Label: TAZVERIK- tazemetostat tablet, film coated

- NDC Code(s): 72607-100-00
- Packager: Epizyme, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated December 21, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TAZVERIK® safely and effectively. See full prescribing information for TAZVERIK.
TAZVERIK (tazemetostat) tablets, for oral use
Initial U.S. Approval: 2020RECENT MAJOR CHANGES
Warnings and Precautions, Secondary Malignancies (5.1) 11/2023 INDICATIONS AND USAGE
TAZVERIK is a methyltransferase inhibitor indicated for the treatment of:
- Adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection. (1.1)
- Adult patients with relapsed or refractory follicular lymphoma whose tumors are positive for an EZH2 mutation as detected by an FDA-approved test and who have received at least 2 prior systemic therapies. (1.2)
- Adult patients with relapsed or refractory follicular lymphoma who have no satisfactory alternative treatment options. (1.2)
These indications are approved under accelerated approval based on overall response rate and duration of response. Continued approval for these indications may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
DOSAGE AND ADMINISTRATION
- Recommended dosage is 800 mg taken orally twice daily with or without food. (2.2)
DOSAGE FORMS AND STRENGTHS
Tablets: 200 mg (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- Secondary Malignancies: TAZVERIK increases the risk of developing secondary malignancies, including T-cell lymphoblastic lymphoma, myelodysplastic syndrome, acute myeloid leukemia, and B-cell acute lymphoblastic leukemia. Monitor patients long-term for the development of secondary malignancies. (5.1)
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of potential risk to a fetus and to use effective non-hormonal contraception. (5.2)
ADVERSE REACTIONS
- The most common (≥20%) adverse reactions in patients with epithelioid sarcoma are pain, fatigue, nausea, decreased appetite, vomiting, and constipation. (6.1)
- The most common (≥20%) adverse reactions in patients with follicular lymphoma are fatigue, upper respiratory tract infection, musculoskeletal pain, nausea, and abdominal pain. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Epizyme at 855-463-5127 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong and Moderate Cytochrome P450 (CYP)3A Inhibitors: Avoid coadministration of strong and moderate CYP3A inhibitors with TAZVERIK. Reduce the dose of TAZVERIK if coadministration of moderate CYP3A inhibitors cannot be avoided. (2.3, 7.1)
- Strong and Moderate CYP3A Inducers: Avoid coadministration with TAZVERIK. (7.1)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Epithelioid Sarcoma
1.2 Relapsed or Refractory Follicular Lymphoma
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage
2.3 Dosage Modifications for Adverse Reactions
2.4 Dosage Modifications for Drug Interactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Secondary Malignancies
5.2 Embryo-Fetal Toxicity
6 ADVERSE REACTIONS
6.1 Clinical Trial Experience
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TAZVERIK
7.2 Effect of TAZVERIK on Other Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Renal Impairment
8.7 Hepatic Impairment
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Epithelioid Sarcoma
14.2 Relapsed or Refractory Follicular Lymphoma
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Epithelioid Sarcoma
TAZVERIK is indicated for the treatment of adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection.
This indication is approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14.1)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
1.2 Relapsed or Refractory Follicular Lymphoma
- TAZVERIK is indicated for the treatment of adult patients with relapsed or refractory (R/R) follicular lymphoma (FL) whose tumors are positive for an EZH2 mutation as detected by an FDA-approved test and who have received at least 2 prior systemic therapies.
- TAZVERIK is indicated for the treatment of adult patients with R/R FL who have no satisfactory alternative treatment options.
These indications are approved under accelerated approval based on overall response rate and duration of response [see Clinical Studies (14.2)]. Continued approval for these indications may be contingent upon verification and description of clinical benefit in a confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
Select patients with R/R FL for treatment with TAZVERIK based on the presence of EZH2 mutation of codons Y646, A682, or A692 in tumor specimens [see Clinical Studies (14.2)]. Information on FDA-approved tests for the detection of EZH2 mutation in relapsed or refractory follicular lymphoma is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage
The recommended dosage of TAZVERIK is 800 mg orally twice daily with or without food until disease progression or unacceptable toxicity.
Swallow tablets whole. Do not cut, crush, or chew tablets.
Do not take an additional dose if a dose is missed or vomiting occurs after TAZVERIK, but continue with the next scheduled dose.
2.3 Dosage Modifications for Adverse Reactions
Table 1 summarizes the recommended dose reductions, and Table 2 summarizes the recommended dosage modifications of TAZVERIK for adverse reactions.
Table 1. Recommended Dose Reductions of TAZVERIK for Adverse Reactions *Permanently discontinue TAZVERIK in patients who are unable to tolerate 400 mg orally twice daily.
Dose Reduction Dosage First 600 mg orally twice daily Second 400 mg orally twice daily* Table 2. Recommended Dosage Modifications of TAZVERIK for Adverse Reactions Adverse Reaction Severity Dosage Modification Neutropenia
[see Adverse Reactions (6.1)]Neutrophil count less than 1 × 109/L - Withhold until neutrophil count is greater than or equal to 1 × 109/L or baseline.
- For first occurrence, resume at same dose.
- For second and third occurrence, resume at reduced dose.
- Permanently discontinue after fourth occurrence.
