancer (BCLC) was stage C in 87% and stage B in 13% of patients. All patients received prior sorafenib and 61% received prior loco-regional transarterial embolization or chemoinfusion procedures.
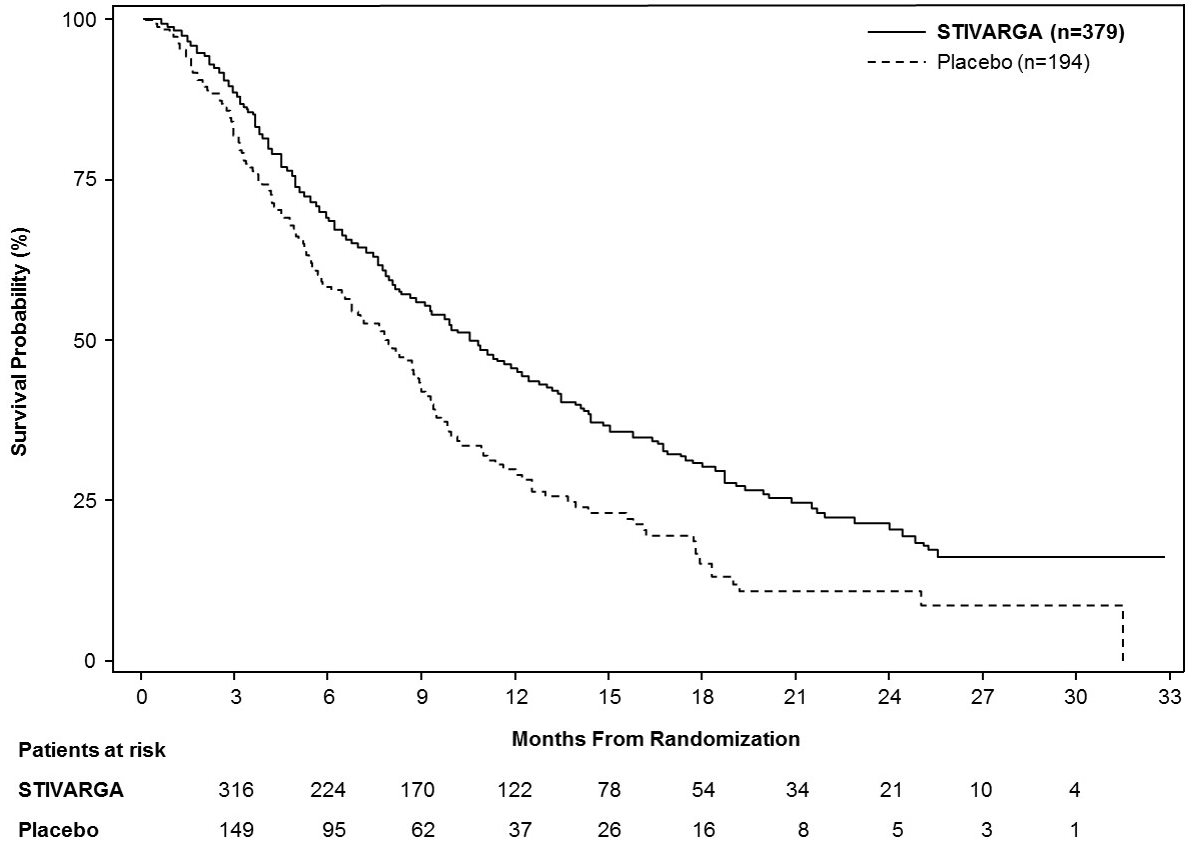
Efficacy results are summarized in Table 9 and Figure 3 below.
