Label: TEPMETKO- tepotinib hydrochloride tablet


- NDC Code(s): 44087-5000-3, 44087-5000-6
- Packager: EMD Serono, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated April 17, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TEPMETKO safely and effectively. See full prescribing information for TEPMETKO.
TEPMETKO® (tepotinib) tablets, for oral use
Initial U.S. Approval: 2021RECENT MAJOR CHANGES
INDICATIONS AND USAGE
TEPMETKO is a kinase inhibitor indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) harboring mesenchymal-epithelial transition (MET) exon 14 skipping alterations. (1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Tablets: 225 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Interstitial Lung Disease (ILD)/Pneumonitis: Immediately withhold TEPMETKO in patients with suspected ILD/pneumonitis. Permanently discontinue TEPMETKO in patients diagnosed with ILD/pneumonitis of any severity. (2.4, 5.1)
- Hepatotoxicity: Monitor liver function tests. Withhold, dose reduce, or permanently discontinue TEPMETKO based on severity. (5.2)
- Pancreatic Toxicity: Monitor amylase and lipase. Withhold, dose reduce, or permanently discontinue TEPMETKO based on severity. (5.3)
- Embryo-fetal toxicity: TEPMETKO can cause fetal harm. Advise of potential risk to a fetus and use of effective contraception. (5.4, 8.1, 8.3)
ADVERSE REACTIONS
Most common adverse reactions (≥ 20%) were edema, nausea, fatigue, musculoskeletal pain, diarrhea, dyspnea, decreased appetite, and rash. The most common Grade 3 to 4 laboratory abnormalities (≥ 2%) were decreased lymphocytes, decreased albumin, decreased sodium, increased gamma-glutamyltransferase, increased amylase, increased lipase, increased ALT, increased AST, and decreased hemoglobin. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact EMD Serono at 1-800-283-8088 ext. 5563 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Certain P-gp substrates: Avoid coadministration of TEPMETKO with P-gp substrates where minimal concentration changes may lead to serious or life-threatening toxicities. (7.1)
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 2/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection for METex14 Skipping Alterations
2.2 Recommended Dosage
2.3 Administration to Patients Who Have Difficulty Swallowing Solids
2.4 Dose Modifications for Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)/Pneumonitis
5.2 Hepatotoxicity
5.3 Pancreatic Toxicity
5.4 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Effects of TEPMETKO on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection for METex14 Skipping Alterations
Select patients for treatment with TEPMETKO based on the presence of MET exon 14 skipping alterations in plasma or tumor specimens. Testing for the presence of MET exon 14 skipping alterations in plasma specimens is recommended only in patients for whom a tumor biopsy cannot be obtained. If an alteration is not detected in a plasma specimen, re-evaluate the feasibility of biopsy for tumor tissue testing. An FDA-approved test for detection of MET exon 14 skipping alterations in NSCLC for selecting patients for treatment with TEPMETKO is not available.
2.2 Recommended Dosage
The recommended dosage of TEPMETKO is 450 mg orally once daily with food [see Clinical Pharmacology (12.3)] until disease progression or unacceptable toxicity.
Instruct patients to take their dose of TEPMETKO at approximately the same time every day and to swallow tablets whole. Do not chew, crush or split tablets. Patients who have difficulty swallowing solids can disperse tablets in water [see Dosage and Administration (2.3)].
Advise patients not to make up a missed dose within 8 hours of the next scheduled dose.
If vomiting occurs after taking a dose of TEPMETKO, advise patients to take the next dose at the scheduled time.
2.3 Administration to Patients Who Have Difficulty Swallowing Solids
Place TEPMETKO tablet(s) in a glass containing 30 mL (1 ounce) of non-carbonated water. No other liquids should be used or added. Stir, without crushing, until the tablet(s) is dispersed into small pieces (tablets will not completely dissolve) and drink immediately or within 1 hour. Swallow the tablet dispersion. Do not chew pieces of the tablet. Rinse the glass with an additional 30 mL and drink immediately ensuring no residue remains in the glass and the full dose is administered.
If an administration via a naso-gastric tube (with at least 8 French gauge) is required, disperse the tablet(s) in 30 mL of non-carbonated water as described above. Administer the 30 mL of liquid immediately or within 1 hour as per naso-gastric tube manufacturer's instructions. Immediately rinse twice with 30 mL each time to ensure that no residue remains in the glass or syringe and the full dose is administered.
2.4 Dose Modifications for Adverse Reactions
The recommended dose reduction of TEPMETKO for the management of adverse reactions is 225 mg orally once daily.
Permanently discontinue TEPMETKO in patients who are unable to tolerate 225 mg orally once daily.