Thrombocytopenia
[see Adverse Reactions (6.1)]Platelet count less than 50 × 109/L - Withhold until platelet count is greater than or equal to 75 × 109/L or baseline.
- For first and second occurrence, resume at reduced dose.
- Permanently discontinue after third occurrence.
Anemia
[see Adverse Reactions (6.1)]Hemoglobin less than 8 g/dL - Withhold until improvement to at least Grade 1 or baseline, then resume at same or reduced dose.
Other adverse reactions
[see Adverse Reactions (6.1)]Grade 3 - Withhold until improvement to at least Grade 1 or baseline.
- For first and second occurrence, resume at reduced dose.
- Permanently discontinue after third occurrence.
Grade 4 - Withhold until improvement to at least Grade 1 or baseline.
- For first occurrence, resume at reduced dose.
- Permanently discontinue after second occurrence.
2.4 Dosage Modifications for Drug Interactions
Strong and Moderate CYP3A Inhibitors
Avoid coadministration of TAZVERIK with strong or moderate CYP3A inhibitors. If coadministration with a moderate CYP3A inhibitor cannot be avoided, reduce the TAZVERIK dose as shown in Table 3 below. After discontinuation of the moderate CYP3A inhibitor for 3 elimination half-lives, resume the TAZVERIK dose that was taken prior to initiating the inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Table 3. Recommended Dose Reductions of TAZVERIK for Moderate CYP3A Inhibitors Current Dosage Adjusted Dosage 800 mg orally twice daily 400 mg orally twice daily 600 mg orally twice daily 400 mg for first dose and 200 mg for second dose 400 mg orally twice daily 200 mg orally twice daily - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Secondary Malignancies
The risk of developing secondary malignancies is increased following treatment with TAZVERIK. Across clinical trials of 758 adults who received TAZVERIK 800 mg twice daily as monotherapy, myelodysplastic syndrome (MDS), acute myeloid leukemia (AML), or B-cell acute lymphoblastic leukemia (B-ALL) occurred in 1.7% of patients. One pediatric patient developed T-cell lymphoblastic lymphoma (T-LBL). Monitor patients long-term for the development of secondary malignancies.
5.2 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, TAZVERIK can cause fetal harm when administered to pregnant women. There are no available data on TAZVERIK use in pregnant women to inform the drug-associated risk. Administration of tazemetostat to pregnant rats and rabbits during organogenesis resulted in dose-dependent increases in skeletal developmental abnormalities in both species beginning at maternal exposures approximately 1.5 times the adult human exposure (area under the plasma concentration time curve [AUC0-45h]) at the 800 mg twice daily dose.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with TAZVERIK and for 6 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with TAZVERIK and for 3 months after the final dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in labeling:
- Secondary Malignancies [see Warnings and Precautions (5.1)].
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Epithelioid Sarcoma
The safety of TAZVERIK was evaluated in a cohort (Cohort 5) of Study EZH-202 that enrolled patients with epithelioid sarcoma [see Clinical Studies (14.1)]. Patients received TAZVERIK 800 mg orally twice daily (n=62). Among patients who received TAZVERIK, 44% were exposed for 6 months or longer and 24% were exposed for greater than one year.
Serious adverse reactions occurred in 37% of patients who received TAZVERIK. Serious adverse reactions in ≥3% of patients who received TAZVERIK were hemorrhage, pleural effusion, skin infection, dyspnea, pain, and respiratory distress.
One patient (2%) permanently discontinued TAZVERIK due to an adverse reaction of altered mood.
Dosage interruptions due to an adverse reaction occurred in 34% of patients who received TAZVERIK. The most frequent adverse reactions requiring dosage interruptions in ≥3% were hemorrhage, increased alanine aminotransferase (ALT), and increased aspartate aminotransferase (AST).
Dose reduction due to an adverse reaction occurred in one (2%) patient who received TAZVERIK; the dose was reduced in this patient for decreased appetite.
The most common adverse reactions (≥20%) were pain, fatigue, nausea, decreased appetite, vomiting, and constipation.
Table 4 presents adverse reactions in patients with epithelioid sarcoma in Cohort 5 of Study EZH-202.
Table 4. Adverse Reactions (≥10%) in Patients with Epithelioid Sarcoma Who Received TAZVERIK in Cohort 5 of Study EZH-202 Adverse Reaction TAZVERIK
N=62All Grades (%) Grade 3 or 4 (%) a Includes tumor pain, pain in extremity, non-cardiac chest pain, flank pain, back pain, arthralgia, bone pain, cancer pain, musculoskeletal pain, myalgia, neck pain
b Includes fatigue and asthenia
c Includes abdominal pain, gastrointestinal pain, abdominal pain lower
d Includes dyspnea and dyspnea exertional
e Includes wound hemorrhage, rectal hemorrhage, pulmonary hemorrhage, hemorrhage intracranial, cerebral hemorrhage, hemoptysis
General Paina 52 7 Fatigueb 47 1.6 Gastrointestinal Nausea 36 0 Vomiting 24 0 Constipation 21 0 Diarrhea 16 0 Abdominal painc 13 1.6 Metabolism and nutrition Decreased appetite 26 4.8 Respiratory, thoracic and mediastinal Cough 18 0 Dyspnead 16 4.8 Vascular Hemorrhagee 18 4.8 Nervous system Headache 18 0 Investigations Weight decreased 16 7 Table 5 summarizes select laboratory abnormalities in patients with epithelioid sarcoma in Cohort 5 of Study EZH-202.
