Label: CARVYKTI- ciltacabtagene autoleucel injection, suspension


- NDC Code(s): 57894-111-01, 57894-111-02
- Packager: Janssen Biotech, Inc
- Category: CELLULAR THERAPY
Drug Label Information
Updated July 5, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use CARVYKTI safely and effectively. See full prescribing information for CARVYKTI.
CARVYKTI ®(ciltacabtagene autoleucel) suspension for intravenous infusion
Initial U.S. Approval: 2022WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, HLH/MAS, PROLONGED and RECURRENT CYTOPENIA, and SECONDARY HEMATOLOGICAL MALIGNANCIES
See full prescribing information for complete boxed warning.
- Cytokine Release Syndrome (CRS), including fatal or life-threatening reactions, occurred in patients following treatment with CARVYKTI. Do not administer CARVYKTI to patients with active infection or inflammatory disorders. Treat severe or life-threatening CRS with tocilizumab or tocilizumab and corticosteroids. ( 2.2, 2.3, 5.2)
- Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), which may be fatal or life-threatening, occurred following treatment with CARVYKTI, including before CRS onset, concurrently with CRS, after CRS resolution, or in the absence of CRS. Monitor for neurologic events after treatment with CARVYKTI. Provide supportive care and/or corticosteroids as needed. ( 2.2, 2.3, 5.3)
- Parkinsonism and Guillain-Barré syndrome and their associated complications resulting in fatal or life-threatening reactions have occurred following treatment with CARVYKTI. ( 5.3)
- Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome (HLH/MAS), including fatal and life-threatening reactions, occurred in patients following treatment with CARVYKTI. HLH/MAS can occur with CRS or neurologic toxicities. ( 5.4)
- Prolonged and/or recurrent cytopenias with bleeding and infection and requirement for stem cell transplantation for hematopoietic recovery occurred following treatment with CARVYKTI. ( 5.6)
- Secondary hematological malignancies, including myelodysplastic syndrome and acute myeloid leukemia, have occurred following treatment with CARVYKTI. T-cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T-cell immunotherapies, including CARVYKTI. ( 5.10)
- CARVYKTI is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the CARVYKTI REMS. ( 5.5)
RECENT MAJOR CHANGES
Boxed Warnings 12/2023 Boxed Warnings 04/2024 Indications and Usage ( 1) 04/2024 Warnings and Precautions ( 5.2, 5.5, 5.6, 5.7, 5.9) 12/2023 Warnings and Precautions ( 5) 04/2024 INDICATIONS AND USAGE
CARVYKTI (ciltacabtagene autoleucel) is a B-cell maturation antigen (BCMA)-directed genetically modified autologous T cell immunotherapy indicated for the treatment of adult patients with relapsed or refractory multiple myeloma who have received at least 1 prior line of therapy, including a proteasome inhibitor and an immunomodulatory agent, and are refractory to lenalidomide. ( 1)
DOSAGE AND ADMINISTRATION
For autologous use only. For intravenous use only.
- Administer a lymphodepleting regimen of cyclophosphamide and fludarabine before infusion of CARVYKTI. ( 2.2)
- Do NOT use a leukodepleting filter. ( 2.2)
- Verify the patient's identity prior to infusion. ( 2.2)
- Premedicate with acetaminophen and an H1-antihistamine. ( 2.2)
- Avoid prophylactic use of systemic corticosteroids. ( 2.2)
- Confirm availability of tocilizumab prior to infusion. ( 2.2, 5.1)
- Dosing of CARVYKTI is based on the number of chimeric antigen receptor (CAR)-positive viable T cells. ( 2.1)
- Recommended dose range is 0.5–1.0×10 6CAR-positive viable T cells per kg of body weight, with a maximum dose of 1×10 8CAR-positive viable T cells per single-dose infusion. ( 2.1)
- Administer CARVYKTI at a REMS-certified healthcare facility. ( 2.2)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
None ( 4)
WARNINGS AND PRECAUTIONS
- Prolonged and Recurrent Cytopenias: Patients may exhibit ≥Grade 3 cytopenias following CARVYKTI infusion. One or more recurrences of Grade 3 or higher cytopenias may occur after partial or complete recovery of cytopenias. Monitor blood counts prior to and after CARVYKTI infusion. Prolonged neutropenia has been associated with increased risk of infection. ( 5.6)
- Infections: Monitor patients for signs and symptoms of infection; treat appropriately. ( 5.7)
- Hypogammaglobulinemia: Monitor and consider immunoglobulin replacement therapy. ( 5.8)
- Hypersensitivity Reactions: Hypersensitivity reactions have occurred. Monitor for hypersensitivity reactions during infusion. ( 5.9)
- Secondary Malignancies: Secondary hematological malignancies, including myelodysplastic syndrome and acute myeloid leukemia, have occurred. T-cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T-cell immunotherapies, including CARVYKTI. In the event that a secondary malignancy occurs after treatment with CARVYKTI, contact Janssen Biotech, Inc. at 1-800-526-7736. ( 5.10)
- Effects on Ability to Drive and Use Machines: Advise patients to refrain from driving and engaging in hazardous occupations or activities, such as operating heavy or potentially dangerous machinery, for at least 8 weeks after receiving CARVYKTI and in the event of any new onset of neurologic toxicities. ( 5.11)
ADVERSE REACTIONS
The most common nonlaboratory adverse reactions (incidence greater than 20%) are pyrexia, cytokine release syndrome, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections-pathogen unspecified, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting. The most common Grade 3 or 4 laboratory adverse reactions (incidence greater than or equal to 50%) include lymphopenia, neutropenia, white blood cell decreased, thrombocytopenia, and anemia. ( 6)
To report SUSPECTED ADVERSE REACTIONS, contact Janssen Biotech, Inc. at 1-800-526-7736 (1-800-JANSSEN) or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 4/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, HLH/MAS, PROLONGED and RECURRENT CYTOPENIA, and SECONDARY HEMATOLOGICAL MALIGNANCIES
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Administration
2.3 Management of Severe Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Increased Early Mortality
5.2 Cytokine Release Syndrome
5.3 Neurologic Toxicities
5.4 Hemophagocytic Lymphohistiocytosis (HLH)/Macrophage Activation Syndrome (MAS)
5.5 CARVYKTI REMS
5.6 Prolonged and Recurrent Cytopenias
5.7 Infections
5.8 Hypogammaglobulinemia
5.9 Hypersensitivity Reactions
5.10 Secondary Malignancies
5.11 Effects on Ability to Drive and Use Machines
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, HLH/MAS, PROLONGED and RECURRENT CYTOPENIA, and SECONDARY HEMATOLOGICAL MALIGNANCIES
Cytokine Release Syndrome (CRS), including fatal or life-threatening reactions, occurred in patients following treatment with CARVYKTI. Do not administer CARVYKTI to patients with active infection or inflammatory disorders. Treat severe or life-threatening CRS with tocilizumab or tocilizumab and corticosteroids [see Dosage and Administration (2.2, 2.3), Warnings and Precautions (5.2)] .
Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), which may be fatal or life-threatening, occurred following treatment with CARVYKTI, including before CRS onset, concurrently with CRS, after CRS resolution, or in the absence of CRS. Monitor for neurologic events after treatment with CARVYKTI. Provide supportive care and/or corticosteroids as needed [see Dosage and Administration (2.2, 2.3), Warnings and Precautions (5.3)] .
Parkinsonism and Guillain-Barré syndrome (GBS) and their associated complications resulting in fatal or life-threatening reactions have occurred following treatment with CARVYKTI [see Warnings and Precautions (5.3)] .
Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome (HLH/MAS), including fatal and life-threatening reactions, occurred in patients following treatment with CARVYKTI. HLH/MAS can occur with CRS or neurologic toxicities [see Warnings and Precautions (5.4)].
Prolonged and/or recurrent cytopenias with bleeding and infection and requirement for stem cell transplantation for hematopoietic recovery occurred following treatment with CARVYKTI [see Warnings and Precautions (5.6)] .
Secondary hematological malignancies, including myelodysplastic syndrome and acute myeloid leukemia, have occurred in patients following treatment with CARVYKTI. T-cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T-cell immunotherapies, including CARVYKTI [see Warnings and Precautions (5.10)] .
CARVYKTI is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the CARVYKTI REMS Program [see Warnings and Precautions (5.5)] .
-
1 INDICATIONS AND USAGE
CARVYKTI (ciltacabtagene autoleucel) is a B-cell maturation antigen (BCMA)-directed genetically modified autologous T cell immunotherapy indicated for the treatment of adult patients with relapsed or refractory multiple myeloma, who have received at least 1 prior line of therapy, including a proteasome inhibitor and an immunomodulatory agent, and are refractory to lenalidomide.
-
2 DOSAGE AND ADMINISTRATION
For autologous use only. For intravenous use only.
2.1 Dose
CARVYKTI is provided as a single dose for infusion containing a suspension of chimeric antigen receptor (CAR)-positive viable T cells in one infusion bag.
The recommended dose range is 0.5–1.0×10 6CAR-positive viable T cells per kg of body weight, with a maximum dose of 1×10 8CAR-positive viable T cells per single infusion.
2.2 Administration
CARVYKTI is for autologous use only. The patient's identity must match the patient identifiers on the CARVYKTI cassette and infusion bag. Do not infuse CARVYKTI if the information on the patient-specific labels does not match the intended patient.
Preparing the Patient for CARVYKTI Infusion
Confirm availability of CARVYKTI prior to starting the lymphodepleting chemotherapy regimen.
Pretreatment
Administer the lymphodepleting chemotherapy regimen: cyclophosphamide 300 mg/m 2intravenously (IV) and fludarabine 30 mg/m 2IV daily for 3 days.
See the prescribing information of cyclophosphamide and fludarabine for information on dose adjustment in renal impairment.
Lymphodepleting regimen must be delayed if a patient has serious adverse reactions from preceding bridging therapies (including clinically significant active infection, cardiac toxicity, and pulmonary toxicity) or active graft versus host disease in patient with prior allogeneic stem cell transplant. Consider repeating lymphodepleting regimen if CARVYKTI dosing is delayed by more than 14 days and patient has recovered from toxicity of the first lymphodepleting regimen.
Administer CARVYKTI infusion 2 to 4 days after the completion of the lymphodepleting chemotherapy regimen.
CARVYKTI infusion should be delayed if a patient has any of the following conditions:
- Clinically significant active infection or inflammatory disorders.
- Grade ≥3 non-hematologic toxicities of cyclophosphamide and fludarabine conditioning, except for Grade 3 nausea, vomiting, diarrhea, or constipation. CARVYKTI infusion should be delayed until resolution of these events to Grade ≤1.
Premedication
Administer the following pre-infusion medications to all patients 30 – 60 minutes prior to CARVYKTI infusion:
- Antipyretics (oral or intravenous acetaminophen 650 to 1000 mg).
- Antihistamine (oral or intravenous diphenhydramine 25 to 50 mg or equivalent).
Avoid prophylactic use of systemic corticosteroids because their use may interfere with the activity of CARVYKTI.
Receipt of CARVYKTI
- All sites approved for infusion will support required storage conditions for vapor phase of liquid nitrogen.
- CARVYKTI is shipped directly to the cell laboratory or clinical pharmacy associated with the infusion center in the vapor phase of a liquid nitrogen shipper.
- Confirm the patient's identity with the patient identifiers on the shipper.
- If the patient is not expected to be ready for same-day administration, before the shipper expires, transfer CARVYKTI to onsite vapor phase of liquid nitrogen storage.
Preparation of CARVYKTI for Infusion
Do not thaw the product until it is ready to be used. Coordinate the timing of CARVYKTI thaw and infusion. Confirm the infusion time in advance and adjust the start time for thaw so that CARVYKTI is available for infusion when the patient is ready. Once thawed, the CARVYKTI infusion must be completed within 2.5 hours at room/ambient temperature (20°C to 25°C).
Prior to thawing the product, confirm that tocilizumab and emergency equipment are available prior to the infusion and during the recovery period.