The recommended dosage modifications of TEPMETKO for adverse reactions are provided in Table 1.
Table 1: Recommended TEPMETKO Dosage Modifications for Adverse Reactions Adverse Reaction Severity* Dose Modification - *
- Severity as defined by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) Version 5.
Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.1)] Any Grade Withhold TEPMETKO if ILD is suspected.
Permanently discontinue TEPMETKO if ILD is confirmed.Increased ALT and/or AST without increased total bilirubin [see Warnings and Precautions (5.2)] Grade 3 Withhold TEPMETKO until recovery to baseline ALT/AST.
If recovered to baseline within 7 days, then resume TEPMETKO at the same dose; otherwise resume TEPMETKO at a reduced dose.Grade 4 Permanently discontinue TEPMETKO. Increased ALT and/or AST with increased total bilirubin in the absence of cholestasis or hemolysis [see Warnings and Precautions (5.2)] ALT and/or AST greater than 3 times ULN with total bilirubin greater than 2 times ULN Permanently discontinue TEPMETKO. Increased total bilirubin without concurrent increased ALT and/or AST [see Warnings and Precautions (5.2)] Grade 3 Withhold TEPMETKO until recovery to baseline bilirubin.
If recovered to baseline within 7 days, then resume TEPMETKO at a reduced dose; otherwise permanently discontinue.Grade 4 Permanently discontinue TEPMETKO. Increased lipase or amylase [see Warnings and Precautions (5.3)] Grade 3 Withhold TEPMETKO until ≤ Grade 2 or baseline.
If recovered to baseline or ≤ Grade 2 within 14 days, resume TEPMETKO at a reduced dose; otherwise permanently discontinue TEPMETKO.Grade 4 Permanently discontinue TEPMETKO. Pancreatitis [see Warnings and Precautions (5.3)] Grade 3 or 4 Permanently discontinue TEPMETKO. Other adverse reactions [see Adverse Reactions (6.1)] Grade 2 Maintain dose level. If intolerable, consider withholding TEPMETKO until resolved, then resume TEPMETKO at a reduced dose. Grade 3 Withhold TEPMETKO until resolved, then resume TEPMETKO at a reduced dose. Grade 4 Permanently discontinue TEPMETKO. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Interstitial Lung Disease (ILD)/Pneumonitis
ILD/pneumonitis, which can be fatal, occurred in patients treated with TEPMETKO [see Adverse Reactions (6.1)]. ILD/pneumonitis occurred in 2% patients treated with TEPMETKO, with one patient experiencing a Grade 3 or higher event; this event resulted in death. Five patients (1%) discontinued TEPMETKO due to ILD/pneumonitis.
Monitor patients for new or worsening pulmonary symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, fever). Immediately withhold TEPMETKO in patients with suspected ILD/pneumonitis and permanently discontinue if no other potential causes of ILD/pneumonitis are identified [see Dosage and Administration (2.4)].
5.2 Hepatotoxicity
Hepatotoxicity occurred in patients treated with TEPMETKO [see Adverse Reactions (6.1)]. Increased alanine aminotransferase (ALT)/increased aspartate aminotransferase (AST) occurred in 18% of patients treated with TEPMETKO. Grade 3 or 4 increased ALT/AST occurred in 4.7% of patients. A fatal adverse reaction of hepatic failure occurred in one patient (0.2%). Four patients (0.8%) discontinued TEPMETKO due to increased ALT/AST. The median time-to-onset of Grade 3 or higher increased ALT/AST was 47 days (range 1 to 262).
Monitor liver function tests (including ALT, AST, and total bilirubin) prior to the start of TEPMETKO, every 2 weeks during the first 3 months of treatment, then once a month or as clinically indicated, with more frequent testing in patients who develop increased transaminases or bilirubin. Based on the severity of the adverse reaction, withhold, dose reduce, or permanently discontinue TEPMETKO [see Dosage and Administration (2.4)].
5.3 Pancreatic Toxicity
Elevations in amylase and lipase levels occurred in patients treated with TEPMETKO [see Adverse Reactions (6.1)]. Increased amylase and/or lipase occurred in 13% of patients treated with TEPMETKO. Grade 3 and 4 increased amylase and/or lipase occurred in 5% and 1.2% of patients, respectively. Monitor amylase and lipase at baseline and regularly during treatment with TEPMETKO. Based on the severity of the adverse drug reaction, temporarily withhold, dose reduce, or permanently discontinue TEPMETKO [see Dosage and Administration (2.4)].