Table 5. Select Laboratory Abnormalities (≥ 10%) Worsening from Baseline in Patients with Epithelioid Sarcoma Who Received TAZVERIK in Cohort 5 of Study EZH-202 *The denominator used to calculate the rate varied from 39 to 61 based on the number of patients with a baseline value and at least one post-treatment value.
Laboratory Abnormality TAZVERIK* All Grades (%) Grade 3 or 4 (%) Hematology Decreased hemoglobin 49 15 Decreased lymphocytes 36 13 Decreased white blood cell count 19 0 Chemistry Increased triglycerides 36 3.3 Increased glucose 33 1.6 Decreased sodium 30 1.7 Decreased phosphate 28 1.7 Decreased albumin 23 0 Increased alkaline phosphatase 23 1.7 Decreased potassium 20 1.7 Increased aspartate aminotransferase 18 3.5 Decreased calcium 16 0 Decreased glucose 16 0 Increased partial thromboplastin time 15 5 Increased alanine aminotransferase 14 3.4 Increased creatinine 12 0 Increased potassium 12 0 Relapsed or Refractory Follicular Lymphoma
The safety of TAZVERIK was evaluated in two cohorts (Cohorts 4 and 5) of Study E7438-G000-101 that enrolled patients with relapsed or refractory follicular lymphoma [see Clinical Studies (14.2)]. Patients received TAZVERIK 800 mg orally twice daily (n=99). Among patients who received TAZVERIK, 68% were exposed for 6 months or longer, 39% were exposed for 12 months or longer, and 21% were exposed for 18 months or longer.
The median age was 62 years (range 36 to 87 years), 54% were male, and 95% had an Eastern Cooperative Oncology Group (ECOG) performance status of 0-1. The median number of prior therapies was 3 (range 1 to 11). Patients were required have a creatinine clearance ≥40 mL/min per the Cockcroft and Gault formula.
Serious adverse reactions occurred in 30% of patients who received TAZVERIK. Serious adverse reactions in ≥2% of patients who received TAZVERIK were general physical health deterioration, abdominal pain, pneumonia, sepsis, and anemia.
Permanent discontinuation due to an adverse reaction occurred in 8% of patients who received TAZVERIK. Adverse reaction resulting in permanent discontinuation in ≥2% of patients was second primary malignancy.
Dosage interruptions due to an adverse reaction occurred in 28% of patients who received TAZVERIK. Adverse reactions requiring dosage interruptions in ≥3% of patients were thrombocytopenia and fatigue.
Dose reduction due to an adverse reaction occurred in 9% of patients who received TAZVERIK.
The most common adverse reactions (≥20%) were fatigue, upper respiratory tract infection, musculoskeletal pain, nausea, and abdominal pain.
Table 6 presents adverse reactions in patients with relapsed or refractory follicular lymphoma in Cohorts 4 and 5 of Study E7438-G000-101.
Table 6. Adverse Reactions (≥10%) in Patients with Relapsed or Refractory Follicular Lymphoma Who Received TAZVERIK in Cohorts 4 and 5 of Study E7438-G000-101 Adverse Reaction TAZVERIK
N=99All Grades (%) Grade 3 or 4 (%) a Includes fatigue and asthenia
b Includes laryngitis, nasopharyngitis, pharyngitis, rhinitis, sinusitis, upper repiratory tract infection, viral upper respiratory tract infection
c Includes bronchitis, lower respiratory tract infection, tracheobronchitis
d Includes cystitis, urinary tract infection, urinary tract infection staphylococcal
e Includes abdominal discomfort, abdominal pain, abdominal pain lower, abdominal pain upper
f Includes back pain, limb discomfort, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal pain, myalgia, neck pain, non-cardiac chest pain, pain in extremity, pain in jaw, spinal pain
g Includes erythema, rash, rash erythematous, rash generalized, rash maculo-papular, rash pruritic, rash pustular, skin exfoliation
h Includes cough and productive cough
i Includes headache, migraine, sinus headache
General Fatiguea 36 5 Pyrexia 10 0 Infections Upper respiratory tract infectionb 30 0 Lower respiratory tract infectionc 17 0 Urinary tract infectiond 11 2 Gastrointestinal Nausea 24 1 Abdominal paine 20 3 Diarrhea 18 0 Vomiting 12 1 Musculoskeletal and connective tissue Musculoskeletal painf 22 1 Skin and subcutaneous tissue Alopecia 17 0 Rashg 15 0 Respiratory and mediastinal system Coughh 17 0 Nervous system Headachei 13 0 Clinically relevant adverse reactions occurring in <10% of patients who received TAZVERIK included:
- Infection: sepsis (2%), pneumonia (2%), and herpes zoster (2%)
Table 7 summarizes select laboratory abnormalities in patients with follicular lymphoma in Cohorts 4 and 5 of Study E7438-G000-101.
Table 7. Select Laboratory Abnormalities (≥10%) Worsening from Baseline in Patients with Relapsed or Refractory Follicular Lymphoma Who Received TAZVERIK in Cohorts 4 and 5 of Study E7438-G000-101 Laboratory Abnormality TAZVERIK* All Grades (%) Grade 3 or 4 (%) *The denominator used to calculate the rate varied from 88 to 96 based on the number of patients with a baseline value and at least one post-treatment value.