- Confirm patient identity: Prior to CARVYKTI preparation, match the patient's identity with the patient identifiers on the CARVYKTI cassette. Do not remove the CARVYKTI infusion bag from the cassette if the information on the patient-specific label does not match the intended patient. Contact Janssen Biotech, Inc.at 1-800-526-7736if there are any discrepancies between the labels and the patient identifiers.
- Once patient identification is confirmed, remove the CARVYKTI product bag from the cassette and check that the patient information on the cassette label matches the patient information on the bag label.
- Inspect the product bag for any breaches of container integrity, such as breaks or cracks before and after thawing. Do not administer if the bag is compromised, and contact Janssen Biotech, Inc.at 1-800-526-7736.
- Place the infusion bag inside a sealable plastic bag (preferably sterile) prior to thawing.
- Thaw CARVYKTI at 37°C±2°C using either a water bath or dry thaw method until there is no visible ice in the infusion bag. Total time from start of thaw until completion of thawing should be no more than 15 minutes.
- Remove the infusion bag from the sealable plastic bag and wipe dry. Gently mix the contents of the bag to disperse clumps of cellular material. If visible cell clumps remain, continue to gently mix the contents of the bag. Small clumps of cellular material should disperse with gentle manual mixing. Do not pre-filter into a different container, wash, spin down, or resuspend CARVYKTI in new media prior to infusion.
- Do not re-freeze or refrigerate thawed product.
Administration
- For autologous infusion only.
- Do NOT use a leukocyte-depleting filter.
- Ensure that a minimum of two doses of tocilizumab and emergency equipment are available prior to infusion and during the recovery period.
- Central venous access may be utilized for the infusion of CARVYKTI and is encouraged in patients with poor peripheral access.
- Confirm the patient's identity with the patient identifiers on the infusion bag. Do not infuse CARVYKTI if the information on the patient-specific label does not match the intended patient.
- Prime the tubing of the infusion set with normal saline prior to infusion.
- Once thawed, administer the entire contents of the CARVYKTI bag by intravenous infusion within 2.5 hours using infusion sets fitted with an in-line filter.
- Gently mix the contents of the bag during CARVYKTI infusion to disperse cell clumps.
- After the entire content of the product bag is infused, flush the administration line, inclusive of the in-line filter, with normal saline with a volume equal or greater to the total hold up volume of the primary administration set used inclusive of the drip tube, to ensure that all product is delivered.
CARVYKTI contains human blood cells that are genetically modified with replication-incompetent, self-inactivating, lentiviral vector. Follow universal precautions and local biosafety guidelines for handling and disposal of CARVYKTI to avoid potential transmission of infectious diseases.
Monitoring After Infusion
Administer CARVYKTI at a REMS-certified healthcare facility.
Monitor patients at least daily for 10 days following CARVYKTI infusion at a certified healthcare facility for signs and symptoms of cytokine release syndrome (CRS) and neurologic toxicities. Monitor periodically for 4 weeks for signs and symptoms of delayed neurologic toxicity.
Instruct patients to remain within proximity of a certified healthcare facility for at least 4 weeks following infusion.
Instruct patients to refrain from driving or hazardous activities for at least 8 weeks following infusion.
2.3 Management of Severe Adverse Reactions
Cytokine Release Syndrome (CRS)
Identify CRS based on clinical presentation [see Warnings and Precautions (5.2)] . Evaluate for and treat other causes of fever, hypoxia and hypotension. Consider laboratory testing to monitor for disseminated intravascular coagulation, hematology parameters, as well as pulmonary, cardiac, renal, and hepatic function. If CRS is suspected, manage according to the recommendations in Table 1.
Patients who experience CRS should be closely monitored for cardiac and other organ function until resolution of symptoms. Consider anti-seizure prophylaxis with levetiracetam in patients who experience CRS.
Patients who experience Grade 2 or higher CRS (e.g., hypotension not responsive to fluids, or hypoxia requiring supplemental oxygenation) should be monitored with continuous telemetry and pulse oximetry.
For severe or life-threatening CRS, consider intensive care unit level monitoring and supportive therapy.
For CRS refractory to first line interventions such as tocilizumab or tocilizumab and corticosteroids, consider alternate treatment options (i.e., higher corticosteroid dose, alternative anti-cytokine agents, e.g., anti-IL1 and/or anti-TNFα, anti-T cell therapies). Refractory CRS is characterized by fevers, end-organ toxicity (e.g., hypoxia, hypotension) not improving within 12 hours of first line interventions or development of HLH/MAS.
If concurrent neurologic toxicity is suspected during CRS, administer:
- Corticosteroids according to the more aggressive intervention based on the CRS and neurologic toxicity grades in Tables 1 and 2
- Tocilizumab according to the CRS grade in Table 1
- Anti-seizure medication according to the neurologic toxicity in Table 2
Table 1: CRS grading and management guidance CRS Grade * Tocilizumab †/ Corticosteroids ‡ - *
- Based on ASTCT 2019 grading system (Lee et.al, 2019), modified to include organ toxicity.
- †
- Refer to tocilizumab prescribing information for details.
- ‡
- Continue corticosteroids use until the event is Grade 1 or less; taper steroids if total corticosteroid exposure is greater than 3 days.
- §
- Attributed to CRS. Fever may not always be present concurrently with hypotension or hypoxia, as it may be masked by interventions such as antipyretics or anti-cytokine therapy (e.g., tocilizumab or steroids). Absence of fever does not impact CRS management decision. In this case, CRS management is driven by hypotension and/or hypoxia and by the more severe symptom not attributable to any other cause.
- ¶
- Organ toxicity grading based on National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 5.0.
- #
- Monoclonal antibodies targeting cytokines may be considered based on institutional practice for unresponsive CRS.
- Þ
- Low-flow nasal cannula is ≤6 L/min; high-flow nasal cannula is >6 L/min.
Grade 1 Temperature ≥38°C § In patients with: - Early onset of fever (if onset less than 72 hours after infusion)
Corticosteroids: N/AGrade 2 Symptoms require and respond to moderate intervention.
Temperature ≥38°C §with:
Hypotension not requiring vasopressors,
and/or,
Hypoxia requiring oxygen via cannula Þor blow-by,
or,
Grade 2 organ toxicity. ¶Administer tocilizumab 8 mg/kg IV over 1 hour (not to exceed 800 mg).
Repeat tocilizumab every 8 hours as needed if not responsive to intravenous fluids up to 1 liter or increasing supplemental oxygen.
Consider dexamethasone 10 mg IV every 12–24 hours.
If no improvement within 24 hours or rapid progression, repeat tocilizumab and escalate dose and frequency of dexamethasone (20 mg IV every 6 to 12 hours).
If no improvement within 24 hours or continued rapid progression, switch to methylprednisolone 2 mg/kg IV every 12 hours.
After 2 doses of tocilizumab, consider alternative anti-cytokine agents. #
Do not exceed 3 doses of tocilizumab in 24 hours, or 4 doses in total.Grade 3 Symptoms require and respond to aggressive intervention.
Temperature ≥38°C §with:
Hypotension requiring one vasopressor with or without vasopressin,
and/or,
Hypoxia requiring oxygen via high-flow nasal cannula Þ, facemask, non-rebreather mask, or Venturi mask,
or,
Grade 3 organ toxicity or Grade 4 transaminitis.Administer tocilizumab 8 mg/kg IV over 1 hour (not to exceed 800 mg).
Repeat tocilizumab every 8 hours as needed if not responsive to intravenous fluids up to 1 liter or increasing supplemental oxygen.
Consider dexamethasone 10 mg IV every 12 hours.
If no improvement within 24 hours or rapid progression, repeat tocilizumab and escalate dose and frequency of dexamethasone (20 mg IV every 6 to 12 hours).
If no improvement within 24 hours or continued rapid progression, switch to methylprednisolone 2 mg/kg IV every 12 hours.
After 2 doses of tocilizumab, consider alternative anti-cytokine agents. #
Do not exceed 3 doses of tocilizumab in 24 hours, or 4 doses in total.Grade 4 Life-threatening symptoms.
Requirements for ventilator support, continuous veno-venous hemodialysis (CVVHD).
Temperature ≥38°C §with:
Hypotension requiring multiple vasopressors (excluding vasopressin),
and/or,
Hypoxia requiring positive pressure (e.g., CPAP, BiPAP, intubation, and mechanical ventilation),
or,
Grade 4 organ toxicity (excluding transaminitis).Administer tocilizumab 8 mg/kg IV over 1 hour (not to exceed 800 mg).
Repeat tocilizumab every 8 hours as needed if not responsive to intravenous fluids up to 1 liter or increasing supplemental oxygen.
Administer dexamethasone 20 mg IV every 6 hours.
After 2 doses of tocilizumab, consider alternative anti-cytokine agents #.
Do not exceed 3 doses of tocilizumab in 24 hours, or 4 doses in total.
If no improvement within 24 hours, consider methylprednisolone (1–2 g IV, repeat every 24 hours if needed; taper as clinically indicated) or other immunosuppressants (e.g. other anti-T cell therapies).Neurologic Toxicities
Monitor patients for signs and symptoms of neurologic toxicities (ICANS and other neurologic toxicities) (Table 2). Rule out other causes of neurologic signs or symptoms. Provide intensive care and supportive therapy for severe or life-threatening neurologic toxicities. Please see section 5.3for non ICANS neurologic toxicities. If ICANS is suspected, manage according to the recommendations in Table 2.
If concurrent CRS is suspected during the neurologic toxicity event, administer:
- Corticosteroids according to the more aggressive intervention based on the CRS and neurologic toxicity grades in Tables 1 and 2
- Tocilizumab according to CRS grade in Table 1
- Anti-seizure medication according to neurologic toxicity in Table 2
Table 2: Guideline for management of ICANS ICANS Grade * Corticosteroids Note: ICANS grade and management is determined by the most severe event (ICE score, level of consciousness, seizure, motor findings, raised ICP/cerebral edema), not attributable to any other cause. - *
- ASTCT 2019 criteria for grading Neurologic Toxicity (Lee et.al, 2019).
- †
- If patient is arousable and able to perform Immune Effector Cell-Associated Encephalopathy (ICE) Assessment, assess: Orientation (oriented to year, month, city, hospital = 4 points); Naming (name 3 objects, e.g., point to clock, pen, button = 3 points); Following Commands (e.g., "show me 2 fingers" or "close your eyes and stick out your tongue" = 1 point); Writing (ability to write a standard sentence = 1 point); and Attention (count backwards from 100 by ten = 1 point). If patient is unarousable and unable to perform ICE Assessment (Grade 4 ICANS) = 0 points.
- ‡
- All references to dexamethasone administration are dexamethasone or equivalent.
- §
- Intracranial hemorrhage with or without associated edema is not considered a neurotoxicity feature and is excluded from ICANS grading. It may be graded according to NCI CTCAE v5.0.
- ¶
- Tremors and myoclonus associated with immune effector cell therapies may be graded according to NCI CTCAE v5.0, but they do not influence ICANS grading.
Grade 1
ICE score 7–9 †
or depressed level of consciousness: awakens spontaneously.Consider dexamethasone ‡10 mg IV every 12 to 24 hours for 2 to 3 days.
Consider non-sedating, anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis.Grade 2
ICE score-3–6 †
or depressed level of consciousness: awakens to voiceAdminister dexamethasone ‡10 mg IV every 12 hours for 2–3 days, or longer for persistent symptoms.
Consider steroid taper if total corticosteroid exposure is greater than 3 days.
If no improvement after 24 hours or worsening of neurologic toxicity, increase the dose and/or frequency of dexamethasone up to a maximum of 20 mg IV every 6 hours.
Consider non-sedating, anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis.Grade 3
ICE score-0–2 †
(If ICE score is 0, but the patient is arousable (e.g., awake with global aphasia) and able to perform assessment)
or depressed level of consciousness: awakens only to tactile stimulus,
or seizures, either:- any clinical seizure, focal or generalized, that resolves rapidly, or
- non-convulsive seizures on EEG that resolve with intervention,
Administer dexamethasone ‡10 mg–20 mg IV every 6 hours.