5.4 Embryo-Fetal Toxicity
Based on findings in animal studies and its mechanism of action TEPMETKO can cause fetal harm when administered to a pregnant woman. Oral administration of tepotinib to pregnant rabbits during the period of organogenesis resulted in malformations (teratogenicity) and anomalies at exposures less than the human exposure based on area under the curve (AUC) at the 450 mg daily clinical dose. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential or males with female partners of reproductive potential to use effective contraception during treatment with TEPMETKO and for one week after the last dose. [See Use in Specific Populations (8.1, 8.3)]
-
6 ADVERSE REACTIONS
The following adverse reactions are described in greater detail elsewhere in the labeling:
- Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Pancreatic Toxicity [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the WARNINGS AND PRECAUTIONS reflect exposure to TEPMETKO in 506 patients with solid tumors enrolled in five open-label, single-arm studies receiving TEPMETKO as single agent at a dose of 450 mg once daily. This included 313 patients with NSCLC positive for METex14 skipping alterations, who received TEPMETKO in VISION. Among 506 patients who received TEPMETKO, 44% were exposed for 6 months or longer, and 22% were exposed for more than one year.
The data described below reflect exposure to TEPMETKO 450 mg once daily in 313 patients with metastatic non-small cell lung cancer (NSCLC) with METex14 skipping alterations in VISION [see Clinical Studies (14)].
Serious adverse reactions occurred in 51% of patients who received TEPMETKO. Serious adverse reactions in > 2% of patients included pleural effusion (6%), pneumonia (6%), edema (5%), general health deterioration (3.8%), dyspnea (3.5%), musculoskeletal pain (2.9%), and pulmonary embolism (2.2%). Fatal adverse reactions occurred in 1.9% of patients who received TEPMETKO, including pneumonitis (0.3%), hepatic failure (0.3%), dyspnea from fluid overload (0.3%), pneumonia (0.3%), sepsis (0.3%), and death of unknown cause (0.3%).
Permanent discontinuation due to an adverse reaction occurred in 25% of patients who received TEPMETKO. The most frequent adverse reactions (> 1%) leading to permanent discontinuations of TEPMETKO were edema (8%), pleural effusion (1.6%), and general health deterioration (1.6%).
Dosage interruptions due to an adverse reaction occurred in 53% of patients who received TEPMETKO. Adverse reactions which required dosage interruption in > 2% of patients who received TEPMETKO included edema (28%), increased blood creatinine (6%), pleural effusion (3.5%), nausea (3.2%), increased ALT (2.9%), pneumonia (2.6%), decreased appetite (2.2%), and dyspnea (2.2%).
Dose reductions due to an adverse reaction occurred in 36% of patients who received TEPMETKO. Adverse reactions which required dose reductions in > 2% of patients who received TEPMETKO included edema (22%), increased blood creatinine (2.9%), fatigue (2.2%), and pleural effusion (2.2%).
The most common adverse reactions (≥ 20%) in patients who received TEPMETKO were edema, nausea, fatigue, musculoskeletal pain, diarrhea, dyspnea, decreased appetite, and rash. The most common Grade 3 to 4 laboratory abnormalities (≥ 2%) were decreased lymphocytes, decreased albumin, decreased sodium, increased gamma-glutamyltransferase, increased amylase, increased lipase, increased ALT, increased AST, and decreased hemoglobin.
Table 2 summarizes the adverse reactions in VISION.
Table 2: Adverse Reactions in ≥ 10% of Patients with NSCLC with METex14 Skipping Alterations Who Received TEPMETKO in VISION Adverse Reactions TEPMETKO
(N=313)All Grades*
(%)Grades 3 to 4*
(%)- *
- Severity as defined by National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
- †
- Edema includes eye edema, face edema, generalized edema, localized edema, edema, genital edema, peripheral edema, peripheral swelling, periorbital edema, and scrotal edema.
- ‡
- Fatigue includes asthenia and fatigue.
- §
- Abdominal pain includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper, gastrointestinal pain, and hepatic pain.
- ¶
- Vomiting includes retching and vomiting.
- #
- Musculoskeletal pain includes arthralgia, arthritis, back pain, bone pain, musculoskeletal chest pain, musculoskeletal pain, myalgia, non-cardiac chest pain, pain in extremity, and spinal pain.
- Þ
- Dyspnea includes dyspnea, dyspnea at rest, and dyspnea exertional.
- ß
- Cough includes cough, and productive cough.
- à
- Rash includes rash, palmar-plantar erythrodysesthesia syndrome, rash maculo-papular, eczema, exfoliative rash, rash erythematous, rash pustular, skin exfoliation, dermatitis acneiform, drug eruption, dermatitis, rash pruritic, dermatitis bullous, toxic skin eruption.
