Label: HEPSERA- adefovir dipivoxil tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 46014-0501-1 - Packager: Excella GmbH
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated October 27, 2009
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use HEPSERA safely and effectively. See full prescribing information for HEPSERA.
HEPSERA (adefovir dipivoxil) tablet for oral use
Initial U.S. Approval: 2002WARNING: SEVERE ACUTE EXACERBATIONS OF HEPATITIS, NEPHROTOXICITY, HIV RESISTANCE, LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS
See full prescribing information for complete boxed warning.
- Severe acute exacerbations of hepatitis may occur in patients who discontinue HEPSERA. Monitor hepatic function closely in these patients. (5.1)
- Chronic use of HEPSERA may result in nephrotoxicity in patients at risk of renal dysfunction or having underlying renal dysfunction. Monitor renal function closely in these patients. Dose adjustment may be required. (5.2)
- HIV resistance may emerge in chronic hepatitis B patients with unrecognized or untreated HIV infection. (5.3)
- Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogues. (5.4)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
HEPSERA is a nucleotide analogue indicated for the treatment of chronic hepatitis B in patients ≥12 years of age. (1)
DOSAGE AND ADMINISTRATION
- One tablet containing 10 mg adefovir dipivoxil once daily orally with or without food. (2.1)
- Dose adjustment in renal impairment for adults (2.2)
Creatinine Clearance (mL/min)* ≥50 30–49 10–29 Hemodialysis Patients - *
- Creatinine clearance calculated by Cockcroft-Gault method using lean or ideal body weight.
Recommended dose and dosing interval 10 mg every 24 hours 10 mg every 48 hours 10 mg every 72 hours 10 mg every 7 days following dialysis - No dose recommendations for (2.1):
- Non-hemodialysis patients with creatinine clearance <10mL/min.
- Adolescent patients with renal impairment.
DOSAGE FORMS AND STRENGTHS
Tablets: 10 mg (3)
CONTRAINDICATIONS
HEPSERA is contraindicated in patients with previously demonstrated hypersensitivity to any of the components of the product. (4)
WARNINGS AND PRECAUTIONS
- Severe acute exacerbations of hepatitis: Monitor hepatic function closely at repeated intervals for at least several months in patients who discontinue HEPSERA. (5.1)
- Nephrotoxicity: Monitor renal function during therapy for all patients, particularly those with pre-existing or other risks for renal impairment. Dose adjustment may be required. (5.2)
- HIV Resistance: Offer HIV testing to all patients prior to initiating HEPSERA. Untreated HIV may result in HIV resistance. (5.3)
- Lactic acidosis and severe hepatomegaly with steatosis: If suspected, suspend treatment. (5.4)
- Coadministration with Other Products: Do not administer HEPSERA concurrently with VIREAD or other tenofovir-containing products. (5.5)
- Clinical Resistance: For patients with lamivudine-resistant HBV use adefovir dipivoxil in combination with lamivudine. For all patients, consider modifying treatment in case serum HBV DNA remains above 1000 copies/mL with continued treatment. (5.6)
ADVERSE REACTIONS
Most common adverse reaction (>10%) in compensated disease patients is asthenia and in pre- and post-transplantation lamivudine-resistant liver disease patients is increased creatinine. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Gilead at (1-800-GILEAD-5) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
- Co-administration with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of adefovir or the co-administered drug. Monitor for HEPSERA associated adverse events. (7)
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2009
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: SEVERE ACUTE EXACERBATIONS OF HEPATITIS, NEPHROTOXICITY, HIV RESISTANCE, LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Chronic Hepatitis B
2.2 Dose Adjustment in Renal Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Exacerbation of Hepatitis after Discontinuation of Treatment
5.2 Nephrotoxicity
5.3 HIV Resistance
5.4 Lactic Acidosis/Severe Hepatomegaly with Steatosis
5.5 Coadministration with Other Products
5.6 Clinical Resistance
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Special Risk Patients
6.3 Pediatric Patients
6.4 Post-Marketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Labor and Delivery
8.3 Nursing Mothers
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Patients with Impaired Renal Function
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Studies 437 and 438 (Pivotal Studies)
14.2 Study 435 (Pre- and Post- Liver Transplantation Patients)
14.3 Study 461 (Clinical Evidence of Lamivudine Resistance)
14.4 Study 518 (Pediatric Study)
16 HOW SUPPLIED / STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
17.1 Instructions for Safe Use
17.2 Pregnancy and Breastfeeding
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: SEVERE ACUTE EXACERBATIONS OF HEPATITIS, NEPHROTOXICITY, HIV RESISTANCE, LACTIC ACIDOSIS AND SEVERE HEPATOMEGALY WITH STEATOSIS
Severe acute exacerbations of hepatitis have been reported in patients who have discontinued anti-Hepatitis B therapy including HEPSERA. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-Hepatitis B therapy. If appropriate, resumption of anti-Hepatitis B therapy may be warranted [see Warnings and Precautions (5.1)].
In patients at risk of or having underlying renal dysfunction, chronic administration of HEPSERA may result in nephrotoxicity. These patients should be monitored closely for renal function and may require dose adjustment [see Warnings and Precautions (5.2) and Dosage and Administration (2.2)].
HIV resistance may emerge in chronic hepatitis B patients with unrecognized or untreated Human Immunodeficiency Virus (HIV) infection treated with anti-hepatitis B therapies, such as therapy with HEPSERA, that may have activity against HIV [see Warnings and Precautions (5.3)].
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs alone or in combination with other antiretrovirals [see Warnings and Precautions (5.4)].
-
1 INDICATIONS AND USAGE
HEPSERA is indicated for the treatment of chronic hepatitis B in patients 12 years of age and older with evidence of active viral replication and either evidence of persistent elevations in serum aminotransferases (ALT or AST) or histologically active disease.
This indication is based on histological, virological, biochemical, and serological responses in adult patients with HBeAg+ and HBeAg- chronic hepatitis B with compensated liver function, and with clinical evidence of lamivudine-resistant hepatitis B virus with either compensated or decompensated liver function.
For patients 12 to <18 years of age, the indication is based on virological and biochemical responses in patients with HBeAg+ chronic hepatitis B virus infection with compensated liver function.
-
2 DOSAGE AND ADMINISTRATION
2.1 Chronic Hepatitis B
The recommended dose of HEPSERA in chronic hepatitis B patients for patients ≥12 years of age with adequate renal function is 10 mg, once daily, taken orally, without regard to food. The optimal duration of treatment is unknown.
HEPSERA is not recommended for use in children less than 12 years of age.
2.2 Dose Adjustment in Renal Impairment
Significantly increased drug exposures were seen when HEPSERA was administered to adult patients with renal impairment [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.3)]. Therefore, the dosing interval of HEPSERA should be adjusted in adult patients with baseline creatinine clearance <50 mL/min using the following suggested guidelines (see Table 1). The safety and effectiveness of these dosing interval adjustment guidelines have not been clinically evaluated.
Additionally, it is important to note that these guidelines were derived from data in patients with pre-existing renal impairment at baseline. They may not be appropriate for patients in whom renal insufficiency evolves during treatment with HEPSERA. Therefore, clinical response to treatment and renal function should be closely monitored in these patients.
Table 1. Dosing Interval Adjustment of HEPSERA in Adult Patients with Renal Impairment Creatinine Clearance (mL/min)* ≥50 30–49 10–29 Hemodialysis Patients - *
- Creatinine clearance calculated by Cockcroft-Gault method using lean or ideal body weight.