If no improvement after 24 hours or worsening of neurologic toxicity, escalate dexamethasone ‡dose to at least 20 mg IV every 6 hours,
OR escalate to high-dose methylprednisolone (1–2 g/day, repeat every 24 hours if needed; taper as clinically indicated)
Consider non-sedating, anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis.
If cerebral edema is suspected, consider hyperventilation and hyperosmolar therapy. Give high-dose methylprednisolone (1–2 g, repeat every 24 hours if needed; taper as clinically indicated).Grade 4
ICE score-0 †(Patient is unarousable and unable to perform ICE assessment)
or depressed level of consciousness either:- patient is unarousable or requires vigorous or repetitive tactile stimuli to arouse, or
- stupor or coma,
- life-threatening prolonged seizure (>5 min), or
- repetitive clinical or electrical seizures without return to baseline in between,
- deep focal motor weakness such as hemiparesis or paraparesis,
- diffuse cerebral edema on neuroimaging, or
- decerebrate or decorticate posturing, or
- cranial nerve VI palsy, or
- papilledema, or
- Cushing's triad
Administer dexamethasone ‡20 mg IV every 6 hours.
If no improvement after 24 hours or worsening of neurologic toxicity, escalate to high-dose methylprednisolone (1–2 g/day, repeated every 24 hours if needed; taper as clinically indicated).
Consider non-sedating, anti-seizure medicines (e.g., levetiracetam) for seizure prophylaxis.
If raised ICP/cerebral edema is suspected, consider hyperventilation and hyperosmolar therapy. Give high-dose methylprednisolone (1–2 g/day, repeat every 24 hours if needed; taper as clinically indicated), and consider neurology and/or neurosurgery consultation. -
3 DOSAGE FORMS AND STRENGTHS
CARVYKTI is a cell suspension for intravenous infusion.
A single dose of CARVYKTI contains a cell suspension of 0.5–1.0×10 6CAR-positive viable T cells per kg body weight in one infusion bag up to a maximum of 1×10 8CAR-positive viable T cells [see How Supplied/Storage and Handling (16)] .
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Increased Early Mortality
In CARTITUDE-4, a randomized (1:1), controlled trial, there was a numerically higher percentage of early deaths in patients randomized to the CARVYKTI treatment arm compared to the control arm. Among patients with deaths occurring within the first 10 months from randomization, a greater proportion (29/208; 14%) occurred in the CARVYKTI arm compared to (25/211; 12%) in the control arm [see Clinical Studies (14)]. Of the 29 deaths that occurred in the CARVYKTI arm within the first 10 months of randomization, 10 deaths occurred prior to CARVYKTI infusion, and 19 deaths occurred after CARVYKTI infusion. Of the 10 deaths that occurred prior to CARVYKTI infusion, all occurred due to disease progression, and none occurred due to adverse events. Of the 19 deaths that occurred after CARVYKTI infusion, 3 occurred due to disease progression, and 16 occurred due to adverse events. The most common adverse events were due to infection (n=12).
5.2 Cytokine Release Syndrome
Cytokine release syndrome (CRS), including fatal or life-threatening reactions, occurred following treatment with CARVYKTI. Among patients receiving CARVYKTI for relapsed or refractory multiple myeloma in the CARTITUDE-1 and CARTITUDE-4 studies (N=285), CRS occurred in 84% (238/285), including ≥ Grade 3 CRS (ASTCT 2019) in 4% (11/285) of patients. The median time to onset of CRS, any grade, was 7 days (range: 1 to 23 days). Cytokine release syndrome resolved in 82% with a median duration of 4 days (range: 1 to 97 days). The most common manifestations of CRS in all patients combined (≥ 10%) included fever (84%), hypotension (29%) and aspartate aminotransferase increased (11%). Serious events that may be associated with CRS include pyrexia, hemophagocytic lymphohistiocytosis, respiratory failure, disseminated intravascular coagulation, capillary leak syndrome, and supraventricular and ventricular tachycardia [see Adverse Reactions (6.1].
Cytokine release syndrome occurred in 78% of patients in CARTITUDE-4 (3% Grade 3 to 4) and in 95% of patients in CARTITUDE-1 (4% Grade 3 to 4).
Identify CRS based on clinical presentation. Evaluate for and treat other causes of fever, hypoxia, and hypotension. CRS has been reported to be associated with findings of HLH/MAS, and the physiology of the syndromes may overlap. HLH/MAS is a potentially life-threatening condition. In patients with progressive symptoms of CRS or refractory CRS despite treatment, evaluate for evidence of HLH/MAS. Please see Section 5.4; Hemophagocytic Lymphohistiocytosis (HLH)/Macrophage Activation Syndrome (MAS).
Ensure that a minimum of two doses of tocilizumab are available prior to infusion of CARVYKTI.
Of the 285 patients who received CARVYKTI in clinical trials, 53% (150/285) patients received tocilizumab; 35% (100/285) received a single dose, while 18% (50/285) received more than 1 dose of tocilizumab. Overall, 14% (39/285) of patients received at least one dose of corticosteroids for treatment of CRS.
Monitor patients at least daily for 10 days following CARVYKTI infusion at a REMS-certified healthcare facility for signs and symptoms of CRS. Monitor patients for signs or symptoms of CRS for at least 4 weeks after infusion. At the first sign of CRS, immediately institute treatment with supportive care, tocilizumab, or tocilizumab and corticosteroids, as indicated in Table 1 [see Dosing and Administration (2.3)] .
Counsel patients to seek immediate medical attention should signs or symptoms of CRS occur at any time [see Patient Counseling information (17)].
5.3 Neurologic Toxicities
Neurologic toxicities, which may be severe, life-threatening or fatal, occurred following treatment with CARVYKTI. Neurologic toxicities included ICANS, neurologic toxicity with signs and symptoms of parkinsonism, GBS, immune mediated myelitis, peripheral neuropathies and cranial nerve palsies. Counsel patients on the signs and symptoms of these neurologic toxicities, and on the delayed nature of onset of some of these toxicities. Instruct patients to seek immediate medical attention for further assessment and management if signs or symptoms of any of these neurologic toxicities occur at any time [see Patient Counseling Information (17)] .
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies for relapsed and refractory multiple myeloma, one or more neurologic toxicities occurred in 24% (69/285), including ≥ Grade 3 cases in 7% (19/285) of patients. The median time to onset was 10 days (range: 1 to 101) with 63/69 (91%) of cases developing by 30 days. Neurologic toxicities resolved in 72% (50/69) of patients with a median duration to resolution of 23 days (range: 1 to 544). Of patients developing neurotoxicity, 96% (66/69) also developed CRS. Subtypes of neurologic toxicities included ICANS in 13%, peripheral neuropathy in 7%, cranial nerve palsy in 7%, parkinsonism in 3%, and immune mediated-myelitis in 0.4% of the patients [see Adverse Reactions (6.1)] .
Immune Effector Cell-associated Neurotoxicity Syndrome (ICANS)
Patients receiving CARVYKTI may experience fatal or life-threatening ICANS following treatment with CARVYKTI, including before CRS onset, concurrently with CRS, after CRS resolution, or in the absence of CRS.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, ICANS occurred in 13% (36/285), including Grade ≥ 3 in 2% (6/285) of the patients. The median time to onset of ICANS was 8 days (range: 1 to 28 days). ICANS resolved in 30 of 36 (83%) of patients with a median time to resolution of 3 days (range: 1 to 143 days). The median duration of ICANS was 6 days (range: 1 to 1229 days) in all patients including those with ongoing neurologic events at the time of death or data cut off. Of patients with ICANS 97% (35/36) had CRS. The onset of ICANS occurred during CRS in 69% of patients, before and after the onset of CRS in 14% of patients respectively.
Immune Effector Cell-associated Neurotoxicity Syndrome occurred in 7% of patients in CARTITUDE-4 (0.5% Grade 3) and in 23% of patients in CARTITUDE-1 (3% Grade 3).
The most frequent ≥2% manifestations of ICANS included encephalopathy (12%), aphasia (4%), headache (3%), motor dysfunction (3%), ataxia (2%) and sleep disorder (2%) [see Adverse Reactions (6.1)] .
Monitor patients at least daily for 10 days following CARVYKTI infusion at the REMS-certified healthcare facility for signs and symptoms of ICANS. Rule out other causes of ICANS symptoms. Monitor patients for signs or symptoms of ICANS for at least 4 weeks after infusion and treat promptly. Neurologic toxicity should be managed with supportive care and/or corticosteroids as needed [see Dosage and Administration (2.3)] .
Parkinsonism
Neurologic toxicity with parkinsonism has been reported in clinical trials of CARVYKTI.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, parkinsonism occurred in 3% (8/285), including Grade ≥ 3 in 2% (5/285) of the patients. The median time to onset of parkinsonism was 56 days (range: 14 to 914 days). Parkinsonism resolved in 1 of 8 (13%) of patients with a median time to resolution of 523 days. The median duration of parkinsonism was 243.5 days (range: 62 to 720 days) in all patients including those with ongoing neurologic events at the time of death or data cut off. The onset of parkinsonism occurred after CRS for all patients and after ICANS for 6 patients.
Parkinsonism occurred in 1% of patients in CARTITUDE-4 (no Grade 3 to 4) and in 6% of patients in CARTITUDE-1 (4% Grade 3 to 4).
The manifestations of parkinsonism included movement disorders, cognitive impairment, and personality changes [see Adverse Reactions (6.1)] .
Monitor patients for signs and symptoms of parkinsonism that may be delayed in onset and managed with supportive care measures. There is limited efficacy information with medications used for the treatment of Parkinson's disease for the improvement or resolution of parkinsonism symptoms following CARVYKTI treatment.
Guillain-Barré Syndrome
A fatal outcome following GBS occurred following treatment with CARVYKTI despite treatment with intravenous immunoglobulins. Symptoms reported include those consistent with Miller-Fisher variant of GBS, encephalopathy, motor weakness, speech disturbances, and polyradiculoneuritis.
Monitor for GBS. Evaluate patients presenting with peripheral neuropathy for GBS. Consider treatment of GBS with supportive care measures and in conjunction with immunoglobulins and plasma exchange, depending on severity of GBS.
Immune Mediated Myelitis
Grade 3 myelitis occurred 25 days following treatment with CARVYKTI in CARTITUDE-4 in a patient who received CARVYKTI as subsequent therapy. Symptoms reported included hypoesthesia of the lower extremities and the lower abdomen with impaired sphincter control. Symptoms improved with the use of corticosteroids and intravenous immune globulin. Myelitis was ongoing at the time of death from other cause [see Adverse Reactions (6.1)] .
Peripheral Neuropathy
Peripheral neuropathy occurred following treatment with CARVYKTI.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, peripheral neuropathy occurred in 7% (21/285), including Grade ≥ 3 in 1% (3/285) of the patients. The median time to onset of peripheral neuropathy was 57 days (range: 1 to 914 days). Peripheral neuropathy resolved in 11 of 21 (52%) of patients with a median time to resolution of 58 days (range: 1 to 215 days). The median duration of peripheral neuropathy was 149.5 days (range: 1 to 692 days) in all patients including those with ongoing neurologic events at the time of death or data cut off [see Adverse Reactions (6.1)] .
Peripheral neuropathies occurred in 7% of patients in CARTITUDE-4 (0.5% Grade 3 to 4) and in 7% of patients in CARTITUDE-1 (2% Grade 3 to 4).
Monitor patients for signs and symptoms of peripheral neuropathies.
Patients who experience peripheral neuropathy may also experience cranial nerve palsies or GBS.
Cranial Nerve Palsies
Cranial nerve palsies occurred following treatment with CARVYKTI.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, cranial nerve palsies occurred in 7% (19/285), including Grade ≥ 3 in 1% (1/285) of the patients. The median time to onset of cranial nerve palsies was 21 days (range: 17 to 101 days). Cranial nerve palsies resolved in 17 of 19 (89%) of patients with a median time to resolution of 66 days (range: 1 to 209 days). The median duration of cranial nerve palsies was 70 days (range: 1 to 262 days) in all patients including those with ongoing neurologic events at the time of death or data cut off [see Adverse Reactions (6.1)] .