Hematology Decreased lymphocytes 57 18 Decreased hemoglobin 50 8 Decreased platelets 50 7 Decreased white blood cells 41 9 Decreased neutrophils 20 7 Chemistry Increased glucose 53 10 Increased aspartate aminotransferase 24 0 Increased alanine aminotransferase 21 2.3 Increased alkaline phosphatase 18 1.0 Increased creatinine 17 0 -
7 DRUG INTERACTIONS
7.1 Effect of Other Drugs on TAZVERIK
Strong and Moderate CYP3A Inhibitors
Coadministration of TAZVERIK with a strong or moderate CYP3A inhibitor increases tazemetostat plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the frequency or severity of adverse reactions. Avoid coadministration of strong or moderate CYP3A inhibitors with TAZVERIK. If coadministration of moderate CYP3A inhibitors cannot be avoided, reduce TAZVERIK dose [see Dosage and Administration (2.3)].
Strong and Moderate CYP3A Inducers
Coadministration of TAZVERIK with a strong or moderate CYP3A inducer may decrease tazemetostat plasma concentrations [see Clinical Pharmacology (12.3)], which may decrease the efficacy of TAZVERIK. Avoid coadministration of moderate and strong CYP3A inducers with TAZVERIK.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], TAZVERIK can cause fetal harm when administered to pregnant women. There are no available data on TAZVERIK use in pregnant women to inform the drug-associated risk. Administration of tazemetostat to pregnant rats and rabbits during organogenesis resulted in dose-dependent increases in skeletal developmental abnormalities in both species beginning at maternal exposures approximately 1.5 times the adult human exposure [AUC0-45h] at the 800 mg twice daily dose (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In pregnant rats, once daily oral administration of tazemetostat during the period of organogenesis from gestation day (GD) 7 through 17 resulted in no maternal adverse effects at doses up to 100 mg/kg/day (approximately 6 times the adult human exposure at 800 mg twice daily). Skeletal malformations and variations occurred in fetuses at doses of ≥50 mg/kg (approximately 2 times the adult human exposure at the 800 mg twice daily dose). At 200 mg/kg (approximately 14 times the adult human exposure at the 800 mg twice daily dose), major findings included increased post implantation loss, missing digits, fused vertebrae, domed heads and fused bones of the skull, and reduced fetal body weights.
In pregnant rabbits, no adverse maternal effects were observed after once daily oral administration of 400 mg/kg/day tazemetostat (approximately 7 times the adult human exposure at the 800 mg twice daily dose) from GD 7 through 19. Skeletal variations were present at doses ≥100 mg/kg/day (approximately 1.5 times the adult human exposure at the 800 mg twice daily dose), with skeletal malformations at ≥200 mg/kg/day (approximately 5.6 times the adult human exposure at the 800 mg twice daily dose). At 400 mg/kg (approximately 7 times the adult human exposure at the 800 mg twice daily dose), major findings included increased post implantation loss and cleft palate and snout.
8.2 Lactation
Risk Summary
There are no animal or human data on the presence of tazemetostat in human milk or on its effects on the breastfed child or milk production. Because of the potential risk for serious adverse reactions from TAZVERIK in the breastfed child, advise women not to breastfeed during treatment with TAZVERIK and for one week after the final dose.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating TAZVERIK [see Use in Specific Populations (8.1)].
Risk Summary
TAZVERIK can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Contraception
Females
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with TAZVERIK and for 6 months after the final dose. TAZVERIK can render some hormonal contraceptives ineffective [see Drug Interactions (7.2)].
8.4 Pediatric Use
The safety and effectiveness of TAZVERIK have been established in pediatric patients aged 16 years and older (adolescents) with metastatic or locally advanced epithelioid sarcoma. Use of TAZVERIK for this indication is supported by evidence from adequate and well-controlled studies in adults (including 3 adolescent patients aged 16 years) [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.1)].
The safety and effectiveness of TAZVERIK in pediatric patients aged less than 16 years have not been established.
Juvenile Animal Toxicity Data
In a 13-week juvenile rat toxicology study, animals were dosed daily from post-natal day 7 to day 97 (approximately equivalent to neonate to adulthood). Tazemetostat resulted in:
- T-LBL at doses ≥50 mg/kg (approximately 2.8 times the adult human exposure at the 800 mg twice daily dose)
- Increased trabecular bone at doses ≥100 mg/kg (approximately 10 times the adult human exposure at the 800 mg twice daily dose)
- Increased body weight at doses ≥50 mg/kg (approximately equal to the adult human exposure at the 800 mg twice daily dose)
- Distended testicles in males at doses ≥50 mg/kg (approximately equal to the adult human exposure at the 800 mg twice daily dose)
8.5 Geriatric Use
Clinical studies of TAZVERIK did not include sufficient numbers of patients with epithelioid sarcoma or relapsed or refactory follicular lymphoma aged 65 and over to determine whether they respond differently from younger subjects.