- è
- Pneumonia includes pneumonia, pneumonia aspiration, and pneumonia bacterial.
General disorders and administration-site conditions Edema† 81 16 Fatigue‡ 30 1.9 Gastrointestinal disorders Nausea 31 1.3 Diarrhea 29 0.6 Abdominal pain§ 19 0.6 Constipation 19 0.3 Vomiting¶ 15 1 Musculoskeletal and Connective Tissue Disorders Musculoskeletal pain# 30 3.2 Respiratory, thoracic, and mediastinal disorders DyspneaÞ 24 2.6 Coughß 18 0.3 Pleural effusion 14 4.2 Metabolism and nutrition disorders Decreased appetite 21 1.9 Skin and Subcutaneous Tissue Disorders Rashà 21 1.3 Infections and Infestations Pneumoniaè 12 3.8 Clinically relevant adverse reactions in < 10% of patients who received TEPMETKO included ILD/pneumonitis, fever, dizziness, pruritus, and headache.
Table 3 summarizes the laboratory abnormalities observed in VISION.
Table 3: Select Laboratory Abnormalities (≥ 20%) That Worsened from Baseline in Patients Who Received TEPMETKO in VISION Laboratory Abnormalities TEPMETKO* Grades 1 to 4†
(%)Grades 3 to 4†
(%)Chemistry Decreased albumin 81 9 Increased creatinine 60 1 Increased alkaline phosphatase aminotransferase 52 1.6 Increased alanine aminotransferase 50 4.9 Increased aspartate aminotransferase 40 3.6 Decreased sodium 36 9 Increased gamma-glutamyltransferase 29 6 Increased potassium 26 1.9 Increased amylase 25 5 Increased lipase 21 5 Hematology Decreased lymphocytes 57 15 Decreased hemoglobin 31 3.6 Decreased leukocytes 25 1.9 Decreased platelets 24 0.6 -
7 DRUG INTERACTIONS
7.1 Effects of TEPMETKO on Other Drugs
Certain P-gp Substrates
Tepotinib is a P-gp inhibitor. Concomitant use of TEPMETKO increases the concentration of P-gp substrates [see Clinical Pharmacology (12.3)], which may increase the incidence and severity of adverse reactions of these substrates. Avoid concomitant use of TEPMETKO with certain P-gp substrates where minimal concentration changes may lead to serious or life-threatening toxicities. If concomitant use is unavoidable, reduce the P-gp substrate dosage if recommended in its approved product labeling.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings in animal studies and the mechanism of action [see Clinical Pharmacology (12.1)], TEPMETKO can cause fetal harm when administered to a pregnant woman. There are no available data on the use of TEPMETKO in pregnant women. Oral administration of tepotinib to pregnant rabbits during the period of organogenesis resulted in malformations (teratogenicity) and anomalies at maternal exposures less than the human exposure based on area under the curve (AUC) at the 450 mg daily clinical dose (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In embryo-fetal development studies, pregnant rabbits received oral doses of 0.5, 5, 25, 50, 150, or 450 mg/kg tepotinib hydrochloride hydrate daily during organogenesis. Severe maternal toxicity occurred at the 450 mg/kg dose (approximately 0.75 times the human exposure at the 450 mg clinical dose). At 150 mg/kg (approximately 0.5 times the human exposure by AUC at the 450 mg clinical dose), two animals aborted and one animal died prematurely; mean fetal body weight was also decreased. A dose-dependent increase of skeletal malformations, including malrotations of fore and/or hind paws with concomitant misshapen scapula and/or malpositioned clavicle and/or calcaneus and/or talus, occurred at doses ≥ 5 mg/kg (approximately 0.003 times the human exposure by AUC at the 450 mg clinical dose); there was also an incidence of spina bifida at the 5 mg/kg dose level.
8.3 Females and Males of Reproductive Potential
Based on animal data, TEPMETKO can cause malformations at doses less than the human exposure based on AUC at the 450 mg clinical dose [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating TEPMETKO [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and efficacy of TEPMETKO in pediatric patients have not been established.
8.5 Geriatric Use
Of 313 patients with NSCLC positive for METex14 skipping alterations in VISION who received 450 mg TEPMETKO once daily, 79% were 65 years or older, and 41% were 75 years or older. No clinically important differences in safety or efficacy were observed between patients aged 65 years or older and younger patients.