Cranial nerve palsies occurred in 9% of patients in CARTITUDE-4 (1% Grade 3 to 4) and in 3% of patients in CARTITUDE-1 (1% Grade 3 to 4).
The most frequent cranial nerve affected was the 7 thcranial nerve. Additionally, cranial nerves III, V, and VI have been reported to be affected.
Monitor patients for signs and symptoms of cranial nerve palsies. Consider management with systemic corticosteroids, depending on the severity and progression of signs and symptoms.
5.4 Hemophagocytic Lymphohistiocytosis (HLH)/Macrophage Activation Syndrome (MAS)
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, HLH/MAS occurred in 1% (3/285) of patients. All events of HLH/MAS had onset within 99 days of receiving CARVYKTI, with a median onset of 10 days (range: 8 to 99 days) and all occurred in the setting of ongoing or worsening CRS. The manifestations of HLH/MAS included hyperferritinemia, hypotension, hypoxia with diffuse alveolar damage, coagulopathy and hemorrhage, cytopenia and multi-organ dysfunction, including renal dysfunction and respiratory failure.
Patients who develop HLH/MAS have an increased risk of severe bleeding. Monitor hematologic parameters in patients with HLH/MAS and transfuse per institutional guidelines. Fatal cases of HLH/MAS occurred following treatment with CARVYKTI [see Adverse Reactions (6.1)].
HLH is a life-threatening condition with a high mortality rate if not recognized and treated early. Treatment of HLH/MAS should be administered per institutional standards.
5.5 CARVYKTI REMS
Because of the risk of CRS and neurologic toxicities, CARVYKTI is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS) called the CARVYKTI REMS [see Boxed Warning, Warnings and Precautions (5.2, 5.3)] . The required components of the CARVYKTI REMS are:
- Healthcare facilities that dispense and administer CARVYKTI must be enrolled and comply with the REMS requirements.
- Certified healthcare facilities must have on-site, immediate access to tocilizumab.
- Ensure that a minimum of 2 doses of tocilizumab are available for each patient for infusion within 2 hours after CARVYKTI infusion, if needed for treatment of CRS.
Further information is available at www.carvyktirems.com or 1-844-672-0067.
5.6 Prolonged and Recurrent Cytopenias
Patients may exhibit prolonged and recurrent cytopenias following lymphodepleting chemotherapy and CARVYKTI infusion.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, Grade 3 or higher cytopenias not resolved by day 30 following CARVYKTI infusion occurred in 62% (176/285) of the patients and included thrombocytopenia 33% (94/285), neutropenia 27% (76/285), lymphopenia 24% (67/285) and anemia 2% (6/285). After Day 60 following CARVYKTI infusion 22%, 20%, 5%, and 6% of patients had a recurrence of Grade 3 or 4 lymphopenia, neutropenia, thrombocytopenia, and anemia respectively, after initial recovery of their Grade 3 or 4 cytopenia. Seventy-seven percent (219/285) of patients had one, two or three or more recurrences of Grade 3 or 4 cytopenias after initial recovery of Grade 3 or 4 cytopenia. Sixteen and 25 patients had Grade 3 or 4 neutropenia and thrombocytopenia, respectively, at the time of death [see Adverse Reactions (6.1)].
Monitor blood counts prior to and after CARVYKTI infusion. Manage cytopenias with growth factors and blood product transfusion support according to local institutional guidelines.
5.7 Infections
CARVYKTI should not be administered to patients with active infection or inflammatory disorders. Severe, life-threatening, or fatal infections, occurred in patients after CARVYKTI infusion.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, infections occurred in 57% (163/285), including ≥ Grade 3 in 24% (69/285) of patients. Grade 3 or 4 infections with an unspecified pathogen occurred in 12%, viral infections in 6%, bacterial infections in 5%, and fungal infections in 1% of patients. Overall, 5% (13/285) of patients had Grade 5 infections, 2.5% of which were due to COVID-19. Patients treated with CARVYKTI had an increased rate of fatal COVID-19 infections compared to the standard therapy arm [see Adverse Reactions (6.1)].
Monitor patients for signs and symptoms of infection before and after CARVYKTI infusion and treat patients appropriately. Administer prophylactic, pre-emptive and/or therapeutic antimicrobials according to the standard institutional guidelines. Febrile neutropenia was observed in 5% of patients after CARVYKTI infusion and may be concurrent with CRS. In the event of febrile neutropenia, evaluate for infection and manage with broad-spectrum antibiotics, fluids and other supportive care, as medically indicated.
Counsel patients on the importance of prevention measures. Follow institutional guidelines for the vaccination and management of immunocompromised patients with COVID-19.
Viral Reactivation
Hepatitis B virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure and death, can occur in patients with hypogammaglobulinemia.
Perform screening for Cytomegalovirus (CMV), HBV, hepatitis C virus (HCV), and human immunodeficiency virus (HIV) or any other infectious agents if clinically indicated in accordance with clinical guidelines before collection of cells for manufacturing.
Consider antiviral therapy to prevent viral reactivation per local institutional guidelines/clinical practice.
5.8 Hypogammaglobulinemia
Hypogammaglobulinemia can occur in patients receiving treatment with CARVYKTI.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, hypogammaglobulinemia adverse event was reported in 36% (102/285) of patients; laboratory IgG levels fell below 500 mg/dl after infusion in 93% (265/285) of patients. Hypogammaglobulinemia either as an adverse reaction or laboratory IgG level below 500 mg/dl, after infusion occurred in 94% (267/285) of patients treated. Fifty six percent (161/285) of patients received intravenous immunoglobulin (IVIG) post CARVYKTI for either an adverse reaction or prophylaxis [see Adverse Reactions (6.1)] .
Monitor immunoglobulin levels after treatment with CARVYKTI and administer IVIG for IgG <400 mg/dL. Manage per local institutional guidelines, including infection precautions and antibiotic or antiviral prophylaxis.
Use of Live Vaccines
The safety of immunization with live viral vaccines during or following CARVYKTI treatment has not been studied. Vaccination with live virus vaccines is not recommended for at least 6 weeks prior to the start of lymphodepleting chemotherapy, during CARVYKTI treatment, and until immune recovery following treatment with CARVYKTI.
5.9 Hypersensitivity Reactions
Hypersensitivity reactions occurred following treatment with CARVYKTI.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, hypersensitivity reactions occurred in 5% (13/285), all of which were ≤ Grade 2. Manifestations of hypersensitivity reactions included flushing, chest discomfort, tachycardia, wheezing, tremor, burning sensation, non-cardiac chest pain, and pyrexia.
Serious hypersensitivity reactions, including anaphylaxis, may be due to the dimethyl sulfoxide (DMSO) in CARVYKTI. Patients should be carefully monitored for 2 hours after infusion for signs and symptoms of severe reaction. Treat promptly and manage patients appropriately according to the severity of the hypersensitivity reaction.
5.10 Secondary Malignancies
Patients treated with CARVYKTI may develop secondary malignancies.
Among patients receiving CARVYKTI in the CARTITUDE-1 and CARTITUDE-4 studies, myeloid neoplasms occurred in 5% (13/285) of patients (9 cases of myelodysplastic syndrome, 3 cases of acute myeloid leukemia, and 1 case of myelodysplastic syndrome followed by acute myeloid leukemia). The median time to onset of myeloid neoplasms was 447 days (range: 56 to 870 days) after treatment with CARVYKTI. Ten of these 13 patients died following the development of myeloid neoplasms; 2 of the 13 cases of myeloid neoplasm occurred after initiation of subsequent antimyeloma therapy. Cases of myelodysplastic syndrome and acute myeloid leukemia have also been reported in the post marketing setting.
T-cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T-cell immunotherapies, including CARVYKTI. Mature T-cell malignancies, including CAR-positive tumors, may present as soon as weeks following infusions, and may include fatal outcomes [see Boxed Warning, Adverse Reactions (6.1, 6.3), Patient Counseling Information (17)] .
Monitor life-long for secondary malignancies. In the event that a secondary malignancy occurs, contact Janssen Biotech, Inc. at 1-800-526-7736 for reporting and to obtain instructions on collection of patient samples.
5.11 Effects on Ability to Drive and Use Machines
Due to the potential for neurologic events, including altered mental status, seizures, neurocognitive decline or neuropathy, patients receiving CARVYKTI are at risk for altered or decreased consciousness or coordination in the 8 weeks following CARVYKTI infusion. Advise patients to refrain from driving and engaging in hazardous occupations or activities, such as operating heavy or potentially dangerous machinery during this initial period, and in the event of new onset of any neurologic toxicities.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are also described elsewhere in the labeling:
- Increased Early Mortality [see Warnings and Precautions (5.1), Clinical Studies (14)] .
- Cytokine Release Syndrome [see Warnings and Precautions (5.2)] .
- Neurologic Toxicities [see Warnings and Precautions (5.3)] .
- Hemophagocytic Lymphohistiocytosis (HLH)/Macrophage Activation Syndrome (MAS) [see Warnings and Precautions (5.4)] .
- Prolonged and Recurrent Cytopenias [see Warnings and Precautions (5.6)] .
- Infections [see Warnings and Precautions (5.7)] .
- Hypogammaglobulinemia [see Warnings and Precautions (5.8)] .
- Hypersensitivity Reactions [see Warnings and Precautions (5.9)] .
- Secondary Malignancies [see Warnings and Precautions (5.10)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described in the WARNINGS and PRECAUTIONS section reflect exposure to CARVYKTI in 285 patients with relapsed or refractory multiple myeloma: one randomized, open label with 188 patients in CARTITUDE-4 and one single-arm, open label study with 97 patients in CARTITUDE-1.
CARTITUDE-4
The safety of CARVYKTI was evaluated in CARTITUDE-4, a randomized, open label multicenter study, in which patients with relapsed and lenalidomide refractory multiple myeloma received CARVYKTI meeting the product specifications (N=188) or standard therapy (N=211) [see Clinical Studies (14)] . Patients with known active or prior history of central nervous system involvement, patients who exhibit clinical signs of meningeal involvement of multiple myeloma and patients with a history of Parkinson's disease or other neurodegenerative disorder, were excluded from the trial. Patients received CARVYKTI at a median dose of 0.71×10 6CAR-positive viable T-cells/kg (range: 0.41 to 1.08×10 6cells/kg). The median age of the 188 participants was 62 years (range: 27 to 78 years); 40% were 65 years or older, and 57% were male; 76% were White, were 9% Hispanic or Latino, 8% were Asian, and 3% were Black.
The Eastern Cooperative Oncology Group (ECOG) performance status at baseline was 0 in 56%, 1 in 44%. For the details about the study population, see Clinical Studies (14).
The most common nonlaboratory adverse reactions (≥20%) included pyrexia, CRS, hypogammaglobulinemia, musculoskeletal pain, fatigue, diarrhea, upper respiratory tract infection, viral infections, headache, hypotension, and nausea.
Serious adverse reactions occurred in 34% of patients. The most common nonlaboratory serious adverse reactions (≥5%) were pneumonia (9%), viral infection (6%), CRS (6%), and cranial nerve palsies (5%).
Table 3 summarizes the adverse reactions that occurred in at least 10% of patients treated with CARVYKTI.
Table 3: Adverse reactions observed in at least 10% of patients treated with CARVYKTI (N=188) and standard therapy (N=208) in CARTITUDE-4 CARVYKTI
N=188Standard Therapy
N=208System Organ Class (SOC)
Preferred termAny Grade
(%)Grade 3 or higher (%) Any Grade
(%)Grade 3 or higher (%) Adverse reactions are reported using MedDRA version 25.0 - *
- Diarrhea includes Colitis, and Diarrhea.
- †
- Fatigue includes Asthenia, Fatigue, and Malaise.
- ‡
- Edema includes Face edema, Generalized edema, Localized edema, Edema peripheral, Periorbital edema, Peripheral swelling, Pulmonary edema, and Scrotal edema.
