Label: XOLAIR- omalizumab injection, solution
XOLAIR PFS- omalizumab injection, solution
XOLAIR- omalizumab injection, solution









-
NDC Code(s):
50242-040-62,
50242-040-86,
50242-214-01,
50242-214-03, view more50242-214-55, 50242-214-83, 50242-214-86, 50242-214-99, 50242-215-01, 50242-215-03, 50242-215-55, 50242-215-83, 50242-215-86, 50242-215-99, 50242-227-01, 50242-227-55, 50242-227-86, 50242-227-99
- Packager: Genentech, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated December 2, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use XOLAIR safely and effectively. See full prescribing information for XOLAIR.
XOLAIR® (omalizumab) injection, for subcutaneous use
XOLAIR® (omalizumab) for injection, for subcutaneous use
Initial U.S. Approval: 2003WARNING: ANAPHYLAXIS
See full prescribing information for complete boxed warning.
Anaphylaxis, presenting as bronchospasm, hypotension, syncope, urticaria, and/or angioedema of the throat or tongue, has been reported to occur after administration of XOLAIR. Anaphylaxis has occurred after the first dose of XOLAIR but also has occurred beyond 1 year after beginning treatment. Initiate XOLAIR therapy in a healthcare setting, closely observe patients for an appropriate period of time after XOLAIR administration and be prepared to manage anaphylaxis which can be life-threatening. Inform patients of the signs and symptoms of anaphylaxis and have them seek immediate medical care should symptoms occur. Selection of patients for self-administration of XOLAIR should be based on criteria to mitigate risk from anaphylaxis. (2.6, 5.1, 6.1, 6.2)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
XOLAIR is an anti-IgE antibody indicated for:
- Moderate to severe persistent asthma in adults and pediatric patients 6 years of age and older with a positive skin test or in vitro reactivity to a perennial aeroallergen and symptoms that are inadequately controlled with inhaled corticosteroids (1.1)
- Chronic rhinosinusitis with nasal polyps (CRSwNP) in adult patients 18 years of age and older with inadequate response to nasal corticosteroids, as add-on maintenance treatment (1.2)
- IgE-mediated food allergy in adult and pediatric patients aged 1 year and older for the reduction of allergic reactions (Type I), including anaphylaxis, that may occur with accidental exposure to one or more foods. To be used in conjunction with food allergen avoidance (1.3)
- Chronic spontaneous urticaria (CSU) in adults and adolescents 12 years of age and older who remain symptomatic despite H1 antihistamine treatment (1.4)
Limitations of Use:
DOSAGE AND ADMINISTRATION
For subcutaneous (SC) administration only. (2.2, 2.3, 2.4, 2.5)
See full prescribing information for administration instructions (2.6, 2.7, 2.8).
- Asthma: XOLAIR 75 to 375 mg SC every 2 or 4 weeks. Determine dose (mg) and dosing frequency by serum total IgE level (IU/mL), measured before the start of treatment, and body weight (kg). See the dose determination charts. (2.2)
- Chronic Rhinosinusitis with Nasal Polyps: XOLAIR 75 to 600 mg SC every 2 or 4 weeks. Determine dose (mg) and dosing frequency by serum total IgE level (IU/mL), measured before the start of treatment, and body weight (kg). See the dose determination charts. (2.3)
- IgE-Mediated Food Allergy: XOLAIR 75 mg to 600 mg SC every 2 or 4 weeks. Determine dose (mg) and dosing frequency by serum total IgE level (IU/mL), measured before the start of treatment, and body weight (kg). See the dose determination chart. (2.4)
- Chronic Spontaneous Urticaria: XOLAIR 150 or 300 mg SC every 4 weeks. Dosing in CSU is not dependent on serum IgE level or body weight. (2.5)
DOSAGE FORMS AND STRENGTHS
WARNINGS AND PRECAUTIONS
- Anaphylaxis: Initiate XOLAIR therapy in a healthcare setting prepared to manage anaphylaxis which can be life-threatening and observe patients for an appropriate period of time after administration. (5.1)
- Malignancy: Malignancies have been observed in clinical studies. (5.2)
- Acute Asthma Symptoms: Do not use for the treatment of acute bronchospasm or status asthmaticus. (5.3)
- Corticosteroid Reduction: Do not abruptly discontinue corticosteroids upon initiation of XOLAIR therapy. (5.4)
- Eosinophilic Conditions: Be alert to eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy, especially upon reduction of oral corticosteroids. (5.5)
- Fever, Arthralgia, and Rash: Stop XOLAIR if patients develop signs and symptoms similar to serum sickness. (5.6)
- Potential Medication Error Related to Emergency Treatment of Anaphylaxis: XOLAIR should not be used for emergency treatment of allergic reactions, including anaphylaxis. (5.9)
ADVERSE REACTIONS
- Asthma: The most common adverse reactions (≥ 1% of patients) in clinical studies with adult and adolescent patients ≥12 years of age were arthralgia, pain (general), leg pain, fatigue, dizziness, fracture, arm pain, pruritus, dermatitis, and earache. In clinical studies with pediatric patients 6 to <12 years of age, the most common adverse reactions (≥3% of patients) were nasopharyngitis, headache, pyrexia, upper abdominal pain, pharyngitis streptococcal, otitis media, viral gastroenteritis, arthropod bites, and epistaxis. (6.1)
- Chronic Rhinosinusitis with Nasal Polyps: The most common adverse reactions (≥ 3% of patients) in clinical studies with adult patients included the following: headache, injection site reaction, arthralgia, upper abdominal pain, and dizziness. (6.1)
- IgE-Mediated Food Allergy: The most common adverse reactions (≥3% of patients) were injection site reactions and pyrexia. (6.1)
- Chronic Spontaneous Urticaria: The most common adverse reactions (≥2% of patients) included the following: nausea, nasopharyngitis, sinusitis, upper respiratory tract infection, viral upper respiratory tract infection, arthralgia, headache, and cough. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Genentech at 1-888-835-2555 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
No formal drug interaction studies have been performed. (7)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 2/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: ANAPHYLAXIS
1 INDICATIONS AND USAGE
1.1 Asthma
1.2 Chronic Rhinosinusitis with Nasal Polyps
1.3 IgE-Mediated Food Allergy
1.4 Chronic Spontaneous Urticaria
2 DOSAGE AND ADMINISTRATION
2.1 Overview of Dosage Determination
2.2 Recommended Dosage for Asthma
2.3 Recommended Dosage for Chronic Rhinosinusitis with Nasal Polyps
2.4 Recommended Dosage for IgE-Mediated Food Allergy
2.5 Recommended Dosage for Chronic Spontaneous Urticaria
2.6 Administration Overview
2.7 XOLAIR Prefilled Syringe and Autoinjector
2.8 Preparation for Use and Injection of XOLAIR Lyophilized Powder
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis
5.2 Malignancy
5.3 Acute Asthma Symptoms and Deteriorating Disease
5.4 Corticosteroid Reduction
5.5 Eosinophilic Conditions
5.6 Fever, Arthralgia, and Rash
5.7 Parasitic (Helminth) Infection
5.8 Laboratory Tests
5.9 Potential Medication Error Related to Emergency Treatment of Anaphylaxis
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Asthma
14.2 Chronic Rhinosinusitis with Nasal Polyps
14.3 IgE-Mediated Food Allergy
14.4 Chronic Spontaneous Urticaria
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: ANAPHYLAXIS
Anaphylaxis presenting as bronchospasm, hypotension, syncope, urticaria, and/or angioedema of the throat or tongue, has been reported to occur after administration of XOLAIR. Anaphylaxis has occurred as early as after the first dose of XOLAIR, but also has occurred beyond 1 year after beginning regularly administered treatment. Because of the risk of anaphylaxis, initiate XOLAIR therapy in a healthcare setting and closely observe patients for an appropriate period of time after XOLAIR administration. Health care providers administering XOLAIR should be prepared to manage anaphylaxis which can be life-threatening. Inform patients of the signs and symptoms of anaphylaxis and instruct them to seek immediate medical care should symptoms occur. Selection of patients for self-administration of XOLAIR should be based on criteria to mitigate risk from anaphylaxis [see Dosage and Administration (2.6), Warnings and Precautions (5.1) and Adverse Reactions (6.1, 6.2)].
-
1 INDICATIONS AND USAGE
1.1 Asthma
XOLAIR is indicated for adults and pediatric patients 6 years of age and older with moderate to severe persistent asthma who have a positive skin test or in vitro reactivity to a perennial aeroallergen and whose symptoms are inadequately controlled with inhaled corticosteroids.
1.2 Chronic Rhinosinusitis with Nasal Polyps
XOLAIR is indicated for add-on maintenance treatment of chronic rhinosinusitis with nasal polyps (CRSwNP) in adult patients 18 years of age and older with inadequate response to nasal corticosteroids.
1.3 IgE-Mediated Food Allergy
XOLAIR is indicated for the reduction of allergic reactions (Type I), including anaphylaxis, that may occur with accidental exposure to one or more foods in adult and pediatric patients aged 1 year and older with IgE-mediated food allergy.
XOLAIR is to be used in conjunction with food allergen avoidance.
-
2 DOSAGE AND ADMINISTRATION
2.1 Overview of Dosage Determination
Asthma, and Chronic Rhinosinusitis with Nasal Polyps, and IgE-Mediated Food Allergy
- Determine dosage of XOLAIR by serum total IgE level (IU/mL) measured before the start of treatment, and by body weight (kg).
- For patients with asthma, chronic rhinosinusitis with nasal polyps (CRSwNP), and IgE-mediated food allergy, dosage determination should be based on the primary diagnosis for which XOLAIR is being prescribed.
- Adjust doses for significant changes in body weight during treatment.
- Refer to Tables 1 and 2 for the recommended dosage for treatment of asthma, Table 3 for treatment of CRSwNP, and Table 4 for treatment of IgE-mediated food allergy.
-
Total IgE levels are elevated during treatment and remain elevated for up to one year after the discontinuation of treatment. Therefore, re-testing of IgE levels during XOLAIR treatment cannot be used as a guide for dose determination.
- Interruptions lasting less than one year: Dose based on serum IgE levels obtained at the initial dose determination.
- Interruptions lasting one year or more: Re-test total serum IgE levels for dose determination (Table 1 or 2 for treatment of asthma, based on the patient's age, Table 3 for treatment of CRSwNP, and Table 4 for treatment of IgE-mediated food allergy).
Chronic Spontaneous Urticaria
Dosage of XOLAIR in patients with chronic spontaneous urticaria (CSU) is not dependent on serum IgE (free or total) level or body weight [see Dosage and Administration (2.5)].
2.2 Recommended Dosage for Asthma
The recommended dosage for asthma is XOLAIR 75 mg to 375 mg by subcutaneous injection every 2 or 4 weeks based on serum total IgE level (IU/mL) measured before the start of treatment and by body weight (kg) [see Dosage and Administration (2.1)].
- Adult and adolescent patients 12 years of age and older: Initiate dosing according to Table 1.
- Pediatric patients 6 to <12 years of age: Initiate dosing according to Table 2.
Table 1. Subcutaneous XOLAIR Doses Every 2 or 4 Weeks* for Patients 12 Years of Age and Older with Asthma 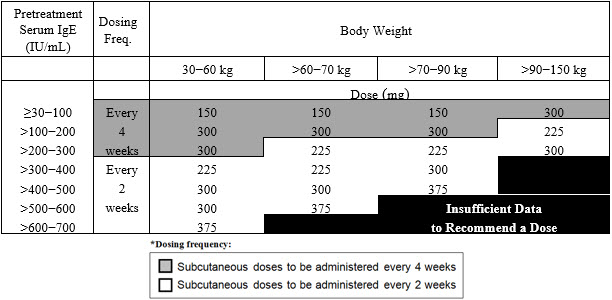
Table 2. Subcutaneous XOLAIR Doses Every 2 or 4 Weeks* for Pediatric Patients with Asthma Who Begin XOLAIR Between the Ages of 6 to <12 Years 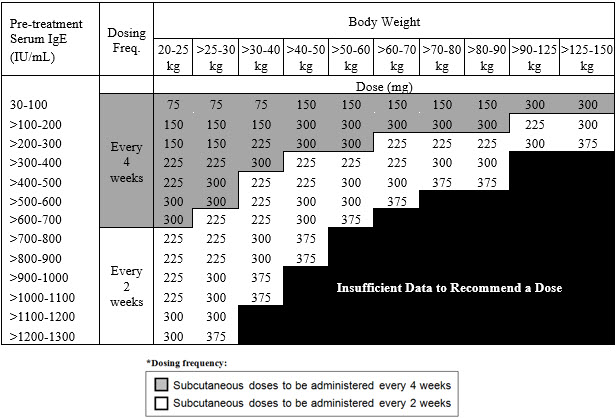
2.3 Recommended Dosage for Chronic Rhinosinusitis with Nasal Polyps
The recommended dosage for chronic rhinosinusitis with nasal polyps (CRSwNP) is XOLAIR 75 mg to 600 mg by subcutaneous injection every 2 or 4 weeks based on serum total IgE level (IU/mL) measure before the start of treatment and by body weight (kg) [see Dosage and Administration (2.1)]. Refer to Table 3 for recommended dosage based on serum total IgE level and body weight for patients with CRSwNP.
Table 3. Subcutaneous XOLAIR Doses Every 2 or 4 Weeks* for Adult Patients with CRSwNP 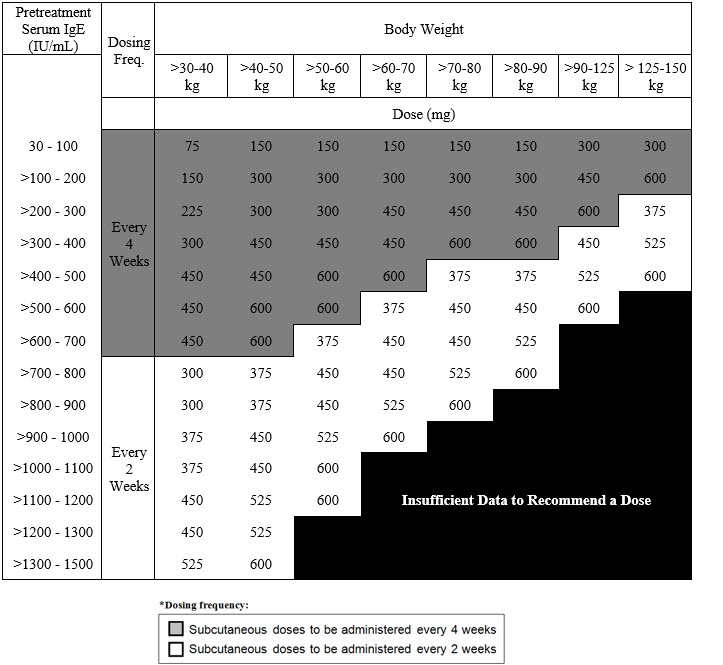
2.4 Recommended Dosage for IgE-Mediated Food Allergy
The recommended dosage for IgE-mediated food allergy is XOLAIR 75 mg to 600 mg by subcutaneous injection every 2 or 4 weeks based on serum total IgE level (IU/mL), measured before the start of treatment, and by body weight [see Dosage and Administration (2.1)]. Refer to Table 4 for recommended dosage based on serum IgE level and body weight for patients with IgE-mediated food allergy.
Table 4. Subcutaneous XOLAIR Doses Every 2 or 4 Weeks* for Adult and Pediatric Patients 1 Year of Age and Older with IgE-Mediated Food Allergy 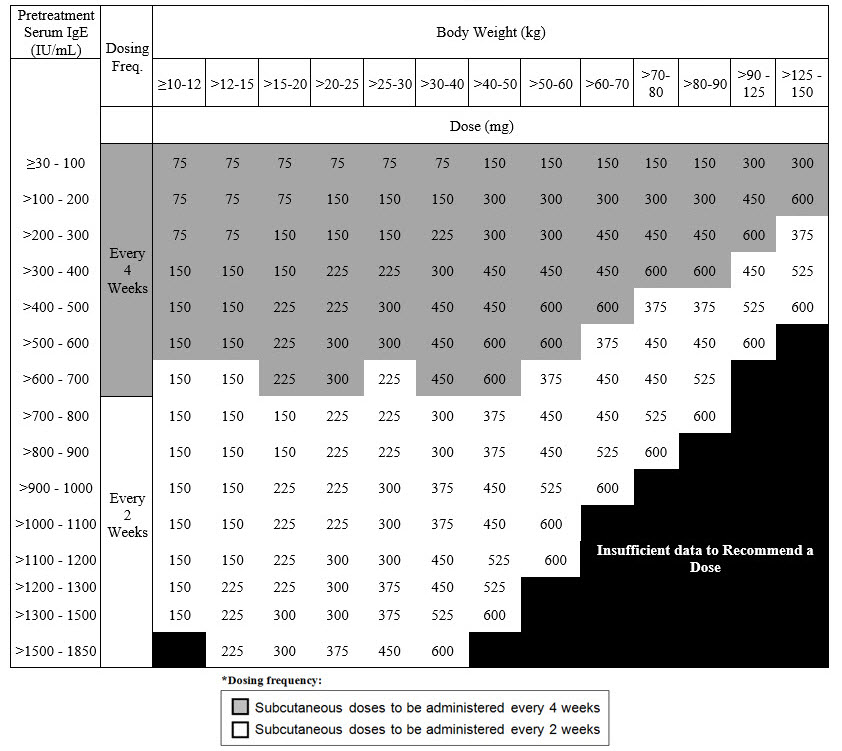
2.5 Recommended Dosage for Chronic Spontaneous Urticaria
The recommended dosage for chronic spontaneous urticaria (CSU) is XOLAIR 150 mg or 300 mg by subcutaneous injection every 4 weeks.
- The 300 mg dose may be administered as one subcutaneous injection of 300 mg/2 mL or as two subcutaneous injections of 150 mg/mL.
- Dosing of XOLAIR in CSU patients is not dependent on serum IgE (free or total) level or body weight.
2.6 Administration Overview
- Administer XOLAIR by subcutaneous injection.
- XOLAIR is intended for use under the guidance of a healthcare provider.
- Initiate therapy in a healthcare setting and once therapy has been safely established, the healthcare provider may determine whether self-administration of XOLAIR prefilled syringe or autoinjector by the patient or caregiver is appropriate, based on careful assessment of risk for anaphylaxis and mitigation strategies.
Selection of Patients for Self-Administration of XOLAIR Prefilled Syringe or Autoinjector
Healthcare providers should consider known risk factors for anaphylaxis to XOLAIR [see Warnings and Precautions (5.1)] and mitigation strategies when selecting patients for self-administration. Patient-specific factors including the following criteria should be considered:
- 1a)
- Asthma, CRSwNP and CSU: Patient should have no prior history of anaphylaxis to XOLAIR or other agents, such as foods, drugs, biologics, etc.
- 1b)
- IgE-Mediated Food Allergy: Patient should have no prior history of anaphylaxis to XOLAIR or other agents (except foods), such as drugs, biologics, etc.
- 2)
- Patient should receive at least 3 doses of XOLAIR under the guidance of a healthcare provider with no hypersensitivity reactions
- 3)
- Patient or caregiver is able to recognize symptoms of anaphylaxis
- 4)
- Patient or caregiver is able to treat anaphylaxis appropriately
- 5)
- Patient or caregiver is able to perform subcutaneous injections with XOLAIR prefilled syringe or autoinjector with proper technique according to the prescribed dosing regimen and Instructions for Use
2.7 XOLAIR Prefilled Syringe and Autoinjector
XOLAIR injection doses are available as a prefilled syringe or as an autoinjector. Instruct patients or caregivers to follow the directions provided in the "Instructions for Use" for preparation and administration of XOLAIR prefilled syringe or autoinjector [see Instructions for Use].