Recommended dose
and dosing interval10 mg every
24 hours10 mg every
48 hours10 mg every
72 hours10 mg every 7 days following
dialysisThe pharmacokinetics of adefovir have not been evaluated in non-hemodialysis patients with creatinine clearance <10 mL/min; therefore, no dosing recommendation is available for these patients.
No clinical data are available to make dosing recommendations in adolescent patients with renal insufficiency [see Warnings and Precautions (5.2)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Exacerbation of Hepatitis after Discontinuation of Treatment
Severe acute exacerbation of hepatitis has been reported in patients who have discontinued anti-hepatitis B therapy, including therapy with HEPSERA. Hepatic function should be monitored at repeated intervals with both clinical and laboratory follow-up for at least several months in patients who discontinue HEPSERA. If appropriate, resumption of anti-hepatitis B therapy may be warranted.
In clinical trials of HEPSERA, exacerbations of hepatitis (ALT elevations 10 times the upper limit of normal or greater) occurred in up to 25% of patients after discontinuation of HEPSERA. These events were identified in studies GS-98-437 and GS-98-438 (N=492). Most of these events occurred within 12 weeks of drug discontinuation. These exacerbations generally occurred in the absence of HBeAg seroconversion, and presented as serum ALT elevations in addition to re-emergence of viral replication. In the HBeAg-positive and HBeAg-negative studies in patients with compensated liver function, the exacerbations were not generally accompanied by hepatic decompensation. However, patients with advanced liver disease or cirrhosis may be at higher risk for hepatic decompensation. Although most events appear to have been self-limited or resolved with re-initiation of treatment, severe hepatitis exacerbations, including fatalities, have been reported. Therefore, patients should be closely monitored after stopping treatment.
5.2 Nephrotoxicity
Nephrotoxicity characterized by a delayed onset of gradual increases in serum creatinine and decreases in serum phosphorus was historically shown to be the treatment-limiting toxicity of adefovir dipivoxil therapy at substantially higher doses in HIV-infected patients (60 and 120 mg daily) and in chronic hepatitis B patients (30 mg daily). Chronic administration of HEPSERA (10 mg once daily) may result in delayed nephrotoxicity. The overall risk of nephrotoxicity in patients with adequate renal function is low. However, this is of special importance in patients at risk of or having underlying renal dysfunction and patients taking concomitant nephrotoxic agents such as cyclosporine, tacrolimus, aminoglycosides, vancomycin and non-steroidal anti-inflammatory drugs [see Adverse Reactions (6.2) and Clinical Pharmacology (12.3)]. It is recommended that creatinine clearance is calculated in all patients prior to initiating therapy with HEPSERA.
It is important to monitor renal function for all patients during treatment with HEPSERA, particularly for those with pre-existing or other risks for renal impairment. Patients with renal insufficiency at baseline or during treatment may require dose adjustment [see Dosage and Administration (2.2)]. The risks and benefits of HEPSERA treatment should be carefully evaluated prior to discontinuing HEPSERA in a patient with treatment-emergent nephrotoxicity.
Pediatric Patients
The efficacy and safety of HEPSERA have not been studied in patients less than 18 years of age with different degrees of renal impairment and no data are available to make dosage recommendations in these patients [see Dosage and Administration (2.2)]. Caution should be exercised when prescribing HEPSERA to adolescents with underlying renal dysfunction, and renal function in these patients should be closely monitored.
5.3 HIV Resistance
Prior to initiating HEPSERA therapy, HIV antibody testing should be offered to all patients. Treatment with anti-hepatitis B therapies, such as HEPSERA, that have activity against HIV in a chronic hepatitis B patient with unrecognized or untreated HIV infection may result in emergence of HIV resistance. HEPSERA has not been shown to suppress HIV RNA in patients; however, there are limited data on the use of HEPSERA to treat patients with chronic hepatitis B co-infected with HIV.
5.4 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs alone or in combination with antiretrovirals.
A majority of these cases have been in women. Obesity and prolonged nucleoside exposure may be risk factors. Particular caution should be exercised when administering nucleoside analogs to any patient with known risk factors for liver disease; however, cases have also been reported in patients with no known risk factors. Treatment with HEPSERA should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.5 Coadministration with Other Products
HEPSERA should not be used concurrently with VIREAD (tenofovir disoproxil fumarate) or tenofovir disoproxil fumarate-containing products including TRUVADA (emtricitabine/tenofovir disoproxil fumarate combination tablet) and ATRIPLA (efavirenz/emtricitabine/tenofivir disoproxil fumarate combination tablet).
5.6 Clinical Resistance
Resistance to adefovir dipivoxil can result in viral load rebound which may result in exacerbation of hepatitis B and, in the setting of diminished hepatic function, lead to liver decompensation and possible fatal outcome.
In order to reduce the risk of resistance in patients with lamivudine resistant HBV, adefovir dipivoxil should be used in combination with lamivudine and not as adefovir dipivoxil monotherapy.
In order to reduce the risk of resistance in all patients receiving adefovir dipivoxil monotherapy, a modification of treatment should be considered if serum HBV DNA remains above 1000 copies/mL with continued treatment.
Long-term (144 week) data from Study 438 (n=124) show that patients with HBV DNA levels greater than 1000 copies/mL at Week 48 of treatment with HEPSERA were at greater risk of developing resistance than patients with serum HBV DNA levels below 1000 copies/mL at Week 48 of therapy.
-
6 ADVERSE REACTIONS
The following adverse reactions are discussed in other sections of the labeling:
- Severe acute exacerbations of Hepatitis [see Boxed Warning, Warnings and Precautions (5.1)]
- Nephrotoxicity [see Boxed Warning , Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical and laboratory evidence of exacerbations of hepatitis have occurred after discontinuation of treatment with HEPSERA.
Adverse reactions to HEPSERA identified from placebo-controlled and open label studies include the following: asthenia, headache, abdominal pain, diarrhea, nausea, dyspepsia, flatulence, increased creatinine, and hypophosphatemia.
The incidence of these adverse reactions in studies 437 and 438, where 522 patients with chronic hepatitis B and compensated liver disease received double-blind treatment with HEPSERA (n=294) or placebo (n=228) for 48 weeks is presented in Table 2. Patients who received open-label HEPSERA for up to 240 weeks in Study 438 reported adverse reactions similar in nature and severity to those reported in the first 48 weeks.
Table 2 Adverse Reactions (Grades 1–4) Reported in ≥3% of All HEPSERA-Treated Patients in Pooled Studies 437–438 Studies (0–48 Weeks)* Adverse Reaction HEPSERA
10mg
(N=294)
Placebo
(N=228)
- *
- In these studies, the overall incidence of adverse reactions with HEPSERA was similar to that reported with placebo. The incidence of adverse reactions is derived from treatment-related events as identified by the study investigators.