8.6 Renal Impairment
No dose adjustment of TAZVERIK is recommended for patients with mild to severe renal impairment or end stage renal disease [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dose adjustment of TAZVERIK is recommended for patients with mild hepatic impairment (total bilirubin > 1 to 1.5 times upper limit of normal [ULN] or AST > ULN). TAZVERIK has not been studied in patients with moderate (total bilirubin > 1.5 to 3 times ULN) or severe (total bilirubin > 3 times ULN) hepatic impairment [see Clinical Pharmacology (12.3)].
-
11 DESCRIPTION
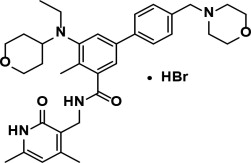
Tazemetostat is a methyltransferase inhibitor. Tazemetostat hydrobromide has the following chemical name: [1,1'-Biphenyl]-3-carboxamide, N-[(1,2-dihydro-4,6-dimethyl-2-oxo-3-pyridinyl)methyl]-5-[ethyl(tetrahydro-2H-pyran-4-yl)amino]-4-methyl-4'-(4-morpholinylmethyl)-, hydrobromide (1:1). The molecular formula of tazemetostat hydrobromide is C34H44N4O4∙HBr. Tazemetostat hydrobromide has a molecular weight of 653.66 g/mol and the following structural formula:
Tazemetostat hydrobromide is a white to off-white solid that is slightly soluble in water and has pKa values of 5.26, 6.88, and 12.62. A saturated aqueous solution of tazemetostat hydrobromide has a pH of approximately 5 at ambient conditions.
TAZVERIK (tazemetostat) tablets for oral use contain 200 mg tazemetostat, equivalent to 228 mg tazemetostat hydrobromide.
Each tablet is film-coated and contains the following inactive ingredients in the tablet core: hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, magnesium stearate, and sodium starch glycolate. The film-coat contains hypromellose, polyethylene glycol, red iron oxide, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Tazemetostat is an inhibitor of the methyltransferase, EZH2, and some EZH2 gain-of-function mutations including Y646X, A682G, and A692V. Tazemetostat also inhibited EZH1 with a half-maximal inhibitory concentration (IC50) of 392 nM, approximately 36 times higher than the IC50 for inhibition of EZH2.
The most well-characterized function of EZH2 is as the catalytic subunit of the polycomb repressive complex 2 (PRC2), catalyzing mono-, di-, and trimethylation of lysine 27 of histone H3. Trimethylation of histone H3 leads to transcriptional repression.
SWItch/Sucrose Non-Fermentable (SWI/SNF) complexes can antagonize PRC2 function in the regulation of the expression of certain genes of patients with epithelioid sarcoma. Preclinical in vitro and in vivo models with the loss or dysfunction of certain SWI/SNF complex members (e.g., integrase interactor 1 [INI1/SNF5/SMARCB1/BAF47], SMARCA4, and SMARCA2) can lead to aberrant EZH2 activity or expression and a resulting oncogenic dependence on EZH2.
Tazemetostat suppressed proliferation of B-cell lymphoma cell lines in vitro and demonstrated antitumor activity in a mouse xenograft model of B-cell lymphoma with or without EZH2 gain-of-function mutations. Tazemetostat demonstrated greater effects on the inhibition of proliferation of lymphoma cell lines with mutant EZH2.
12.2 Pharmacodynamics
Tazemetostat exposure-response relationships and the time course of pharmacodynamic responses are unknown.
Cardiac Electrophysiology
The effect of orally administered TAZVERIK, at doses ranging from 100 mg to 1600 mg twice daily (0.125 to 2 times the approved recommended dosage) for 15 days, on the heart-rate corrected QT (QTc) interval was evaluated in a dose-finding study in 38 patients with advanced malignancies. Tazemetostat and its metabolite EPZ-6930 did not cause a large mean increase (i.e. >20 ms) on the QTc interval at the 800 mg twice daily dose. The largest mean increase (upper bound of 90% confidence interval) in QTc were 6.1 ms (8.5 ms) and 9.3 ms (12.5 ms) at a dose of 800 mg twice daily and 1600 mg twice daily, respectively.
12.3 Pharmacokinetics
The systemic exposure of tazemetostat is approximately dose proportional over the dose range of 200 mg to 1600 mg twice daily of TAZVERIK (0.25 to 2 times the approved recommended dosage). Following TAZVERIK 800 mg orally twice daily, steady-state was reached by Day 15. The mean (coefficient of variation [CV]%) steady-state peak plasma concentration (Cmax) was 829 (56%) ng/mL and AUC0-12h was 3340 (49%) ng•h/mL. Tazemetostat exhibited time-dependent pharmacokinetics (PK). The mean accumulation ratio (measured by AUC) was 0.58.
Absorption
The mean absolute oral bioavailability of tazemetostat is approximately 33%. The median time to reach the peak plasma concentration of tazemetostat is 1 to 2 hours.
Distribution
The mean (CV%) apparent volume of distribution at steady-state (Vss/F) is 1230 L (46%). Tazemetostat is 88% bound to human plasma proteins in vitro. The blood-to-plasma ratio is 0.73.
Elimination
At steady-state, the estimated mean (CV%) terminal elimination half-life of tazemetostat is 3.1 hours (14%) and the apparent total clearance (CLss/F) is 274 L/h (49%).
Metabolism
In vitro, tazemetostat is metabolized by CYP3A to form the inactive major metabolites M5 (EPZ-6930) and M3 (EPZ006931). M5 undergoes further metabolism by CYP3A.