XOLAIR Prefilled Syringe
- Adolescents 12 years of age and older: XOLAIR prefilled syringe may be self-administered under adult supervision.
- Pediatric Patients 1 to 11 years of age: XOLAIR prefilled syringe should be administered by a caregiver.
XOLAIR Autoinjector
- Adolescents 12 years of age and older: XOLAIR autoinjector may be self-administered under adult supervision. The XOLAIR autoinjectors (all doses) are intended for use only in adults and adolescents aged 12 years and older.
- Pediatric Patients 1 to 11 years of age: The XOLAIR autoinjectors (all doses) are not intended for use in pediatric patients under 12 years of age.
Administration Instructions for Prefilled Syringe and Autoinjector
- Persons with latex allergies should not handle XOLAIR prefilled syringe because the needle cap of the XOLAIR 75 mg/0.5 mL and 150 mg/mL prefilled syringes contains a derivative of natural rubber latex which may cause allergic reactions in latex sensitive individuals [see How Supplied/Storage and Handling (16)].
- Visually inspect the contents of the prefilled syringe or autoinjector for particulate matter and discoloration prior to administration. XOLAIR prefilled syringe or autoinjector solution should be clear and colorless to pale brownish yellow. Do not use the prefilled syringe or autoinjector if the medicine is cloudy, discolored, or contains particles.
- Determine the number of prefilled syringes or autoinjectors needed for patient's dosage (see Table 5). For pediatric patients 1 to 11 years of age, consideration should be given to the number of prefilled syringe injections needed and volume to be injected relative to the patient's bodyweight.
- For patients requiring more than 1 injection to complete a full dose, administer each injection at least 1 inch apart from other injection sites.
- Administer subcutaneous injection into the thigh or abdomen, avoiding the 2-inch (5 cm) area directly around the navel. The outer area of the upper arms may be used only if the injection is being given by a caregiver or healthcare provider [see Instructions for Use]. The injection may take up to 15 seconds to administer.
Table 5. Number of XOLAIR Prefilled Syringes or Autoinjectors*, Injections and Total Injection Volumes† XOLAIR Dose‡ 75 mg 150 mg 300mg‡ Total Volume Injected - *
- The autoinjector (all doses) are not intended for use in patients under 12 years of age.
- †
- This table represents the fewest number of injections for the patient, however, there are other syringe/autoinjector dosing combinations to achieve desired dose.
- ‡
- The 75 mg, 150 mg, 225 mg, 300 mg, and 375 mg XOLAIR doses are approved for use in asthma patients. All doses in the table are approved for use in CRSwNP and IgE-mediated food allergy patients. The 150 mg and 300 mg XOLAIR doses are also approved for use in CSU patients.
75 mg 1 0 0 0.5 mL 150 mg 0 1 0 1 mL 225 mg 1 1 0 1.5 mL 300 mg 0 0 1 2 mL 375 mg 1 0 1 2.5 mL 450 mg 0 1 1 3 mL 525 mg 1 1 1 3.5 mL 600 mg 0 0 2 4 mL 2.8 Preparation for Use and Injection of XOLAIR Lyophilized Powder
XOLAIR lyophilized powder should only be prepared and injected by a healthcare provider. The supplied XOLAIR lyophilized powder must be reconstituted with Sterile Water for Injection (SWFI) USP, using the following instructions:
- 1)
- Before reconstitution, determine the number of vials that will need to be reconstituted (each vial delivers 150 mg of XOLAIR in 1.2 mL) (see Table 6).
Table 6. Number of Vials, Injections and Total Injection Volumes XOLAIR Dose* Number of Vials Number of Injections Total Volume Injected - *
- The 75 mg, 150 mg, 225 mg, 300 mg, and 375 mg XOLAIR doses are approved for use in asthma patients. All doses in the table are approved for use in CRSwNP and IgE-mediated food allergy patients. The 150 mg and 300 mg XOLAIR doses are also approved for use in CSU patients.
75 mg 1 1 0.6 mL 150 mg 1 1 1.2 mL 225 mg 2 2 1.8 mL 300 mg 2 2 2.4 mL 375 mg 3 3 3.0 mL 450 mg 3 3 3.6 mL 525 mg 4 4 4.2 mL 600 mg 4 4 4.8 mL - 2)
- Draw 1.4 mL of SWFI, USP, into a 3 mL syringe equipped with a 1-inch, 18-gauge needle.
- 3)
- Place the vial upright on a flat surface and using standard aseptic technique, insert the needle and inject the SWFI, USP, directly onto the product.
- 4)
- Keeping the vial upright, gently swirl the upright vial for approximately 1 minute to evenly wet the powder. Do not shake.
- 5)
- Gently swirl the vial for 5 to 10 seconds approximately every 5 minutes in order to dissolve any remaining solids. The lyophilized product takes 15 to 20 minutes to dissolve. If it takes longer than 20 minutes to dissolve completely, gently swirl the vial for 5 to 10 seconds approximately every 5 minutes until there are no visible gel-like particles in the solution. Do not use if the contents of the vial do not dissolve completely by 40 minutes.
- 6)
- After reconstitution, XOLAIR solution is somewhat viscous and will appear clear or slightly opalescent. It is acceptable if there are a few small bubbles or foam around the edge of the vial; there should be no visible gel-like particles in the reconstituted solution. Do not use if foreign particles are present.
- 7)
- Invert the vial for 15 seconds in order to allow the solution to drain toward the stopper.
- 8)
- Use the XOLAIR solution within 8 hours following reconstitution when stored in the vial at 2ºC to 8ºC (36ºF to 46ºF), or within 4 hours of reconstitution when stored at room temperature. Reconstituted XOLAIR vials should be protected from sunlight.
- 9)
- Using a new 3 mL syringe equipped with a 1-inch, 18-gauge needle, insert the needle into the inverted vial. Position the needle tip at the very bottom of the solution in the vial stopper when drawing the solution into the syringe. The reconstituted product is somewhat viscous. Withdraw all of the product from the vial before expelling any air or excess solution from the syringe. Before removing the needle from the vial, pull the plunger all the way back to the end of the syringe barrel in order to remove all of the solution from the inverted vial.
- 10)
- Replace the 18-gauge needle with a 25-gauge needle for subcutaneous injection.
- 11)
- Expel air, large bubbles, and any excess solution in order to obtain a volume of 1.2 mL corresponding to a dose of 150 mg of XOLAIR. To obtain a volume of 0.6 mL corresponding to a dose of 75 mg of XOLAIR, expel air, large bubbles and discard 0.6 mL from the syringe. A thin layer of small bubbles may remain at the top of the solution in the syringe.
- 12)
- Administer XOLAIR by subcutaneous injection. The injection may take 5-10 seconds to administer because the solution is slightly viscous. Do not administer more than 150 mg (contents of one vial) per injection site. Divide doses of more than 150 mg between two or more injection sites. Choose a different injection site for each new injection at least 1 inch from the area used for other injections.
-
3 DOSAGE FORMS AND STRENGTHS
Injection:
- 75 mg/0.5 mL is a clear to slightly opalescent and colorless to pale brownish-yellow solution in a single-dose prefilled syringe with needle shield or single-dose prefilled autoinjector
- 150 mg/mL is a clear to slightly opalescent and colorless to pale brownish-yellow solution in a single-dose prefilled syringe with needle shield or single-dose prefilled autoinjector
- 300 mg/2 mL is a clear to slightly opalescent and colorless to pale brownish-yellow solution in a single-dose prefilled syringe with needle shield or single-dose prefilled autoinjector
- For injection: 150 mg white lyophilized powder in a single-dose vial for reconstitution
-
4 CONTRAINDICATIONS
XOLAIR is contraindicated in patients with severe hypersensitivity reaction to XOLAIR or any ingredient of XOLAIR [see Warnings and Precautions (5.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Anaphylaxis
Anaphylaxis has been reported to occur after administration of XOLAIR in premarketing clinical trials and in postmarketing spontaneous reports [see Boxed Warning and Adverse Reactions (6.2)]. Signs and symptoms in these reported cases have included bronchospasm, hypotension, syncope, urticaria, and/or angioedema of the throat or tongue. Some of these events have been life-threatening. In premarketing clinical trials in patients with asthma, anaphylaxis was reported in 3 of 3507 (0.1%) patients. Anaphylaxis occurred with the first dose of XOLAIR in two patients and with the fourth dose in one patient. The time to onset of anaphylaxis was 90 minutes after administration in two patients and 2 hours after administration in one patient.
A case-control study in asthma patients showed that, among XOLAIR users, patients with a history of anaphylaxis to foods, medications, or other causes were at increased risk of anaphylaxis associated with XOLAIR, compared to those with no prior history of anaphylaxis [see Adverse Reactions (6.1)].
In postmarketing spontaneous reports, the frequency of anaphylaxis attributed to XOLAIR use was estimated to be at least 0.2% of patients based on an estimated exposure of about 57,300 patients from June 2003 through December 2006. Anaphylaxis has occurred as early as after the first dose of XOLAIR, but also has occurred beyond one year after beginning regularly scheduled treatment. Approximately 60% to 70% of anaphylaxis cases have been reported to occur within the first three doses of XOLAIR, with additional cases occurring sporadically beyond the third dose.
Initiate XOLAIR only in a healthcare setting equipped to manage anaphylaxis, which can be life-threatening. Observe patients closely for an appropriate period of time after administration of XOLAIR, taking into account the time to onset of anaphylaxis seen in premarketing clinical trials and postmarketing spontaneous reports [see Adverse Reactions (6.1, 6.2)]. Inform patients of the signs and symptoms of anaphylaxis, and instruct them to seek immediate medical care should signs or symptoms occur.
Once XOLAIR therapy has been established, administration of XOLAIR prefilled syringe or autoinjector outside of a healthcare setting by a patient or a caregiver may be appropriate for selected patients. Patient selection, determined by the healthcare provider in consultation with the patient, should take into account the pattern of anaphylaxis events seen in premarketing clinical trials and postmarketing spontaneous reports, as well as individual patient risk factors (e.g., prior history of anaphylaxis), ability to recognize signs and symptoms of anaphylaxis, and ability to perform subcutaneous injections with XOLAIR prefilled syringe or autoinjector with proper technique according to the prescribed dosing regimen and Instructions for Use [see Dosage and Administration (2.6), Adverse Reactions (6.1, 6.2)].
Discontinue XOLAIR in patients who experience a severe hypersensitivity reaction [see Contraindications (4)].
5.2 Malignancy
Malignant neoplasms were observed in 20 of 4127 (0.5%) XOLAIR-treated patients compared with 5 of 2236 (0.2%) control patients in clinical studies of adults and adolescents ≥12 years of age with asthma and other allergic disorders. The observed malignancies in XOLAIR-treated patients were a variety of types, with breast, non-melanoma skin, prostate, melanoma, and parotid occurring more than once, and five other types occurring once each. The majority of patients were observed for less than 1 year. The impact of longer exposure to XOLAIR or use in patients at higher risk for malignancy (e.g., elderly, current smokers) is not known.
In a subsequent observational study of 5007 XOLAIR-treated and 2829 non-XOLAIR-treated adolescent and adult patients with moderate to severe persistent asthma and a positive skin test reaction or in vitro reactivity to a perennial aeroallergen, patients were followed for up to 5 years. In this study, the incidence rates of primary malignancies (per 1000 patient years) were similar among XOLAIR-treated (12.3) and non-XOLAIR-treated patients (13.0) [see Adverse Reactions (6.1)]. However, study limitations preclude definitively ruling out a malignancy risk with XOLAIR. Study limitations include: the observational study design, the bias introduced by allowing enrollment of patients previously exposed to XOLAIR (88%), enrollment of patients (56%) while a history of cancer or a premalignant condition were study exclusion criteria, and the high study discontinuation rate (44%).
5.3 Acute Asthma Symptoms and Deteriorating Disease
XOLAIR has not been shown to alleviate asthma exacerbations acutely. Do not use XOLAIR to treat acute bronchospasm or status asthmaticus. Patients should seek medical advice if their asthma remains uncontrolled or worsens after initiation of treatment with XOLAIR.
5.4 Corticosteroid Reduction
Do not discontinue systemic or inhaled corticosteroids abruptly upon initiation of XOLAIR therapy for asthma or CRSwNP. Decrease corticosteroids gradually under the direct supervision of a physician. In CSU patients, the use of XOLAIR in combination with corticosteroids has not been evaluated.
5.5 Eosinophilic Conditions
In rare cases, patients with asthma on therapy with XOLAIR may present with serious systemic eosinophilia sometimes presenting with clinical features of vasculitis consistent with Churg-Strauss syndrome, a condition which is often treated with systemic corticosteroid therapy. These events usually, but not always, have been associated with the reduction of oral corticosteroid therapy. Physicians should be alert to eosinophilia, vasculitic rash, worsening pulmonary symptoms, cardiac complications, and/or neuropathy presenting in their patients. A causal association between XOLAIR and these underlying conditions has not been established.
5.6 Fever, Arthralgia, and Rash
In post-approval use, some patients have experienced a constellation of signs and symptoms including arthritis/arthralgia, rash, fever, and lymphadenopathy with an onset 1 to 5 days after the first or subsequent injections of XOLAIR. These signs and symptoms have recurred after additional doses in some patients. Although circulating immune complexes or a skin biopsy consistent with a Type III reaction were not seen with these cases, these signs and symptoms are similar to those seen in patients with serum sickness. Physicians should stop XOLAIR if a patient develops this constellation of signs and symptoms [see Adverse Reactions (6.2)].
5.7 Parasitic (Helminth) Infection
Monitor patients at high risk of geohelminth infection while on XOLAIR therapy. Insufficient data are available to determine the length of monitoring required for geohelminth infections after stopping XOLAIR treatment.
In a one-year clinical trial conducted in Brazil in adult and adolescent patients at high risk for geohelminthic infections (roundworm, hookworm, whipworm, threadworm), 53% (36/68) of XOLAIR-treated patients experienced an infection, as diagnosed by standard stool examination, compared to 42% (29/69) of placebo controls. The point estimate of the odds ratio for infection was 1.96, with a 95% confidence interval (0.88, 4.36) indicating that in this study a patient who had an infection was anywhere from 0.88 to 4.36 times as likely to have received XOLAIR than a patient who did not have an infection. Response to appropriate anti-geohelminth treatment of infection as measured by stool egg counts was not different between treatment groups.
5.8 Laboratory Tests
Serum total IgE levels increase following administration of XOLAIR due to formation of XOLAIR:IgE complexes [see Clinical Pharmacology (12.2)]. Elevated serum total IgE levels may persist for up to 1 year following discontinuation of XOLAIR. Do not use serum total IgE levels obtained less than 1 year following discontinuation to reassess the dosing regimen for asthma, CRSwNP or IgE-mediated food allergy patients, because these levels may not reflect steady-state free IgE levels [see Dosage and Administration (2.2, 2.3, 2.4)].
5.9 Potential Medication Error Related to Emergency Treatment of Anaphylaxis
XOLAIR should not be used for the emergency treatment of allergic reactions, including anaphylaxis. In studies to simulate use, some patients and caregivers did not understand that XOLAIR is not intended for the emergency treatment of allergic reactions, including anaphylaxis. The safety and effectiveness of XOLAIR for emergency treatment of allergic reactions, including anaphylaxis, have not been established. Instruct patients that XOLAIR is for maintenance use to reduce allergic reactions, including anaphylaxis, while avoiding food allergens.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Anaphylaxis [see Boxed Warning and Warnings and Precautions (5.1)]
- Malignancies [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adverse Reactions from Clinical Studies in Adult and Adolescent Patients 12 Years of Age and Older with Asthma
The data described below reflect XOLAIR exposure for 2076 adult and adolescent patients ages 12 and older, including 1687 patients exposed for six months and 555 exposed for one year or more, in either placebo-controlled or other controlled asthma studies. The mean age of patients receiving XOLAIR was 42 years, with 134 patients 65 years of age or older; 60% were women, and 85% Caucasian. Patients received XOLAIR 150 mg to 375 mg every 2 or 4 weeks or, for patients assigned to control groups, standard therapy with or without a placebo.
The adverse reactions most frequently resulting in clinical intervention (e.g., discontinuation of XOLAIR, or the need for concomitant medication to treat an adverse reaction) were injection site reaction (45%), viral infections (23%), upper respiratory tract infection (20%), sinusitis (16%), headache (15%), and pharyngitis (11%). These reactions were observed at similar rates in XOLAIR-treated patients and control patients.
Table 7 shows adverse reactions from four placebo-controlled asthma trials that occurred ≥1% and more frequently in adult and adolescent patients 12 years of age and older receiving XOLAIR than in those receiving placebo. Adverse reactions were classified using preferred terms from the International Medical Nomenclature (IMN) dictionary. Injection site reactions were recorded separately from the reporting of other adverse reactions.
Table 7. Adverse Reactions ≥1% More Frequent in XOLAIR-Treated Adult or Adolescent Patients 12 years of Age and Older in Four Placebo-controlled Asthma Trials Adverse reaction XOLAIR
n=738Placebo
n=717Body as a whole Pain 7% 5% Fatigue 3% 2% Musculoskeletal system Arthralgia 8% 6% Fracture 2% 1% Leg pain 4% 2% Arm pain 2% 1% Nervous system Dizziness 3% 2% Skin and appendages Pruritus 2% 1% Dermatitis 2% 1% Special senses Earache 2% 1% There were no differences in the incidence of adverse reactions based on age (among patients under 65), gender or race.
Anaphylaxis Case Control Study
A retrospective case-control study investigated risk factors for anaphylaxis to XOLAIR among patients treated with XOLAIR for asthma. Cases with an adjudicated history of anaphylaxis to XOLAIR were compared to controls with no such history. The study found that a self-reported history of anaphylaxis to foods, medications or other causes was more common among patients with XOLAIR anaphylaxis (57% of 30 cases) compared to controls (23% of 88 controls) [OR 8.1, 95% CI 2.7 to 24.3]. Because this is a case-control study, the study cannot provide the incidence of anaphylaxis among XOLAIR users. From other sources, anaphylaxis to XOLAIR was observed in 0.1% of patients in clinical trials and at least 0.2% of patients based upon postmarketing reports. Approximately 60% to 70% of cases were reported to occur within the first three doses of XOLAIR, with additional cases occurring sporadically beyond the third dose. The time to onset for anaphylaxis was reported to occur within 2 hours for the majority of cases (approximately 75%) [see Warnings and Precautions (5.1), Adverse Reactions (6.2)].
Injection Site Reactions
In adults and adolescents, injection site reactions of any severity occurred at a rate of 45% in XOLAIR-treated patients compared with 43% in placebo-treated patients. The types of injection site reactions included: bruising, redness, warmth, burning, stinging, itching, hive formation, pain, indurations, mass, and inflammation.
Severe injection site reactions occurred more frequently in XOLAIR-treated patients compared with patients in the placebo group (12% versus 9%).
The majority of injection site reactions occurred within 1 hour post injection, lasted less than 8 days, and generally decreased in frequency at subsequent dosing visits.