Asthenia 13% 14% Headache 9% 10% Abdominal Pain 9% 11% Nausea 5% 8% Flatulence 4% 4% Diarrhea 3% 4% Dyspepsia 3% 2% No patients treated with HEPSERA developed a confirmed serum creatinine increase ≥0.5 mg/dL or confirmed phosphorus decrease ≤2 mg/dL from baseline by Week 48. By Week 96, 2% of HEPSERA-treated patients, by Kaplan-Meier estimate, had increases in serum creatinine ≥0.5 mg/dL from baseline (no placebo-controlled results were available for comparison beyond Week 48). For patients who chose to continue HEPSERA for up to 240 weeks in Study 438, 4 of 125 patients (3%) had a confirmed increase of 0.5 mg/dL from baseline. The creatinine elevation resolved in 1 patient who permanently discontinued treatment and remained stable in 3 patients who continued treatment. For 65 patients who chose to continue HEPSERA for up to 240 weeks in Study 437, 6 had a confirmed increase in serum creatinine of ≥0.5 mg/dL from baseline with 2 patients discontinuing from the study due to the elevated serum creatinine concentration. See Adverse Reactions (6.2) for changes in serum creatinine in patients with underlying renal insufficiency at baseline.
6.2 Special Risk Patients
Pre- and Post-Liver Transplantation Patients
Additional adverse reactions observed from an open-label study (Study 435) in pre- and post- liver transplantation patients with chronic hepatitis B and lamivudine-resistant hepatitis B administered HEPSERA once daily for up to 203 weeks include: abnormal renal function, renal failure, vomiting, rash, and pruritus.
Changes in renal function occurred in pre-and post-liver transplantation patients with risk factors for renal dysfunction, including concomitant use of cyclosporine and tacrolimus, renal insufficiency at baseline, hypertension, diabetes, and on-study transplantation. Therefore, the contributory role of HEPSERA to these changes in renal function is difficult to assess.
Increases in serum creatinine ≥0.3 mg/dL from baseline were observed in 37% and 53% of pre-liver transplantation patients by Weeks 48 and 96, respectively, by Kaplan-Meier estimates. Increases in serum creatinine ≥0.3 mg/dL from baseline were observed in 32% and 51% of post-liver transplantation patients by Weeks 48 and 96, respectively, by Kaplan-Meier estimates. Serum phosphorus values <2 mg/dL were observed in 3/226 (1.3%) of pre-liver transplantation patients and in 6/241 (2.5%) of post-liver transplantation patients by last study visit. Four percent (19 of 467) of patients discontinued treatment with HEPSERA due to renal adverse events.
6.3 Pediatric Patients
Assessment of adverse reactions is based on a placebo-controlled study (Study 518) in which 173 pediatric patients aged 2 to <18 years with chronic hepatitis B and compensated liver disease received double-blind treatment with HEPSERA (n=115), or placebo (n=58) for 48 weeks [see Clinical Studies (14.4) and Use In Specific Populations (8.4)].
The safety profile of HEPSERA in patients ≥12 to <18 years of age (n=56) was similar to that observed in adults. No pediatric patients treated with HEPSERA developed a confirmed serum creatinine increase ≥ 0.5 mg/dL or confirmed phosphorus decrease to <2 mg/dL from baseline by Week 48.
6.4 Post-Marketing Experience
In addition to adverse reaction reports from clinical trials, the following possible adverse reactions have also been identified during post-approval use of adefovir dipivoxil. Because these events have been reported voluntarily from a population of unknown size, estimates of frequency cannot be made.
Metabolism and Nutrition Disorders: hypophosphatemia
Gastrointestinal Disorders: pancreatitis
Musculoskeletal System and Connective Tissue Disorders: myopathy, osteomalacia (both associated with proximal renal tubulopathy)
Renal and Urinary Disorders: renal failure, Fanconi syndrome, proximal renal tubulopathy
-
7 DRUG INTERACTIONS
Since adefovir is eliminated by the kidney, co-administration of HEPSERA with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of either adefovir and/or these co-administered drugs [see Clinical Pharmacology (12.3)].
Patients should be monitored closely for adverse events when HEPSERA is co-administered with drugs that are excreted renally or with other drugs known to affect renal function [see Warnings and Precautions (5.2)].
HEPSERA should not be administered in combination with VIREAD [see Warnings and Precautions (5.5)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Teratogenic Effects: Pregnancy Category C
There are no adequate and well-controlled studies of HEPSERA in pregnant women. Chronic hepatitis B is a serious condition that requires treatment. HEPSERA should be used during pregnancy only if the potential benefit to the mother justifies the potential risk to the fetus.
Reproduction studies with oral administration of adefovir dipivoxil to pregnant rats and rabbits showed no evidence of embryotoxicity or teratogenicity at systemic exposures equivalent to 23 times (rats) and 40 times (rabbits) that achieved in humans at the therapeutic dose. However, embryotoxicity and an increased incidence of fetal malformations (anasarca, depressed eye bulge, umbilical hernia and kinked tail) occurred when adefovir was administered intravenously to pregnant rats at 38 times the human therapeutic exposure. These adverse reproductive effects did not occur following an intravenous dose where exposure was 12 times the human therapeutic exposure.
Because animal reproduction studies are not always predictive of human response, HEPSERA should be used during pregnancy only if clearly needed and after careful consideration of the risks and benefits [see Nonclinical Toxicology (13.2)].
8.2 Labor and Delivery
There are no studies in pregnant women and no data on the effect of HEPSERA on transmission of HBV from mother to infant. Therefore, appropriate infant immunizations should be used to prevent neonatal acquisition of hepatitis B virus.
8.3 Nursing Mothers
It is not known whether adefovir is excreted in human milk.
Because many drugs are excreted into human milk and because of the potential for serious adverse reactions in nursing infants from HEPSERA, a decision should be made whether to discontinue nursing or to discontinue drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
Pediatric patients 12 to <18 years: The safety, efficacy, and pharmacokinetics of HEPSERA in pediatric patients (aged ≥12 to <18 years) were evaluated in a double-blind, randomized, placebo-controlled study (GS-US-103-518, Study 518) in 83 pediatric patients with chronic hepatitis B and compensated liver disease. The proportion of patients treated with HEPSERA who achieved the primary efficacy endpoint of serum HBV DNA <1,000 copies/mL and normal ALT levels at the end of 48 weeks blinded treatment was significantly greater (23%) when compared to placebo-treated patients (0%). [see Clinical Studies (14.4), Dosage And Administration (2) and Adverse Reactions (6.3)].
Pediatric patients 2 to <12 years: Patients 2 to <12 years of age were also evaluated in Study 518. The efficacy of adefovir dipivoxil was not significantly different from placebo in patients less than 12 years of age.
Hepsera is not recommended for use in children below 12 years of age.
8.5 Geriatric Use
Clinical studies of HEPSERA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger patients. In general, caution should be exercised when prescribing to elderly patients since they have greater frequency of decreased renal or cardiac function due to concomitant disease or other drug therapy.
8.6 Patients with Impaired Renal Function
It is recommended that the dosing interval for HEPSERA be modified in adult patients with baseline creatinine clearance <50 mL/min. The pharmacokinetics of adefovir have not been evaluated in non-hemodialysis patients with creatinine clearance <10 mL/min or in adolescent patients with renal insufficiency; therefore, no dosing recommendations are available for these patients. [see Dosage And Administration (2.2) and Warning And Precautions (5.2)].
-
10 OVERDOSAGE
Doses of adefovir dipivoxil 500 mg daily for 2 weeks and 250 mg daily for 12 weeks have been associated with gastrointestinal side effects. If overdose occurs the patient must be monitored for evidence of toxicity, and standard supportive treatment applied as necessary.
Following a 10 mg single dose of HEPSERA, a four-hour hemodialysis session removed approximately 35% of the adefovir dose.
-
11 DESCRIPTION
HEPSERA® is the tradename for adefovir dipivoxil, a diester prodrug of adefovir. Adefovir is an acyclic nucleotide analog with activity against human hepatitis B virus (HBV).