- §
- Pain includes Anorectal discomfort, Catheter site pain, Flank pain, Inflammatory pain, Pain, Pain in jaw, Pain of skin, Pelvic pain, Rhinalgia, and Sacral pain.
- ¶
- Hypogammaglobulinemia includes subjects with adverse event of hypogammaglobulinemia and/or laboratory IgG levels that fell below 500 mg/dL following CARVYKTI infusion or standard therapy.
- #
- Upper respiratory tract infection includes Bronchitis, Nasal congestion, Nasopharyngitis, Pharyngitis, Respiratory tract infection, Rhinitis, Rhinorrhea, Rhinovirus infection, Sinusitis, Upper respiratory tract infection, and Viral pharyngitis.
- Þ
- Viral infection includes Adenovirus infection, Asymptomatic COVID-19, COVID-19, Cytomegalovirus infection, Cytomegalovirus infection reactivation, Cytomegalovirus viremia, Hepatitis B reactivation, Herpes simplex reactivation, Herpes virus infection, Herpes zoster, Human herpesvirus 6 infection, Influenza, Lymphadenitis viral, Metapneumovirus infection, Parainfluenza virus infection, Parvovirus B19 infection, Parvovirus infection, Respiratory syncytial virus infection, Respiratory tract infection viral, and Rotavirus infection.
- ß
- Bacterial infection includes Bordetella infection, Bronchitis bacterial, Campylobacter infection, Catheter site infection, Cellulitis, Chalazion, Citrobacter infection, Clostridium difficile colitis, Device related infection, Gingivitis, Perichondritis, Pyelonephritis acute, Salmonellosis, Skin infection, Staphylococcal infection, Superinfection bacterial, Vascular access site infection, and Vascular device infection.
- à
- Pneumonia includes COVID-19 pneumonia, Lower respiratory tract infection, Metapneumovirus pneumonia, Pneumonia, Pneumonia moraxella, Pneumonia pseudomonal, and Pneumonia streptococcal.
- è
- Musculoskeletal pain includes Arthralgia, Back pain, Bone pain, Bursitis, Musculoskeletal chest pain, Musculoskeletal pain, Myalgia, Myositis, Neck pain, Non-cardiac chest pain, Osteoarthritis, Pain in extremity, Plantar fasciitis, Rotator cuff syndrome, Spinal pain, and Tendonitis.
- ð
- Headache includes Headache and Tension headache.
- ø
- Encephalopathy includes Amnesia, Bradyphrenia, Confusional state, Depressed level of consciousness, Disturbance in attention, Immune effector cell-associated neurotoxicity syndrome, Lethargy, and Psychomotor retardation.
- ý
- Cough includes Cough, Productive cough, and Upper-airway cough syndrome.
- £
- Hypotension includes Hypotension, and Orthostatic hypotension.
Gastrointestinal disorders - - - - Diarrhea * 27 3 27 2 Nausea 20 0 18 1 Constipation 10 0 21 1 General disorders and administrative site conditions - - - - Pyrexia 79 5 16 1 Fatigue † 28 3 50 3 Edema ‡ 11 1 20 1 Pain § 10 1 14 <1 Immune system disorders - - - - Hypogammaglobulinemia ¶ 94 9 72 <1 Cytokine release syndrome 78 3 <1 0 Infections and infestations - - - - Upper respiratory tract infection # 25 1 40 5 Viral infection Þ 23 4 31 6 Bacterial infection ß 15 6 17 4 Pneumonia à 14 9 18 11 Metabolism and nutrition disorders - - - - Decreased appetite 10 0 5 0 Musculoskeletal and connective tissue disorders - - - - Musculoskeletal pain è 34 2 47 4 Nervous system disorders - - - - Headache ð 23 0 13 0 Encephalopathy ø 11 2 4 1 Respiratory, thoracic and mediastinal disorders - - - - Cough ý 15 0 18 0 Hypoxia 12 3 1 1 Vascular disorders - - - - Hypotension £ 23 4 3 0 Other clinically important adverse reactions that occurred in less than 10% of patients treated with CARVYKTI include the following:
- Blood and lymphatic system disorders: coagulopathy 1(5%), febrile neutropenia (2%), lymphocytosis (2%),
- Cardiac disorders: tachycardia 2(5%), cardiac arrhythmias 3(3%)
- Gastrointestinal disorders: abdominal pain 4(6%), vomiting (5%)
- General disorders and administration site conditions: chills (6%)
- Immune system disorders:HLH (1%)
- Infections and Infestations:gastroenteritis 5(7%), sepsis 6(9%), urinary tract infection 7(5%), fungal infection 8(3%)
- Investigations: c-reactive protein increased (6%)
- Metabolism and Nutrition Disorders: hypophosphatemia (10%), hyperferritinemia (7%)
- Neoplasms benign, malignant, and unspecified (incl cysts and polyps): hematologic malignancy 9(3%)
- Nervous system disorders: dizziness 10(9%), cranial nerve palsies 11(9%), motor dysfunction 12(9%), peripheral neuropathy 13(7%), sleep disorder 14(6%), tremor (4%), aphasia 15(3%), ataxia 16(3%),
- Psychiatric disorders: delirium 17(2%) personality changes 18(2%)
- Renal and urinary disorders: renal failure 19(5%)
- Respiratory, thoracic and mediastinal disorders: dyspnea 20(10%)
- Skin and subcutaneous tissues: rash 21(7%)
- Vascular Disorders: hemorrhage 22(9%), hypertension (7%), thrombosis 23(3%), capillary leak syndrome (1%)
- 1
- Coagulopathy includes Blood fibrinogen decreased, Coagulation test abnormal, Coagulopathy, Disseminated intravascular coagulation, and Hypofibrinogenemia.
- 2
- Tachycardia includes Sinus tachycardia, and Tachycardia.
- 3
- Cardiac arrhythmias includes Atrial fibrillation, and Atrioventricular block second degree.
- 4
- Abdominal pain includes Abdominal discomfort, Abdominal pain, Abdominal pain lower, Abdominal pain upper, and Dyspepsia.
- 5
- Gastroenteritis includes Enterocolitis viral, Enterovirus infection, Gastroenteritis, Gastroenteritis rotavirus, Gastroenteritis salmonella, Gastrointestinal infection, and Large intestine infection.
- 6
- Sepsis includes Bacteremia, Candida sepsis, Device related bacteremia, Enterococcal bacteremia, Hemophilus sepsis, Neutropenic sepsis, Pseudomonal sepsis, Sepsis, Septic shock, Staphylococcal bacteremia, Systemic candida, and Urosepsis.
- 7
- Urinary tract infection includes Cystitis, Escherichia urinary tract infection, and Urinary tract infection.
- 8
- Fungal infection includes Candida infection, Oral candidiasis, Tongue fungal infection, and Vulvovaginal candidiasis.
- 9
- Hematologic malignancy includes Myelodysplastic syndrome, Acute myeloid leukemia, and T-cell lymphoma. Incidence based on cutoff date of 01 November 2022 (median follow-up time of 15.9 months).
- 10
- Dizziness includes Dizziness, Dizziness postural, Presyncope, Syncope, and Vertigo.
- 11
- Cranial nerve palsies includes Facial paralysis, Facial paresis, IIIrd nerve paralysis, and Trigeminal palsy.
- 12
- Motor dysfunction includes Bradykinesia, Coordination abnormal, Dysgraphia, Extrapyramidal disorder, Micrographia, Muscle spasms, Muscular weakness, and Parkinsonism.
- 13
- Neuropathy peripheral includes Peripheral motor neuropathy, Peripheral sensory neuropathy, and Polyneuropathy.
- 14
- Sleep disorder includes Insomnia, Sleep disorder, and Somnolence.
- 15
- Aphasia includes Aphasia, and Dysarthria.
- 16
- Ataxia includes Ataxia, Balance disorder, Dysmetria, and Gait disturbance.
- 17
- Delirium includes Agitation, Disorientation, and Hallucination.
- 18
- Personality changes includes Personality change, and Reduced facial expression.
- 19
- Renal failure includes Acute kidney injury, Blood creatinine increased, Chronic kidney disease, Renal failure, and Renal impairment.
- 20
- Dyspnea includes Dyspnea, Dyspnea exertional, Respiratory failure, Tachypnea, and Wheezing.
- 21
- Rash includes Dermatitis psoriasiform, Drug eruption, Erythema, Pityriasis lichenoides et varioliformis acuta, Rash, Rash erythematous, Rash maculo-papular, Rash papular, and Urticaria.
- 22
- Hemorrhage includes Catheter site hemorrhage, Conjunctival hemorrhage, Contusion, Epistaxis, Hematemesis, Hematoma, and Hematuria.
- 23
- Thrombosis includes Deep vein thrombosis, Pulmonary embolism, and Venous thrombosis limb.
Laboratory Abnormalities
Table 4 presents the most common Grade 3 or 4 laboratory abnormalities based on laboratory data, occurring in at least 10% of patients.
Table 4: Grade 3 or 4 laboratory abnormalities in at least 10% of patients treated with CARVYKTI (N=188) and standard therapy (N=208) in CARTITUDE-4 CARVYKTI
(N=188)Standard Therapy
(N=208)Laboratory Abnormality Grade 3 or 4 (%) Grade 3 or 4 (%) Laboratory abnormalities graded using NCI Common Terminology Criteria for Adverse Events version 5.0. Laboratory abnormalities are sorted by decreasing frequency in the Grade column. Lymphocyte count decreased 99 62 Neutrophil count decreased 95 88 White blood cell decreased 94 69 Platelet count decreased 47 20 Hemoglobin decreased 34 17 Other clinically important Grade 3 or 4 laboratory abnormalities (based on laboratory data) that occurred in less than 10% of patients treated with CARVYKTI include fibrinogen decreased, gamma glutamyl transferase increased, hypokalemia, alanine aminotransferase increased, aspartate aminotransferase increased, alkaline phosphatase increased, hypoalbuminemia, hyponatremia, hypertriglyceridemia, hypomagnesemia, hypocalcemia, and blood bilirubin increased.
CARTITUDE-1
The safety data described in this section reflect the exposure of 97 adult patients with relapsed/refractory multiple myeloma in the CARTITUDE-1 study (USA cohort) to CARVYKTI and includes 17 patients (18%) with manufacturing failures either because they received CARVYKTI that did not meet product release specifications or there were insufficient data to confirm product release specifications for CARVYKTI. Patients received CARVYKTI across a dose range of 0.51 to 0.95×10 6CAR-positive viable T cells/kg body weight [see Clinical Studies (14)] . Patients with a history of CNS disease (such as seizure or cerebrovascular ischemia) or requiring ongoing treatment with chronic immunosuppression were excluded. The median duration of follow-up was 18 months. The median age of the study population was 61 years (range: 43 to 78 years); 36% were 65 years or older, and 59% were men. The Eastern Cooperative Oncology Group (ECOG) performance status at baseline was 0 in 40%, 1 in 56%, and 2 in 4% of patients. Three of the patients treated with CARVYKTI had a creatinine clearance of <45 mL/min at baseline. For the details about the study population, see Clinical Studies (14).
The most common (greater or equal to 10%) Grade 3 or higher nonlaboratory adverse reactions were infections-pathogen unspecified (19%), pneumonia (13%), hematologic malignancy (10%) and hypotension (10%).
The most common nonlaboratory adverse reactions (incidence greater than or equal to 20%) included pyrexia, CRS, hypogammaglobulinemia, hypotension, musculoskeletal pain, fatigue, infections of unspecified pathogen, cough, chills, diarrhea, nausea, encephalopathy, decreased appetite, upper respiratory tract infection, headache, tachycardia, dizziness, dyspnea, edema, viral infections, coagulopathy, constipation, and vomiting.
Serious adverse reactions occurred in 55% of patients. The most common non-laboratory (greater than or equal to 5%) serious adverse reactions included CRS (21%), sepsis (7%), encephalopathy (10%), and pneumonia (8%). Fatal adverse reactions occurred in 9% of patients.