Adverse Reactions from Clinical Studies in Pediatric Patients 6 to <12 Years of Age with Asthma
The data described below reflect XOLAIR exposure for 926 patients 6 to <12 years of age, including 583 patients exposed for six months and 292 exposed for one year or more, in either placebo-controlled or other controlled asthma studies. The mean age of pediatric patients receiving XOLAIR was 8.8 years; 69% were male, and 64% were Caucasian. Pediatric patients received XOLAIR 75 mg to 375 mg every 2 or 4 weeks or, for patients assigned to control groups, standard therapy with or without a placebo. No cases of malignancy were reported in patients treated with XOLAIR in these trials.
The most common adverse reactions occurring at ≥3% in the pediatric patients receiving XOLAIR and more frequently than in patients treated with placebo were nasopharyngitis, headache, pyrexia, upper abdominal pain, pharyngitis streptococcal, otitis media, viral gastroenteritis, arthropod bite, and epistaxis.
The adverse reactions most frequently resulting in clinical intervention (e.g., discontinuation of XOLAIR, or the need for concomitant medication to treat an adverse event) were bronchitis (0.2%), headache (0.2%) and urticaria (0.2%). These reactions were observed at similar rates in XOLAIR-treated patients and control patients.
Adverse Reactions from Clinical Studies in Adult Patients with Chronic Rhinosinusitis with Nasal Polyps
The data described below reflect XOLAIR exposure for 135 patients ≥ 18 years of age, exposed for six months in two placebo-controlled studies. The mean age of patients receiving XOLAIR was 49.7 years; 64% were male, and 94% were Caucasian. Patients received XOLAIR or placebo SC every 2 or 4 weeks, with dosage and frequency according to Table 3. All patients received background nasal mometasone therapy throughout the study. Table 8 lists the adverse reactions occurring in ≥3% of XOLAIR-treated patients and more frequently than in patients treated with placebo in chronic rhinosinusitis with nasal polyps (CRSwNP) Trials 1 and 2; results were pooled.
Table 8. Adverse Reactions Occurring in ≥3% of XOLAIR-Treated Patients and More Frequently than in Patients Treated with Placebo in CRSwNP Trials 1 and 2 Adverse reaction XOLAIR
n=135Placebo
n=130CRSwNP = Chronic Rhinosinusitis with Nasal Polyps. - *
- Injection site reactions terms: 'injection site reaction', 'injection related reaction' and 'injection site pain'. All injection site reactions were mild to moderate severity and none resulted in study discontinuation.
Gastrointestinal disorder Upper abdominal pain 4 (3.0%) 1 (0.8%) General disorders and administration site conditions Injection site reactions* 7 (5.2%) 2 (1.5%) Musculoskeletal system and connective tissue disorders Arthralgia 4 (3.0%) 2 (1.5%) Nervous system disorders Headache 11 (8.1%) 7 (5.4%) Dizziness 4 (3.0%) 1 (0.8%) Adverse Reactions from a Clinical Study in Patients with IgE-Mediated Food Allergy
The safety of XOLAIR in patients with IgE-mediated allergic reactions (Type I), including anaphylaxis, that may occur with accidental exposure to one or more foods, was based on data from the Food Allergy (FA) Trial, a randomized, double-blind, placebo-controlled trial in 168 patients (165 pediatric patients and 3 adults) who were allergic to peanut and at least two other foods [see Clinical Studies (14.3)]. Patients received a dosage of XOLAIR or placebo subcutaneously every 2 or 4 weeks for 16 to 20 weeks according to the recommended dosage based on IgE level (IU/mL), measured before the start of treatment, and by body weight (kg) provided in Table 4 [see Dosage and Administration (2.4)]. Safety data provided in Table 9 are from the primary analysis population of pediatric patients aged 1 year to 17 years. Safety data obtained from adults (n=3) in this trial was limited. Table 9 lists the adverse reactions occurring in ≥3% of XOLAIR-treated pediatric patients and more frequently than in patients treated with placebo in the FA trial. There were no discontinuations due to adverse reactions.
Table 9. Adverse Reactions Occurring in ≥3% of XOLAIR-Treated Pediatric Patients 1 Year of Age and Older and More Frequently than in Patients Treated with Placebo in FA Trial Adverse Reaction XOLAIR
n=110Placebo
n=55- *
- Injection site reactions terms: 'injection site reaction','injection site urticaria','injection site discomfort','injection site erythema','injection site pain' and 'injection site rash'. All injection site reactions were mild to moderate severity and none resulted in study discontinuation.
General disorders and administration site conditions Injection site reactions* 17 (15.5%) 6 (10.9%) Pyrexia 7 (6.4%) 2 (3.6%) Adverse Reactions from Clinical Studies in Patients with Chronic Spontaneous Urticaria
The safety of XOLAIR for the treatment of chronic spontaneous urticaria (CSU) was assessed in three placebo-controlled, multiple-dose clinical trials of 12 weeks' (CSU Trial 2) and 24 weeks' duration (CSU Trials 1 and 3). In CSU Trials 1 and 2, patients received XOLAIR 75 mg, 150 mg, or 300 mg or placebo every 4 weeks in addition to their baseline level of H1 antihistamine therapy throughout the treatment period. In CSU Trial 3 patients were randomized to XOLAIR 300 mg or placebo every 4 weeks in addition to their baseline level of H1 antihistamine therapy. The data described below reflect XOLAIR exposure for 733 patients enrolled and receiving at least one dose of XOLAIR in the three clinical trials, including 684 patients exposed for 12 weeks and 427 exposed for 24 weeks. The mean age of patients receiving XOLAIR 300 mg was 43 years, 75% were women, and 89% were white. The demographic profiles for patients receiving XOLAIR 150 mg and 75 mg were similar.
Table 10 shows adverse reactions that occurred in ≥2% of patients receiving XOLAIR (150 or 300 mg) and more frequently than those receiving placebo. Adverse reactions are pooled from CSU Trial 2 and the first 12 weeks of CSU Trials 1 and 3.
Table 10. Adverse Reactions Occurring in ≥2% in XOLAIR-Treated Patients and More Frequently than in Patients Treated with Placebo (Day 1 to Week 12) in CSU Trials Adverse Reactions* CSU Trials 1, 2 and 3 Pooled 150mg
(n=175)300mg
(n=412)Placebo
(n=242)- *
- by MedDRA (15.1) System Organ Class and Preferred Term
Gastrointestinal disorders Nausea 2 (1.1%) 11 (2.7%) 6 (2.5%) Infections and infestations Nasopharyngitis 16 (9.1%) 27 (6.6%) 17 (7.0%) Sinusitis 2 (1.1%) 20 (4.9%) 5 (2.1%) Upper respiratory tract infection 2 (1.1%) 14 (3.4%) 5 (2.1%) Viral upper respiratory tract infection 4 (2.3%) 2 (0.5%) (0.0%) Musculoskeletal and connective tissue disorders Arthralgia 5 (2.9%) 12 (2.9%) 1 (0.4%) Nervous system disorders Headache 21 (12.0%) 25 (6.1%) 7 (2.9%) Respiratory, thoracic, and mediastinal disorders Cough 2 (1.1%) 9 (2.2%) 3 (1.2%) Additional reactions reported during the 24-week treatment period in CSU Trials 1 and 3 [≥2% of patients receiving XOLAIR (150 mg or 300 mg) and more frequently than those receiving placebo] included: toothache, fungal infection, urinary tract infection, myalgia, pain in extremity, musculoskeletal pain, peripheral edema, pyrexia, migraine, sinus headache, anxiety, oropharyngeal pain, asthma, urticaria, and alopecia.
Injection Site Reactions in Patients with CSU
Injection site reactions of any severity occurred during the studies in more XOLAIR-treated patients [11 patients (2.7%) at 300 mg, 1 patient (0.6%) at 150 mg] compared with 2 placebo-treated patients (0.8%). The types of injection site reactions included: swelling, erythema, pain, bruising, itching, bleeding, and urticaria. None of the events resulted in study discontinuation or treatment interruption.
Cardiovascular and Cerebrovascular Events from Clinical Studies in Patients with Asthma
A 5-year observational cohort study was conducted in patients ≥12 years of age with moderate to severe persistent asthma and a positive skin test reaction to a perennial aeroallergen to evaluate the long-term safety of XOLAIR, including the risk of malignancy [see Warnings and Precautions (5.2)]. A total of 5007 XOLAIR-treated and 2829 non–XOLAIR-treated patients enrolled in the study. Similar percentages of patients in both cohorts were current (5%) or former smokers (29%). Patients had a mean age of 45 years and were followed for a mean of 3.7 years. More XOLAIR-treated patients were diagnosed with severe asthma (50%) compared to the non–XOLAIR-treated patients (23%) and 44% of patients prematurely discontinued the study. Additionally, 88% of patients in the XOLAIR-treated cohort had been previously exposed to XOLAIR for a mean of 8 months.
A higher incidence rate (per 1000 patient-years) of overall cardiovascular and cerebrovascular serious adverse events (SAEs) was observed in XOLAIR-treated patients (13.4) compared to non–XOLAIR-treated patients (8.1). Increases in rates were observed for transient ischemic attack (0.7 versus 0.1), myocardial infarction (2.1 versus 0.8), pulmonary hypertension (0.5 versus 0), pulmonary embolism/venous thrombosis (3.2 versus 1.5), and unstable angina (2.2 versus 1.4), while the rates observed for ischemic stroke and cardiovascular death were similar among both study cohorts. The results suggest a potential increased risk of serious cardiovascular and cerebrovascular events in patients treated with XOLAIR. However, the observational study design, the inclusion of patients previously exposed to XOLAIR (88%), baseline imbalances in cardiovascular risk factors between the treatment groups, an inability to adjust for unmeasured risk factors, and the high study discontinuation rate limit the ability to quantify the magnitude of the risk.
A pooled analysis of 25 randomized double-blind, placebo-controlled clinical trials of 8 to 52 weeks in duration was conducted to further evaluate the imbalance in cardiovascular and cerebrovascular SAEs noted in the above observational cohort study. A total of 3342 XOLAIR-treated patients and 2895 placebo-treated patients were included in the pooled analysis. The patients had a mean age of 38 years, and were followed for a mean duration of 6.8 months. No notable imbalances were observed in the rates of cardiovascular and cerebrovascular SAEs listed above. However, the results of the pooled analysis were based on a low number of events, slightly younger patients, and shorter duration of follow-up than the observational cohort study; therefore, the results are insufficient to confirm or reject the findings noted in the observational cohort study.
Adverse Reactions from Clinical Study in Healthy Adults
Injection Site Reactions in Healthy Adults
In an open label trial in healthy adults, in which the 300 mg/2 mL autoinjector was compared to the 300 mg/2 mL prefilled syringe, injection site reactions (e.g., induration, pain, erythema, hemorrhage, swelling, discomfort, bruising, hypoesthesia, edema, pruritus) were observed in 24% (16/66) of subjects treated with the autoinjector compared with 14% (9/64) of subjects treated with the prefilled syringe.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of XOLAIR. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Anaphylaxis: Based on spontaneous reports and an estimated exposure of about 57,300 patients from June 2003 through December 2006, the frequency of anaphylaxis attributed to XOLAIR use was estimated to be at least 0.2% of patients.
Diagnostic criteria of anaphylaxis were skin or mucosal tissue involvement, and, either airway compromise, and/or reduced blood pressure with or without associated symptoms, and a temporal relationship to XOLAIR administration with no other identifiable cause. Signs and symptoms in these reported cases included bronchospasm, hypotension, syncope, urticaria, angioedema of the throat or tongue, dyspnea, cough, chest tightness, and/or cutaneous angioedema. Pulmonary involvement was reported in 89% of the cases. Hypotension or syncope was reported in 14% of cases. Fifteen percent of the reported cases resulted in hospitalization. A previous history of anaphylaxis unrelated to XOLAIR was reported in 24% of the cases.
Of the reported cases of anaphylaxis attributed to XOLAIR, 39% occurred with the first dose, 19% occurred with the second dose, 10% occurred with the third dose, and the rest after subsequent doses. One case occurred after 39 doses (after 19 months of continuous therapy, anaphylaxis occurred when treatment was restarted following a 3-month gap). The time to onset of anaphylaxis in these cases was up to 30 minutes in 35%, greater than 30 and up to 60 minutes in 16%, greater than 60 and up to 90 minutes in 2%, greater than 90 and up to 120 minutes in 6%, greater than 2 hours and up to 6 hours in 5%, greater than 6 hours and up to 12 hours in 14%, greater than 12 hours and up to 24 hours in 8%, and greater than 24 hours and up to 4 days in 5%. In 9% of cases the times to onset were unknown.
Twenty-three patients who experienced anaphylaxis were rechallenged with XOLAIR and 18 patients had a recurrence of similar symptoms of anaphylaxis. In addition, anaphylaxis occurred upon rechallenge with XOLAIR in 4 patients who previously experienced urticaria only.
Eosinophilic Conditions: Eosinophilic conditions have been reported [see Warnings and Precautions (5.5)].
Fever, Arthralgia, and Rash: A constellation of signs and symptoms including arthritis/arthralgia, rash (urticaria or other forms), fever and lymphadenopathy similar to serum sickness have been reported in post-approval use of XOLAIR [see Warnings and Precautions (5.6)].
-
7 DRUG INTERACTIONS
No formal drug interaction studies have been performed with XOLAIR.
In patients with asthma, CRSwNP, and IgE-mediated food allergy the concomitant use of XOLAIR and allergen immunotherapy has not been evaluated.
In patients with CSU, the use of XOLAIR in combination with immunosuppressive therapies has not been studied.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
A registry study of XOLAIR exposure during pregnancy showed no increase in the rate of major birth defects or miscarriage. There was an increased rate of low birth weight among registry infants compared to infants in the other cohorts, despite average gestational age at birth; however, women taking XOLAIR during pregnancy also had more severe asthma, which makes it difficult to determine whether the low birth weight is due to the drug or the disease severity [see Data]. There are risks associated with poorly or moderately controlled asthma in pregnancy [see Clinical Considerations].
Human IgG antibodies are known to cross the placental barrier; therefore, XOLAIR may be transmitted from the mother to the developing fetus.
In animal reproduction studies, no evidence of fetal harm was observed in Cynomolgus monkeys with subcutaneous doses of omalizumab up to approximately 5 times the maximum recommended human dose (MRHD) [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population(s) is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the US general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
In women with poorly or moderately controlled asthma, evidence demonstrates that there is an increased risk of preeclampsia in the mother and prematurity, low birth weight, and small for gestational age in the neonate. The level of asthma control should be closely monitored in pregnant women and treatment adjusted as necessary to maintain optimal control.
Data
Human Data
A prospective cohort pregnancy exposure registry study conducted in the US from 2006 to 2018, included 250 pregnant women with asthma treated with XOLAIR. Of these, 246 patients were exposed to XOLAIR in the first trimester of pregnancy, and the median exposure duration was 8.7 months.
The registry findings for applicable mother and infant subgroups were compared to age-adjusted frequencies in a disease-matched external cohort of 1,153 pregnant women with asthma (without exposure to XOLAIR) identified from healthcare databases of residents in the Canadian province of Quebec, and referred to as the Quebec External Comparator Cohort ("comparator cohort").
Among applicable registry infants, the prevalence of major congenital anomalies (8.1%) was similar to that for infants in the comparator cohort (8.9%). Among applicable registry pregnancies, 99.1% led to live births, similar to 99.3% for the comparator cohort. There was an increased rate of low birth weight among registry infants (13.7%) as compared to the comparator cohort (9.8%); however, women taking XOLAIR during pregnancy also had more severe asthma, which makes it difficult to determine whether the low birth weight is due to the drug or the disease severity.
The registry study cannot definitively establish the absence of any risk because of methodological limitations, including the observational nature of the registry, small sample size, and potential differences between the registry population and the comparator cohort.
Animal Data
Reproductive studies have been performed in Cynomolgus monkeys. There was no evidence of maternal toxicity, embryotoxicity, or teratogenicity when omalizumab was administered throughout the period of organogenesis at doses that produced exposures approximately 5 times the MRHD (on a mg/kg basis with maternal subcutaneous doses up to 75 mg/kg/week). Omalizumab did not elicit adverse effects on fetal or neonatal growth when administered throughout late gestation, delivery, and nursing.
8.2 Lactation
Risk Summary
There is no information regarding the presence of omalizumab in human milk, or the effects on milk production. However, omalizumab is a human monoclonal antibody (IgG1 kappa), and immunoglobulin (IgG) is present in human milk in small amounts.
The majority of infants (80.9%, 186/230) in the pregnancy exposure registry were breastfed. Events categorized as "infections and infestations" were not significantly increased in infants who were exposed to XOLAIR through breastfeeding compared with infants who were not breastfed, or infants who were breastfed without exposure to XOLAIR.
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for XOLAIR and any potential adverse effects on the breastfed child from omalizumab or from the underlying maternal condition.
8.4 Pediatric Use
Asthma
Safety and effectiveness of XOLAIR for moderate to severe persistent asthma who had a positive skin test or in vitro reactivity to a perennial aeroallergen and whose symptoms are inadequately controlled with inhaled corticosteroids, have been established in pediatric patients aged 6 years and older. Use of XOLAIR for this indication is supported by evidence from adequate and well-controlled studies. XOLAIR was evaluated in 2 trials in 926 (XOLAIR 624; placebo 302) pediatric patients 6 to <12 years of age with moderate to severe persistent asthma who had a positive skin test or in vitro reactivity to a perennial aeroallergen. One trial was a pivotal trial of similar design and conduct to that of adult and adolescent Asthma Trials 1 and 2. The other trial was primarily a safety study and included evaluation of efficacy as a secondary outcome. In the pivotal trial, XOLAIR-treated patients had a statistically significant reduction in the rate of exacerbations (exacerbation was defined as worsening of asthma that required treatment with systemic corticosteroids or a doubling of the baseline ICS dose) [see Clinical Studies (14.1)].
Safety and efficacy in pediatric patients with asthma below 6 years of age have not been established.
Chronic Rhinosinusitis with Nasal Polyps
Safety and effectiveness in pediatric patients with chronic rhinosinusitis with nasal polyps (CRSwNP) below 18 years of age have not been established.
IgE-Mediated Food Allergy
The safety and effectiveness of XOLAIR for the reduction of allergic reactions (Type I), including anaphylaxis, that may occur with accidental exposure to one or more foods have been established in pediatric patients aged 1 year and older with IgE-mediated food allergy. Use of XOLAIR for this indication is supported by evidence from an adequate and well-controlled study that included a total of 165 pediatric patients; 61 patients aged 1 year to less than 6 years of age and 104 patients aged 6 to less than 18 years of age. A significantly greater percentage of XOLAIR-treated patients compared to placebo-treated patients was able to consume a single dose of food (peanut, cashew, milk, egg) without dose- limiting symptoms [see Clinical Studies (14.3)].
Safety and effectiveness in pediatric patients with IgE-mediated food allergy below 1 year of age have not been established.
Chronic Spontaneous Urticaria
The safety and effectiveness of XOLAIR for chronic spontaneous urticaria (CSU) who remain symptomatic despite H1 antihistamine treatment have been established in pediatric patients aged 12 years and older. Use of XOLAIR in this population is supported by evidence from adequate and well-controlled studies. Adolescent patients with CSU were evaluated in 39 patients 12 to 17 years of age (XOLAIR 29, placebo 10) included in three randomized, placebo-controlled CSU trials. A numerical decrease in weekly itch score was observed, and adverse reactions were similar to those reported in patients 18 years and older.
Safety and effectiveness in pediatric patients with CSU below 12 years of age have not been established.