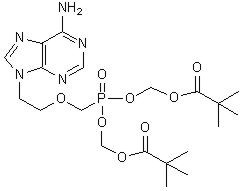
The chemical name of adefovir dipivoxil is 9-[2-[[bis[(pivaloyloxy)methoxy]-phosphinyl]-methoxy]ethyl]adenine. It has a molecular formula of C20H32N5O8P, a molecular weight of 501.48 and the following structural formula:
Adefovir dipivoxil is a white to off-white crystalline powder with an aqueous solubility of 19 mg/mL at pH 2.0 and 0.4 mg/mL at pH 7.2. It has an octanol/aqueous phosphate buffer (pH 7) partition coefficient (log p) of 1.91.
HEPSERA tablets are for oral administration. Each tablet contains 10 mg of adefovir dipivoxil and the following inactive ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, pregelatinized starch, and talc.
-
12 CLINICAL PHARMACOLOGY
12.3 Pharmacokinetics
Adult Subjects
The pharmacokinetics of adefovir have been evaluated in healthy volunteers and patients with chronic hepatitis B. Adefovir pharmacokinetics are similar between these populations.
Absorption
Adefovir dipivoxil is a diester prodrug of the active moiety adefovir. Based on a cross study comparison, the approximate oral bioavailability of adefovir from HEPSERA is 59%.
Following oral administration of a 10 mg single dose of HEPSERA to chronic hepatitis B patients (N=14), the peak adefovir plasma concentration (Cmax) was 18.4 ± 6.26 ng/mL (mean ± SD) and occurred between 0.58 and 4.00 hours (median=1.75 hours) post dose. The adefovir area under the plasma concentration-time curve (AUC0–∞) was 220 ± 70.0 ng∙h/mL. Plasma adefovir concentrations declined in a biexponential manner with a terminal elimination half-life of 7.48 ± 1.65 hours.
The pharmacokinetics of adefovir in subjects with adequate renal function were not affected by once daily dosing of 10 mg HEPSERA over seven days. The impact of long-term once daily administration of 10 mg HEPSERA on adefovir pharmacokinetics has not been evaluated.
Effects of Food on Oral Absorption
Adefovir exposure was unaffected when a 10 mg single dose of HEPSERA was administered with food (an approximately 1000 kcal high-fat meal). HEPSERA may be taken without regard to food.
Distribution
In vitro binding of adefovir to human plasma or human serum proteins is ≤4% over the adefovir concentration range of 0.1 to 25 µg/mL. The volume of distribution at steady-state following intravenous administration of 1.0 or 3.0 mg/kg/day is 392 ± 75 and 352 ± 9 mL/kg, respectively.
Metabolism and Elimination
Following oral administration, adefovir dipivoxil is rapidly converted to adefovir. Forty-five percent of the dose is recovered as adefovir in the urine over 24 hours at steady state following 10 mg oral doses of HEPSERA. Adefovir is renally excreted by a combination of glomerular filtration and active tubular secretion [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
Assessment of Drug Interactions
Adefovir dipivoxil is rapidly converted to adefovir in vivo. At concentrations substantially higher (>4000-fold) than those observed in vivo, adefovir did not inhibit any of the common human CYP450 enzymes, CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4. Adefovir is not a substrate for these enzymes. However, the potential for adefovir to induce CYP450 enzymes is unknown. Based on the results of these in vitro experiments and the renal elimination pathway of adefovir, the potential for CYP450 mediated interactions involving adefovir as an inhibitor or substrate with other medicinal products is low.
The pharmacokinetics of adefovir have been evaluated in healthy adult volunteers following multiple dose administration of HEPSERA (10 mg once daily) in combination with lamivudine (100 mg once daily) (N=18), trimethoprim/sulfamethoxazole (160/800 mg twice daily) (N=18), acetaminophen (1000 mg four times daily) (N=20), ibuprofen (800 mg three times daily) (N=18), and enteric coated didanosine (400 mg) (N=21). The pharmacokinetics of adefovir have also been evaluated in post-liver transplantation patients following multiple dose administration of HEPSERA (10 mg once daily) in combination with tacrolimus (N=16). The pharmacokinetics of adefovir have been evaluated in healthy volunteers following single dose pegylated interferon α-2a (PEG-IFN) (180 µg) (N=15).
Adefovir did not alter the pharmacokinetics of lamivudine, trimethoprim/sulfamethoxazole, acetaminophen, ibuprofen, enteric coated didanosine (didanosine EC), or tacrolimus. The evaluation of the effect of adefovir on the pharmacokinetics of pegylated interferon α-2a was inconclusive due to the high variability of pegylated interferon alpha-2a.
The pharmacokinetics of adefovir were unchanged when HEPSERA was coadministered with lamivudine, trimethoprim/sulfamethoxazole, acetaminophen, didanosine EC, tacrolimus (based on cross study comparison), and pegylated interferon α-2a. When HEPSERA was coadministered with ibuprofen (800 mg three times daily) increases in adefovir Cmax (33%), AUC (23%) and urinary recovery were observed. This increase appears to be due to higher oral bioavailability, not a reduction in renal clearance of adefovir.
Apart from lamivudine, trimethoprim/sulfamethoxazole, and acetaminophen, the effects of co-administration of HEPSERA with drugs that are excreted renally, or other drugs known to affect renal function have not been evaluated.
The effect of adefovir on cyclosporine concentrations is not known.
No drug interaction studies have been performed in adolescent patients aged ≥12 years to <18 years.
Special Populations
Race
The pharmacokinetics of adefovir have been shown to be comparable in Caucasians and Asians. Pharmacokinetic data are not available for other racial groups.
Pediatric Patients
The pharmacokinetics of adefovir were assessed from drug plasma concentrations in 53 HBeAg positive hepatitis B pediatric patients with compensated liver disease. The exposure of adefovir following a 48 week daily treatment with adefovir dipivoxil 10 mg tablet in pediatric patients aged ≥ 12 to <18 years (Cmax = 23.3 ng/ml and AUC0–24 = 248.8 ng∙h/ml) was comparable to that observed in adult patients.
Renal Impairment
In adults with moderately or severely impaired renal function or with end-stage renal disease (ESRD) requiring hemodialysis, Cmax, AUC, and half-life (T1/2) were increased compared to adults with normal renal function. It is recommended that the dosing interval of HEPSERA be modified in these patients [see Dosage and Administration (2.2)].
The pharmacokinetics of adefovir in non-chronic hepatitis B patients with varying degrees of renal impairment are described in Table 3. In this study, subjects received a 10 mg single dose of HEPSERA.
Table 3. Pharmacokinetic Parameters (Mean ± SD) of Adefovir in Patients with Varying Degrees of Renal Function Renal Function Group Unimpaired Mild Moderate Severe Baseline creatinine clearance (mL/min)
>80
(N=7)50–80
(N=8)30–49
(N=7)10–29
(N=10)Cmax (ng/mL) 17.8 ± 3.22 22.4 ± 4.04 28.5 ± 8.57 51.6 ± 10.3 AUC 0–∞ (ng∙h/mL) 201 ± 40.8 266 ± 55.7 455 ± 176 1240 ± 629 CL/F (mL/min) 469 ± 99.0 356 ± 85.6 237 ± 118 91.7 ± 51.3 CLrenal (mL/min) 231 ± 48.9 148 ± 39.3 83.9 ± 27.5 37.0 ± 18.4 A four-hour period of hemodialysis removed approximately 35% of the adefovir dose. The effect of peritoneal dialysis on adefovir removal has not been evaluated.