Table 5 summarizes the adverse reactions that occurred in at least 10% of patients treated with CARVYKTI.
Table 5: Adverse reactions observed in at least 10% of patients treated with CARVYKTI in CARTITUDE-1 (N=97) System Organ Class (SOC)
Preferred termAny Grade (%) Grade 3 or higher (%) Adverse reactions are reported using MedDRA version 23.0 - *
- Coagulopathy includes Activated partial thromboplastin time prolonged, Coagulopathy, Disseminated intravascular coagulation, Hypofibrinogenemia, International normalized ratio increased, and Prothrombin time prolonged. Also includes terms reported under investigation SOC.
- †
- Tachycardia includes Sinus tachycardia, and Tachycardia.
- ‡
- Diarrhea includes Colitis, and Diarrhea.
- §
- Fatigue includes Asthenia, Fatigue, and Malaise.
- ¶
- Edema includes Face edema, Generalized edema, Localized edema, Edema peripheral, Periorbital edema, Peripheral swelling, Pulmonary edema, and Scrotal edema.
- #
- Cytokine release syndrome includes CRS, and Systemic inflammatory response syndrome.
- Þ
- Hypogammaglobulinemia includes subjects with adverse event of hypogammaglobulinemia (12%) and/or laboratory IgG levels that fell below 500 mg/dL following CARVYKTI infusion (92%).
- ß
- Infections and infestations System Organ Class Adverse Events are grouped by pathogen type and selected clinical syndromes.
- à
- Infections - pathogen unspecified includes Abscess limb, Atypical pneumonia, Bacteremia, Bronchitis, Conjunctivitis, Enterocolitis infectious, Folliculitis, Gastroenteritis, Lung abscess, Lung opacity, Osteomyelitis, Otitis media, Parotitis, Perirectal abscess, Pneumonia, Rash pustular, Rhinitis, Sepsis, Septic shock, Sinusitis, Skin infection, Soft tissue infection, Upper respiratory tract infection, and Urinary tract infection.
- è
- Upper respiratory tract infection includes Human rhinovirus test positive, Rhinitis, Rhinovirus infection, Sinusitis, Upper respiratory tract infection, and Viral upper respiratory tract infection. Also includes terms reported under investigation SOC. Upper respiratory tract infections may also be included under pathogen categories.
- ð
- Viral infection includes Adenovirus test positive, Coronavirus infection, Cytomegalovirus syndrome, Cytomegalovirus viremia, Enterovirus infection, Gastroenteritis viral, Herpes zoster, Herpes zoster disseminated, Influenza, Influenza like illness, Oral herpes, Parainfluenza virus infection, Rhinovirus infection, Urinary tract infection viral, and Viral upper respiratory tract infection.
- ø
- Pneumonia includes Atypical pneumonia, Lung abscess, Lung opacity, Pneumocystis jirovecii pneumonia, Pneumonia, and Pneumonia aspiration.
- ý
- Sepsis includes Bacteremia, Bacterial sepsis, Pseudomonal bacteremia, Sepsis, Septic shock, and Staphylococcal bacteremia.
- £
- Musculoskeletal pain includes Arthralgia, Back pain, Bone pain, Joint stiffness, Muscle strain, Musculoskeletal chest pain, Musculoskeletal discomfort, Musculoskeletal pain, Musculoskeletal stiffness, Myalgia, Neck pain, Non-cardiac chest pain, and Pain in extremity.
- ¥
- Encephalopathy includes Amnesia, Bradyphrenia, Confusional state, Depressed level of consciousness, Disturbance in attention, Encephalopathy, Immune effector cell-associated neurotoxicity syndrome, Lethargy, Memory impairment, Mental impairment, Mental status changes, Noninfective encephalitis, and Somnolence.
- Œ
- Dizziness includes Dizziness, Presyncope, and Syncope.
- œ
- Motor dysfunction includes Motor dysfunction, Muscle spasms, Muscle tightness, Muscular weakness, and Myoclonus.
- Ɖ
- Cough includes Cough, Productive cough, and Upper-airway cough syndrome.
- A
- Dyspnea includes Acute respiratory failure, Dyspnea, Dyspnea exertional, Respiratory failure, and Tachypnea.
- B
- Hematologic malignancy includes Myelodysplastic syndrome and Acute myeloid leukemia.
- C
- Hypotension includes Hypotension, and Orthostatic hypotension.
- D
- Hemorrhage includes Conjunctival hemorrhage, Contusion, Ecchymosis, Epistaxis, Eye contusion, Hematochezia, Hemoptysis, Infusion site hematoma, Oral contusion, Petechiae, Post procedural hemorrhage, Pulmonary hemorrhage, Retinal hemorrhage, and Subdural hematoma.
Blood and lymphatic system disorders - - Coagulopathy * 22 2 Febrile Neutropenia 10 9 Cardiac disorders - - Tachycardia † 27 1 Gastrointestinal disorders - - Diarrhea ‡ 33 1 Nausea 31 1 Constipation 22 0 Vomiting 20 0 General disorders and administrative site conditions - - Pyrexia 96 5 Fatigue § 47 7 Chills 33 0 Edema ¶ 23 0 Immune system disorders - - Cytokine release syndrome # 95 5 Hypogammaglobulinemia Þ 93 2 Infections and infestations ß - - Infections-pathogen unspecified à 41 19 Upper respiratory tract infection è 28 3 Viral infections ð 23 7 Pneumonia ø 14 13 Sepsis ý 10 7 Metabolism and nutrition disorders - - Decreased appetite 29 1 Musculoskeletal and connective tissue disorders - - Musculoskeletal pain £ 48 2 Nervous system disorders - - Encephalopathy ¥ 30 6 Headache 27 0 Dizziness Œ 23 1 Motor dysfunction œ 16 3 Psychiatric disorders - - Insomnia 13 0 Respiratory, thoracic and mediastinal disorders - - Cough Ɖ 39 0 Dyspnea A 23 3 Nasal congestion 15 0 Hypoxia 12 4 Neoplasms benign, malignant, and unspecified (incl cysts and polyps) Hematologic malignancy B 10 10 Vascular disorders - - Hypotension C 51 10 Hypertension 19 6 Hemorrhage D 16 4 Other clinically important adverse reactions that occurred in less than 10% of patients treated with CARVYKTI include the following:
- Cardiac disorders: cardiac arrhythmias 24(8%), chest pain 25(7%)
- Eye disorders: diplopia (1%)
- Gastrointestinal disorders: dysphagia (1%)
- Immune system disorders:HLH (1%), hypersensitivity reaction (5%)
- Infections and Infestations:bacterial infections 26(9%), urinary tract infection 27(4.1%)
- Injury, Poisoning and Procedural complications: fall (3.1%)
- Metabolism and Nutrition Disorders: tumor lysis syndrome (1%)
- Musculoskeletal and Connective tissue disorders: posture abnormal (1%)
- Nervous system disorders: aphasia 28(8%), ataxia 29(8%), peripheral neuropathy 30(7%), tremor (6%), parkinsonism (4.1%), micrographia (4.1%), dysgraphia (3.1%), reduced facial expression (3.1%), cranial nerve palsies (3.1%), bradykinesia (2.1%), paresis 31(1%), cogwheel rigidity (1%), cerebrovascular accident (1%), seizure (1%), slow speech (1%), nystagmus (1%)
- Psychiatric disorders: delirium 32(5%) depression 33(4.1%), psychomotor retardation (1%)
- Renal and urinary disorders: renal failure 34(7%)
- Skin and subcutaneous tissues: rash 35(8%)
- Vascular Disorders: thrombosis 36(5%)
- 24
- Cardiac arrhythmias includes atrial fibrillation, atrial flutter, supraventricular tachycardia, ventricular extrasystoles, ventricular tachycardia.
- 25
- Chest pain includes Angina pectoris, Chest discomfort, and Chest pain.
- 26
- Bacterial infection includes Abscess limb, Cholecystitis, Cholecystitis acute, Clostridium difficile colitis, Clostridium difficile infection, Enterocolitis bacterial, Osteomyelitis, Perirectal abscess, Soft tissue infection, Staphylococcal infection.
- 27
- Urinary tract infection includes Urinary tract infection, and Urinary tract infection viral.
- 28
- Aphasia includes Aphasia, Dysarthria, and Speech disorder.
- 29
- Ataxia includes Ataxia, Balance disorder, and Gait disturbance.
- 30
- Peripheral neuropathy includes Peripheral neuropathy, Peripheral motor neuropathy and Peripheral sensory neuropathy.
- 31
- Paresis includes Facial paralysis, and Peroneal nerve palsy.
- 32
- Delirium includes Agitation, Hallucination, Irritability, Personality change, and Restlessness.
- 33
- Depression includes Depression, and Flat affect.
- 34
- Renal failure includes Acute kidney injury, Blood creatinine increased, Chronic kidney disease, and Renal impairment.
- 35
- Rash includes Erythema, Rash, Rash maculo-papular, and Rash pustular.
- 36
- Thrombosis includes Deep vein thrombosis, and Device related thrombosis.
Laboratory Abnormalities
Table 6 presents the most common Grade 3 or 4 laboratory abnormalities based on laboratory data, occurring in at least 10% of patients.
Table 6: Grade 3 or 4 laboratory abnormalities in at least 10% of patients treated with CARVYKTI in CARTITUDE-1 (N=97) Laboratory Abnormality Grade 3 or 4 (%) Laboratory abnormalities graded using NCI Common Terminology Criteria for Adverse Events version 5.0. Laboratory abnormalities are sorted by decreasing frequency in the Grade column. Lymphopenia 99 Neutropenia 98 White blood cell decreased 98 Anemia 72 Thrombocytopenia 63 Aspartate aminotransferase increased 21 Other clinically important Grade 3 or 4 laboratory abnormalities (based on laboratory data) that occurred in less than 10% of patients treated with CARVYKTI include the following: fibrinogen decreased, hypoalbuminemia, alanine aminotransferase increased, hyponatremia, hypocalcemia, gamma glutamyl transferase increased, alkaline phosphatase increased, hypokalemia, blood bilirubin increased.
6.2 Immunogenicity
The immunogenicity of CARVYKTI has been evaluated using a validated assay for the detection of binding antibodies against the extracellular portion of the anti-BCMA CAR pre-dose, and at multiple timepoints post-infusion. In CARTITUDE-1, 19 of 97 (19.6%) patients were positive for anti-product antibodies. In CARTITUDE-4, 39 of 186 patients (21%) were positive for anti-CAR antibodies.
There was no clear evidence that the observed anti-product antibodies impact CARVYKTI kinetics of initial expansion and persistence, efficacy, or safety.
6.3 Postmarketing Experience
Because adverse events to marketed products are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure.
The following adverse event has been identified during postmarketing use of CARVYKTI.
Neoplasms:T cell malignancies
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data on the use of CARVYKTI in pregnant women. No reproductive and developmental toxicity studies in animals have been conducted with CARVYKTI to assess whether it can cause fetal harm when administered to a pregnant woman. It is not known whether CARVYKTI has the potential to be transferred to the fetus and cause fetal toxicity. Based on the mechanism of action, if the transduced cells cross the placenta, they may cause fetal toxicity, including B-cell lymphocytopenia and hypogammaglobulinemia. Therefore, CARVYKTI is not recommended for women who are pregnant, or for women of childbearing potential not using contraception. Pregnant women should be advised that there may be risks to the fetus. Pregnancy after CARVYKTI therapy should be discussed with the treating physician.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2%–4% and 15%–20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of CARVYKTI in human milk, the effect on the breastfed infant, and the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for CARVYKTI and any potential adverse effects on the breastfed infant from CARVYKTI or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy status for females of child-bearing age should be verified prior to starting treatment with CARVYKTI.
Contraception
There are insufficient data to provide a recommendation concerning duration of contraception following treatment with CARVYKTI.
In clinical trials, female patients of childbearing potential were advised to practice a highly effective method of contraception and male patients with partners of childbearing potential or whose partners were pregnant were instructed to use a barrier method of contraception, until one year after the patient has received CARVYKTI infusion.