8.5 Geriatric Use
In clinical studies, 134 asthma patients, 20 CRSwNP patients, 37 CSU patients and no IgE-mediated food allergy patients 65 years of age or older were treated with XOLAIR. Although there were no apparent age-related differences observed in these studies, the number of patients aged 65 and over is not sufficient to determine whether they respond differently from younger patients.
-
11 DESCRIPTION
Omalizumab is a recombinant DNA-derived humanized IgG1κ monoclonal antibody that selectively binds to human immunoglobulin E (IgE). The antibody has a molecular weight of approximately 149 kiloDaltons. XOLAIR is produced by a Chinese hamster ovary cell suspension culture.
XOLAIR (omalizumab) is administered as a subcutaneous (SC) injection and is available in prefilled syringe, autoinjector and in vials.
XOLAIR Injection (Prefilled Syringe or Autoinjector)
XOLAIR (omalizumab) injection is supplied as a sterile, preservative-free, clear to slightly opalescent and colorless to pale brownish-yellow solution for subcutaneous injection. XOLAIR (omalizumab) injection is available as a single-dose prefilled syringe or a single-dose autoinjector.
Each 75 mg prefilled syringe or autoinjector delivers 75 mg omalizumab in 0.5 mL and contains arginine hydrochloride (21.05 mg), histidine (0.68 mg), L-histidine hydrochloride monohydrate (1.17 mg), and polysorbate 20 (0.2 mg) in Sterile Water for Injection (SWFI), USP.
Each 150 mg prefilled syringe or autoinjector delivers 150 mg omalizumab in 1 mL and contains arginine hydrochloride (42.1 mg), histidine (1.37 mg), L-histidine hydrochloride monohydrate (2.34 mg), and polysorbate 20 (0.4 mg) in SWFI, USP.
Each 300 mg prefilled syringe or autoinjector delivers 300 mg omalizumab in 2 mL and contains arginine hydrochloride (84.2 mg), histidine (2.74 mg), L-histidine hydrochloride monohydrate (4.68 mg), and polysorbate 20 (0.8 mg) in SWFI, USP.
The needle cap of the XOLAIR 75 mg/0.5 mL and 150 mg/mL prefilled syringe with 26-gauge staked needle contains a derivative of natural rubber latex which may cause allergic reactions in latex sensitive individuals [see How Supplied/Storage and Handling (16)].
The XOLAIR autoinjector is not made with natural rubber latex.
XOLAIR for Injection (Vial)
XOLAIR (omalizumab) for injection is a sterile, white, preservative free, lyophilized powder in a single-dose vial. After reconstitution with 1.4 mL of Sterile Water for Injection, USP, the vial contains 150 mg of omalizumab per 1.2 mL of reconstituted solution for subcutaneous injection. Each 1.2 mL of reconstituted solution also contains histidine (1.3 mg), L-histidine hydrochloride monohydrate (2.1 mg), polysorbate 20 (0.4 mg) and sucrose (108 mg).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Asthma, Chronic Rhinosinusitis with Nasal Polyps, and IgE-Mediated Food Allergy
Omalizumab inhibits the binding of IgE to the high-affinity IgE receptor (FcεRI) on the surface of mast cells, basophils, and dendritic cells, resulting in FcεRI down-regulation on these cells.
In allergic asthmatics, treatment with omalizumab inhibits IgE-mediated inflammation, as evidenced by reduced blood and tissue eosinophils and reduced inflammatory mediators, including IL-4, IL-5, and IL-13.
12.2 Pharmacodynamics
Asthma
In clinical studies, serum free IgE levels were reduced in a dose-dependent manner within 1 hour following the first dose and maintained between doses. Mean serum free IgE decrease was greater than 96% using recommended doses. Serum total IgE levels (i.e., bound and unbound) increased after the first dose due to the formation of omalizumab:IgE complexes, which have a slower elimination rate compared with free IgE. At 16 weeks after the first dose, average serum total IgE levels were five-fold higher compared with pre-treatment when using standard assays. After discontinuation of XOLAIR dosing, the XOLAIR-induced increase in total IgE and decrease in free IgE were reversible, with no observed rebound in IgE levels after drug washout. Total IgE levels did not return to pre-treatment levels for up to one year after discontinuation of XOLAIR.
Chronic Rhinosinusitis with Nasal Polyps
In clinical studies in chronic rhinosinusitis with nasal polyps (CRSwNP) patients, omalizumab treatment led to a reduction in serum free IgE and an increase in serum total IgE levels, similar to the observations in asthma patients. The mean total IgE concentrations at baseline were 168 IU/mL and 218 IU/mL in CRSwNP Trial 1 and 2, respectively. After repeated dosing every 2 or 4 weeks, with dosage and frequency according to Table 3, the mean predose free IgE concentrations at Week 16 were 10.0 IU/mL in CRSwNP Trial 1 and 11.7 IU/mL in CRSwNP Trial 2 and remained stable at 24 weeks of treatment. Total IgE levels in serum increased due to the formation of omalizumab-IgE complexes, which have a slower elimination rate compared with free IgE. After repeated dosing every 2 or 4 weeks, with dosage and frequency according to Table 3, mean and median predose serum total IgE levels at Week 16 were 3- to 4- fold higher compared with pre-treatment levels, and remained stable between 16 and 24 weeks of treatment.
IgE-Mediated Food Allergy
In a clinical study in patients with IgE-mediated food allergy, omalizumab treatment led to a reduction in serum free IgE and an increase in serum total IgE levels, similar to the observations in asthma patients. The mean total IgE concentration at baseline was 810 IU/mL. After repeated dosing every 2 or 4 weeks, with dosage and frequency according to Table 4, the mean pre-dose free IgE concentration at Week 16 was 10.0 IU/mL. Mean total IgE levels in serum increased about 2.4-fold due to the formation of omalizumab-IgE complexes, which have a longer half-life compared with free IgE.
Chronic Spontaneous Urticaria
In clinical studies in chronic spontaneous urticaria (CSU) patients, XOLAIR treatment led to a dose-dependent reduction of serum free IgE and an increase of serum total IgE levels, similar to the observations in asthma patients. Maximum suppression of free IgE was observed 3 days following the first subcutaneous dose. After repeat dosing once every 4 weeks, predose serum free IgE levels remained stable between 12 and 24 weeks of treatment. Total IgE levels in serum increased after the first dose due to the formation of omalizumab-IgE complexes which have a slower elimination rate compared with free IgE. After repeat dosing once every 4 weeks at 75 mg up to 300 mg, average predose serum total IgE levels at Week 12 were two- to three-fold higher compared with pre-treatment levels, and remained stable between 12 and 24 weeks of treatment. After discontinuation of XOLAIR dosing, free IgE levels increased and total IgE levels decreased towards pre-treatment levels over a 16-week follow-up period.
12.3 Pharmacokinetics
After SC administration, omalizumab was absorbed with an average absolute bioavailability of 62%. Following a single SC dose in adult and adolescent patients with asthma, omalizumab was absorbed slowly, reaching peak serum concentrations after an average of 7–8 days. In patients with CSU, the peak serum concentration was reached at a similar time after a single SC dose. The pharmacokinetics of omalizumab was linear at doses greater than 0.5 mg/kg. In patients with asthma, following multiple doses of XOLAIR, areas under the serum concentration-time curve from Day 0 to Day 14 at steady state were up to 6-fold of those after the first dose. In patients with CSU, omalizumab exhibited linear pharmacokinetics across the dose range of 75 mg to 600 mg given as single subcutaneous dose. Following repeat dosing from 75 to 300 mg every 4 weeks, trough serum concentrations of omalizumab increased proportionally with the dose levels.
In vitro, omalizumab formed complexes of limited size with IgE. Precipitating complexes and complexes larger than 1 million daltons in molecular weight were not observed in vitro or in vivo. Tissue distribution studies in Cynomolgus monkeys showed no specific uptake of 125I-omalizumab by any organ or tissue. The apparent volume of distribution of omalizumab in patients with asthma following SC administration was 78 ± 32 mL/kg. In patients with CSU, based on population pharmacokinetics, distribution of omalizumab was similar to that in patients with asthma.
Clearance of omalizumab involved IgG clearance processes as well as clearance via specific binding and complex formation with its target ligand, IgE. Liver elimination of IgG included degradation in the liver reticuloendothelial system (RES) and endothelial cells. Intact IgG was also excreted in bile. In studies with mice and monkeys, omalizumab:IgE complexes were eliminated by interactions with Fcγ receptors within the RES at rates that were generally faster than IgG clearance. In asthma patients omalizumab serum elimination half-life averaged 26 days, with apparent clearance averaging 2.4 ± 1.1 mL/kg/day. Doubling body weight approximately doubled apparent clearance. In CSU patients, at steady state, based on population pharmacokinetics, omalizumab serum elimination half-life averaged 24 days and apparent clearance averaged 240 mL/day (corresponding to 3.0 mL/kg/day for an 80 kg patient).
Specific Populations
Asthma
The population pharmacokinetics of omalizumab was analyzed to evaluate the effects of demographic characteristics in patients with asthma. Analyses of these data suggested that no dose adjustments are necessary for age (6 to 76 years), race, ethnicity, or gender.
Chronic Rhinosinusitis with Nasal Polyps
The population pharmacokinetics analyses of omalizumab suggested that the pharmacokinetics of omalizumab in chronic rhinosinusitis with nasal polyps (CRSwNP) were consistent with that in asthma. Graphical covariate analyses were performed to evaluate the effects of demographic characteristics and other factors on omalizumab exposure and clinical responses. These analyses demonstrate that no dose adjustments are necessary for age (18 to 75 years) or gender. Race and ethnicity data are too limited in CRSwNP studies to inform dose adjustment.
IgE-Mediated Food Allergy
Population pharmacokinetic (PK) analyses of omalizumab suggested that the PK of omalizumab in patients with IgE-mediated food allergy were generally consistent with that in patients with asthma. Covariate analyses were performed to evaluate the effects of demographic characteristics and other factors on omalizumab exposure and clinical responses. These analyses demonstrate that no dose adjustments are necessary for age (1 year and older), race, ethnicity, or gender.
Chronic Spontaneous Urticaria
The population pharmacokinetics of omalizumab was analyzed to evaluate the effects of demographic characteristics and other factors on omalizumab exposure in patients with chronic spontaneous urticaria (CSU). Covariate effects were evaluated by analyzing the relationship between omalizumab concentrations and clinical responses. These analyses demonstrate that no dose adjustments are necessary for age (12 to 75 years), race/ethnicity, gender, body weight, body mass index, or baseline IgE level.
12.6 Immunogenicity
The observed incidence of anti-drug antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-drug antibodies in the studies described below with the incidence of anti-drug antibodies in other studies, including those of XOLAIR or of other omalizumab products.
Antibodies to XOLAIR were detected in approximately 1/1723 (<0.1%) of patients treated with XOLAIR in the clinical studies evaluated for asthma in patients 12 years of age and older. In three pediatric studies, antibodies to XOLAIR were detected in one patient out of 581 patients 6 to <12 years of age treated with XOLAIR and evaluated for antibodies. There were no detectable antibodies in the patients treated in the CSU clinical trials, but due to levels of XOLAIR at the time of anti-therapeutic antibody sampling and missing samples for some patients, antibodies to XOLAIR could only have been determined in 88% of the 733 patients treated in these clinical studies. The data reflect the percentage of patients whose test results were considered positive for antibodies to XOLAIR in ELISA assays and are highly dependent on the sensitivity and specificity of the assays.
Anti-drug antibodies were not measured in the CRSwNP or IgE-mediated food allergy trials.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No long-term studies have been performed in animals to evaluate the carcinogenic potential of XOLAIR.
There were no effects on fertility and reproductive performance in male and female Cynomolgus monkeys that received XOLAIR at subcutaneous doses up to 75 mg/kg/week (approximately 5 times the maximum recommended human dose on a mg/kg basis).
-
14 CLINICAL STUDIES
14.1 Asthma
Adult and Adolescent Patients 12 Years of Age and Older
The safety and efficacy of XOLAIR were evaluated in three randomized, double-blind, placebo-controlled, multicenter trials.
The trials enrolled patients 12 to 76 years old, with moderate to severe persistent (NHLBI criteria) asthma for at least one year, and a positive skin test reaction to a perennial aeroallergen. In all trials, XOLAIR dosing was based on body weight and baseline serum total IgE concentration. All patients were required to have a baseline IgE between 30 and 700 IU/mL and body weight not more than 150 kg. Patients were treated according to a dosing table to administer at least 0.016 mg/kg/IU (IgE/mL) of XOLAIR or a matching volume of placebo over each 4-week period. The maximum XOLAIR dose per 4 weeks was 750 mg.
In all three trials an exacerbation was defined as a worsening of asthma that required treatment with systemic corticosteroids or a doubling of the baseline ICS dose. Most exacerbations were managed in the outpatient setting and the majority were treated with systemic steroids. Hospitalization rates were not significantly different between XOLAIR and placebo-treated patients; however, the overall hospitalization rate was small. Among those patients who experienced an exacerbation, the distribution of exacerbation severity was similar between treatment groups.
Asthma Trials 1 and 2
At screening, patients in Asthma Trials 1 and 2 had a forced expiratory volume in one second (FEV1) between 40% and 80% predicted. All patients had a FEV1 improvement of at least 12% following beta2-agonist administration. All patients were symptomatic and were being treated with inhaled corticosteroids (ICS) and short-acting beta2-agonists. Patients receiving other concomitant controller medications were excluded, and initiation of additional controller medications while on study was prohibited. Patients currently smoking were excluded.
Each trial was comprised of a run-in period to achieve a stable conversion to a common ICS (beclomethasone dipropionate), followed by randomization to XOLAIR or placebo. Patients received XOLAIR for 16 weeks with an unchanged corticosteroid dose unless an acute exacerbation necessitated an increase. Patients then entered an ICS reduction phase of 12 weeks during which ICS dose reduction was attempted in a step-wise manner.
The distribution of the number of asthma exacerbations per patient in each group during a study was analyzed separately for the stable steroid and steroid-reduction periods.
In both Asthma Trials 1 and 2 the number of exacerbations per patient was reduced in patients treated with XOLAIR compared with placebo (Table 11).
Measures of airflow (FEV1) and asthma symptoms were also evaluated in these trials. The clinical relevance of the treatment-associated differences is unknown. Results from the stable steroid phase Asthma Trial 1 are shown in Table 12. Results from the stable steroid phase of Asthma Trial 2 and the steroid reduction phases of both Asthma Trials 1 and 2 were similar to those presented in Table 12.
Table 11. Frequency of Asthma Exacerbations per Patient by Phase in Asthma Trials 1 and 2 Stable Steroid Phase (16 wks) Asthma Trial 1 Asthma Trial 2 Exacerbations per patient XOLAIR
N=268Placebo
N=257XOLAIR
N=274Placebo
N=2720 85.8% 76.7% 87.6% 69.9% 1 11.9% 16.7% 11.3% 25.0% ≥2 2.2% 6.6% 1.1% 5.1% p-Value 0.005 <0.001 Mean number exacerbations/patient 0.2 0.3 0.1 0.4 Steroid Reduction Phase (12 wks) Exacerbations per patient XOLAIR
N=268Placebo
N=257XOLAIR
N=274Placebo
N=2720 78.7% 67.7% 83.9% 70.2% 1 19.0% 28.4% 14.2% 26.1% ≥2 2.2% 3.9% 1.8% 3.7% p-Value 0.004 <0.001 Mean number exacerbations/patient 0.2 0.4 0.2 0.3 Table 12. Asthma Symptoms and Pulmonary Function During Stable Steroid Phase of Asthma Trial 1 XOLAIR
N=268*Placebo
N=257*Endpoint Mean Baseline Median Change (Baseline to Wk 16) Mean Baseline Median Change (Baseline to Wk 16) Asthma symptom scale: total score from 0 (least) to 9 (most); nocturnal and daytime scores from 0 (least) to 4 (most symptoms). Total asthma symptom score 4.3 –1.5† 4.2 –1.1† Nocturnal asthma score 1.2 –0.4† 1.1 –0.2† Daytime asthma score 2.3 –0.9† 2.3 –0.6† FEV1 % predicted 68 3† 68 0† Asthma Trial 3
In Asthma Trial 3, there was no restriction on screening FEV1, and unlike Asthma Trials 1 and 2, long-acting beta2-agonists were allowed. Patients were receiving at least 1000 µg/day fluticasone propionate and a subset was also receiving oral corticosteroids. Patients receiving other concomitant controller medications were excluded, and initiation of additional controller medications while on study was prohibited. Patients currently smoking were excluded.
The trial was comprised of a run-in period to achieve a stable conversion to a common ICS (fluticasone propionate), followed by randomization to XOLAIR or placebo. Patients were stratified by use of ICS-only or ICS with concomitant use of oral steroids. Patients received XOLAIR for 16 weeks with an unchanged corticosteroid dose unless an acute exacerbation necessitated an increase. Patients then entered an ICS reduction phase of 16 weeks during which ICS or oral steroid dose reduction was attempted in a step-wise manner.
The number of exacerbations in patients treated with XOLAIR was similar to that in placebo-treated patients (Table 13). The absence of an observed treatment effect may be related to differences in the patient population compared with Asthma Trials 1 and 2, study sample size, or other factors.
Table 13. Percentage of Patients with Asthma Exacerbations by Subgroup and Phase in Asthma Trial 3 Stable Steroid Phase (16 wks) Inhaled Only Oral + Inhaled XOLAIR
N=126Placebo
N=120XOLAIR
N=50Placebo
N=45% Patients with ≥1 exacerbations 15.9% 15.0% 32.0% 22.2% Difference
(95% CI)0.9
(–9.7, 13.7)9.8
(–10.5, 31.4)Steroid Reduction Phase (16 wks) XOLAIR
N=126Placebo
N=120XOLAIR
N=50Placebo
N=45% Patients with ≥1 exacerbations 22.2% 26.7% 42.0% 42.2% Difference
(95% CI)–4.4
(–17.6, 7.4)–0.2
(–22.4, 20.1)In all three of the trials, a reduction of asthma exacerbations was not observed in the XOLAIR-treated patients who had FEV1>80% at the time of randomization. Reductions in exacerbations were not seen in patients who required oral steroids as maintenance therapy.
Pediatric Patients 6 to <12 Years of Age
The safety and efficacy of XOLAIR in pediatric patients 6 to <12 years of age with moderate to severe asthma is based on one randomized, double-blind, placebo controlled, multi-center trial (Asthma Trial 4 [NCT00079937]) and an additional supportive study (Asthma Trial 5).
Asthma Trial 4 was a 52-week study that evaluated the safety and efficacy of XOLAIR as add-on therapy in 628 pediatric patients ages 6 to <12 years with moderate to severe asthma inadequately controlled despite the use of inhaled corticosteroids (fluticasone propionate DPI ≥200 mcg/day or equivalent) with or without other controller asthma medications. Eligible patients were those with a diagnosis of asthma >1 year, a positive skin prick test to at least one perennial aeroallergen, and a history of clinical features such as daytime and/or night-time symptoms and exacerbations within the year prior to study entry. During the first 24 weeks of treatment, steroid doses remained constant from baseline. This was followed by a 28-week period during which inhaled corticosteroid adjustment was allowed.
The primary efficacy variable was the rate of asthma exacerbations during the 24-week, fixed steroid treatment phase. An asthma exacerbation was defined as a worsening of asthma symptoms as judged clinically by the investigator, requiring doubling of the baseline inhaled corticosteroid dose for at least 3 days and/or treatment with rescue systemic (oral or IV) corticosteroids for at least 3 days. At 24 weeks, the XOLAIR group had a statistically significantly lower rate of asthma exacerbations (0.45 vs. 0.64) with an estimated rate ratio of 0.69 (95% CI: 0.53, 0.90).