The pharmacokinetics of adefovir have not been studied in adolescent patients with renal dysfunction [see Use in Specific Populations (8.4)].
Hepatic Impairment
The pharmacokinetics of adefovir following a 10 mg single dose of HEPSERA have been studied in non-chronic hepatitis B patients with hepatic impairment. There were no substantial alterations in adefovir pharmacokinetics in patients with moderate and severe hepatic impairment compared to unimpaired patients. No change in HEPSERA dosing is required in patients with hepatic impairment.
12.4 Microbiology
Mechanism of Action
Adefovir is an acyclic nucleotide analog of adenosine monophosphate which is phosphorylated to the active metabolite adefovir diphosphate by cellular kinases. Adefovir diphosphate inhibits HBV DNA polymerase (reverse transcriptase) by competing with the natural substrate deoxyadenosine triphosphate and by causing DNA chain termination after its incorporation into viral DNA. The inhibition constant (Ki) for adefovir diphosphate for HBV DNA polymerase was 0.1 µM. Adefovir diphosphate is a weak inhibitor of human DNA polymerases α and γ with Ki values of 1.18 µM and 0.97 µM, respectively.
Antiviral Activity
The concentration of adefovir that inhibited 50% of viral DNA synthesis (EC50) in HBV transfected human hepatoma cell lines ranged from 0.2 to 2.5 µM. The combination of adefovir with lamivudine showed additive anti-HBV activity.
Resistance
Clinical isolates with genotypic changes conferring reduced susceptibility in cell culture to nucleoside analog reverse transcriptase inhibitors for the treatment of HBV infection have been observed. Long-term resistance analyses performed by genotyping samples from all adefovir dipivoxil-treated patients with detectable serum HBV DNA demonstrated that amino acid substitutions rtN236T and rtA181T/V have been observed in association with adefovir resistance. In cell culture, the rtN236T substitution demonstrated 4- to 14-fold, the rtA181V substitution 2.5- to 4.2-fold, and the rtA181T substitution 1.3- to 1.9-fold reduced susceptibility to adefovir.
In HBeAg-positive nucleoside-naïve patient isolates (Study GS-98-437, N=171), no adefovir resistance-associated substitutions were observed at Week 48. Sixty-five patients continued on long term treatment after a median duration on adefovir dipivoxil of 235 weeks (range 110–279 weeks). Isolates from 16 of 38 (42%) patients developed adefovir resistance-associated substitutions in the setting of virologic failure (confirmed increase of ≥1 log10 HBV DNA copies/mL above nadir or never suppressed below 103 copies/mL). The substitutions included rtN236T (n=2), rtA181V (n=4), rtA181T (n=3), rtA181T+rtN236T (n=5), and rtA181V+rtN236T (n=2). In HBeAg-negative nucleoside-naïve patients (Study GS-98-438), isolates from 30 patients were identified with adefovir resistance-associated substitutions with a cumulative probability of 0%, 3%, 11%, 19%, and 30% at 48, 96, 144, 192, and 240 weeks, respectively. Of those 30 patients, 22 had a confirmed increase of ≥1 log10 HBV DNA copies/mL above nadir or never achieved HBV DNA levels below 103 copies/mL; an additional 8 patients had adefovir resistance-associated substitutions without virologic failure. In addition, the long term (4 to 5 years) development of resistance to adefovir dipivoxil was significantly lower in patients who had serum HBV DNA below the limit of quantification (less than 1,000 copies/mL) at Week 48 as compared to patients who had serum HBV DNA above 1,000 copies/mL at Week 48.
In an open-label study of pre- and post-liver transplantation patients (Study GS-98-435), isolates from 129 patients with clinical evidence of lamivudine-resistant hepatitis B virus at baseline were evaluated for adefovir resistance-associated substitutions. The incidence of adefovir resistance-associated (rtN236T or rtA181T/V) substitutions was 0% at 48 weeks. Isolates from four patients developed the rtN236T substitution after 72 weeks of adefovir dipivoxil therapy. Development of the rtN236T substitution was associated with serum HBV DNA rebound. All 4 patients who developed the rtN236T substitution in their HBV had discontinued lamivudine therapy before the development of genotypic resistance and all 4 lost the lamivudine resistance-associated substitutions present at baseline. In a study of 35 HIV/HBV co-infected patients with lamivudine-resistant HBV (Study 460i) who added adefovir dipivoxil to lamivudine, no adefovir resistance-associated substitutions were observed in HBV isolates from 15/35 patients tested up to 144 weeks of therapy.
Clinical resistance in pediatric patients
In a Phase 3 pediatric Study GS-US-103-518, HBV isolates from 49 of 56 pediatric subjects (aged 12 to 17 years) had serum HBV DNA >169 copies/mL and were evaluated for adefovir resistance-associated substitutions. rtN236T and/or rtA181V adefovir resistance-associated substitutions were not observed at Week 48. However, the rtA181T substitution was present in baseline and Week 48 isolates from 2 pediatric patients.
Cross-resistance
Recombinant HBV variants containing lamivudine-resistance-associated substitutions (rtL180M, rtM204I, rtM204V, rtL180M + rtM204V, rtV173L + rtL180M + rtM204V) were susceptible to adefovir in cell culture. Adefovir dipivoxil has also demonstrated anti-HBV activity (median reduction in serum HBV DNA of 4.1 log10 copies/mL) in patients with HBV containing lamivudine-resistance-associated substitutions (Study 435). Adefovir also demonstrated in cell culture activity against HBV variants with entecavir resistance-associated substitutions (rtT184G, rtS202I, rtM250V). HBV variants with DNA polymerase substitutions rtT128N and rtR153Q or rtW153Q associated with resistance to hepatitis B virus immunoglobulin were susceptible to adefovir in cell culture.
HBV variants expressing the adefovir resistance-associated substitution rtN236T showed no change in susceptibility to entecavir in cell culture, and a 2- to 3-fold decrease in lamivudine susceptibility. HBV mutants with the adefovir resistance-associated substitution rtA181V showed a range of decreased susceptibilities to lamivudine of 1- to 14-fold and a 12-fold decrease in susceptibility to entecavir. In patients whose HBV expressed the rtA181V substitution (n=2) or the rtN236T substitution (n=3), a reduction in serum HBV DNA of 2.4 to 3.1 and 2.0 to 5.1 log10 copies/mL, respectively, was observed when treatment with lamivudine was added to treatment with adefovir dipivoxil.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Long-term oral carcinogenicity studies of adefovir dipivoxil in mice and rats were carried out at exposures up to approximately 10 times (mice) and 4 times (rats) those observed in humans at the therapeutic dose for HBV infection. In both mouse and rat studies, adefovir dipivoxil was negative for carcinogenic findings. Adefovir dipivoxil was mutagenic in the in vitro mouse lymphoma cell assay (with or without metabolic activation). Adefovir induced chromosomal aberrations in the in vitro human peripheral blood lymphocyte assay without metabolic activation. Adefovir dipivoxil was not clastogenic in the in vivo mouse micronucleus assay and adefovir was not mutagenic in the Ames bacterial reverse mutation assay using S. typhimurium and E. coli strains in the presence or absence of metabolic activation. In reproductive toxicology studies, no evidence of impaired fertility was seen in male or female rats at systemic exposure approximately 19 times that achieved in humans at the therapeutic dose.