See the prescribing information for lymphodepleting chemotherapy for information on the need for contraception in patients who receive the lymphodepleting chemotherapy.
8.4 Pediatric Use
Safety and effectiveness of CARVYKTI in pediatric patients have not been established.
8.5 Geriatric Use
Of the 97 patients in CARTITUDE-1 that received CARVYKTI, 28% were 65 to 75 years of age, and 8% were 75 years of age or older. CARTITUDE-1 did not include sufficient numbers of patients aged 65 and older to determine whether the effectiveness differs compared with that of younger patients. In 62 patients less than 65 years of age, all grade and Grade 3 and higher neurologic toxicities occurred in 19% (12/62) and 6% (4/62), respectively. Of the 35 patients ≥65 years of age, all grade and Grade 3 and higher neurologic toxicities occurred in 37% (13/35) and 20% (7/35), respectively.
Of the 188 patients in CARTITUDE-4 that received CARVYKTI, 38% were 65 to 75 years of age, and 2% were 75 years of age or older. In 112 patients less than 65 years of age, all grade and Grade 3 and higher neurologic toxicities occurred in 16% (18/112) and 3% (3/112) respectively. Of the 76 patients ≥65 years of age, all grade and Grade 3 and higher neurologic toxicities occurred in 34% (26/76) and 7% (5/76) respectively.
-
11 DESCRIPTION
CARVYKTI ®(ciltacabtagene autoleucel) is a BCMA-directed genetically modified autologous T cell immunotherapy. CARVYKTI is prepared from the patient's peripheral blood mononuclear cells, which are obtained via a standard leukapheresis procedure. The mononuclear cells are enriched for T cells and genetically modified ex vivoby transduction with a replication-incompetent lentiviral vector to express a CAR comprising an anti-BCMA targeting domain, which consists of two single-domain antibodies linked to a 4-1BB costimulatory domain and a CD3-zeta signaling domain.
The transduced anti-BCMA CAR T cells are expanded in cell culture, washed, formulated into a suspension and cryopreserved. The product must pass a sterility test before release for shipping as a frozen suspension in a patient-specific infusion bag. The product is thawed and then infused back into the patient, where the anti-BCMA CAR T cells can recognize and eliminate BCMA-expressing target cells. [see Dosage and Administration (2.2), How Supplied/Storage and Handling (16)].
In addition to T cells, CARVYKTI may contain Natural Killer (NK) cells. The formulation contains 5% dimethyl sulfoxide (DMSO).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
CARVYKTI is a BCMA-directed, genetically modified autologous T cell immunotherapy, which involves reprogramming a patient's own T cells with a transgene encoding a CAR that identifies and eliminates cells that express BCMA. The CARVYKTI CAR protein features two BCMA-targeting single-domain antibodies designed to confer high avidity against human BCMA, a 4-1BB co-stimulatory domain and a CD3-zeta (CD3ζ) signaling cytoplasmic domain. Upon binding to BCMA-expressing cells, the CAR promotes T cell activation, expansion, and elimination of target cells.
12.2 Pharmacodynamics
After a single infusion of CARVYKTI, expansion of CAR-positive T cells coincided with decreases of serum soluble BCMA, serum M-protein, and/or free light chains. Across all patients, levels of IL-6, IL-10, IFN-γ and IL-2 receptor alpha increased post-infusion and peaked at Days 7–14. The serum levels of all cytokines generally returned to baseline levels within 2–3 months post-infusion.
12.3 Pharmacokinetics
The pharmacokinetics (PK) of CARVYKTI was assessed in 285 adult patients with relapsed or refractory multiple myeloma in CARTITUDE-1 and CARTITUDE-4 receiving a single infusion at the median dose of 0.71×10 6CAR-positive viable T cells/kg (range: 0.41×10 6to 1.08×10 6cells/kg).
Following a single infusion, CARVYKTI exhibited an initial expansion phase followed by a rapid decline, and then a slower decline. However, high inter-individual variability was observed.
Table 7: Pharmacokinetic parameters of CARVYKTI in patients with multiple myeloma Parameter Summary Statistics CARTITUDE-1
N=97CARTITUDE-4
N=188C max(copies/µg genomic DNA) Median (range), n 47806 (7189 – 115234), 97 34891 (935 – 104861), 185 t max(day) Median (range), n 12.7 (8.7 – 329.8), 97 12.8 (7.8 – 222.8), 185 AUC 0–28d(copies*day/µg genomic DNA) Median (range), n 371569 (58691 – 2024126), 97 293490 (9215 – 1738455), 184 t 1/2(day) Median (range), n 15.3 (3.0 – 95.4), 42 11.7 (4.1 – 179.6), 49 After the cell expansion, the persistence phase of CARVYKTI was observed for all patients. At the time of analysis in CARTITUDE-1 (n=65) and CARTITUDE-4 (n=87) studies, the median time for CAR transgene levels in peripheral blood to return to the pre-dose baseline level was approximately 100 days (range: 28 to 365 days) and 109 days (range: 29 to 366 days) post-infusion respectively.
Detectable CARVYKTI exposures in bone marrow indicate a distribution of CARVYKTI from systemic circulation to bone marrow. Similar to blood transgene levels, bone marrow transgene levels declined over time and exhibited high inter-individual variability.
Patients with higher CAR-T cell expansion tended to have higher rates of CRS. Some patients required tocilizumab, corticosteroids, and anakinra for the management of CRS. CARVYKTI continues to expand and persist following administration of tocilizumab, corticosteroids, and anakinra. In CARTITUDE-1, CARVYKTI median C maxand AUC 0–28din patients treated with tocilizumab (n=68) for CRS were 168% and 209% of those in patients (n=29) who did not receive tocilizumab for CRS, respectively. The median C maxand AUC 0–28dof CARVYKTI in patients who received corticosteroids (n=21) for CRS were 186% and 307% of those in patients who did not receive corticosteroids (n=76) for CRS, respectively. In addition, the median C maxand AUC 0–28dof CARVYKTI in patients who received anakinra (n=18) for CRS were 139% and 232% of those in patients who did not receive anakinra (n=79) for CRS, respectively. In CARTITUDE-4, the results related to tocilizumab and corticosteroid were consistent with CARTITUDE-1.
Specific Populations
The pharmacokinetics of CARVYKTI (C maxand AUC 0–28d) were not impacted by age (27 to 78 years), gender, body weight, race, mild hepatic dysfunction [(total bilirubin ≤ upper limit of normal (ULN) and aspartate aminotransferase > ULN) or (ULN < total bilirubin ≤1.5 times ULN)], mild renal dysfunction (60 mL/min ≤ creatinine clearance [CRCL] <90 mL/min) or moderate renal dysfunction (30 mL/min ≤ creatinine clearance <60 mL/min). Formal renal and hepatic impairment studies of CARVYKTI were not conducted.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No genotoxicity or carcinogenicity studies have been performed with CARVYKTI as they were not indicated. In vitrostudies with CARVYKTI manufactured from healthy donors and patients with multiple myeloma showed no evidence of cytokine independent growth and no preferential integration near genes associated with oncogenic transformation.
No studies have been conducted to evaluate the effects of CARVYKTI on fertility.
-
14 CLINICAL STUDIES
CARTITUDE-4
Efficacy of CARVYKTI was evaluated in CARTITUDE-4 (NCT04181827) a randomized, open label, multicenter controlled study in adult patients with relapsed and lenalidomide-refractory multiple myeloma, who previously received at least 1 prior line of therapy including a proteasome inhibitor and an immunomodulatory agent. A total of 419 patients were randomized 1:1 to receive either a sequence of apheresis, bridging therapy, lymphodepletion and CARVYKTI (n=208) or standard therapy which included daratumumab, pomalidomide and dexamethasone (DPd) or bortezomib, pomalidomide and dexamethasone (PVd) selected by physician prior to randomization based on patient's prior antimyeloma therapy (n=211). Randomization was stratified by physician's choice of treatment (DPd vs. PVd), ISS (I vs. II vs. III) and number of prior lines of therapy (1 vs. 2 or 3).
Patients with known active or prior history of central nervous system involvement, patients who exhibit clinical signs of meningeal involvement of multiple myeloma and patients with a history of Parkinson's disease or other neurodegenerative disorder, were excluded from the trial.
In the overall study population (N=419), the median age was 61 years (range: 27 to 80 years), 57% were male, 75% were White, 3% were Black or African American, 9% were Asian, and 7% were Hispanic or Latino. Most patients (94%) were International Staging System (ISS) Stage I or II. High-risk cytogenetics [presence of t(4:14), (14:16), and 17p13 del] were present in 34% of patients. Nineteen percent of patients had presence of soft tissue plasmacytoma.
Patients had received a median of 2 (range: 1 to 3) prior lines of therapy and 85% of patients had received prior autologous stem cell transplantation (ASCT). Ninety-nine percent of patients were refractory to their last line of prior therapy. Forty-seven percent were refractory to a proteasome inhibitor (PI) and 100% were refractory to an immunomodulatory agent.
All 208 patients randomized to the CARVYKTI arm underwent apheresis, twelve (6%) were not treated with CARVYKTI due to progressive disease (n=10) or death (n=2), and twenty (10%) progressed prior to infusion with CARVYKTI but were able to receive CARVYKTI as subsequent therapy. Eight (4%) patients received CAR-T positive T cells that did not meet product release specification for CARVYKTI (non-conforming product).
Patients randomized to CARVYKTI were to receive lymphodepleting chemotherapy consisting of fludarabine 30 mg/m 2/day and cyclophosphamide 300 mg/m 2/day concurrently for 3 days followed by CARVYKTI infusion 5 to 7 days after start of lymphodepleting chemotherapy. At least one cycle of DPd or PVd bridging therapy was received for disease control between leukapheresis and the start of the lymphodepleting chemotherapy.
CARVYKTI was administered as a single IV infusion 5 to 7 days after the start of a lymphodepleting chemotherapy at a median dose of 0.71×10 6CAR-positive viable T-cells/kg (range: 0.39 to 1.07×10 6cells/kg).
In the 176 patients that received CARVYKTI as study treatment, the median time from the day after receipt of apheresis material at manufacturing facility to release of product for infusion was 44 days (range: 25 to 127 days) and the median time from first apheresis to CARVYKTI infusion was 79 days (range: 45 days to 246 days).
The primary efficacy measure was progression-free survival (PFS) analyzed based on the Intent-To-Treat Analysis Set (see Table 8and Figure 1). After a median follow-up of 15.9 months, median PFS was 12 months (95% CI: 9.8, 14) for standard therapy arm and NE (95% CI: 22.8, NE) for CARVYKTI arm (Hazard ratio: 0.41 [95% CI: 0.30, 0.56]). The estimated PFS rate at 12 months was 75.9% (95% CI: 69.4%, 81.1%) in the CARVYKTI arm and 49.5% (95% CI: 42.3%, 56.3%) in the standard therapy arm.
Table 8: Efficacy results for CARTITUDE-4 (Intent-To-Treat Analysis Set) CARVYKTI
(N=208)Standard Therapy
(N=211)NE=not estimable; CI=confidence interval
Notes: Based on a median duration of follow up of 15.9 months- *
- Per the International Myeloma Working Group (IMWG) consensus, as assessed by IRC
- †
- Kaplan-Meier estimate
- ‡
- Based on a stratified Cox proportional hazards model. A hazard ratio <1 indicates an advantage for CARVYKTI Arm. For all stratified analyses, stratification was based on investigator's choice (PVd or DPd), ISS staging (I, II, III) and number of prior lines (1 vs. 2 or 3) as randomized.