8.6 Renal Impairment
No dosage modification is recommended in patients with mild or moderate renal impairment (creatinine clearance [CLcr] 30 to 89 mL/min, estimated by Cockcroft-Gault). The recommended dosage has not been established for patients with severe renal impairment (CLcr < 30 mL/min) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage modification is recommended in patients with mild (Child Pugh Class A) or moderate (Child Pugh Class B) hepatic impairment. The pharmacokinetics and safety of tepotinib in patients with severe hepatic impairment (Child Pugh Class C) have not been studied [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
Tepotinib is a kinase inhibitor. TEPMETKO (tepotinib) tablets for oral use are formulated with tepotinib hydrochloride hydrate. The chemical name for tepotinib hydrochloride hydrate is 3-{1-[(3-{5-[(1-methylpiperidin-4-yl)methoxy]pyrimidin-2-yl}phenyl)methyl]-6-oxo-1,6-dihydropyridazin-3-yl}benzonitrile hydrochloride hydrate. The molecular formula is C29H28N6O2∙HCl∙H2O and the molecular weight is 547.05 g/mol for tepotinib hydrochloride hydrate and 492.58 g/mol for tepotinib (free base). The chemical structure is shown below:
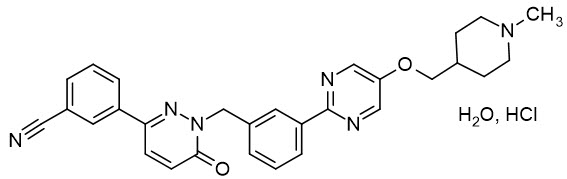
Tepotinib hydrochloride hydrate is a white to off-white powder with a pKa of 9.5.
TEPMETKO is supplied as film-coated tablets containing 225 mg of tepotinib (equivalent to 250 mg tepotinib hydrochloride hydrate). Inactive ingredients in the tablet core are mannitol, microcrystalline cellulose, crospovidone, magnesium stearate, and colloidal silicon dioxide. The tablet coating consists of hypromellose, titanium dioxide, lactose monohydrate, polyethylene glycol, triacetin, and red iron oxides.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tepotinib is a kinase inhibitor that targets MET, including variants with exon 14 skipping alterations. Tepotinib inhibits hepatocyte growth factor (HGF)-dependent and -independent MET phosphorylation and MET-dependent downstream signaling pathways. Tepotinib also inhibited melatonin 2 and imidazoline 1 receptors at clinically achievable concentrations.
In vitro, tepotinib inhibited tumor cell proliferation, anchorage-independent growth, and migration of MET-dependent tumor cells. In mice implanted with tumor cell lines with oncogenic activation of MET, including METex14 skipping alterations, tepotinib inhibited tumor growth, led to sustained inhibition of MET phosphorylation, and, in one model, decreased the formation of metastases.
12.2 Pharmacodynamics
12.3 Pharmacokinetics
The pharmacokinetics of tepotinib were evaluated in patients with cancer administered 450 mg once daily unless otherwise specified. Tepotinib exposure (AUC0-12h and Cmax) increases dose-proportionally over the dose range of 27 mg (0.06 times the recommended daily dosage) to 450 mg. At the recommended dosage, the geometric mean (coefficient of variation [CV] %) steady state Cmax was 1,291 ng/mL (48.1%) and the AUC0-24h was 27,438 ng∙h/mL (51.7%). The oral clearance of tepotinib did not change with respect to time. The median accumulation was 2.5-fold for Cmax and 3.3-fold for AUC0-24h after multiple daily doses of tepotinib.
Absorption
The median Tmax of tepotinib is 8 hours (range from 6 to 12 hours). The geometric mean (CV%) absolute bioavailability of TEPMETKO in the fed state was 71.6% (10.8%) in healthy subjects.
Effect of Food
The mean AUC0-INF of tepotinib increased by 1.6-fold and Cmax increased by 2-fold, following administration of a high-fat, high-calorie meal (approximately 800 to 1,000 calories, 150 calories from protein, 250 calories from carbohydrate, and 500 to 600 calories from fat). The median Tmax shifted from 12 hours to 8 hours.
Distribution
The geometric mean (CV%) apparent volume of distribution (VZ/F) of tepotinib is 1,038 L (24.3%). Protein binding of tepotinib is 98% and is independent of drug concentration at clinically relevant exposures.
Elimination
The apparent clearance (CL/F) of tepotinib is 23.8 L/h (87.5%) and the half-life is 32 hours following oral administration of TEPMETKO in patients with cancer.