13.2 Animal Toxicology and/or Pharmacology
Toxicology Studies
Animal reproduction studies were conducted in rats and rabbits with orally administered adefovir dipivoxil and intravenously administered adefovir.
In rats and rabbits, no embryotoxicity or teratogenicity was shown from oral administration of adefovir dipivoxil at maternal doses producing systemic exposures approximately 23 times (rats) and 40 times (rabbits) that achieved in humans at the therapeutic dose of 10 mg/day.
When pregnant rats were administered intravenous adefovir at maternally toxic doses associated with systemic exposure 38 times that in humans, embryotoxicity and an increased incidence of fetal malformations (anasarca, depressed eye bulge, umbilical hernia, and kinked tail) were observed. No adverse effects on development were seen with intravenous adefovir administered to pregnant rats at a systemic exposure 12 times that in humans.
Animal Toxicology Studies
Renal tubular nephropathy characterized by histological alterations and/or increases in BUN and serum creatinine was the primary dose-limiting toxicity associated with administration of adefovir dipivoxil in animals. Nephrotoxicity was observed in animals at systemic exposures approximately 3–10 times higher than those in humans at the recommended therapeutic dose of 10 mg/day.
-
14 CLINICAL STUDIES
14.1 Studies 437 and 438 (Pivotal Studies)
HBeAg-Positive Chronic Hepatitis B
Study 437 was a randomized, double-blind, placebo-controlled, three-arm study in patients with HBeAg-positive chronic hepatitis B that allowed for a comparison between placebo and HEPSERA. The median age of patients was 33 years. Seventy-four percent were male, 59% were Asian, 36% were Caucasian, and 24% had prior interferon-α treatment. At baseline, patients had a median total Knodell Histology Activity Index (HAI) score of 10, a median serum HBV DNA level as measured by the Roche Amplicor Monitor polymerase chain reaction (PCR) assay (LLOQ = 1000 copies/mL) of 8.36 log10 copies/mL and a median ALT level of 2.3 times the upper limit of normal.
HBeAg-Negative (Anti-HBe Positive/HBV DNA Positive) Chronic Hepatitis B
Study 438 was a randomized, double-blind, placebo-controlled study in patients who were HBeAg-negative at screening, and anti-HBe positive. The median age of patients was 46 years. Eighty-three percent were male, 66% were Caucasian, 30% were Asian and 41% had prior interferon-α treatment. At baseline, the median total Knodell HAI score was 10, the median serum HBV DNA level as measured by the Roche Amplicor Monitor PCR assay (LLOQ = 1000 copies/mL) was 7.08 log10 copies/mL, and the median ALT was 2.3 times the upper limit of normal.
The primary efficacy endpoint in both studies was histological improvement at Week 48; results of which are shown in Table 4.
Table 4. Histological Response at Week 48* Study 437 Study 438 HEPSERA
10 mg
(N=168)Placebo
(N=161)HEPSERA
10 mg
(N=121)Placebo
(N=57)Improvement† 53% 25% 64% 35% No Improvement 37% 67% 29% 63% Missing/Unassessable Data 10% 7% 7% 2% Table 5 illustrates the changes in Ishak Fibrosis Score by treatment group.
Table 5. Changes in Ishak Fibrosis Score at Week 48 Study 437 Study 438 Number of Adequate
Biopsy PairsHEPSERA
10 mg
(N=152)Placebo
(N=149)HEPSERA
10 mg
(N=113)Placebo
(N=56)
- *
- Change of 1 point or more in Ishak Fibrosis Score.
Ishak Fibrosis Score
Improved*34% 19% 34% 14% Unchanged 55% 60% 62% 50% Worsened* 11% 21% 4% 36% At Week 48, improvement was seen with respect to mean change in serum HBV DNA (log10 copies/mL), normalization of ALT, and HBeAg seroconversion as compared to placebo in patients receiving HEPSERA (Table 6).
Table 6. Change in Serum HBV DNA, ALT Normalization, and HBeAg Seroconversion at Week 48 Study 437 Study 438 HEPSERA
10 mg
(N=171)Placebo
(N=167)HEPSERA
10 mg
(N=123)Placebo
(N=61)
- *
- Patients with HBeAg-negative disease cannot undergo HBeAg seroconversion.
Mean change ± SD in serum HBV DNA from baseline
(log10 copies/mL)–3.57 ± 1.64 –0.98 ± 1.32 –3.65 ± 1.14 –1.32 ± 1.25 ALT normalization 48% 16% 72% 29% HBeAg seroconversion 12% 6% NA* NA* Treatment Beyond 48 Weeks
In Study 437, continued treatment with HEPSERA to 72 weeks resulted in continued maintenance of mean reductions in serum HBV DNA observed at Week 48. An increase in the proportion of patients with ALT normalization was also observed in Study 437. The effect of continued treatment with HEPSERA on seroconversion is unknown.
In Study 438, patients who received HEPSERA during the first 48 weeks were re-randomized in a blinded manner to continue on HEPSERA or receive placebo for an additional 48 weeks. At Week 96, 50 of 70 (71%) of patients who continued treatment with HEPSERA had undetectable HBV DNA levels (<1000 copies/mL), and 47 of 64 (73%) of patients had ALT normalization. HBV DNA and ALT levels returned towards baseline in most patients who stopped treatment with HEPSERA.
From 141 eligible patients, there were 125 (89%) patients in Study 438 who chose to continue HEPSERA for up to 192 weeks or 240 weeks (4 years or 5 years). As these patients had already received HEPSERA for at least 48 weeks and appeared to be experiencing a benefit, they are not necessarily representative of patients initiating HEPSERA. Of these patients, 89/125 (71%) and 47/70 (67%) had an undetectable HBV DNA level (<1000 copies/mL) at Week 192 and Week 240, respectively. Of the patients who had an elevated ALT at baseline, 77/104 (74%) and 42/64 (66%) had a normal ALT at Week 192 and Week 240, respectively. Six (5%) patients experienced HBsAg loss.
14.2 Study 435 (Pre- and Post- Liver Transplantation Patients)
HEPSERA was also evaluated in an open-label, uncontrolled study of 467 chronic hepatitis B patients pre- (N=226) and post- (N=241) liver transplantation with clinical evidence of lamivudine- resistant hepatitis B virus (Study 435). At baseline, 60% of pre-liver transplantation patients were classified as Child-Pugh-Turcotte score of Class B or C. The median baseline HBV DNA as measured by the Roche Amplicor Monitor PCR assay (LLOQ = 1000 copies/mL) was 7.4 and 8.2 log10 copies/mL, and the median baseline ALT was 1.8 and 2.0 times the upper limit of normal in pre- and post-liver transplantation patients, respectively. Results of this study are displayed in Table 5. Treatment with HEPSERA resulted in a similar reduction in serum HBV DNA regardless of the patterns of lamivudine-resistant HBV DNA polymerase mutations at baseline. The significance of the efficacy results listed in Table 7 as they relate to clinical outcomes is not known.
Table 7. Efficacy in Pre- and Post-Liver Transplantation Patients at Week 48 Efficacy Parameter* Pre-Liver Transplantation
(N=226)Post-Liver Transplantation
(N=241)- *
- Data are missing for 29% (HBV DNA) and 37% to 45% (CPT Score, Normalization of ALT, Albumin, Bilirubin, and PT) of total patients enrolled in the study.
- †
- Denominator is the number of patients with serum HBV DNA ≥1000 copies/mL at baseline using the Roche Amplicor Monitor PCR Assay (LLOQ = 1000 copies/mL) and non-missing value at Week 48.