The XOLAIR group also had a lower rate of asthma exacerbations compared to placebo over the full 52-week double-blind treatment period (0.78 vs. 1.36; rate ratio: 0.57; 95% CI: 0.45, 0.72). Other efficacy variables such as nocturnal symptom scores, beta-agonist use, and measures of airflow (FEV1) were not significantly different in XOLAIR-treated patients compared to placebo.
Asthma Trial 5 was a 28-week randomized, double blind, placebo-controlled study that primarily evaluated safety in 334 pediatric patients, 298 of whom were 6 to <12 years of age, with moderate to severe asthma who were well-controlled with inhaled corticosteroids (beclomethasone dipropionate 168-420 mcg/day). A 16-week steroid treatment period was followed by a 12-week steroid dose reduction period. Patients treated with XOLAIR had fewer asthma exacerbations compared to placebo during both the 16-week fixed steroid treatment period (0.18 vs. 0.32; rate ratio: 0.58; 95% CI: 0.35, 0.96) and the 28-week treatment period (0.38 vs. 0.76; rate ratio: 0.50; 95% CI: 0.36, 0.71).
14.2 Chronic Rhinosinusitis with Nasal Polyps
Adult Patients 18 Years of Age and Older
The safety and efficacy of XOLAIR was evaluated in two, randomized, multicenter, double-blind, placebo-controlled clinical trials (CRSwNP Trial 1 [NCT03280550] and CRSwNP Trial 2 [NCT03280537]) that enrolled patients with chronic rhinosinusitis with nasal polyps (CRSwNP) with inadequate response to nasal corticosteroids (CRSwNP Trial 1, n=138; CRSwNP Trial 2, n=127). Patients received XOLAIR or placebo SC every 2 or 4 weeks, with XOLAIR dosage and frequency according to Table 3, for 24 weeks followed by a 4-week follow-up period. All patients received background nasal mometasone therapy during both the treatment period and during a 5-week run-in period. Prior to randomization, patients were required to have evidence of bilateral polyps as determined by a nasal polyp score (NPS) ≥ 5 with NPS ≥ 2 in each nostril, despite use of nasal mometasone during the run-in period. NPS was measured via endoscopy and scored (range 0-4 per nostril: 0= no polyps; 1=small polyps in the middle meatus not reaching below the inferior border of the middle turbinate; 2=polyps reaching below the lower border of the middle turbinate; 3=large polyps reaching the lower border of the inferior turbinate or polyps medial to the middle turbinate; 4=large polyps causing complete obstruction of the inferior nasal cavity) for a total NPS (range 0-8). Patients were furthermore required to have a weekly average of nasal congestion score (NCS) > 1 prior to randomization, despite use of nasal mometasone. Nasal congestion was measured by a daily assessment on a 0 to 3 point severity scale (0=none, 1=mild, 2=moderate, 3=severe). Prior sino-nasal surgery or prior systemic corticosteroid usage were not required for inclusion in the trials and sinus CT scans were not performed to evaluate for sinus opacification. Demographics and baseline characteristics, including allergic comorbidities, are described in Table 14.
Table 14. Demographics and Baseline Characteristics of CRSwNP Trials 1 and 2 Parameter CRSwNP Trial 1
(n=138)CRSwNP Trial 2
(n=127)CRSwNP= chronic rhinosinusitis with nasal polyps; SD=standard deviation; NPS=nasal polyp score; IgE = Immunoglobulin E; IU=international units. For NPS, NCS, sense of smell, post nasal drip, and runny nose, higher scores indicate greater disease severity. Mean age (years) (SD) 51 (13) 50 (12) % Male 64 65 Patients with systemic corticosteroid use in the previous year (%) 19 26 Patients with prior surgery for nasal polyps (%) 79 (57) 79 (62) Mean bilateral endoscopic NPS (SD), range 0-8 6.2 (1.0) 6.3 (0.9) Mean nasal congestion score (SD) range 0-3 2.4 (0.6) 2.3 (0.7) Mean sense of smell score (SD) range 0-3 2.7 (0.7) 2.7 (0.7) Mean post nasal drip score (SD) range 0-3 1.8 (0.9) 1.7 (0.9) Mean runny nose score (SD) range 0-3 2.0 (0.8) 1.9 (0.9) Mean blood eosinophils (cells/mcL) (SD) 346 (284) 335 (188) Mean total IgE IU/mL (SD) 161 (140) 190 (201) Asthma (%) 54 61 Aspirin exacerbated respiratory disease (%) 20 35 The co-primary endpoints in CRSwNP Trials 1 and 2 were NPS and average daily NCS at Week 24. In both trials, patients who received XOLAIR had a statistically significant greater improvement from baseline at Week 24 in NPS and weekly average NCS, than patients who received placebo. Results from CRSwNP Trials 1 and 2 are shown in Table 15.
The greater improvements in NPS and NCS in the XOLAIR group compared to the placebo group were observed as early as the first assessment at Week 4 in both studies, as seen in Figure 1.
Table 15. Change from Baseline at Week 24 in Nasal Polyp Score and 7-day Average of Daily Nasal Congestion Score in CRSwNP Trials 1 and 2 Trial 1 Trial 2 Placebo XOLAIR Placebo XOLAIR CRSwNP= chronic rhinosinusitis with nasal polyps; LS=least-square. Change from baseline was analyzed using a mixed-effect model of repeated measures (MMRM) model with baseline score, baseline score/timepoint (week) interaction as covariates, and the following factors: geographic region, asthma/aspirin sensitivity comorbidity status, timepoint, treatment group, treatment/timepoint interaction. Number of patients 65 72 65 62 Nasal Polyp Score Mean Baseline Score 6.3 6.2 6.1 6.4 LS Mean Change From Baseline at Week 24 0.1 -1.1 -0.3 -0.9 Difference in LS means vs. placebo -1.1 -0.6 95% CI for difference -1.6, -0.7 -1.1, -0.1 p-value <0.0001 0.0140 7-day Average of Daily Nasal Congestion Score Mean Baseline Score 2.5 2.4 2.3 2.3 LS Mean Change From Baseline at Week 24 -0.4 -0.9 -0.2 -0.7 Difference in LS means vs. placebo -0.6 -0.5 95% CI for difference -0.8, -0.3 -0.8, -0.2 p-value 0.0004 0.0017 The mean NPS and NCS at each study week by treatment group is shown in Figure 1.
Figure 1. Mean Change from Baseline in Nasal Congestion Score and Mean Change from Baseline in Nasal Polyp Score by Treatment Group in CRSwNP Trials 1 and 2 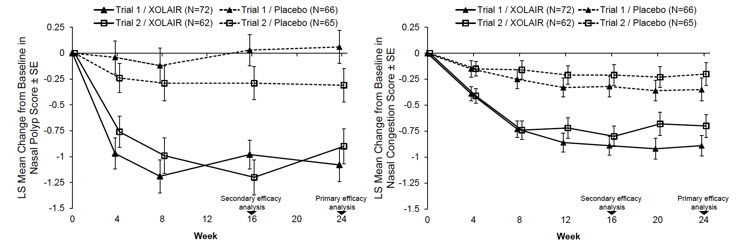
XOLAIR had statistically significant improvements on sense of smell score compared to placebo. Sense of smell was measured by a daily assessment on a 0 to 3 point severity scale (0=no symptoms, 1=mild symptoms, 2=moderate symptoms, 3=severe symptoms). The LS mean difference for change from baseline at Week 24 in sense of smell score in XOLAIR compared to placebo was -0.3 (95% CI: -0.6, -0.1) in CRSwNP Trial 1 and -0.5 (95% CI: -0.7, -0.2) in CRSwNP Trial 2.
XOLAIR had statistically significant improvements on post-nasal drip compared to placebo. The LS mean difference for change from baseline at Week 24 in post-nasal drip score in XOLAIR compared to placebo was -0.6 (95% CI: -0.8, -0.3) in CRSwNP Trial 1 and -0.5 (95% CI: -0.8, -0.3) in CRSwNP Trial 2.
XOLAIR had statistically significant improvements on runny nose compared to placebo. The LS mean difference for change from baseline at Week 24 in runny nose score in XOLAIR compared to placebo was -0.4 (95% CI: -0.7, -0.2) in CRSwNP Trial 1 and -0.6 (95% CI: -0.9, -0.4) in CRsWNP Trial 2.
In a pre-specified pooled analysis of systemic corticosteroid use during the 24-week treatment period, there was no significant reduction in systemic corticosteroid use between the treatment arms. The proportion of patients taking systemic corticosteroid in XOLAIR was 2.3% compared to 6.2% in placebo. The odds-ratio of systemic corticosteroid use with XOLAIR compared to placebo was 0.4 (95% CI: 0.1, 1.5).
There were no sino-nasal surgeries reported, in either placebo or XOLAIR arms, in either Trial.
14.3 IgE-Mediated Food Allergy
The safety and efficacy of XOLAIR was evaluated in a multi-center, randomized, double-blind, placebo-controlled Food Allergy (FA) trial [NCT03881696] in 168 adult patients and pediatric patients 1 year of age to less than 56 years who were allergic to peanut and at least two other foods, including milk, egg, wheat, cashew, hazelnut, or walnut (i.e., studied foods). The FA trial enrolled patients who experienced dose-limiting symptoms (e.g., moderate to severe skin, respiratory or gastrointestinal symptoms) to a single dose of ≤100 mg of peanut protein and ≤300 mg protein for each of the other two foods (milk, egg, wheat, cashew, hazelnut, or walnut) during the screening double-blind placebo-controlled food challenge (DBPCFC). Patients with a history of severe anaphylaxis (defined as neurological compromise or requiring intubation) were excluded from the study. Patients were randomized 2:1 to receive a subcutaneous dosage of XOLAIR or placebo based on serum total IgE level (IU/mL), measured before the start of treatment, and by body weight according to Table 4 [see Dosage and Administration (2.4)] for 16 to 20 weeks. After 16 to 20 weeks of treatment, each patient completed a DBPCFC consisting of placebo and each of their 3 studied foods. Following the DBPCFC, the first 60 patients that included 59 pediatric patients and one adult patient who completed the double-blind, placebo-controlled phase of the study could continue to receive XOLAIR in a 24 to 28 week open-label extension.
Efficacy of XOLAIR is based on 165 pediatric patients who were included in the efficacy analyses provided below. The mean age of the pediatric patients was 8 years (age range: 1 to 17 years); 37% were less than 6 years of age, 38% were 6 to less than 12 years of age, and 25% were 12 to less than 18 years of age. Patient population were 56% male, 63% White, 13% Asian, 7% Black, 16% Other, and 55% of patients had a history of asthma.
The primary efficacy endpoint was the percentage of patients who were able to consume a single dose of ≥600 mg of peanut protein without dose-limiting symptoms (e.g., moderate to severe skin, respiratory or gastrointestinal symptoms) during DBPCFC. Table 16 shows XOLAIR treatment led to a statistically higher response rate (68%) than placebo (5%).
The secondary efficacy endpoints were the percentage of patients who were able to consume a single dose of ≥1000 mg of cashew, milk, or egg protein without dose-limiting symptoms during DBPCFC. The study met the secondary endpoints and demonstrated that XOLAIR treatment led to statistically higher response rates than placebo for all three foods. See Table 16 for details.
Table 16. DBPCFC Response Rates in Pediatric Patients for Single Dose of Peanut, Cashew, Milk or Egg Protein in FA Trial Food, Challenge Dose Response Rate* (%)
(n/N)Treatment Difference (%)
(XOLAIR-Placebo)
(95% CI)XOLAIR Placebo CI = Confidence interval; DBPCFC = Double-blind placebo-controlled food challenge; n = Number of responders; N = Total number of patients receiving food, challenge dose.
Notes: Subjects without an exit DBPCFC or evaluable exit DBPCFC were counted as non-responders; P-values from two-sided Fisher's exact tests were <0.0001 for all the food challenge doses.- *
- Response defined as consumption of a single dose of the specified amount of food without dose-limiting symptoms.
- †
- Consumption of a single dose of ≥1000 mg of peanut protein was an additional secondary endpoint. The key secondary efficacy endpoints were the percentage of patients who were able to consume a single dose of ≥1000 mg of cashew, milk, or egg protein.
Peanut, ≥600 mg 68%
(75/110)5%
(3/55)63%
(50%, 73%)Peanut, ≥1000 mg† 65%
(72/110)0%
(0/55)65%
(56%, 74%)Cashew, ≥1000 mg 42%
(27/64)3%
(1/30)39%
(20%, 53%)Milk, ≥1000 mg 66%
(25/38)11%
(2/19)55%
(29%, 73%)Egg, ≥1000 mg 67%
(31/46)0%
(0/19)67%
(49%, 80%)Seventeen percent of XOLAIR treated patients were not able to consume >100 mg of peanut protein without moderate to severe dose-limiting symptoms. Eighteen, 22, and 41 percent of XOLAIR-treated patients were not able to consume >300 mg of milk, egg, or cashew protein, respectively, without moderate to severe dose-limiting symptoms.
Additional secondary analyses included the percentage of patients who were able to consume at least two or all three foods during DBPCFC. For two foods, 71% of XOLAIR treated patients were able to consume a single dose of ≥600 mg versus 5% in the placebo group and 67% were able to consume a single dose of ≥1000 mg versus 4% in the placebo group. For a single dose of ≥600 mg of three foods, the response rates were 48% in the XOLAIR group versus 4% in the placebo group and for a single dose of ≥1000 mg of three foods, the response rate in the XOLAIR group was 39% while none of the placebo patients were able to consume the challenge dose without symptoms.
The effectiveness of XOLAIR in adults is supported by the adequate and well-controlled trial of XOLAIR in pediatric patients, disease similarity in pediatric and adult patients, and pharmacokinetic (PK) similarity [see Clinical Pharmacology (12.3)].
While efficacy cannot be established from uncontrolled, open-label studies, for 38 pediatric patients who continued XOLAIR for 24-28 weeks in an open-label extension, the percentage of patients who were able to consume ≥600 mg of peanut protein and ≥1000 mg of egg, milk, and/or cashew protein without moderate to severe dose-limiting symptoms was maintained.
14.4 Chronic Spontaneous Urticaria
Adult and Adolescent Patients 12 Years of Age and Older
The safety and efficacy of XOLAIR for the treatment of chronic spontaneous urticaria (CSU), previously referred to as chronic idiopathic urticaria (CIU) was assessed in two placebo-controlled, multiple-dose clinical trials of 24 weeks' duration (CSU Trial 1; n= 319, [NCT01287117]) and 12 weeks' duration (CSU Trial 2; n=322, [NCT01292473]). Patients received XOLAIR 75 mg, 150 mg, or 300 mg or placebo by SC injection every 4 weeks in addition to their baseline level of H1 antihistamine therapy for 24 or 12 weeks, followed by a 16-week washout observation period. A total of 640 patients (165 males, 475 females) were included for the efficacy analyses. Most patients were white (84%) and the median age was 42 years (range 12–72).
Disease severity was measured by a weekly urticaria activity score (UAS7, range 0–42), which is a composite of the weekly itch severity score (range 0–21) and the weekly hive count score (range 0–21). All patients were required to have a UAS7 of ≥16, and a weekly itch severity score of ≥8 for the 7 days prior to randomization, despite having used an H1 antihistamine for at least 2 weeks.
The mean weekly itch severity scores at baseline were fairly balanced across treatment groups and ranged between 13.7 and 14.5 despite use of an H1 antihistamine at an approved dose. The reported median durations of CSU at enrollment across treatment groups were between 2.5 and 3.9 years (with an overall subject-level range of 0.5 to 66.4 years).
In both CSU Trials 1 and 2, patients who received XOLAIR 150 mg or 300 mg had greater decreases from baseline in weekly itch severity scores and weekly hive count scores than placebo at Week 12. Representative results from CSU Trial 1 are shown (Table 17); similar results were observed in CSU Trial 2. The 75-mg dose did not demonstrate consistent evidence of efficacy and is not approved for use.
Table 17. Change from Baseline to Week 12 in Weekly Itch Severity Score and Weekly Hive Count Score in CSU Trial 1* XOLAIR
75mgXOLAIR
150mgXOLAIR
300mgPlacebo N 77 80 81 80 Weekly Itch Severity Score Mean Baseline Score (SD) 14.5 (3.6) 14.1 (3.8) 14.2 (3.3) 14.4 (3.5) Mean Change Week 12 (SD) −6.46 (6.14) −6.66 (6.28) −9.40 (5.73) −3.63 (5.22) Difference in LS means vs. placebo −2.96 −2.95 −5.80 95% CI for difference −4.71, −1.21 −4.72, −1.18 −7.49, −4.10 - Weekly Hive Count Score † Mean Baseline Score (SD) 17.2 (4.2) 16.2 (4.6) 17.1 (3.8) 16.7 (4.4) Mean Change Week 12 (SD) −7.36 (7.52) −7.78 (7.08) −11.35 (7.25) −4.37 (6.60) Difference in LS means vs. placebo −2.75 −3.44 −6.93 95% CI for difference −4.95, −0.54 −5.57, −1.32 −9.10, −4.76 - The mean weekly itch severity score at each study week by treatment groups is shown in Figure 2. Representative results from CSU Trial 1 are shown; similar results were observed in CSU Trial 2. The appropriate duration of therapy for CSU with XOLAIR has not been determined.
Figure 2. Mean Weekly Itch Severity Score by Treatment Group Modified Intent to Treat Patients in CSU Trial 1 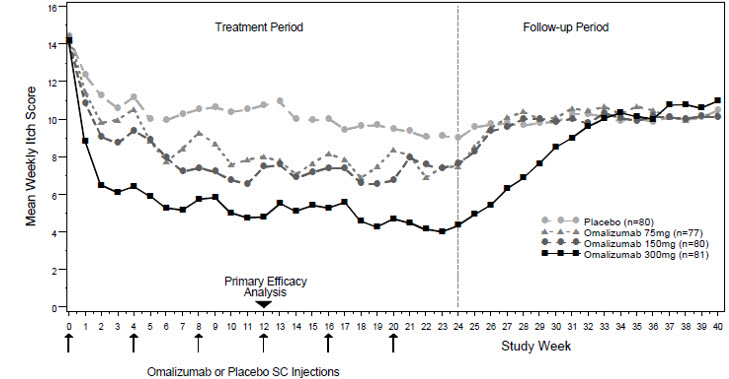
In CSU Trial 1, a larger proportion of patients treated with XOLAIR 300 mg (36%) reported no itch and no hives (UAS7=0) at Week 12 compared to patients treated with XOLAIR 150 mg (15%), XOLAIR 75 mg (12%), and placebo group (9%). Similar results were observed in CSU Trial 2.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
Injection (Prefilled Syringe or Autoinjector)
XOLAIR (omalizumab) injection is a clear to slightly opalescent and colorless to pale brownish-yellow solution for subcutaneous use.
XOLAIR injection is provided as either a single-dose prefilled syringe with staked needle, rigid needle cap, and needle shield or a single-dose prefilled autoinjector with staked needle, needle cap and needle guard.
XOLAIR is available as prefilled syringe and autoinjector as described in Tables 18 and 19.
Table 18. XOLAIR Prefilled Syringe Strengths and Package Configurations Package Configuration Strength NDC - *
- The needle cap of the XOLAIR prefilled syringe contains a derivative of natural rubber latex which may cause allergic reactions in latex sensitive individuals.