Specific Populations
No clinically significant effects on tepotinib pharmacokinetics were observed based on age (18 to 89 years), race/ethnicity (White, Black, Asian, Japanese, and Hispanic), sex, body weight (35.5 to 136 kg), mild to moderate renal impairment (CLcr 30 to 89 mL/min), or mild to moderate hepatic impairment (Child-Pugh A and B). The effect of severe renal impairment (CLcr < 30 mL/min) and severe hepatic impairment (Child-Pugh C) on the pharmacokinetics of tepotinib has not been studied.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
No clinically significant differences in the pharmacokinetics of tepotinib were observed when coadministered with the following drugs: itraconazole (strong CYP3A and P-gp inhibitor), carbamazepine (strong CYP3A inducer) or omeprazole (proton pump inhibitor/acid reducing agent) under fed conditions.
No clinically significant differences in the pharmacokinetics of the following drugs were observed or predicted when coadministered with tepotinib: midazolam (sensitive CYP3A substrate) or CYP2C9 substrates.
In Vitro Studies
Cytochrome P450 Enzymes: Tepotinib is a substrate of CYP3A4 and CYP2C8. Tepotinib and M506 do not inhibit CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C19, CYP2D6 or CYP2E1, and do not induce CYP1A2 or 2B6 at clinically relevant concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been performed with tepotinib. Tepotinib and its major circulating metabolite were not mutagenic in vitro in the bacterial reverse mutation (Ames) assay, or a mouse lymphoma assay. In vivo, tepotinib was not genotoxic in a rat micronucleus test.
Fertility studies of tepotinib have not been performed. There were no morphological changes in male or female reproductive organs in repeat-dose toxicity studies in dogs.
-
14 CLINICAL STUDIES
The efficacy of TEPMETKO was evaluated in a single-arm, open-label, multicenter, non-randomized, multicohort study (VISION, NCT02864992). Eligible patients were required to have advanced or metastatic NSCLC harboring METex14 skipping alterations, epidermal growth factor receptor (EGFR) wild-type and anaplastic lymphoma kinase (ALK) negative status, at least one measurable lesion as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, and Eastern Cooperative Oncology Group (ECOG) Performance Status of 0 to 1. Patients with symptomatic CNS metastases, clinically significant uncontrolled cardiac disease, or who received treatment with any MET or hepatocyte growth factor (HGF) inhibitor were not eligible for the study.
Identification of METex14 skipping alterations was prospectively determined using central laboratories employing either a PCR-based or next-generation sequencing-based clinical trial assay using tissue (66%) and/or plasma (57%) samples.
Patients received TEPMETKO 450 mg once daily until disease progression or unacceptable toxicity. The major efficacy outcome measure was confirmed overall response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST v1.1) as evaluated by a Blinded Independent Review Committee (BIRC). An additional efficacy outcome measure was duration of response (DOR) by BIRC.
The efficacy population included 164 treatment naïve patients and 149 previously treated patients. The median age was 72 years (range 41 to 94 years); 51% female; 62% White, 34% Asian; 26% had Eastern Cooperative Oncology Group (ECOG) Performance Status (PS) 0 and 74% had ECOG PS 1; 49% never smoked; 81% had adenocarcinoma; 94% had metastatic disease; and 13% had CNS metastases. Amongst previously treated patients, 84% received prior platinum-based chemotherapy.
Efficacy results are presented in Table 4.
Table 4: Efficacy Results in the VISION study Efficacy parameter Treatment-Naïve
(N=164)Previously Treated
(N=149)CI: Confidence interval, + denotes ongoing response. Overall response rate (95% CI) *,† 57% (49, 65) 45% (37, 53) Duration of Response Range in months‡ 1.3+, 56.6+ 1.4+, 67.6+ Patients with DOR ≥ 6 months‡ 66% 66% Patients with DOR ≥ 12 months‡ 40% 36% -
16 HOW SUPPLIED/STORAGE AND HANDLING
TEPMETKO (tepotinib) tablets: 225 mg tepotinib, white-pink, oval, biconvex film-coated tablet with embossment "M" on one side and plain on the other side.
NDC number Size 44087-5000-3 Box of 30 tablets: 3 blister cards each containing 10 tablets 44087-5000-6 Box of 60 tablets: 6 blister cards each containing 10 tablets The blister cards consist of a child-resistant blister foil.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Interstitial Lung Disease (ILD)/Pneumonitis
Inform patients of the risk of severe or fatal ILD/pneumonitis. Advise patients to contact their healthcare provider immediately to report new or worsening respiratory symptoms [see Warnings and Precautions (5.1)].
Hepatotoxicity
Inform patients that they will need to undergo lab tests to monitor liver function. Advise patients to immediately contact their healthcare provider for signs and symptoms of liver dysfunction [see Warnings and Precautions (5.2)].