Excretion
Following a single oral dose of radiolabeled tazemetostat, 94% of the total radioactivity was recovered over 12 days, with 15% excreted into urine and 79% into feces.
Specific Populations
Age (16 to 91 years), sex, race (White, Black, Asian), body weight (37.3 to 173 kg), mild hepatic impairment (total bilirubin > 1 to 1.5 times ULN or AST > ULN) and renal impairment, including end stage renal disease, have no clinically meaningful effect on the pharmacokinetics of tazemetostat. The effect of moderate to severe hepatic impairment has not been studied.
Drug Interaction Studies
Effect of CYP3A Inhibitors on Tazemetostat:
Coadministration of fluconazole (a moderate CYP3A inhibitor) with TAZVERIK 400 mg twice daily in patients increased tazemetostat steady-state AUC0-8h by 3.1-fold and Cmax by 2.3-fold.
Effect of Gastric Acid Reducing Agents on Tazemetostat:
Coadministration of omeprazole (a proton pump inhibitor) with TAZVERIK 800 mg twice daily in patients increased tazemetostat steady-state AUC0-8h by 26% and Cmax by 25%, which is not expected to have clinically relevant impact.
Effect of Tazemetostat on CYP3A Substrate:
Coadministration of TAZVERIK 800 mg twice daily with oral midazolam (a sensitive CYP3A substrate) in patients decreased midazolam AUC0-12h by 40% and Cmax by 21%.
Effect of Tazemetostat on CYP2C8 and CYP2C19 Substrates:
Coadministration of TAZVERIK 800 mg twice daily with repaglinide (a sensitive CYP2C8 substrate) and omeprazole (a sensitive CYP2C19 substrate) in patients increased repaglinide AUC0-8h by 80% and Cmax by 51%; and had no effect on the exposure of omeprazole.
Metabolic Enzymes:
Tazemetostat does not inhibit CYP1A2, CYP2B6, CYP2C9, and CYP2D6 at clinically relevant concentrations.
Drug Transporters:
Tazemetostat is a substrate of p-glycoprotein (P-gp). Tazemetostat is not a substrate of breast cancer resistance protein (BCRP); renal transporters organic cation transporter 2 (OCT2), organic anion transporter 3 (OAT3), and multidrug and toxin extrusion transporter 1 (MATE1); or hepatic transporters organic anion transporting polypeptide 1B1 (OATP1B1) and organic anion transporting polypeptide 1B3 (OATP1B3).
Tazemetostat is an inhibitor of MATE1 and multidrug and toxin extrusion transporter 2-K (MATE2-K). Tazemetostat does not inhibit P-gp, BCRP, OATP1B1, OATP1B3, organic cation transporter 1 (OCT1), OCT2, organic anion transporter 1 (OAT1), OAT3, or bile salt export pump (BSEP) at clinically relevant concentrations.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Dedicated carcinogenicity studies were not conducted with tazemetostat, but T-LBL, MDS, AML, and B-ALL have been reported clinically and T-LBL occurred in juvenile and adult rats after ~9 or more weeks of tazemetostat administration during 13-week toxicity studies. Based on nonclinical studies in rats, the risk of T-LBL appears to be greater with longer duration dosing.
Tazemetostat did not cause genetic damage in a standard battery of studies including a screening and pivotal bacterial reverse mutation (Ames) assay, an in vitro micronucleus assessment in human peripheral blood lymphocytes, and an in vivo micronucleus assessment in rats after oral administration.
Fertility and early embryonic development studies have not been conducted with tazemetostat; however, an assessment of male and female reproductive organs were included in 4- and 13-week repeat-dose toxicity studies in rats and Cynomolgus monkeys. Oral daily administration of tazemetostat did not result in any notable effects in the adult male and female reproductive organs [see Use in Specific Populations (8.3)].
-
14 CLINICAL STUDIES
14.1 Epithelioid Sarcoma
The efficacy of TAZVERIK was evaluated in an open-label, single-arm cohort (Cohort 5) of a multi-center study (Study EZH-202, NCT02601950) in patients with histologically confirmed, metastatic or locally advanced epithelioid sarcoma. Patients were required to have INI1 loss, detected using local tests, and an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0-2. Patients received TAZVERIK 800 mg orally twice daily until disease progression or unacceptable toxicity. Tumor response assessments were performed every 8 weeks. The major efficacy outcome measures were confirmed overall response rate (ORR) according to Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 as assessed by blinded independent central review (BICR) and duration of response (DOR). Median duration of follow-up was 14 months (range 0.4 to 31).
Among the 62 patients who received TAZVERIK, median age was 34 years (range 16 to 79); 63% were male, 76% were White, 11% were Asian, 44% had proximal disease, 92% had an ECOG PS of 0 or 1, and 8% had an ECOG PS of 2. Prior surgery occurred in 77% of patients; 61% received prior systemic chemotherapy.
Efficacy results are summarized in Table 8.
Table 8. Efficacy Results for Patients with Epithelioid Sarcoma Enrolled in Cohort 5 of Study EZH-202 CI = Confidence Interval
*Time to response ranged from 1.4 to 18.4 months.