Table 9: Efficacy Results from Study RESORCE STIVARGA
n=379
Placebo
n=194
Overall Survival
Number of Deaths (%)
233 (62)
140 (72)
Median OS in months (95% CIa)
10.6 (9.1, 12.1)
7.8 (6.3, 8.8)
Hazard Ratiob (95% CIa)
0.63 (0.50, 0.79)
P-valuec
<0.0001
Progression-free Survival (mRECIST)
Number of Events (%)
293 (77)
181(93)
Progressive Disease
274 (72)
173 (89)
Death
19 (5)
8 (4)
Median PFS in months (95% CIa)
3.1 (2.8, 4.2)
1.5 (1.4, 1.6)
Hazard Ratiob (95% CIa)
0.46 (0.37, 0.56)
P-valuec
<0.0001
Progression-free Survival (RECIST 1.1)
Number of Events (%)
288 (76)
184 (95)
Progressive Disease
270 (71)
175 (90)
Death
18 (5)
9 (5)
Median PFS in months (95% CIa)
3.4 (2.9, 4.2)
1.5 (1.4, 1.5)
Hazard Ratiob (95% CIa)
0.43 ( 0.35, 0.52)
Overall Response (mRECIST)
Overall Response Rate
11%
4%
95% CIa
(8%, 14%)
(2%, 8%)
Complete Response
0.5%
0
Partial Response
10%
4%
Overall Response (RECIST 1.1)
Overall Response Rate
7%
3%
95% CIa
(4%, 10%)
(1%, 6%)
Complete Response
0
0
Partial Response
7%
3%
- aCI=confidence interval.
- bEstimated with Cox proportional hazard model stratified by geographic region, ECOG performance status, Alpha-fetoprotein level, presence versus absence of extrahepatic disease, and presence versus absence of macrovascular invasion.
- cLog rank test stratified by geographic region, ECOG performance status, Alpha-fetoprotein level, presence versus absence of extrahepatic disease, and presence versus absence of macrovascular invasion.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
How Supplied
Tablets are supplied in the following:
Packages containing three bottles, with each bottle containing 28 tablets, for a total of 84 tablets per package
(NDC 50419-171-03).Packages containing four bottles, with each bottle containing 21 tablets, for a total of 84 tablets per package
(NDC 50419-171-06).Storage and Handling
Store STIVARGA at 25°C (77°F); excursions are permitted from 15 to 30°C (59 to 86°F) [see USP Controlled Room Temperature].
Store tablets in the original bottle and do not remove the desiccant. Keep the bottle tightly closed after first opening.
Discard any unused tablets 7 weeks after opening the bottle. Dispose of unused tablets in accordance with local requirements.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hepatotoxicity
Advise patients that they will need to undergo monitoring for liver damage and to report immediately any signs or symptoms of severe liver damage to their healthcare provider [see Warnings and Precautions (5.1), Use in Specific Populations (8.6)].
Infections
Advise patients to contact their healthcare provider if they experience signs and symptoms of infection [see Warnings and Precautions (5.2)].
Hemorrhage
Advise patients to contact their healthcare provider for unusual bleeding, bruising, or symptoms of bleeding, such as lightheadedness [see Warnings and Precautions (5.3)].
Gastrointestinal Perforation or Fistula
Advise patients to contact a healthcare provider immediately if they experience severe pains in their abdomen, persistent swelling of the abdomen, high fever, chills, nausea, vomiting, or dehydration [see Warnings and Precautions (5.4)].
Dermatologic Toxicity
Advise patients to contact their healthcare provider if they experience skin changes including HFSR, rash, pain, blisters, bleeding, or swelling [see Warnings and Precautions (5.5)].
Hypertension
Advise patients they will need to undergo blood pressure monitoring and to contact their healthcare provider if blood pressure is elevated or if symptoms from hypertension occur including severe headache, lightheadedness, or neurologic symptoms [see Warnings and Precautions (5.6)].
Cardiac Ischemia and Infarction
Advise patients to seek immediate emergency help if they experience chest pain, shortness of breath, feel dizzy, or feel like passing out [see Warnings and Precautions (5.7)].
Reversible Posterior leukoencephalopathy syndrome
Advise patients to contact their healthcare provider if they experience signs and symptoms of RPLS [see Warnings and Precautions (5.8)].