Pancreatic Toxicity
Inform patients that they will need to undergo lab tests to monitor pancreatic function. Advise patients to immediately contact their healthcare provider for signs and symptoms of pancreatitis [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
Advise males and females of reproductive potential that TEPMETKO can cause fetal harm.
Advise females of reproductive potential to use effective contraception during and for one week after the last dose of TEPMETKO [see Warnings and Precautions (5.4) and Use in Specific Populations (8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with TEPMETKO and for one week after the last dose of TEPMETKO [see Warnings and Precautions (5.4) and Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with TEPMETKO and for one week after the last dose [see Use in Specific Populations (8.2)].
Drug Interactions
Advise patients to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs and herbal products [see Drug Interactions (7)].
Dosing and Administration
Instruct patients to take 450 mg TEPMETKO once daily with food [see Dosage and Administration (2.2)].
Missed Dose
Advise patients that a missed dose of TEPMETKO can be taken as soon as remembered on the same day, unless the next dose is due within 8 hours. If vomiting occurs after taking a dose of TEPMETKO, advise patients to take the next dose at the scheduled time [see Dosage and Administration (2.2)].
- SPL UNCLASSIFIED SECTION
-
PATIENT PACKAGE INSERT
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised 01/2025 PATIENT INFORMATION
TEPMETKO® (tep-MET-co)
(tepotinib)
tablets, for oral useWhat is the most important information I should know about TEPMETKO?
TEPMETKO may cause serious side effects, including:-
Lung problems. TEPMETKO may cause severe or life-threatening swelling (inflammation) of the lungs during treatment that can lead to death. Tell your healthcare provider right away if you develop any new or worsening symptoms of lung problems, including:
- trouble breathing
- shortness of breath
- cough
- fever
What is TEPMETKO?
TEPMETKO is a prescription medicine used to treat adults with non-small cell lung cancer (NSCLC) that:- has spread to other parts of the body (metastatic), and
- whose tumors have an abnormal mesenchymal epithelial transition (MET) gene. Your healthcare provider will perform a test to make sure that TEPMETKO is right for you.
Before you take TEPMETKO, tell your healthcare provider about all of your medical conditions, including if you: - have or have had lung or breathing problems other than your lung cancer
- have or have had liver problems
- have or have had pancreatic problems
- are pregnant or plan to become pregnant. TEPMETKO can harm your unborn baby.
Females who are able to become pregnant:- Your healthcare provider may do a pregnancy test before you start treatment with TEPMETKO.
- You should use effective birth control (contraception) during treatment and for 1 week after the last dose of TEPMETKO. Talk to your healthcare provider about birth control methods that may be right for you.
Males with female partners who are able to become pregnant should use effective birth control (contraception) during treatment with TEPMETKO and for 1 week after the last dose of TEPMETKO. - are breastfeeding or plan to breastfeed. It is not known if TEPMETKO passes into your breast milk. Do not breastfeed during treatment and for 1 week after the last dose of TEPMETKO.
How should I take TEPMETKO? - Take TEPMETKO exactly as your healthcare provider tells you to. Do not change your dose or stop taking TEPMETKO unless your healthcare provider tells you to.
- Take TEPMETKO 1 time a day with food.
- Take your dose of TEPMETKO at about the same time each day.
- Swallow TEPMETKO tablets whole. Do not chew, crush or split the tablets.
If you cannot swallow TEPMETKO tablets whole:- Place your prescribed dose of TEPMETKO tablets in a glass that contains 30 mL (1 ounce) of non-carbonated water. Do not use or add any other liquids.
- Stir the TEPMETKO tablets and water until the TEPMETKO tablets are in small pieces (the tablets will not completely dissolve). Do not crush TEPMETKO tablets.
- Drink the TEPMETKO and water mixture right away or within 1 hour. Make sure to swallow the mixture. Do not chew pieces of the tablet.
- Add another 30 mL of non-carbonated water to the glass and drink it right away to get your full dose of TEPMETKO.
If you have a nasogastric (NG) tube that is 8 French gauge or larger:- Follow the same instructions for mixing TEPMETKO tablets in a glass that contains 30 mL of non-carbonated water.
- Do not use or add any other liquids.
- Stir the TEPMETKO tablets and water until the TEPMETKO tablets are in small pieces (the tablets will not completely dissolve). Do not crush TEPMETKO tablets.
- Give the TEPMETKO tablet and water mixture using the NG tube manufacturer instructions right away or within 1 hour.
- Pieces of the tablet may remain in the glass. Right away, add 30 mL of non-carbonated water into the glass. Give the water and any remaining TEPMETKO through the NG tube right away to make sure that all of the medicine is given. Repeat this step. This will help to ensure that no medicine is left in the glass or syringe and the full prescribed dose of TEPMETKO is given.