- ‡
- Denominator is patients with abnormal values at baseline and non-missing value at Week 48.
Mean change ± SD in HBV DNA from baseline (log10 copies/mL) –3.7 ± 1.6
(n=117)–4.0 ± 1.6
(n=164)Proportion with undetectable HBV DNA (< 1000 copies/mL)† 77/109 (71%) 64/159 (40%) Stable or improved Child-Pugh-Turcotte score 86/90 (96%) 107/115 (93%) Normalization of: ‡ ALT 61/82 (74%) 56/110 (51%) Albumin 43/54 (80%) 21/26 (81%) Bilirubin 38/68 (58%) 29/38 (76%) Prothrombin time 39/46 (85%) 5/9 (56%) 14.3 Study 461 (Clinical Evidence of Lamivudine Resistance)
In Study 461, a double-blind, active controlled study in 59 chronic hepatitis B patients with clinical evidence of lamivudine-resistant hepatitis B virus, patients were randomized to receive either HEPSERA monotherapy or HEPSERA in combination with lamivudine 100 mg or lamivudine 100 mg alone. At Week 48, the mean ± SD decrease in serum HBV DNA as measured by the Roche Amplicor Monitor PCR assay (LLOQ = 1000 copies/mL) was 4.00 ± 1.41 log10 copies/mL for patients treated with HEPSERA and 3.46 ± 1.10 log10 copies/mL for patients treated with HEPSERA in combination with lamivudine. There was a mean decrease in serum HBV DNA of 0.31 ± 0.93 log10 copies/mL in patients receiving lamivudine alone. ALT normalized in 47% of patients treated with HEPSERA, in 53% of patients treated with HEPSERA in combination with lamivudine, and 5% of patients treated with lamivudine alone. The significance of these findings as they relate to clinical outcomes is not known.
14.4 Study 518 (Pediatric Study)
Study 518 was a double-blind, placebo-controlled, study in which 173 pediatric patients (ages 2 to <18 years) with chronic hepatitis B (CHB) infection and elevated ALT were randomized 2:1 (115 receiving adefovir dipivoxil and 58 receiving placebo). Randomization was stratified by prior treatment and age 2 to <7 years old (cohort 1), 7 to <12 years old (cohort 2), and 12 to <18 years old (cohort 3). All patients in cohort 3 received 10 mg tablet formulation; all patients in cohorts 1 and 2 received an investigational suspension formulation (0.3 mg/kg/day cohort 1, 0.25 mg/kg/day cohort 2) once daily. The primary efficacy endpoint was HBV DNA <1000 copies/mL plus normalization of ALT at the end of Week 48.
In cohort 3 (n=83), significantly more patients treated with HEPSERA achieved the primary efficacy endpoint at the end of 48 weeks of blinded treatment (23%) when compared to placebo-treated patients (0%). The proportion of patients from cohorts 1 and 2 who responded to treatment with adefovir dipivoxil was not statistically significant when compared to the placebo arm, although the adefovir plasma concentrations in these patients were comparable to those observed in older patients. Overall, 22 of 115 (19%) of pediatric patients who received adefovir dipivoxil vs. 1 of 58 (2%) of placebo treated patients responded to treatment by Week 48 [see Adverse Reactions (6.3), Use In Special Populations (8.4) and Clinical Pharmacology (12.3, 12.4)].
-
16 HOW SUPPLIED / STORAGE AND HANDLING
HEPSERA is available as tablets. Each tablet contains 10 mg of adefovir dipivoxil. The tablets are white and debossed with "10" and "GILEAD" on one side and the stylized figure of a liver on the other side. They are packaged as follows: Bottles of 30 tablets (NDC 61958-0501-1) containing desiccant (silica gel) and closed with a child-resistant closure.
-
17 PATIENT COUNSELING INFORMATION
17.1 Instructions for Safe Use
See FDA-approved patient labeling
- Physicians should inform patients of the potential risks and benefits of HEPSERA and of alternative modes of therapy.
- Physicians should instruct their patients to
- –
- Read the Patient Package Insert before starting HEPSERA therapy.
- –
- Follow a regular dosing schedule to avoid missing doses.
- –
- Immediately report any severe abdominal pain, muscle pain, yellowing of the eyes, dark urine, pale stools, and/or loss in appetite.
- –
- Inform their doctor or pharmacist if they develop any unusual symptom(s), or if any known symptom persists or worsens.
- Patients should remain under the care of a physician when using HEPSERA.
- Patients should be advised that
- –
- The optimal duration of HEPSERA treatment and the relationship between treatment response and long-term outcomes such as hepatocellular carcinoma or decompensated cirrhosis are not known.
- –
- Patients should not discontinue Hepsera without first informing their physician [See Warnings and Precautions. (5.1)]
- –
- Routine laboratory monitoring and follow-up with a physician is important during HEPSERA therapy.
- –
- Obtaining HIV antibody testing prior to starting HEPSERA is important [See Warnings and Precautions. (5.3)]
- –
- HEPSERA should not be administered concurrently with VIREAD or TRUVADA or ATRIPLA [See Warnings and Precautions. (5.5)]
- –
- Lamivudine-resistant patients should use HEPSERA in combination with lamivudine and not as HEPSERA monotherapy [See Warnings and Precautions. (5.6)]
17.2 Pregnancy and Breastfeeding
- Physicians should inform women of childbearing age about the risks associated with exposure to HEPSERA during pregnancy.
- Patients should inform their physician if they become pregnant while using HEPSERA.
- Pregnant patients using HEPSERA should be informed about the HEPSERA pregnancy registry and offered the opportunity to enroll.
- Patients should be informed that it is not known whether HEPSERA is excreted into human milk or if it can harm a nursing infant. Therefore, a decision should be made whether to discontinue breastfeeding or drug.
-
PATIENT PACKAGE INSERT
FDA-Approved Patient Labeling
PATIENT INFORMATION
HEPSERA® (hep-SER-rah)
Generic Name: (adefovir dipivoxil) tablets
Read this information carefully before you start taking HEPSERA. Read and check for new information each time you get more HEPSERA. This information does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about HEPSERA?
- 1.
- Some people who stop taking HEPSERA get a very serious hepatitis. This usually happens within 12 weeks after stopping. You will need to have regular blood tests to check for liver function and hepatitis B virus levels if you stop taking HEPSERA.
- 2.
- HEPSERA may cause a severe kidney problem called nephrotoxicity. It usually happens in people that already have a kidney problem, but it can happen to anyone that uses HEPSERA. You will need to have regular blood tests to check for kidney function while you are taking HEPSERA.
- 3.
- If you get or have HIV that isn't being treated with medicines, HEPSERA may increase the chances your HIV infection cannot be helped with usual HIV medicines. This can happen if you get or have HIV and don't know it, or if your HIV is not being treated while you are taking HEPSERA. You should get an HIV test before you start taking HEPSERA and anytime after that when there's a chance you were exposed to HIV.
- 4.
-
Some people who have taken medicines like HEPSERA that are called nucleoside or nucleotide analogs have developed a serious condition called lactic acidosis (build up of an acid in the blood). Lactic acidosis is a medical emergency and must be treated in the hospital. Call your doctor right away if you get any of the following signs of lactic acidosis:
- You feel very weak or tired.
- You have unusual (not normal) muscle pain.
- You have trouble breathing.
- You have stomach pain with nausea and vomiting.