1 prefilled syringe with a 26-gauge staked needle* 75 mg/0.5 mL NDC 50242-214-01 1 prefilled syringe with a 26-gauge staked needle* 150 mg/mL NDC 50242-215-01 1 prefilled syringe with a 27-gauge staked needle 75 mg/0.5 mL NDC 50242-214-03 1 prefilled syringe with a 27-gauge staked needle 150 mg/mL NDC 50242-215-03 1 prefilled syringe with a 27-gauge staked needle 300 mg/2 mL NDC 50242-227-01 Table 19. XOLAIR Autoinjector Strengths and Package Configurations Package Configuration Strength NDC 1 autoinjector with a 27-gauge staked needle 75 mg/0.5 mL NDC 50242-214-55 1 autoinjector with a 27-gauge staked needle 150 mg/mL NDC 50242-215-55 1 autoinjector with a 27-gauge staked needle 300 mg/2 mL NDC-50242-227-55 The XOLAIR autoinjector is not made with natural rubber latex.
Storage
XOLAIR prefilled syringe and autoinjector should be shipped and stored under refrigerated conditions 2°C to 8°C (36°F to 46°F) in the original carton. Protect from direct sunlight. XOLAIR prefilled syringe and autoinjector can be removed from and placed back in the refrigerator if needed. The total combined time out of the refrigerator may not be more than 2 days. Do not use if prefilled syringe or autoinjector is left at temperatures above 25°C (77°F).
Do not freeze. Do not use if the prefilled syringe or autoinjector has been frozen.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Anaphylaxis
Inform patients of the risk of life-threatening anaphylaxis with XOLAIR including the following points [see Boxed Warning and Warnings and Precautions (5.1)]:
- There have been reports of anaphylaxis occurring up to 4 days after administration of XOLAIR
- Initiate XOLAIR only in a healthcare setting by healthcare providers
- Observe patients closely following administration
- Inform patients of the signs and symptoms of anaphylaxis
- Instruct patients to seek immediate medical care should such signs or symptoms occur
Potential Medication Error Related to Emergency Treatment of Anaphylaxis
Advise patients, parents, or caregivers that XOLAIR should not be used for the emergency treatment of allergic reactions, including anaphylaxis [see Warnings and Precautions (5.9)].
Continuation of Other Medications
Instruct patients receiving XOLAIR not to decrease the dose of, or stop taking any other asthma, CRSwNP, CSU or IgE-mediated food allergy medications or allergen immunotherapy unless otherwise instructed by their physician. Inform patients that they may not see immediate improvement in their asthma, CRSwNP, CSU or IgE-mediated food allergy symptoms after beginning XOLAIR therapy.
Prefilled Syringe Needle Cover Contains Latex
Inform patients the needle cover on the prefilled syringe contains dry natural rubber (a derivative of latex), which may cause allergic reactions in individuals sensitive to latex [see How Supplied/Storage and Handling (16)].
Instruction on Injection Technique
If a patient or caregiver is to administer subcutaneous XOLAIR prefilled syringe or autoinjector, instruct on injection technique and assess ability to inject subcutaneously to ensure proper administration of XOLAIR. For patients who require more than 1 injection to complete their prescribed dose, instruct patients to administer all injections consecutively and in one sitting [see Dosage and Administration (2.7), Warnings and Precautions (5.1), and Instructions for Use].
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised:2/2024 MEDICATION GUIDE XOLAIR® (ZOHL-air)
(omalizumab)
injection, for subcutaneous useXOLAIR® (ZOHL-air)
(omalizumab)
for injection, for subcutaneous useWhat is the most important information I should know about XOLAIR?
XOLAIR may cause serious side effects, including:
Severe allergic reaction. A severe allergic reaction called anaphylaxis can happen when you receive XOLAIR. The reaction can occur after the first dose, or after many doses. It may also occur right after a XOLAIR injection or days later. Anaphylaxis is a life-threatening condition and can lead to death. Go to the nearest emergency room right away if you have any of these symptoms of an allergic reaction:- wheezing, shortness of breath, cough, chest tightness, or trouble breathing
- low blood pressure, dizziness, fainting, rapid or weak heartbeat, anxiety, or feeling of "impending doom"
- flushing, itching, hives, or feeling warm
- swelling of the throat or tongue, throat tightness, hoarse voice, or trouble swallowing
What is XOLAIR?
XOLAIR is an injectable prescription medicine used to treat:- moderate to severe persistent asthma in people 6 years of age and older whose asthma symptoms are not well controlled with asthma medicines called inhaled corticosteroids. A skin or blood test is performed to see if you have allergies to year-round allergens. It is not known if XOLAIR is safe and effective in people with asthma under 6 years of age.
- chronic rhinosinusitis with nasal polyps (CRSwNP) in people 18 years of age and older when medicines to treat CRSwNP called nasal corticosteroids have not worked well enough. It is not known if XOLAIR is safe and effective in people with CRSwNP under 18 years of age.
- food allergy in people 1 year of age and older to reduce allergic reactions that may occur after accidentally eating one or more foods to which you are allergic. While taking XOLAIR you should continue to avoid all foods to which you are allergic. It is not known if XOLAIR is safe and effective in people with food allergy under 1 year of age.
- chronic spontaneous urticaria (CSU, previously referred to as chronic idiopathic urticaria (CIU), chronic hives without a known cause) in people 12 years of age and older who continue to have hives that are not controlled with H1 antihistamine treatment. It is not known if XOLAIR is safe and effective in people with CSU under 12 years of age.
Who should not receive and use XOLAIR?
Do not receive and use XOLAIR if you:- are allergic to omalizumab or any of the ingredients in XOLAIR. See the end of this Medication Guide for a complete list of ingredients in XOLAIR.
What should I tell my healthcare provider before receiving XOLAIR?
Before receiving XOLAIR, tell your healthcare provider about all of your medical conditions, including if you:- have a latex allergy or any other allergies (such as seasonal allergies). The needle cap on the XOLAIR prefilled syringe contains a type of natural rubber latex.
- have sudden breathing problems (bronchospasm).
- have ever had a severe allergic reaction called anaphylaxis.
- have or have had a parasitic infection.
- have or have had cancer.
- are pregnant or plan to become pregnant. It is not known if XOLAIR may harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if XOLAIR passes into your breast milk. Talk with your healthcare provider about the best way to feed your baby while you receive and use XOLAIR.
How should I receive and use XOLAIR? - When starting treatment XOLAIR should be given by your healthcare provider in a healthcare setting.
- If your healthcare provider decides that you or a caregiver may be able to give your own XOLAIR prefilled syringe or autoinjector injections, you should receive training on the right way to prepare and inject XOLAIR.
- Do not try to inject XOLAIR until you have been shown the right way to give XOLAIR prefilled syringe or autoinjector injections by a healthcare provider. Use XOLAIR exactly as prescribed by your healthcare provider.
- The XOLAIR autoinjector (all doses) is intended for use only in adults and adolescents aged 12 years and older. For children 12 years of age and older, XOLAIR prefilled syringe or autoinjector may be self-injected under adult supervision. For children 1 to 11 years of age, XOLAIR prefilled syringe should be injected by a caregiver.
- See the detailed Instructions for Use that comes with XOLAIR for information on the right way to prepare and inject XOLAIR.
- XOLAIR is given in 1 or more injections under the skin (subcutaneous), 1 time every 2 or 4 weeks.
- In people with asthma,CRSwNP and food allergy, a blood test for a substance called IgE must be performed before starting XOLAIR to determine the appropriate dose and dosing frequency.
- In people with chronic hives, a blood test is not necessary to determine the dose or dosing frequency.
- Do not decrease or stop taking any of your other asthma, CRSwNP, hive medicine, food allergy medicine or allergen immunotherapy unless your healthcare providers tell you to.
- You may not see improvement in your symptoms right away after XOLAIR treatment. If your symptoms do not improve or get worse, call your healthcare provider.
- If you inject more XOLAIR than prescribed, call your healthcare provider right away.
What are the possible side effects of XOLAIR?
XOLAIR may cause serious side effects, including:- See "What is the most important information I should know about XOLAIR?"
- Cancer. Cases of cancer were observed in some people who received XOLAIR.
- Inflammation of your blood vessels. Rarely, this can happen in people with asthma who receive XOLAIR. This usually, but not always, happens in people who also take a steroid medicine by mouth that is being stopped or the dose is being lowered. It is not known whether this is caused by XOLAIR. Tell your healthcare provider right away if you have:
- rash
- shortness of breath
- chest pain
- a feeling of pins and needles or numbness of your arms or legs
- Fever, muscle aches, and rash. Some people get these symptoms 1 to 5 days after receiving a XOLAIR injection. If you have any of these symptoms, tell your healthcare provider.
- Parasitic infection. Some people who are at a high risk for parasite (worm) infections, get a parasite infection after receiving XOLAIR. Your healthcare provider can test your stool to check if you have a parasite infection.
- Heart and circulation problems. Some people who receive XOLAIR have had chest pain, heart attack, blood clots in the lungs or legs, or temporary symptoms of weakness on one side of the body, slurred speech, or altered vision. It is not known whether these are caused by XOLAIR.
- In adults and children 12 years of age and older with asthma: joint pain especially in your arms and legs, dizziness, feeling tired, itching, skin rash, bone fractures, and pain or discomfort of your ears.
- In children 6 to less than 12 years of age with asthma: swelling of the inside of your nose, throat, or sinuses, headache, fever, throat infection, ear infection, abdominal pain, stomach infection, and nose bleeds.
- In adults with chronic rhinosinusitis with nasal polyps: headache, injection site reactions, joint pain, upper abdominal pain, and dizziness.
- In people with chronic spontaneous urticaria: nausea, headaches, swelling of the inside of your nose, throat or sinuses, cough, joint pain, and upper respiratory tract infection.
- In people with food allergy: injection site reactions and fever.
How should I store XOLAIR? - Store XOLAIR in the refrigerator between 36°F to 46°F (2°C to 8°C).
- Keep your unused XOLAIR prefilled syringes or autoinjectors in the original carton until use to protect them from light.
- XOLAIR prefilled syringe or autoinjector can be removed from and placed back in the refrigerator if needed. The total combined time out of refrigerator may not be more than 2 days. Do not use if XOLAIR prefilled syringe or autoinjector is left at temperatures above 77°F (25°C) and discard in a sharps disposal container.
- Do not freeze. Do not use if XOLAIR prefilled syringes or autoinjectors have been frozen.
- Keep XOLAIR out of direct sunlight.
- Do not use XOLAIR past the expiration date.
General information about the safe and effective use of XOLAIR.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use XOLAIR for a condition for which it was not prescribed. Do not give XOLAIR to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about XOLAIR that is written for health professionals.
For more information, go to www.xolair.com or call 1-866-4XOLAIR (1-866-496-5247).What are the ingredients in XOLAIR?
Active ingredient: omalizumab
Inactive ingredients:
Prefilled syringe or Autoinjector: arginine hydrochloride, histidine, L-histidine hydrochloride monohydrate, and polysorbate 20
Vial: histidine, L-histidine hydrochloride monohydrate, polysorbate 20 and sucrose
Manufactured by: Genentech, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990. U.S. License No.: 1048
Jointly marketed by:
Genentech USA, Inc., A Member of the Roche Group, 1 DNA Way, South San Francisco, CA 94080-4990
Novartis Pharmaceuticals Corporation, One Health Plaza, East Hanover, NJ 07936-1080
XOLAIR® is a registered trademark of Novartis AG. ©2024 Genentech USA, Inc. -
INSTRUCTIONS FOR USE
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: 2/2024 Instructions for Use
Xolair® (ZOHL-air)
(omalizumab)
injection, for subcutaneous use
Prefilled SyringeRead this Instructions for Use before you start using the XOLAIR prefilled syringe and each time you get a refill. Before you use the XOLAIR prefilled syringe for the first time, make sure your healthcare provider shows you the right way to use it. Contact your healthcare provider if you have any questions. Do not use XOLAIR for the emergency treatment of any allergic reactions, including anaphylaxis, hives, or sudden breathing problems. Supplies needed to give your injection - XOLAIR prefilled syringes (You may need more than 1 prefilled syringe for your prescribed dose). See "Choose the correct prefilled syringe or combination of prefilled syringes" for more information. Each XOLAIR carton contains 1 prefilled syringe.
- Alcohol swab
- Sterile cotton ball or gauze
- Small bandage
- Sharps disposal container (see step 14 "After the Injection")
Choose the correct prefilled syringe or combination of prefilled syringes
XOLAIR prefilled syringes are available in 2 dose strengths. These instructions are to be used for both dose strengths.
Your prescribed dose may require more than 1 injection. The table below shows the combination of prefilled syringes needed to give your full dose. Check the label on the XOLAIR carton to make sure you have received the correct prefilled syringe or combination of prefilled syringes for your prescribed dose. If your dose requires more than 1 injection, inject the medicine from all of your prescribed prefilled syringes, immediately one after another. Contact your healthcare provider if you have any questions.
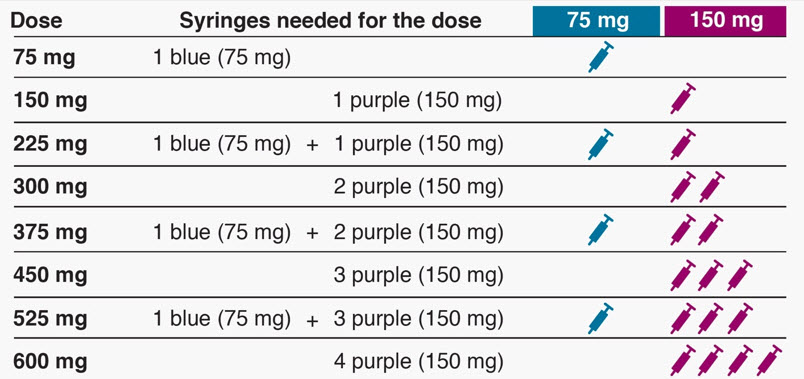
How should I store XOLAIR? - Keep your unused prefilled syringes in the original carton and store the carton in a refrigerator between 36°F to 46°F (2°C to 8°C). Do not remove the prefilled syringe from its original carton during storage.
- Before giving an injection, the carton can be removed from and placed back in the refrigerator if needed. The total combined time out of the refrigerator may not exceed 2 days. If the prefilled syringe is exposed to temperatures above 77 °F (25 °C), do not use it and throw away in a sharps disposal container.
- Keep your XOLAIR prefilled syringes out of direct sunlight.
- Do not freeze. Do not use if the prefilled syringe has been frozen.
Important Information - The needle cap contains a type of natural rubber latex. Tell your healthcare provider if you have a latex allergy.
- Do not open the sealed carton until you are ready to inject XOLAIR.
- Do not take the needle cap off until you are ready to inject XOLAIR.
- Do not try to take the prefilled syringe apart at any time.
- Do not reuse the same prefilled syringe.
- Do not leave the prefilled syringe unattended.
Preparing for the Injection 1 Take the carton containing the prefilled syringe out of the refrigerator. - If your dose requires you to give more than 1 injection, take all cartons out of the refrigerator at the same time. The following steps must be followed for each prefilled syringe.
2 Check the expiration date on the XOLAIR carton. 
-
Do not use if the expiration date has passed.
If the expiration date has passed, safely throw away the prefilled syringe in a sharps disposal container (see step 14) and contact your healthcare provider.
3 Place the carton on a clean, flat surface. - Set aside the carton for at least 15 to 30 minutes so the prefilled syringe can warm up on its own to room temperature. Leave the prefilled syringe in the carton to protect it from light.
- If the prefilled syringe does not reach room temperature, this could cause the injection to feel uncomfortable and make it hard to push the plunger.
- Do not speed up the warming process using any heat sources such as warm water or a microwave.
4 Open the carton. 
- Wash your hands with soap and water.
- Take the blister pack out of the carton.
- Check the expiration date on the blister pack.
-
Do not use it if the expiration date has passed.
If the expiration date has passed, safely throw away the prefilled syringe in a sharps disposal container (see step 14) and contact your healthcare provider.
- Peel off the blister pack cover fully. Be careful when taking out the prefilled syringe.
- Do not flip the blister pack upside down to take out the prefilled syringe. This may damage the prefilled syringe.
- Take the prefilled syringe out of the blister pack by holding the middle part of the prefilled syringe. When holding the prefilled syringe, make sure you always hold the prefilled syringe as shown. Do not touch the needle-shield wings (see "Prefilled Syringe Parts").
- Do not handle the prefilled syringe by holding the plunger or needle cap.
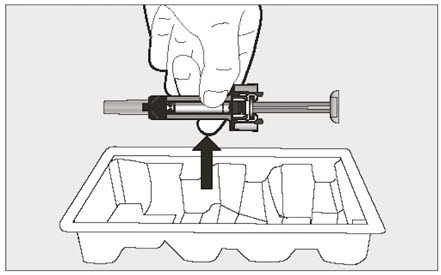
5 Inspect the prefilled syringe closely. - Check the prefilled syringe. The medicine in the prefilled syringe should be clear and colorless to pale brownish-yellow. Do not use the prefilled syringe if the medicine is cloudy, discolored, or contains particles.
- Check the expiration date on the prefilled syringe. Do not use the prefilled syringe if the expiration date has passed.
- If the medicine does not look as described or if the expiration date has passed, safely throw away the prefilled syringe in a sharps disposal container (see step 14) and contact your healthcare provider.
- Do not use if the packaging or prefilled syringe appears damaged, tampered with, or has been dropped.
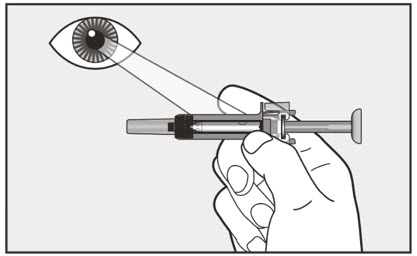
6 Choose an injection site. - If you are giving yourself the injection, you can inject into the front and middle of the thighs and the stomach area (abdomen). The outer area of the upper arms may also be used if the injection is being given by a caregiver. Do not try to inject into the upper arm area by yourself.
- Do not inject within the 2-inch area directly around your belly button (navel).
- Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard, or if there are breaks in the skin.
- Do not inject through clothing. The injection site should be exposed, clean skin.
- If your prescribed dose requires more than 1 injection, choose a different injection site for each new injection, at least 1 inch from other injection sites.
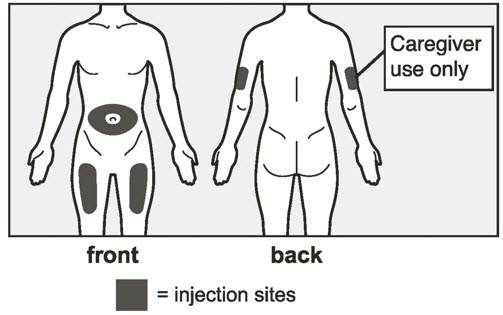
7 Wipe the injection site with an alcohol swab in a circular motion and let it air dry for 10 seconds. - Do not touch the injection site again before giving the injection.
- Do not fan or blow on the cleaned skin.
Giving the Injection 8 Hold the prefilled syringe firmly by the center with 1 hand and pull the needle cap straight off with your other hand. - Do not twist the needle cap.
- Do not hold, push or pull the plunger while you remove the needle cap.
- Do not touch the needle or let it touch any surfaces after removing the needle cap.