- If you miss a dose of TEPMETKO, take it as soon as you remember. If your next dose is due within 8 hours, skip the missed dose and take your next dose at your regular scheduled time.
- If you vomit after taking a dose of TEPMETKO, take your next dose at your regular scheduled time.
What are the possible side effects of TEPMETKO?
TEPMETKO may cause serious side effects, including:- See "What is the most important information I should know about TEPMETKO?"
- Liver problems. TEPMETKO may cause abnormal liver blood test results. Your healthcare provider will do blood tests to check your liver function before you start treatment and during treatment with TEPMETKO. Tell your healthcare provider right away if you develop any signs and symptoms of liver problems, including:
- your skin or the white part of your eyes turns yellow
- dark or "tea colored" urine
- light-colored stools (bowel movements)
- confusion
- tiredness
- loss of appetite for several days or longer
- nausea and vomiting
- pain, aching, or tenderness on the right side of your stomach-area (abdomen)
- weakness
- swelling in your stomach-area
- Pancreas problems. TEPMETKO may cause increases in your blood amylase and lipase levels that may indicate a problem with your pancreas. Your healthcare provider will do blood tests to check your pancreatic function before you start treatment and during treatment with TEPMETKO. Tell your healthcare provider right away if you develop any signs and symptoms of pancreas problems, including:
- upper stomach (abdominal) pain that may spread to your back and get worse with eating
- weight loss
- nausea
- vomiting
The most common side effects of TEPMETKO include: - swelling in your face or other parts of your body
- nausea
- tiredness
- muscle and joint pain
- diarrhea
- shortness of breath
- rash
- changes in certain blood tests
Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with TEPMETKO if you develop serious side effects during treatment.
These are not all of the possible side effects of TEPMETKO.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store TEPMETKO? - Store TEPMETKO at room temperature between 68°F to 77°F (20°C to 25°C).
- TEPMETKO tablets come in blister cards with a child-resistant blister foil.
- Store TEPMETKO in original package.
General information about the safe and effective use of TEPMETKO.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use TEPMETKO for a condition for which it was not prescribed. Do not give TEPMETKO to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TEPMETKO that is written for health professionals.What are the ingredients in TEPMETKO?
Active ingredient: tepotinib
Inactive ingredients: mannitol, microcrystalline cellulose, crospovidone, magnesium stearate, and colloidal silicon dioxide.
Tablet coating: hypromellose, titanium dioxide, lactose monohydrate, polyethylene glycol, triacetin, and red iron oxides.
Manufactured for: EMD Serono, Inc., Boston, MA 02210, U.S.A.
TEPMETKO is a trademark of Merck KGaA, Darmstadt, Germany.
Product of Germany.
For more information, call toll-free 1-844-662-3631 or go to www.TEPMETKO.com. -
Lung problems. TEPMETKO may cause severe or life-threatening swelling (inflammation) of the lungs during treatment that can lead to death. Tell your healthcare provider right away if you develop any new or worsening symptoms of lung problems, including:
- PRINCIPAL DISPLAY PANEL - 30 Tablet Blister Pack Carton
- PRINCIPAL DISPLAY PANEL - 60 Tablet Blister Pack Carton
-
INGREDIENTS AND APPEARANCE
TEPMETKO
tepotinib hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:44087-5000 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Tepotinib Hydrochloride (UNII: VY5YX2TQ1F) (Tepotinib - UNII:1IJV77EI07) Tepotinib 225 mg Inactive Ingredients Ingredient Name Strength Mannitol (UNII: 3OWL53L36A) Microcrystalline Cellulose (UNII: OP1R32D61U) Crospovidone, Unspecified (UNII: 2S7830E561) Magnesium stearate (UNII: 70097M6I30) Silicon Dioxide (UNII: ETJ7Z6XBU4) Hypromellose, Unspecified (UNII: 3NXW29V3WO) Lactose monohydrate (UNII: EWQ57Q8I5X) Polyethylene Glycol, Unspecified (UNII: 3WJQ0SDW1A) Triacetin (UNII: XHX3C3X673) Ferric oxide red (UNII: 1K09F3G675) Titanium dioxide (UNII: 15FIX9V2JP) Product Characteristics Color PINK (White-pink) Score no score Shape OVAL (oval, biconvex) Size 7mm Flavor Imprint Code M Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:44087-5000-3 3 in 1 CARTON 02/03/2021 1 10 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC:44087-5000-6 6 in 1 CARTON 02/03/2021 2 10 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA214096 02/03/2021 Labeler - EMD Serono, Inc. (088514898)