Risk of Impaired Wound Healing
Advise patients that STIVARGA may impair wound healing. Advise patients that temporary interruption of STIVARGA is recommended prior to any elective surgery [see Warnings and Precautions (5.9)].
Embryo-Fetal Toxicity
Advise patients that regorafenib can cause fetal harm. Advise a pregnant woman of the potential risk to a fetus [see Warnings and Precautions (5.10), Use in Specific Populations (8.1, 8.3)].
Females and Males of Reproductive Potential
- •
- Advise women of reproductive potential of the need for effective contraception during STIVARGA treatment and for 2 months after completion of treatment. Instruct women of reproductive potential to immediately contact her healthcare provider if pregnancy is suspected or confirmed during or within 2 months of completing treatment with STIVARGA [see Warnings and Precautions (5.10) and Use in Specific Populations (8.1, 8.3)].
- •
- Advise men of reproductive potential of the need for effective contraception during STIVARGA treatment and for 2 months after completion of treatment [see Use in Specific Populations (8.3)].
Lactation
Advise nursing mothers that it is not known whether regorafenib is present in breast milk and discuss whether to discontinue nursing or to discontinue regorafenib [see Use in Specific Populations (8.2)].
Administration
- •
- Advise patients to swallow the STIVARGA tablet whole with water at the same time each day following a low-fat meal. Inform patients that the low-fat meal should contain less than 600 calories and less than 30% fat [see Dosage and Administration (2.1)].
- •
- Advise patients to store medicine in the original container. Do not place medication in daily or weekly pill boxes. Discard any remaining tablets 7 weeks after opening the bottle. Tightly close bottle after each opening and keep the desiccant in the bottle [see How Supplied (16)].
Dosing Instructions
Advise patients to take STIVARGA after a low fat meal. Advise patients to take any missed dose on the same day, as soon as they remember, and that they must not take two doses on the same day to make up for a dose missed on the previous day [see Dose and Administration (2.1)].
Manufactured for:
Bayer HealthCare Pharmaceuticals Inc.
Whippany, NJ 07981 USA -
Patient Package Insert
Patient Information
STIVARGA (sti-VAR-gah)
(regorafenib)
tablets
What is the most important information I should know about STIVARGA?
STIVARGA can cause serious side effects, including:
Liver problems. STIVARGA can cause liver problems which can be serious and sometimes lead to death. Your healthcare provider will do blood tests to check your liver function before you start taking STIVARGA and during your treatment with STIVARGA to check for liver problems. Tell your healthcare provider right away if you get any of these symptoms of liver problems during treatment:
- •
- yellowing of your skin or the white part of your eyes (jaundice)
- •
- nausea or vomiting
- •
- dark “tea-colored” urine
- •
- change in sleep pattern
What is STIVARGA?
STIVARGA is a prescription medicine used to treat people with:
- •
- colon or rectal cancer that has spread to other parts of the body and for which they have received previous treatment with certain chemotherapy medicines
- •
- a rare stomach, bowel, or esophagus cancer called GIST (gastrointestinal stromal tumors) that cannot be treated with surgery or that has spread to other parts of the body and for which they have received previous treatment with certain medicines
- •
- a type of liver cancer called hepatocellular carcinoma (HCC) in people who have been previously treated with sorafenib
It is not known if STIVARGA is safe and effective in children less than 18 years of age.
Before taking STIVARGA, tell your healthcare provider about all of your medical conditions, including if you:
- •
- have liver problems in addition to liver cancer
- •
- have bleeding problems
- •
- have high blood pressure
- •
- have heart problems or chest pain
- •
- plan to have surgery or have had a recent surgery. You should stop taking STIVARGA at least 2 weeks before planned surgery. See “What are the possible side effects of STIVARGA?”
- •
- are pregnant or plan to become pregnant. STIVARGA can harm your unborn baby.
- •
- Females should use effective birth control during treatment with STIVARGA and for 2 months after your final dose of STIVARGA. Tell your healthcare provider right away if you become pregnant during treatment with STIVARGA or within 2 months after your final dose of STIVARGA.