Efficacy Endpoints TAZVERIK
N=62Overall Response Rate (95% CI)* 15% (7%, 26%) Complete Response 1.6% Partial Response 13% Duration of Response % with duration ≥ 6 months 67% Range in months 3.7, 24.5+ 14.2 Relapsed or Refractory Follicular Lymphoma
The efficacy of TAZVERIK was evaluated in two open-label, single-arm cohorts (Cohorts 4 and 5) of a multi-center study (Study E7438-G000-101, NCT01897571) in patients with histologically confirmed follicular lymphoma after at least 2 prior systemic therapies. Patients were required to have ECOG PS of 0-2 and were enrolled based on EZH2 mutation status. EZH2 mutations were identified prospectively using formalin-fixed, paraffin-embedded tumor samples, which were centrally tested using the cobas® EZH2 Mutation Test; the cobas EZH2 Mutation test is designed to detect the following mutations: Y646X [S,H,C], Y646F, Y646N, A682G, and A692V. Patients received TAZVERIK 800 mg orally twice daily until confirmed disease progression or unacceptable toxicity. Tumor response assessments were performed every 8 weeks through Week 24 and then every 12 weeks. The major efficacy outcome measures were ORR and DOR according to the International Working Group Non-Hodgkin Lymphoma (IWG-NHL) criteria1 as assessed by Independent Review Committee. Median duration of follow-up was 22 months (range 3 months to 44 months) for patients with EZH2 MT positive tumors and 36 months (range 32 months to 39 months) for patients whose tumors did not have an EZH2 mutation detected.
A total of 99 patients were enrolled, including 45 patients whose tumors had one of these EZH2 mutations (mutant) and 54 patients whose tumors did not have one of these mutations (wild-type).
Among the 45 patients with EZH2 mutant follicular lymphoma, median age was 62 years (range 38 to 80), 58% were female, 42% had early progression following front-line therapy (POD24), and all had an ECOG PS of 0
or 1. Race was reported in 84% of patients; of these patients, 82% were White. Based on the cobas EZH2 Mutation test, 36%, 29%, 27%, 11% and 2% of patients had the following mutations: Y646X [S,H,C], Y646F, Y646N, A682G, and A692V, respectively. The median number of lines of prior systemic therapy was 2 (range 1 to 11), with 49% refractory to rituximab, 49% refractory to their last therapy, and 9% had received prior stem cell transplant.Among the 54 patients with EZH2 wild-type follicular lymphoma, median age was 61 years (range 36 to 87), 63% were male, 59% had POD24, and 91% had an ECOG PS of 0 or 1. Race was reported in 57% of patients; of these patients, 48% were White and 3% were Asian. The median number of lines of prior systemic therapy was 3 (range 1 to 8), with 59% refractory to rituximab, 41% refractory to their last therapy, and 39% had received prior stem cell transplant.
The approval of TAZVERIK was based upon the efficacy in 95 patients (42 EZH2 Mutant, 53 EZH2 Wild-Type) who had received at least 2 prior systemic therapies and is presented in Table 9.
Table 9. Efficacy Results for Patients with Relapsed or Refractory Follicular Lymphoma Enrolled into Cohorts 4 and 5 of Study E7438-G000-101 CI = Confidence Interval; NE = Not Estimable.
*Median time to response for patients with EZH2 MT follicular lymphoma was 3.7 months (range 1.6 to 10.9) and for patients with EZH2 WT follicular lymphoma was 3.9 months (range 1.6 to 16.3).
Efficacy Endpoints TAZVERIK
N=95EZH2 Mutant Follicular Lymphoma
N=42EZH2 Wild-Type Follicular Lymphoma
N=53Overall Response Rate (95% CI)* 69% (53%, 82%) 34% (22%, 48%) Complete Response 12% 4% Partial Response 57% 30% Duration of Response Median (95% CI) in months 10.9 (7.2, NE) 13.0 (5.6, NE) Range in months 0.0+, 22.1+ 1, 22.5+ - 15 REFERENCES
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Secondary Malignancies
Advise patients of the increased risk of secondary hematologic malignancies. Advise patients to inform their healthcare provider if they experience fatigue, easy bruising, fever, bone pain, or paleness [see Warnings and Precautions (5.1)].
Embryo-Fetal Toxicity
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with TAZVERIK and for 6 months after the final dose [see Use in Specific Populations (8.3)].
Advise males with female partners of reproductive potential to use effective contraception during treatment with TAZVERIK and for 3 months after the final dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
Advise women not to breastfeed during treatment with TAZVERIK and for 1 week after the final dose [see Use in Special Populations (8.2)].
Drug Interactions
Advise patients and caregivers to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products. Inform patients to avoid St. John's wort, grapefruit, and grapefruit juice while taking TAZVERIK [see Drug Interactions (7.1)].
Manufactured for:
Epizyme, Inc.
An Ipsen Company
One Main St.
Cambridge, MA 02142©2023 Epizyme, Inc.
-
MEDICATION GUIDE
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Issued: November/2023
MEDICATION GUIDE
TAZVERIK® (taz vayr′ ik)
(tazemetostat)
tabletsWhat is the most important information I should know about TAZVERIK?
TAZVERIK can cause serious side effects, including:- Risk of new cancers. An increase in new (second) cancers has happened in people who were treated with TAZVERIK. Talk with your healthcare provider about your risk of developing new cancers. Your healthcare provider will monitor you for new cancers after your treatment with TAZVERIK. Tell your healthcare provider if you are more tired than usual, or have easy bruising, fever, bone pain, or paleness.