- Throw away the needle cap in regular household trash. Do not recap the needle.
- There may be 1 or more small air bubbles in the prefilled syringe. This is normal, and you should not try to remove the air bubbles.
- You may also see a drop of liquid at the end of the needle. This is also normal and will not affect the dose.
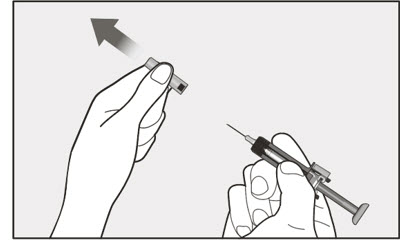
9 Use your other hand and gently pinch the area of skin that was cleaned. Hold the pinched skin tight. - Pinching the skin is important to make sure that you inject under the skin (into the fatty area) but not any deeper (into muscle).
10 Continue holding the prefilled syringe by the center and use a quick, dart-like motion to insert the needle all the way into the pinched skin at an angle between 45 degrees to 90 degrees as shown. 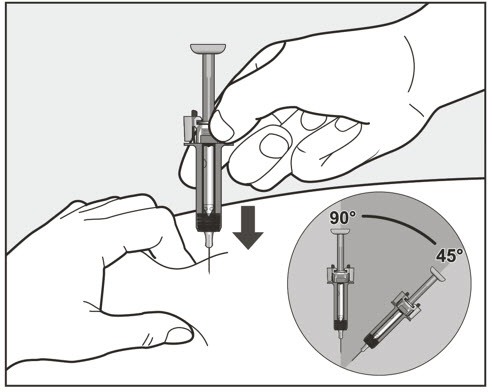
- It is important to use the correct angle to make sure the medicine is delivered under the skin (into the fatty area), or the injection could be uncomfortable and the medicine may not work.
- Do not touch the plunger while inserting the needle into the skin.
- Do not insert the needle through clothing.
- Hold the prefilled syringe tightly in place and do not change the angle of injection or insert the needle again after the needle is inserted.
- You should not move and should avoid sudden movements when giving the injection.
11 Slowly inject all of the medicine by gently pushing the plunger all the way down until the needle-shield wings are pushed apart. - You must press the plunger all the way down to make sure that the full dose of medicine gets injected. If the plunger is not fully pressed, the needle-shield will not extend to cover the needle when it is removed.
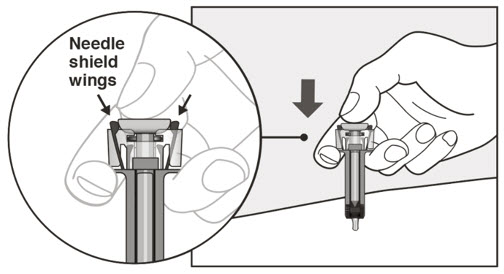
12 Release the plunger and allow the needle to be covered by the needle-shield. - If the needle is not covered by the needle-shield, carefully remove the prefilled syringe from the skin and throw away the prefilled syringe in a sharps disposal container (see step 14).
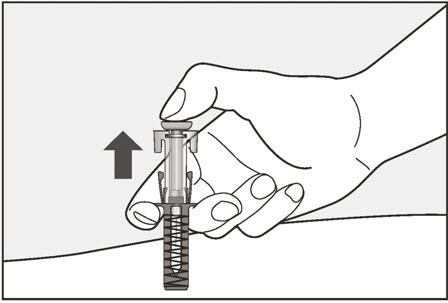
13 There may be a little bleeding at the injection site. You can press a cotton ball or gauze over the injection site. - Do not rub the injection site.
- If needed, cover the injection site with a small bandage.
- In case of skin contact with the medicine, wash the area that touched the medicine with water.
If your prescribed dose requires more than 1 injection: - Throw away the used prefilled syringe as described in step 14.
- Repeat step 2 through step 13 for the next injection using a new prefilled syringe.
- Choose a different injection site for each new injection at least 1 inch from other injection sites.
- Complete all the required injections for your prescribed dose, immediately one after another. Contact your healthcare provider if you have any questions.
After the Injection 14 Throw away (dispose of) your used XOLAIR prefilled syringes in an FDA-cleared sharps disposal container right away after use.
- The XOLAIR prefilled syringe is a single-dose prefilled syringe and should not be used again.
- Do not throw away prefilled syringes in your household trash.
- Do not recap the needle.
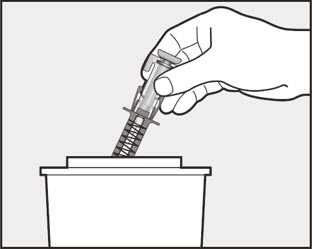
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is: - made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used prefilled syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
Manufactured by:
Genentech, Inc., A Member of the Roche Group,
1 DNA Way, South San Francisco, CA 94080-4990
U.S. License No.: 1048Jointly marketed by:
Genentech USA, Inc., A Member of the Roche Group,
1 DNA Way, South San Francisco, CA 94080-4990
Novartis Pharmaceuticals Corporation,
One Health Plaza, East Hanover, NJ 07936-1080XOLAIR® is a registered trademark of Novartis AG.
©2024 Genentech USA, Inc. -
INSTRUCTIONS FOR USE
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Approved: 2/2024 Instructions for Use
Xolair® (ZOHL-air)
(omalizumab)
injection, for subcutaneous use
Prefilled SyringeRead this Instructions for Use before you start using Xolair prefilled syringe and each time you get a refill. Before you use Xolair prefilled syringe for the first time, make sure your healthcare provider shows you the right way to use it. Contact your healthcare provider if you have any questions. Do not use Xolair for the emergency treatment of any allergic reactions, including anaphylaxis, hives, or sudden breathing problems. Prefilled Syringe Parts
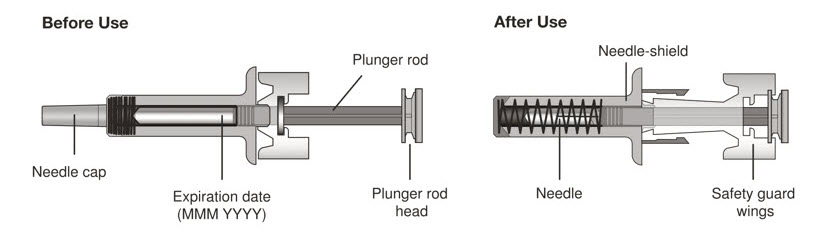
Supplies needed to give your injection - Xolair prefilled syringe .
- Alcohol swab
- Sterile cotton ball or gauze
- Small bandage
- Sharps disposal container (see Step 14)
Note: You may need more than 1 Xolair prefilled syringe for your prescribed dose. See the Dosing Table for more information. Each Xolair carton contains 1 prefilled syringe. Choose the correct prefilled syringe or combination of prefilled syringes
Xolair prefilled syringes are available in 3 dose strengths (1 prefilled syringe in each carton). These instructions are to be used for all 3 dose strengths.
Your prescribed dose may require more than 1 injection. The Dosing Table below shows the combination of prefilled syringes needed to give your full dose. Check the label on the Xolair carton to make sure you have received the correct prefilled syringe or combination of prefilled syringes for your prescribed dose. If your dose requires more than 1 injection, complete all injections for your prescribed dose, immediately one after another. Contact your healthcare provider if you have any questions.Dosing Table 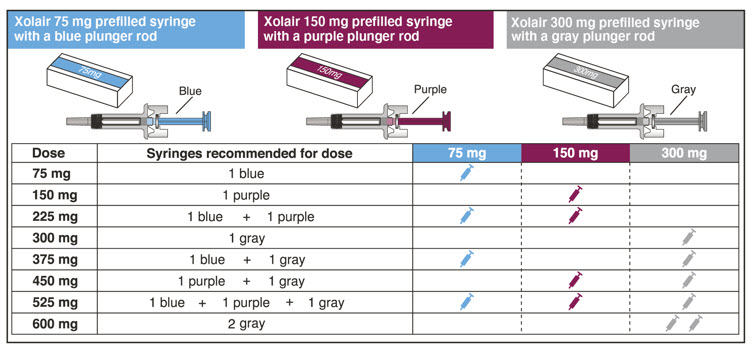
Note: Your healthcare provider may prescribe a different combination of syringes for your complete dose. How should I store Xolair? - Keep your unused prefilled syringes in the original carton and store the carton in a refrigerator between 36°F to 46°F (2°C to 8°C). Do not remove the prefilled syringe from its original carton during storage.
- Before giving an injection, the carton can be removed from and placed back in the refrigerator if needed. The total combined time out of the refrigerator may not be more than 2 days. If the prefilled syringe is left at temperatures above 77°F (25°C), do not use it and throw away in a sharps disposal container.
- Keep your prefilled syringes out of direct light.
- Do not freeze. Do not use if the prefilled syringe has been frozen.
Important Information- Do not use if the carton is damaged or appears to be tampered with.
- Do not open the carton until you are ready to inject.
- Do not use if the prefilled syringe is damaged or appears to be tampered with.
- Do not take the needle cap off until you are ready to inject.
- Do not use if the prefilled syringe has been dropped on a hard surface or dropped after removing the needle cap.
- Do not try to take apart the prefilled syringe at any time.
Preparing for the Injection 1 Take the carton containing the prefilled syringe out of the refrigerator. - If you need more than 1 prefilled syringe to deliver your full dose (see Dosing Table), take all cartons out of the refrigerator at the same time (each carton contains 1 prefilled syringe). The following steps must be followed for each prefilled syringe.
2 Check the expiration date on the Xolair carton. - Do not use if the expiration date has passed.
- If the expiration date has passed, safely throw away the prefilled syringe in a sharps disposal container (see Step 14) and contact your healthcare provider.
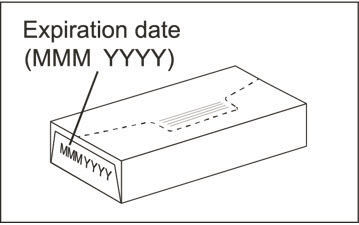
3 Allow the prefilled syringe to reach room temperature. - Set aside the carton on a clean, flat surface for at least 30 to 45 minutes so the prefilled syringe can warm up on its own to room temperature. Leave the prefilled syringe in the carton to protect it from light.
- If the prefilled syringe does not reach room temperature, this could cause the injection to feel uncomfortable and make it hard to push the plunger.
- Do not speed up the warming process using any heat sources such as warm water or a microwave.
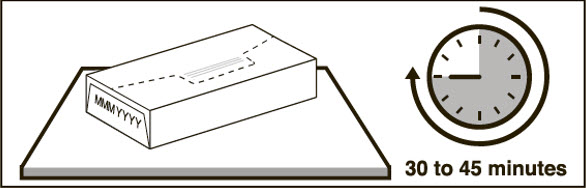
4 Open the carton. - Wash your hands with soap and water.
- Take the blister pack out of the carton.
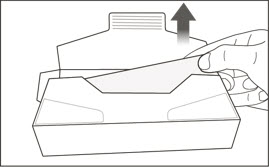
- Check the expiration date on the blister pack.
-
Do not use it if the expiration date has passed.
If the expiration date has passed, safely throw away the prefilled syringe in a sharps disposal container (see Step 14) and contact your healthcare provider.
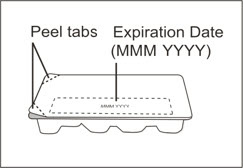
- Peel off the blister pack cover fully. Be careful when taking out the prefilled syringe.
- Do not flip the blister pack upside down to take out the prefilled syringe.
- Take the prefilled syringe out of the blister pack by holding the middle part of the prefilled syringe, as shown.
- Do not handle the prefilled syringe by holding the plunger or needle cap.
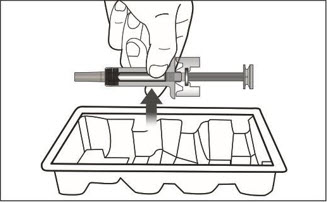
5 Inspect the prefilled syringe closely. - Check the prefilled syringe. The medicine inside should be clear and colorless to pale brownish-yellow. You may see air bubbles in the medicine, which is normal. Do not try to remove the air bubbles.
- Do not use the prefilled syringe if the medicine contains particles, or if the medicine looks cloudy or brown.
- Check the expiration date on the prefilled syringe. Do not use the prefilled syringe if the expiration date has passed.
- Do not use if the prefilled syringe appears to be damaged or tampered with.
- If the medicine does not look as described or if the expiration date has passed, safely throw away the prefilled syringe in a sharps disposal container (see Step 14) and contact your healthcare provider.
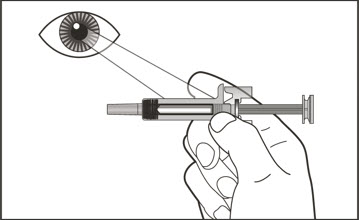
6 Choose where to inject. - If giving yourself the injection, the injection sites are the front of your thigh or stomach area (abdomen). If a caregiver is giving the injection, the injection sites are the outer area of the upper arm, the front of the thigh, or the stomach area (abdomen). Do not try to inject yourself in the upper arm.
- Do not inject within the 2-inch area directly around your belly button (navel).
- Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard, or if there are breaks in the skin.
- Do not inject through clothing. The injection site should be uncovered, clean skin.
- If your prescribed dose requires more than 1 injection, make sure your injections are at least 1 inch apart from each other.
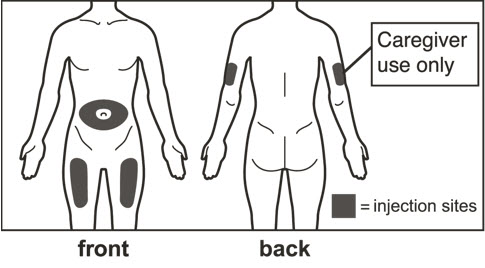
7 Clean the injection site. - Wipe the injection site with an alcohol swab in a circular motion and let air dry for 10 seconds.
- Do not fan or blow on the cleaned skin.
- Do not touch the injection site again before giving the injection.
Giving the Injection 8 Remove the cap. - Hold the prefilled syringe firmly with 1 hand and pull the needle cap straight off with your other hand as shown. Do not twist the cap.
- Do not hold, push or pull the plunger while you remove the needle cap.
- Do not touch the needle or let it touch any surfaces after removing the needle cap.
- Do not put the cap back on the prefilled syringe. Throw away the needle cap in regular household trash.
- You may see a drop of liquid at the end of the needle. This is normal.
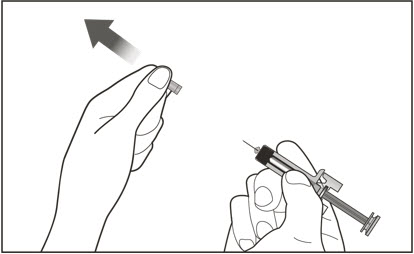
9 Pinch cleaned skin. - Use your other hand and gently pinch the area of skin that was cleaned. Hold the pinched skin tightly until the injection is complete.
- Pinching the skin is important to make sure that you inject under the skin (into the fatty area) but not any deeper (into muscle).
10 Insert the needle using a quick, dart-like motion. - While holding the prefilled syringe by the center, insert the needle all the way into the pinched skin at an angle of about 45 degrees as shown.
- It is important to use the correct angle to make sure the medicine is delivered under the skin (into the fatty area), or the injection could be uncomfortable and the medicine may not work.
- Do not touch the plunger while inserting the needle into the skin.
- Hold the prefilled syringe tightly in place and do not change the angle of injection or insert the needle again after the needle is inserted.
- You should not move and should avoid sudden movements when giving the injection.
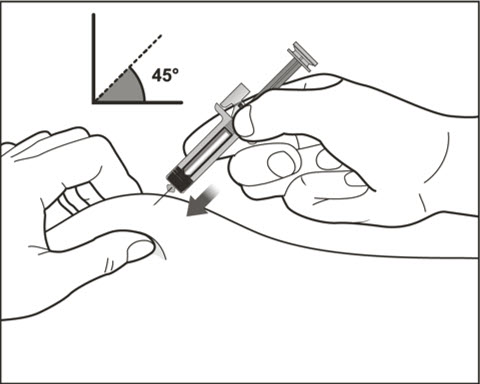
11 Slowly inject all of the medicine. - Gently push the plunger all the way down until the plunger head is between the safety guard wings as shown.
- You must press the plunger all the way down to make sure that the full dose of medicine is injected. If the plunger is not fully pressed, the needle-shield will not extend to cover the needle when it is removed.
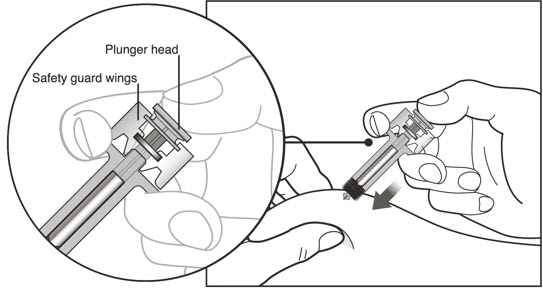
12 Release the plunger. - Release the plunger and allow the needle to be covered by the needle-shield.
- If the needle is not covered by the needle-shield, carefully remove the prefilled syringe from the skin and throw away the prefilled syringe in a sharps disposal container (see Step 14).
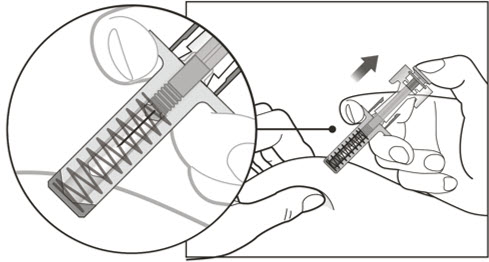
13 Care for the injection site. - Do not rub the injection site.
- There may be a little bleeding or drop of liquid at the injection site. You can press a cotton ball or gauze over the injection site until the bleeding stops.
- In case of skin contact with the medicine, wash the area that touched the medicine with water.
- If needed, cover the injection site with a small bandage.
If your prescribed dose requires more than 1 injection: - Throw away the used prefilled syringe as described in Step 14.
- Repeat Step 2 through Step 13 for the next injection using a new prefilled syringe.
- Make sure each injection is at least 1 inch apart from each other.
- Complete all the required injections for your prescribed dose, immediately one after another. Contact your healthcare provider if you have any questions.
After the Injection 14 Throw away (dispose of) the prefilled syringe. - Put your used prefilled syringes in an FDA-cleared sharps disposal container right away after use.
- The Xolair prefilled syringe is a single-dose prefilled syringe and should not be used again.
- Do not throw away the prefilled syringes in your household trash.
- Do not recap the needle.
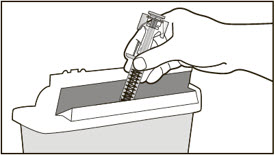
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is: - made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used prefilled syringes. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
Manufactured by:
Genentech, Inc., A Member of the Roche Group,
1 DNA Way, South San Francisco, CA 94080-4990
U.S. License No.: 1048Jointly marketed by:
Genentech USA, Inc., A Member of the Roche Group,
1 DNA Way, South San Francisco, CA 94080-4990
Novartis Pharmaceuticals Corporation,
One Health Plaza, East Hanover, NJ 07936-1080Xolair® is a registered trademark of Novartis AG.