- •
- Males with female partners who can become pregnant should use effective birth control during treatment with STIVARGA and for 2 months after your final dose of STIVARGA.
- •
- are breastfeeding or plan to breastfeed. It is not known if STIVARGA passes into your breast milk. Do not breastfeed during treatment with STIVARGA and for 2 weeks after your final dose of STIVARGA.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. STIVARGA may affect the way other medicines work, and other medicines may affect how STIVARGA works.
How should I take STIVARGA?
- •
- Take STIVARGA exactly as your healthcare provider tells you.
- •
- You will usually take STIVARGA 1 time a day for 21 days (3 weeks) and then stop for 7 days (1 week). This is 1 cycle of treatment. Repeat this cycle for as long as your healthcare provider tells you to.
- •
- Swallow STIVARGA tablets whole with water following a low-fat meal.
- •
- Take STIVARGA at the same time each day following a low-fat meal that contains less than 600 calories and less than 30% fat.
- •
- If you miss a dose, take it as soon as you remember on that day. Do not take two doses on the same day to make up for a missed dose.
If you take too much STIVARGA call your healthcare provider or go to the nearest emergency room right away.
What should I avoid while taking STIVARGA?
Avoid drinking grapefruit juice and taking St. John’s Wort during treatment with STIVARGA. These can affect the way STIVARGA works.
What are the possible side effects of STIVARGA?
STIVARGA can cause serious side effects including:
- •
- See “What is the most important information I should know about STIVARGA?”
- •
- Infection. STIVARGA may lead to a higher risk of infections especially of the urinary tract, nose, throat and lung. STIVARGA may also lead to a higher risk of fungal infections of the mucous membrane, skin or the body. Tell your healthcare provider right away if you get:
- •
- fever
- •
- severe cough with or without an increase in mucus
(sputum) production - •
- severe sore throat
- •
- shortness of breath
- •
- burning or pain when urinating
- •
- unusual vaginal discharge or irritation
- •
- redness, swelling or pain in any part of
the body
- •
- severe bleeding. STIVARGA can cause bleeding which can be serious and sometimes lead to death. Tell your healthcare provider if you have any signs of bleeding during treatment with STIVARGA including:
- •
- vomiting blood or if your vomit looks like coffee-grounds
- •
- pink or brown urine
- •
- red or black (looks like tar) stools
- •
- coughing up blood or blood clots
- •
- menstrual bleeding that is heavier than normal
- •
- unusual vaginal bleeding
- •
- nose bleeds that happen often
- •
- bruising
- •
- lightheadedness
- •
- a tear in your stomach or intestinal wall (bowel perforation). STIVARGA may cause a tear in your stomach or intestinal wall (bowel perforation) that can be serious and sometimes lead to death. Tell your healthcare provider right away if you get:
- •
- severe pain in your stomach-area (abdomen)
- •
- swelling of the abdomen
- •
- fever
- •
- chills
- •
- nausea
- •
- vomiting
- •
- dehydration
- •
- a skin problem called hand-foot skin reaction and severe skin rash. Hand-foot skin reactions are common and sometimes can be severe. Tell your healthcare provider right away if you get redness, pain, blisters, bleeding, or swelling on the palms of your hands or soles of your feet, or a severe rash.
- •
- high blood pressure. Your blood pressure should be checked every week for the first 6 weeks of starting STIVARGA. Your blood pressure should be checked regularly and any high blood pressure should be treated during treatment with STIVARGA. Tell your healthcare provider if you have severe headaches, lightheadedness, or changes in your vision.
- •
- decreased blood flow to the heart and heart attack. Get emergency help right away if you get symptoms such as chest pain, shortness of breath, feel dizzy or feel like passing out.
- •
- a condition called Reversible Posterior Leukoencephalopathy Syndrome (RPLS). Call your healthcare provider right away if you get severe headaches, seizure, confusion, change in vision, or problems thinking.