What is TAZVERIK?
TAZVERIK is a prescription medicine used to treat:
- adults and children aged 16 years and older with epithelioid sarcoma that has spread or grown and cannot be removed by surgery.
- adults with follicular lymphoma when the disease has come back or did not respond to treatment, whose tumors have an abnormal EZH2 gene, and who have been treated with at least two prior medicines. Your healthcare provider will perform a test to make sure TAZVERIK is right for you.
- adults with follicular lymphoma when the disease has come back or did not respond to treatment, who have no other satisfactory treatment options.
Before taking TAZVERIK tell your healthcare provider about all of your medical conditions, including if you:
- are pregnant or plan to become pregnant. TAZVERIK can harm your unborn baby. Your healthcare provider will give you a pregnancy test before you start treatment with TAZVERIK. Tell your healthcare provider right away if you become pregnant or think you may be pregnant.
- Females who are able to become pregnant should use effective non-hormonal birth control (such as condoms) during treatment and for 6 months after the final dose of TAZVERIK. Birth control pills (oral contraceptives) and other hormonal forms of birth control may not be effective if used during treatment with TAZVERIK. Talk to your healthcare provider about birth control options that are right for you.
- Males with female partners who are able to become pregnant should use effective birth control during treatment and for 3 months after the final dose of TAZVERIK.
- are breastfeeding or plan to breastfeed. It is not known if TAZVERIK passes into your breast milk. Do not breastfeed during treatment and for 1 week after the final dose of TAZVERIK.
How should I take TAZVERIK?
- Take TAZVERIK exactly as your healthcare provider tells you.
- Take TAZVERIK 2 times each day.
- Take TAZVERIK with or without food.
- Swallow TAZVERIK tablets whole. Do not cut, crush, or chew tablets.
- If you miss a dose or vomit after taking your dose, just skip that dose and take the next dose at your regular time.
- Your healthcare provider may change your dose, temporarily stop, or completely stop treatment with TAZVERIK if you get certain side effects.
What should I avoid while taking TAZVERIK?
- Avoid eating grapefruit or drinking grapefruit juice during treatment with TAZVERIK.
- Avoid taking St. John's wort during treatment with TAZVERIK.
What are the possible side effects of TAZVERIK?
TAZVERIK can cause serious side effects. See "What is the most important information I should know about TAZVERIK?"
The most common side effects of TAZVERIK in people with epithelioid sarcoma include:- pain
- nausea
- vomiting
- tiredness
- decreased appetite
- constipation
The most common side effects of TAZVERIK in people with follicular lymphoma include: - tiredness
- bone and muscle pain
- cold-like symptoms (upper respiratory infection)
- nausea
- stomach (abdominal) pain
These are not all the possible side effects of TAZVERIK.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store TAZVERIK?
Do not store TAZVERIK tablets above 86°F (30°C).
Keep TAZVERIK and all medicines out of the reach of children.General information about the safe and effective use of TAZVERIK.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use TAZVERIK for a condition for which it was not prescribed. Do not give TAZVERIK to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about TAZVERIK that is written for health professionals.What are the ingredients in TAZVERIK?
Active Ingredient: tazemetostat.
Inactive Ingredients: Tablet core: hydroxypropyl cellulose, lactose monohydrate, low-substituted hydroxypropyl cellulose, magnesium stearate, and sodium starch glycolate. Film-coating: hypromellose, polyethylene glycol, red iron oxide, talc, and titanium dioxide.
Manufactured for: Epizyme, Inc.
An Ipsen Company
One Main St.
Cambridge, MA 02142 For more information, contact Epizyme at 855-463-5127. - Principal Display Panel - 200 mg Bottle Label
-
INGREDIENTS AND APPEARANCE
TAZVERIK
tazemetostat tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72607-100 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength tazemetostat hydrobromide (UNII: 6P89T5M073) (tazemetostat - UNII:Q40W93WPE1) tazemetostat 200 mg Inactive Ingredients Ingredient Name Strength Lactose monohydrate (UNII: EWQ57Q8I5X) Low-substituted hydroxypropyl cellulose, unspecified (UNII: 2165RE0K14) Hydroxypropyl cellulose, unspecified (UNII: 9XZ8H6N6OH) Sodium starch glycolate Type A potato (UNII: 5856J3G2A2) Magnesium stearate (UNII: 70097M6I30) Hypromellose, unspecified (UNII: 3NXW29V3WO) Talc (UNII: 7SEV7J4R1U) Polyethylene glycol 8000 (UNII: Q662QK8M3B) Titanium dioxide (UNII: 15FIX9V2JP) Ferric oxide red (UNII: 1K09F3G675) Product Characteristics Color red (RED) Score no score Shape ROUND (ROUND) Size 10mm Flavor Imprint Code EZM;200 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72607-100-00 240 in 1 BOTTLE; Type 0: Not a Combination Product 01/23/2020 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA211723 01/23/2020 Labeler - Epizyme, Inc. (018795119) Establishment Name Address ID/FEI Business Operations Patheon Pharmaceuticals Inc. 005286822 MANUFACTURE(72607-100) Establishment Name Address ID/FEI Business Operations Sterling Pharma Solutions Limited 349157623 API MANUFACTURE(72607-100)