- You feel cold, especially in your arms and legs.
- You feel dizzy or lightheaded.
- You have a fast or irregular heartbeat.
Some people who have taken medicines like HEPSERA have developed serious liver problems called hepatotoxicity, with liver enlargement (hepatomegaly) and fat in the liver (steatosis). Call your doctor right away if you get any of the following signs of liver problems.
- Your skin or the white part of your eyes turns yellow (jaundice).
- Your urine turns dark.
- Your bowel movements (stools) turn light in color.
- You don't feel like eating food for several days or longer.
- You feel sick to your stomach (nausea).
- You have lower stomach pain.
You may be more likely to get lactic acidosis or serious liver problems if you are very overweight (obese) or have been taking nucleoside analog medicines [Atripla® (efavirenz plus emtricitabine plus tenofovir disoproxil fumarate), Combivir (zidovudine plus lamivudine), Emtriva® (emtricitabine), Epivir, Epivir-HBV (lamivudine), Epzicom (abacavir plus lamivudine), Hivid (zalcitabine), Retrovir (zidovudine), Trizivir (zidovudine plus lamivudine plus abacavir), Truvada® (emtricitabine plus tenofovir disoproxil fumarate), Videx (didanosine), Viread® (tenofovir disoproxil fumarate), Zerit (stavudine), and Ziagen (abacavir)] for a long time.
What is HEPSERA?
HEPSERA is a medicine used to treat patients at least 12 years of age with continuing (chronic) infections with active hepatitis B virus. HEPSERA has not been studied in adults over the age of 65 and is not recommended for use in children less than 12 years of age.
- HEPSERA will not cure your chronic hepatitis B.
- HEPSERA may help lower the amount of hepatitis B virus in your body.
- HEPSERA may lower the ability of the virus to multiply and infect new liver cells.
- We do not know if HEPSERA will reduce your chances of getting liver cancer or liver damage (cirrhosis) from chronic hepatitis B.
- We do not know how long HEPSERA may help your hepatitis. Sometimes viruses change in your body and medicines no longer work. This is called drug resistance.
- HEPSERA does not stop you from spreading hepatitis B virus to others by sex or sharing needles. So practice safe sex and needle use.
Who should not take HEPSERA?
- Do not take HEPSERA if you are allergic to any of the ingredients in HEPSERA. The active ingredient in HEPSERA is adefovir dipivoxil. See the end of this leaflet for a complete list of all the ingredients in HEPSERA.
- Do not take HEPSERA if you are already taking VIREAD, TRUVADA or ATRIPLA.
Tell your doctor if:
- You are pregnant. We do not know if HEPSERA can harm your unborn child. You and your doctor will need to decide if HEPSERA is right for you. If you take HEPSERA and you are pregnant, talk to your doctor about how you can join the HEPSERA pregnancy registry.
- You are breast-feeding. We do not know if HEPSERA can pass into your milk and if it can harm your baby. You will need to choose either to breast feed or take HEPSERA, but not both.
- You have kidney problems now or had them before. Your dose and schedule of HEPSERA may be reduced. Blood tests will need to be done regularly to see how your kidneys are working.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. Some medicines may affect how HEPSERA works, especially medicines that affect how your kidneys work. HEPSERA can affect how your other medicines work. Your dose of HEPSERA and the other medicines may be changed. Do not take any other medicines while you are taking HEPSERA, unless your doctor has told you it is okay.
How should I take HEPSERA?
- Your doctor will tell you how much HEPSERA to take.
- Your doctor will tell you when and how often to take HEPSERA.
- Take HEPSERA the same time each day that your doctor tells you. If you forget to take HEPSERA, take it as soon as you remember that day. Do not take more than 1 dose of HEPSERA in a day. Do not take 2 doses at the same time. Call your doctor or pharmacist if you are not sure what to do.
- Do not change your dose of HEPSERA or stop HEPSERA without talking to your doctor. Your hepatitis may get worse if you change doses or stop.
- You may take HEPSERA with or without food.
- When your HEPSERA supply gets low, call your doctor or pharmacy for a refill. Do not run out of HEPSERA.
- If you take too much HEPSERA, call your local poison control center or emergency room right away.
Some patients get worse or very serious hepatitis B symptoms when they stop taking HEPSERA (see, "What is the most important information I should know about HEPSERA?"). We don't know how long you should use HEPSERA. You and your doctor will need to decide when it is best for you to stop taking HEPSERA. After you stop taking HEPSERA, your doctor will still need to check your health and take blood tests to check your liver for a few months.
What should I avoid while taking HEPSERA?
Avoid doing things that can spread hepatitis B virus since HEPSERA doesn't stop you from passing the infection to others.
- Do not share needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes or razor blades.
- Do not have any kind of sex without protection. Practice "safe sex" using condoms and dental dams.
What are the possible side effects of HEPSERA?
HEPSERA can cause the following serious side effects: (see, "What is the most important information I should know about HEPSERA?")
- 1.
- a very serious hepatitis if you stop taking it.
- 2.
- a severe kidney problem called nephrotoxicity.
- 3.
- increase your chance of developing a form of HIV that cannot be treated with usual HIV medicines
- 4.
- lactic acidosis and liver problems.
The most common side effects of HEPSERA are weakness, headache, stomach pain, nausea, flatulence (intestinal gas), diarrhea, indigestion and changes in the way the kidneys work. Additional side effects in liver transplant patients with chronic hepatitis B are vomiting, rash and itching. Some patients with liver transplants also had undesirable effects on their kidneys, including failure of the kidneys.
Other side effects reported since HEPSERA has been marketed include kidney failure, damage to kidney cells, muscle pain or weakness and weakening of the bone, which could cause them to break (both associated with kidney problems), and inflammation of the pancreas.
These are not all of the possible side effects of HEPSERA. For more information, ask your doctor or pharmacist.
General information about the safe and effective use of HEPSERA:
Medicines are sometimes prescribed for conditions not mentioned in patient information leaflets. Do not use HEPSERA for a condition for which it was not prescribed. Do not give HEPSERA to other people, even if they have the same symptoms that you have.
This leaflet summarizes the most important information about HEPSERA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about HEPSERA that is written for health professionals.
HEPSERA Tablets should be stored at room temperature and should be stored in their original container.
Do not use if seal over bottle opening is broken or missing.
What are the Ingredients of HEPSERA?
Active Ingredient: adefovir dipivoxil
Inactive Ingredients: croscarmellose sodium, lactose monohydrate, magnesium stearate, pregelatinized starch, and talc
-
SPL UNCLASSIFIED SECTION
Manufactured for: Gilead Sciences, Inc.
Foster City, CA 94404VIREAD®, EMTRIVA®, and TRUVADA® are trademarks of Gilead Sciences, Inc. ATRIPLA® is a trademark of Bristol-Myers Squibb & Gilead Sciences, LLC. Other brands listed are the trademarks of their respective owners.
©2009 Gilead Sciences, Inc.
21-449-GS-011
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
HEPSERA
adefovir dipivoxil tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:46014-0501 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength Adefovir Dipivoxil (UNII: U6Q8Z01514) (adefovir - UNII:6GQP90I798) adefovir 10 mg Product Characteristics Color WHITE (white to off-white) Score no score Shape ROUND Size 7mm Flavor Imprint Code GILEAD;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:46014-0501-1 30 in 1 BOTTLE, PLASTIC Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021449 09/20/2002 Labeler - Excella GmbH (329809800)