©2024 Genentech USA, Inc. -
INSTRUCTIONS FOR USE
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Approved: 2/2024 Instructions for Use
Xolair® (ZOHL-air)
(omalizumab)
injection, for subcutaneous use
AutoinjectorRead this Instructions for Use before you start using Xolair autoinjector and each time you get a refill. Before you use Xolair autoinjector for the first time, make sure your healthcare provider shows you the right way to use it. Contact your healthcare provider if you have any questions. The Xolair autoinjector is for use in people 12 years of age and older. Do not use Xolair for the emergency treatment of any allergic reactions, including anaphylaxis, hives, or sudden breathing problems. Xolair Autoinjector Parts 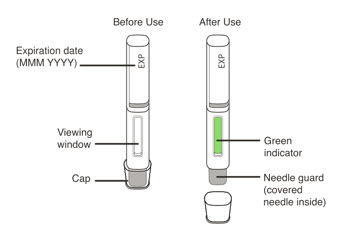
Supplies needed to give your injection - Xolair autoinjector
- Alcohol swab
- Sterile cotton ball or gauze
- Small bandage
- Sharps disposal container (see Step 15)
Note: You may need more than 1 Xolair autoinjector to give your prescribed dose. See the Dosing Table for more information. Each Xolair carton contains 1 autoinjector. Choose the correct autoinjector or combination of autoinjectors Xolair autoinjectors are available in 3 dose strengths (1 autoinjector in each carton). These instructions are to be used for all 3 dose strengths. Your prescribed dose may require more than 1 injection. The Dosing Table below shows the combination of autoinjectors needed to give your full dose. Check the label on the Xolair carton to make sure you have received the correct autoinjector or combination of autoinjectors for your prescribed dose. If your dose requires more than 1 injection, complete all injections for your prescribed dose, immediately one after another. Contact your healthcare provider if you have any questions. Dosing Table 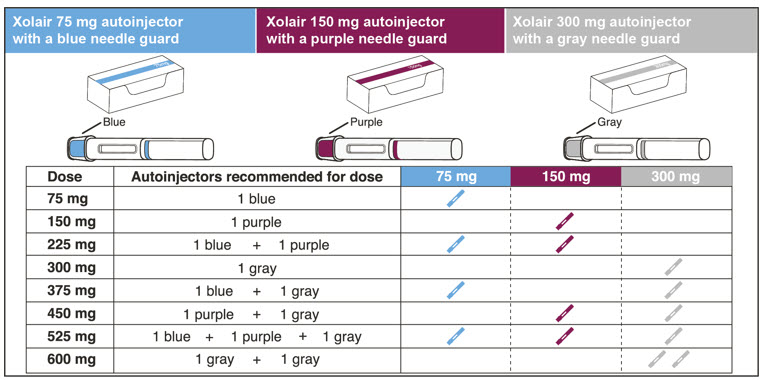
Note: Your healthcare provider may prescribe a different combination of autoinjectors for your complete dose. How should I store Xolair? - Keep your unused autoinjectors in the original carton and store the carton in a refrigerator between 36°F to 46°F (2°C to 8°C).
- Before giving an injection, the carton can be removed from and placed back in the refrigerator if needed. The total combined time out of the refrigerator may not be more than 2 days. If the autoinjector is left at temperatures above 77°F (25°C), do not use it and throw away in a sharps disposal container.
- Do not remove the autoinjector from its original carton during storage.
- Keep your autoinjectors out of direct light.
- Do not freeze. Do not use if the autoinjector has been frozen.
Important Information - Do not use if the carton is damaged or appears to be tampered with.
- Do not open the carton until you are ready to inject.
- Do not use if the autoinjector has been damaged or appears to be tampered with.
- Do not take the cap off the autoinjector until you are ready to inject.
- Do not use if the autoinjector has been dropped on a hard surface or dropped after removing the needle cap.
- Do not take apart the autoinjector at any time.
- Do not clean or touch the needle guard.
Preparing for the Injection 1 Take the carton containing the autoinjector out of the refrigerator. - If you need more than 1 autoinjector to deliver your prescribed dose (see Dosing Table), take all cartons out of the refrigerator at the same time (each carton contains 1 autoinjector). The following steps must be followed for each autoinjector.
2 Check the expiration date on the back of the Xolair carton. - Do not use if the expiration date has passed.
- If the expiration date has passed, safely throw away the autoinjector in a sharps disposal container (see Step 15) and contact your healthcare provider.
3 Allow the autoinjector to reach room temperature. - Set aside the carton on a clean, flat surface for at least 30 to 45 minutes so the autoinjector can warm up on its own to room temperature. Leave the autoinjector in the carton to protect it from light.
- If the autoinjector does not reach room temperature, this could cause the injection to feel uncomfortable.
- Do not speed up the warming process using any heat sources such as warm water or a microwave.
4 Open the carton. - Wash your hands with soap and water.
- Take the autoinjector out of the carton as shown.
- Do not turn the carton upside down to take out the autoinjector, as this may damage the autoinjector.
- Do not hold the autoinjector by the cap. Make sure you hold the autoinjector by the middle as shown.
- Do not remove the cap until you are ready to inject.
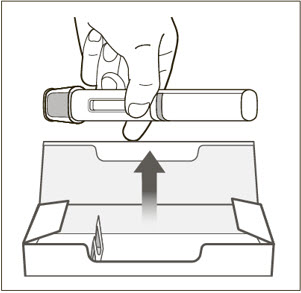
5 Check the autoinjector before use. - Look through the viewing window of the autoinjector. The medicine in the autoinjector viewing window should be clear and colorless to pale brownish-yellow. You may see air bubbles in the medicine, which is normal.
- Do not use the autoinjector if the medicine contains particles, or if the medicine looks cloudy or brown.
- Check the expiration date on the autoinjector. Do not use the autoinjector if the expiration date has passed.
- Do not use the autoinjector if it appears to be damaged or tampered with.
- If the medicine does not look as described or if the expiration date has passed, safely throw away the autoinjector in a sharps disposal container (see Step 15) and contact your healthcare provider.
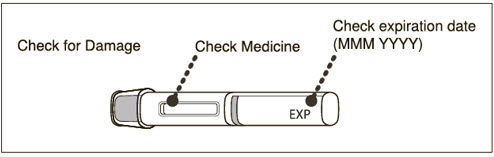
6 Choose where to inject. - If giving yourself the injection, the injection sites are the front of your thigh or stomach area (abdomen). If a caregiver is giving the injection, the injection sites are the outer area of the upper arm, the front of the thigh or stomach area (abdomen). Do not try to inject yourself in the upper arm.
- Do not inject within the 2-inch area directly around your belly button (navel).
- Do not inject into moles, scars, bruises, or areas where the skin is tender, red, hard, or if there are breaks in the skin.
- Do not inject through clothing. The injection site should be uncovered, clean skin.
- If your prescribed dose requires more than 1 injection, make sure your injections are at least 1 inch apart from each other.
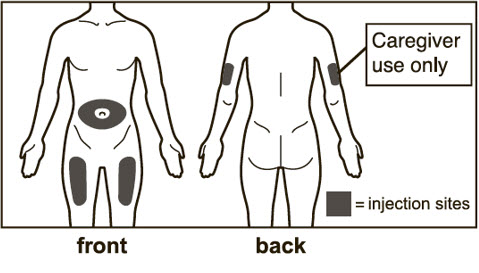
7 Clean the injection site. - Wipe the injection site with an alcohol swab in a circular motion and let it air dry for 10 seconds.
- Do not fan or blow on the cleaned skin.
- Do not touch the injection site again before giving the injection.
Giving the Injection 8 Remove the cap. - Hold the autoinjector firmly with 1 hand and pull the cap straight off with your other hand as shown. Do not twist the cap.
- Do not put the cap back on the autoinjector. Throw away the cap in regular household trash.
- Do not clean or touch the needle guard of the device.
9 Position the autoinjector. - Hold the autoinjector comfortably with the needle guard directly against the skin.
- The autoinjector should be at a 90-degree angle against the skin, as shown.
10 Start the injection. - Press straight down and hold the autoinjector firmly against the skin. The 1st click indicates that the injection has started.
- Hold the autoinjector tightly in place. Do not change the angle of injection or remove the autoinjector until the injection is completed.
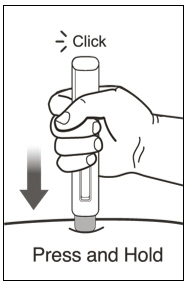
11 Monitor injection using green indicator. - Keep holding the autoinjector against the skin. The green indicator will move within the viewing window.
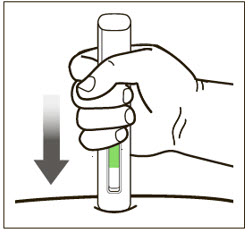
12 Complete injection. - Listen for the 2nd click. This indicates that the injection is almost complete.
- Hold the autoinjector in position until the green indicator has stopped moving and completely fills the viewing window to make sure the injection is complete.
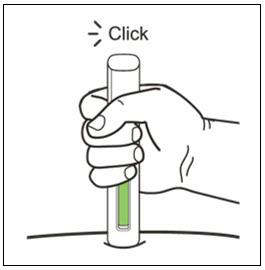
13 Remove from skin and check the green indicator. - After the green indicator has stopped moving and has completely filled the viewing window, lift the autoinjector straight up from the skin. The needle guard will automatically extend and lock over the needle.
- If the green indicator has not completely filled the viewing window, contact your healthcare provider or pharmacist.
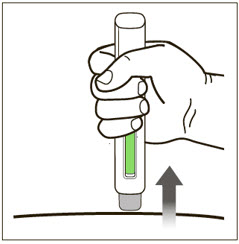
14 Care for the injection site. - Do not rub the injection site.
- There may be a little bleeding or a drop of liquid at the injection site. You can press a cotton ball or gauze pad over the injection site until any bleeding stops.
- In case of skin contact with the medicine, wash the area that touched the medicine with water.
- If needed, cover the injection site with a small bandage.
If your prescribed dose requires more than 1 injection: - Throw away the used autoinjector as described in Step 15.
- Repeat Step 2 through Step 14 for the next injection using a new autoinjector.
- Make sure each injection is at least 1 inch apart from each other.
- Complete all the required injections for your prescribed dose, immediately one after another. Contact your healthcare provider if you have any questions.
After the Injection 15 Throw away (dispose of) autoinjector. - Put your used autoinjectors in an FDA-cleared sharps disposal container right away after use.
- The Xolair autoinjector is a single-dose autoinjector and should not be used again.
- Do not throw away (dispose of) the autoinjectors in your household trash.
- Do not recap the autoinjector.
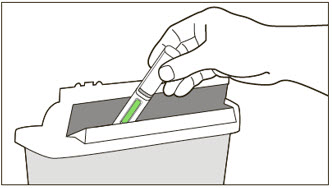
If you do not have an FDA-cleared sharps disposal container, you may use a household container that is: - made of a heavy-duty plastic,
- can be closed with a tight-fitting, puncture-resistant lid, without sharps being able to come out,
- upright and stable during use,
- leak-resistant, and
- properly labeled to warn of hazardous waste inside the container.
- When your sharps disposal container is almost full, you will need to follow your community guidelines for the right way to dispose of your sharps disposal container. There may be state or local laws about how you should throw away used autoinjectors. For more information about safe sharps disposal, and for specific information about sharps disposal in the state that you live in, go to the FDA's website at: http://www.fda.gov/safesharpsdisposal
- Do not dispose of your used sharps disposal container in your household trash unless your community guidelines permit this.
- Do not recycle your used sharps disposal container.
Manufactured by:
Genentech, Inc., A Member of the Roche Group,
1 DNA Way, South San Francisco, CA 94080-4990
U.S. License No.: 1048Jointly marketed by:
Genentech USA, Inc., A Member of the Roche Group,
1 DNA Way, South San Francisco, CA 94080-4990
Novartis Pharmaceuticals Corporation,
One Health Plaza, East Hanover, NJ 07936-1080Xolair® is a registered trademark of Novartis AG.
©2024 Genentech USA, Inc. -
SPL UNCLASSIFIED SECTION
Representative sample of labeling (see the HOW SUPPLIED section for complete listing):
- PRINCIPAL DISPLAY PANEL - 150 mg Vial Carton
-
PRINCIPAL DISPLAY PANEL - 75 mg/0.5 mL Syringe Carton - 214-01
Xolair®
(omalizumab) Injection75 mg/0.5 mL
For Subcutaneous Use. Single-Dose Prefilled Syringe.
Dispense the accompanying
Medication Guide to each patient.1 prefilled syringe
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-214-01
Caution: The needle cap may contain natural rubber latex
which may cause allergic reactions in latex sensitive individuals.Rx only
Genentech
NOVARTIS
11011555

-
PRINCIPAL DISPLAY PANEL - 75 mg/0.5 mL Syringe Carton - 214-03
Xolair®
(omalizumab) Injection75 mg/0.5 mL
For Subcutaneous Use.
1 Single-Dose Prefilled Syringe
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-214-03
Dispense the accompanying Medication Guide to each patient.
Rx only
Must be refrigerated
Genentech
NOVARTIS
11011485

-
PRINCIPAL DISPLAY PANEL - 75 mg/0.5 mL Autoinjector Carton
Xolair®
(omalizumab) Injection75 mg/0.5 mL
For Subcutaneous Use.
1 Single-Dose Autoinjector
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-214-55
Dispense the accompanying Medication Guide to each patient.
Genentech
NOVARTIS
11011505

-
PRINCIPAL DISPLAY PANEL - 150 mg/mL Syringe Carton - 215-01
Xolair®
(omalizumab) Injection150 mg/mL
For Subcutaneous Use. Single-Dose Prefilled Syringe.
Dispense the accompanying
Medication Guide to each patient.1 prefilled syringe
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-215-01
Caution: The needle cap may contain natural rubber latex
which may cause allergic reactions in latex sensitive individuals.Rx only
Genentech
NOVARTIS
11011498

-
PRINCIPAL DISPLAY PANEL - 150 mg/mL Syringe Carton - 215-03
Xolair®
(omalizumab) Injection150 mg/mL
For Subcutaneous Use.
1 Single-Dose Prefilled Syringe
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-215-03
Dispense the accompanying Medication Guide to each patient.
Rx only
Must be refrigerated
Genentech
NOVARTIS
11011522

-
PRINCIPAL DISPLAY PANEL - 150 mg/mL Autoinjector Carton
Xolair®
(omalizumab) Injection150 mg/mL
For Subcutaneous Use.
1 Single-Dose Autoinjector
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-215-55
Dispense the accompanying Medication Guide to each patient.
Genentech
NOVARTIS
11011532

-
PRINCIPAL DISPLAY PANEL - 300 mg/2 mL Syringe Carton
Xolair®
(omalizumab) Injection300 mg/2 mL
For Subcutaneous Use.
1 Single-Dose Prefilled Syringe
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-227-01
Dispense the accompanying Medication Guide to each patient.
Rx only
Must be refrigerated
Genentech
NOVARTIS
11011496

-
PRINCIPAL DISPLAY PANEL - 300 mg/2 mL Autoinjector Carton
Xolair®
(omalizumab) Injection300 mg/2 mL
For Subcutaneous Use.
1 Single-Dose Autoinjector
Do not use for emergency treatment.
Your dose may require more than 1 injection.
For questions, contact your healthcare provider.NDC 50242-227-55
Dispense the accompanying Medication Guide to each patient.
Genentech
NOVARTIS
11011548

-
INGREDIENTS AND APPEARANCE
XOLAIR
omalizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-040 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMALIZUMAB (UNII: 2P471X1Z11) (OMALIZUMAB - UNII:2P471X1Z11) OMALIZUMAB 150 mg in 1.2 mL Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 108 mg in 1.2 mL HISTIDINE HYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 2.1 mg in 1.2 mL HISTIDINE (UNII: 4QD397987E) 1.3 mg in 1.2 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.4 mg in 1.2 mL Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-040-62 1 in 1 CARTON 06/20/2003 1 1.2 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product 2 NDC:50242-040-86 1 in 1 CARTON 12/20/2013 2 1.2 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103976 06/20/2003 XOLAIR PFS
omalizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-214 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMALIZUMAB (UNII: 2P471X1Z11) (OMALIZUMAB - UNII:2P471X1Z11) OMALIZUMAB 75 mg in 0.5 mL Inactive Ingredients Ingredient Name Strength ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 21.05 mg in 0.5 mL HISTIDINE HYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 1.17 mg in 0.5 mL HISTIDINE (UNII: 4QD397987E) 0.68 mg in 0.5 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.2 mg in 0.5 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-214-01 1 in 1 CARTON 09/28/2018 1 0.5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC:50242-214-86 1 in 1 CARTON 09/28/2018 2 0.5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC:50242-214-03 1 in 1 CARTON 08/18/2023 3 0.5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 4 NDC:50242-214-55 1 in 1 CARTON 08/18/2023 4 0.5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 5 NDC:50242-214-99 1 in 1 CARTON 08/18/2023 5 0.5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 6 NDC:50242-214-83 1 in 1 CARTON 08/18/2023 6 0.5 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103976 09/28/2018 XOLAIR PFS
omalizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-215 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMALIZUMAB (UNII: 2P471X1Z11) (OMALIZUMAB - UNII:2P471X1Z11) OMALIZUMAB 150 mg in 1 mL Inactive Ingredients Ingredient Name Strength ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 42.1 mg in 1 mL HISTIDINE HYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 2.34 mg in 1 mL HISTIDINE (UNII: 4QD397987E) 1.37 mg in 1 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.4 mg in 1 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-215-86 1 in 1 CARTON 09/28/2018 1 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC:50242-215-01 1 in 1 CARTON 09/28/2018 2 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC:50242-215-03 1 in 1 CARTON 08/18/2023 3 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 4 NDC:50242-215-55 1 in 1 CARTON 08/18/2023 4 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 5 NDC:50242-215-99 1 in 1 CARTON 08/18/2023 5 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 6 NDC:50242-215-83 1 in 1 CARTON 08/18/2023 6 1 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103976 09/28/2018 XOLAIR
omalizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:50242-227 Route of Administration SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMALIZUMAB (UNII: 2P471X1Z11) (OMALIZUMAB - UNII:2P471X1Z11) OMALIZUMAB 300 mg in 2 mL Inactive Ingredients Ingredient Name Strength ARGININE HYDROCHLORIDE (UNII: F7LTH1E20Y) 84.2 mg in 2 mL HISTIDINE HYDROCHLORIDE MONOHYDRATE (UNII: X573657P6P) 4.68 mg in 2 mL HISTIDINE (UNII: 4QD397987E) 2.74 mg in 2 mL POLYSORBATE 20 (UNII: 7T1F30V5YH) 0.8 mg in 2 mL WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:50242-227-01 1 in 1 CARTON 08/18/2023 1 2 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 2 NDC:50242-227-55 1 in 1 CARTON 08/18/2023 2 2 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 3 NDC:50242-227-99 1 in 1 CARTON 08/18/2023 3 2 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 4 NDC:50242-227-86 1 in 1 CARTON 08/18/2023 4 2 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103976 08/18/2023 Labeler - Genentech, Inc. (080129000) Registrant - Genentech, Inc. (080129000) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 080129000 ANALYSIS(50242-040, 50242-214, 50242-215, 50242-227) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 146373191 ANALYSIS(50242-040, 50242-214, 50242-215, 50242-227) , API MANUFACTURE(50242-040, 50242-214, 50242-215, 50242-227) Establishment Name Address ID/FEI Business Operations Roche Diagnostics GmbH 323105205 ANALYSIS(50242-040, 50242-214, 50242-215, 50242-227) Establishment Name Address ID/FEI Business Operations Genentech, Inc. 833220176 PACK(50242-040) , LABEL(50242-040) , ANALYSIS(50242-040)