- •
- risk of wound healing problems. Wounds may not heal properly during STIVARGA treatment. Tell your healthcare provider if you plan to have any surgery before starting or during treatment with STIVARGA.
- o
- You should stop taking STIVARGA at least 2 weeks before planned surgery.
- o
- Your healthcare provider should tell you when you may start taking STIVARGA again after surgery.
The most common side effects of STIVARGA include:
- •
- pain, including stomach-area (abdomen)
- •
- tiredness, weakness, fatigue
- •
- frequent or loose bowel movements (diarrhea)
- •
- decreased appetite
- •
- infection
- •
- voice changes or hoarseness
- •
- increase in certain liver function test
- •
- fever
- •
- swelling, pain and redness of the lining in your mouth, throat, stomach and bowel (mucositis)
- •
- weight loss
Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with STIVARGA if you have certain side effects.
These are not all of the possible side effects of STIVARGA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088
How do I store STIVARGA?
- •
- Store STIVARGA tablets at room temperature between 68° F to 77° F (20° C to 25°C).
- •
- Keep STIVARGA in the bottle that it comes in. Do not put STIVARGA tablets in a daily or weekly pill box.
- •
- The STIVARGA bottle contains a desiccant to help keep your medicine dry. Keep the desiccant in the bottle.
- •
- Keep the bottle of STIVARGA tightly closed.
- •
- Safely throw away (discard) any unused STIVARGA tablets after 7 weeks of opening the bottle.
Keep STIVARGA and all medicines out of the reach of children.
General information about the safe and effective use of STIVARGA.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use STIVARGA for a condition for which it was not prescribed. Do not give STIVARGA to other people even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about STIVARGA that is written for health professionals.
What are the ingredients in STIVARGA?
Active ingredient: regorafenib
Inactive ingredients: cellulose microcrystalline, croscarmellose sodium, magnesium stearate, povidone and colloidal silicon dioxide.
Film coat: ferric oxide red, ferric oxide yellow, lecithin (soy), polyethylene glycol 3350, polyvinyl alcohol, talc and titanium dioxide.
Manufactured for Bayer HealthCare Pharmaceuticals Inc., Whippany, NJ 07981 USA. © 2017 Bayer HealthCare Pharmaceuticals Inc.
For more information, go to www.STIVARGA-US.com or call 1-888-842-2937.- This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 02/2020
- PACKAGE/LABEL PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
STIVARGA
regorafenib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50419-171 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength REGORAFENIB (UNII: MGN125FS9D) (REGORAFENIB ANHYDROUS - UNII:24T2A1DOYB) REGORAFENIB ANHYDROUS 40 mg Inactive Ingredients Ingredient Name Strength MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) FERRIC OXIDE RED (UNII: 1K09F3G675) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) LECITHIN, SOYBEAN (UNII: 1DI56QDM62) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) Product Characteristics Color PINK (light) Score no score Shape OVAL Size 16mm Flavor Imprint Code BAYER Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50419-171-03 3 in 1 BOX 09/27/2012 1 NDC:50419-171-01 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 2 NDC:50419-171-00 210 in 1 BOX 09/27/2012 2 28 in 1 BOTTLE; Type 0: Not a Combination Product 3 NDC:50419-171-04 1 in 1 CARTON 09/27/2012 3 28 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 4 NDC:50419-171-06 4 in 1 CARTON 05/01/2021 4 NDC:50419-171-05 21 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA203085 09/27/2012 Labeler - Bayer HealthCare Pharmaceuticals Inc. (005436809) Establishment Name Address ID/FEI Business Operations Sharp Corporation 143696495 PACK(50419-171) , LABEL(50419-171) Establishment Name Address ID/FEI Business Operations Bayer AG 314947622 ANALYSIS(50419-171) , MANUFACTURE(50419-171) , PACK(50419-171) , LABEL(50419-171) Establishment Name Address ID/FEI Business Operations Bayer AG 323208116 API MANUFACTURE(50419-171) , PACK(50419-171) , ANALYSIS(50419-171)