- §
- Stratified log-rank test
- ¶
- Stratified Cochran-Mantel-Haenszel Chi-Squared test
Progression-Free Survival* - - Number of events, n (%) 65 (31.3) 119 (56.4) Median, months [95% CI] † NE [22.8, NE] 12 [9.8, 14.0] Hazard ratio [95% CI] ‡ 0.41 [0.30, 0.56] - p-value § <0.0001 - Complete Response or Better Rate *, % [95% CI] 74.0 [67.5, 79.9] 22.3 [16.8, 28.5] p-value ¶ <0.0001 - Stringent Complete Response *(sCR), n (%) 137 (65.9) 38 (18.0) Complete Response *(CR), n (%) 17 (8.2) 9 (4.3) Overall Response Rate, ORR (sCR + CR + VGPR + PR)*, % [95% CI] 84.6 [79.0, 89.2] 67.8 [61.0, 74.0] p-value ¶ <0.0001 - Very Good Partial Response *(VGPR), n (%) 16 (7.7) 49 (23.2) Partial Response *(PR), n (%) 6 (2.9) 47 (22.3) Figure 1. Kaplan-Meier Curve of PFS in CARTITUDE-4 (Intent-To-Treat Analysis Set) Note: Intent-to-treat analysis set consists of subjects who were randomized in the study.
Data cutoff date: November 1, 2022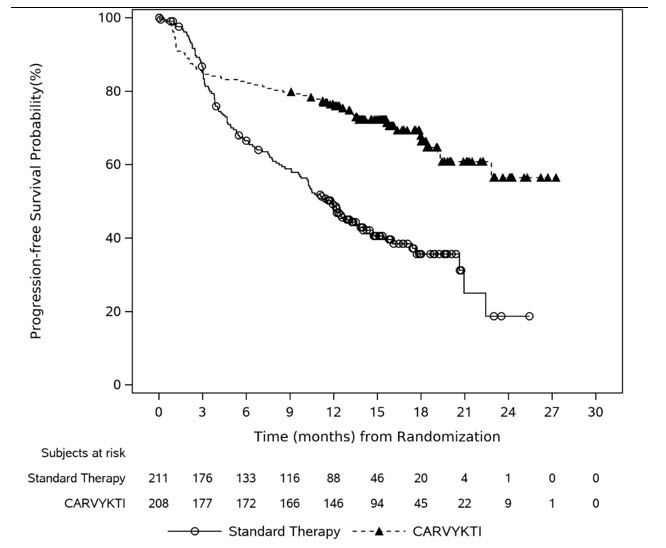
In the CARVYKTI arm, the estimated median duration of response (DOR) has not been reached in patients who achieved PR or better or in patients who achieved CR or better. In the standard therapy arm, the estimated median DOR was 16.6 months (95% CI: 12.9, NE).
A higher proportion of patients in the CARVYKTI arm compared to the standard therapy arm died within the first 10 months of randomization as shown in Figure 2.
Figure 2. Kaplan-Meier Curve of Overall Survival in CARTITUDE-4 (Intent-to-Treat Analysis Set) Note: Intent-to-treat analysis set consists of subjects who were randomized to the study.
Data cutoff date: November 1, 2022.
34% of the planned OS events have occurred.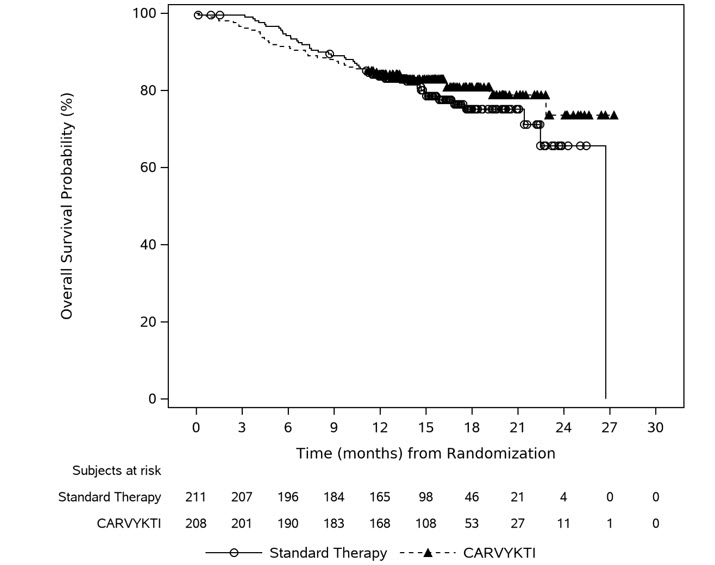
CARTITUDE-1
The efficacy of CARVYKTI was evaluated in CARTITUDE-1 (NCT03548207), an open-label, single-arm, multicenter trial in adult patients with relapsed or refractory multiple myeloma, who previously received at least 3 prior lines of therapy including a proteasome inhibitor, an immunomodulatory agent, and an anti-CD38 monoclonal antibody [see Adverse Reactions (6.1)] .
Patients with known active or prior history of significant central nervous system (CNS) disease, including CNS multiple myeloma, plasma cell leukemia, allogeneic stem cell transplant within 6 months before apheresis or ongoing treatment with immunosuppressants, creatinine clearance <40 mL/min, absolute lymphocyte concentration <300/µL, absolute neutrophil count <750 cells/mm 3, platelet count <50,000/mm 3, hepatic transaminases >3 times the upper limit of normal, cardiac ejection fraction <45%, or with active serious infection were excluded from the trial.
Of the 113 patients who underwent leukapheresis, 16 patients did not receive CARVYKTI due to progressive disease (n=2), death (n=9), or withdrawal from study (n=5). There were 97 patients in the efficacy evaluable population who received CARVYKTI, including 17 patients (18%) with manufacturing failures either because they received CARVYKTI that did not meet product release specifications for CARVYKTI or received CARVYKTI for which there were insufficient data to confirm product release specifications for CARVYKTI.
Of the 97 efficacy-evaluable patients, the median age was 61 years (range: 43 to 78 years), 59% were male, 71% were white, and 18% were black. Most patients (86%) were ISS Stage I or II. Of the 91 patients for whom baseline cytogenetic data were available, high-risk cytogenetics (presence of t(4:14), t(14:16), or 17p13 del) were present in 24% of patients. Thirteen percent of the patients had extramedullary disease.
The median number of prior lines of therapy was 6 (range: 3 to 18), with 82% of patients receiving 4 or more prior lines of therapy, 90% of patients had received prior autologous stem cell transplantation (ASCT) and 8% of patients received an allogeneic transplant. Ninety-nine percent of patients were refractory to their last line of prior therapy, and 88% were refractory to a proteasome inhibitor (PI), immunomodulatory agent, and anti-CD38 antibody.
Most patients (75%) treated with CARVYKTI received bridging therapy for control of their multiple myeloma during the manufacturing process. The median time from leukapheresis to product availability was 32 days (range: 27 to 66 days).
The most commonly used agents as bridging therapies (≥20% of patients) included dexamethasone: 62 patients (64%), bortezomib: 26 patients (27%), cyclophosphamide: 22 patients (23%), and pomalidomide: 21 patients (22%).
Efficacy was established on the basis of overall response rate, complete response rate and duration of response as assessed by the Independent Review Committee (IRC) using International Myeloma Working Group (IMWG) criteria (see Table 9and 10). The median time to first response was 1 month (range: 0.9 to 10.7 months).
Table 9: Summary of efficacy results for CARTITUDE-1 based on IRC using IMWG criteria CARVYKTI treated
(N=97)Notes: Based on a median duration of follow-up of 28 months. CI=confidence interval; IRC=Independent Review Committee; IMWG=International Myeloma Working Group; NE=not estimable. Overall Response Rate (sCR *+ VGPR + PR) n (%) 95 (97.9) 95% CI (%) (92.7, 99.7) Stringent complete response (sCR) *n (%) 78 (80.4) 95% CI †(%) (71.1, 87.8) Very good partial response (VGPR) n (%) 14 (14.4) 95% CI †(%) (8.1, 23.0) Partial response (PR) n (%) 3 (3.1) 95% CI †(%) (0.6, 8.8) Table 10: Duration of Response (DOR) CARVYKTI treated
(N=97)Notes: Based on a median duration of follow-up of 28 months. CI=confidence interval; NE=not estimable. Duration of Response (DOR)* - Number of responders
DOR (Months):Median (95% CI) †95
NE (23.3, NE)Number of responders with sCR ‡
DOR if best response is sCR ‡(Months):Median (95% CI) †78
NE (28.3, NE)Number of responders with VGPR or better
DOR if best response is VGPR or better (Months):Median (95% CI) †92
NE (24.4, NE)The IRC assessed overall response in the 113 patients that underwent leukapheresis was 84% (95% CI: 76, 90) with stringent CR rate of 69% (95% CI: 60, 77), VGPR rate of 12% (95% CI: 7, 20) and PR rate of 3% (95% CI: 1, 8).
-
15 REFERENCES
- Lee DW, Santomasso BD, Locke FL, et al. ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019; 25: 625–638.
- National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) v 5.0; 2017.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
CARVYKTI ®is supplied in one infusion bag containing a frozen suspension of genetically modified autologous T cells in 5% DMSO, either as a:
- 70 mL suspension in an infusion bag and metal cassette (NDC 57894-111-01)
or
- 30 mL suspension in an infusion bag and metal cassette (NDC 57894-111-02)
Each CARVYKTI infusion bag is individually packed in an aluminum cryo-cassette.
Match the identity of the patient with the patient identifiers on the cassette and infusion bag upon receipt.
Store and transport below -120°C, e.g., in a container for cryogenic storage in the vapor phase of liquid nitrogen.
Store CARVYKTI in the original packaging containing the cassette protecting the infusion bag.
Thaw CARVYKTI prior to infusion [see Dosage and Administration (2)] .
- 70 mL suspension in an infusion bag and metal cassette (NDC 57894-111-01)
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients of the risk of manufacturing failure [18%, (17/97 in the clinical study)]. In case of a manufacturing failure, a second manufacturing of CARVYKTI may be attempted. In addition, while the patient awaits the product, additional anticancer treatment (other than lymphodepletion) may be necessary and may increase the risk of adverse reactions during the pre-infusion period, which could delay or prevent the administration of CARVYKTI.
Advise patients that they will be monitored daily for the first 10 days following the infusion at a REMS-certified healthcare facility, and instruct patients to remain within proximity of a certified healthcare facility for at least 4 weeks following the infusion.
Prior to infusion, advise patients of the following risks and to seek immediate medical attention in the event of the following signs or symptoms:
Increased Early Mortality
Inform patients of the risk of early mortality. In a clinical study, treatment in the CARVYKTI arm was associated with a higher rate of death (14%) compared to the control arm (12%) in the first 10 months from randomization. This higher rate of death was observed before receiving CARVYKTI and after treatment with CARVYKTI. The reasons for death were progression of multiple myeloma and adverse events [see Warnings and Precautions (5.1), Clinical Studies (14)] .
Cytokine Release Syndrome (CRS)
Signs or symptoms of CRS, including fever, chills, fatigue, headache, tachycardia, hypotension, hypoxia, dizziness/lightheadedness or organ toxicities [see Warnings and Precautions (5.2), Adverse Reactions (6.1)].
Neurologic Toxicities
Signs or symptoms associated with neurologic events, some of which occur days, weeks or months following the infusion including [see Warnings and Precautions (5.3), Adverse Reactions (6.1)] :
ICANS:e.g., aphasia, encephalopathy, depressed level of consciousness, seizures, delirium, dysgraphia
Parkinsonism:e.g., tremor, micrographia, bradykinesia, rigidity, shuffling gait, stooped posture, masked facies, apathy, flat affect, lethargy, somnolence
Guillain Barré Syndrome:e.g., motor weakness and polyradiculoneuritis
Peripheral neuropathy:e.g., peripheral motor and/or sensory nerve dysfunction
Cranial Nerve Palsies:e.g., facial paralysis, facial numbness
Prolonged and Recurrent Cytopenias
Signs or symptoms associated with bone marrow suppression including neutropenia, thrombocytopenia, anemia, or febrile neutropenia for several weeks or months. Signs or symptoms associated with bone marrow suppression may recur [see Warnings and Precautions (5.6), Adverse Reactions (6.1)] .
Infections
Signs or symptoms associated with infection [see Warnings and Precautions (5.7), Adverse Reactions (6.1)] .
Hypersensitivity Reactions
Signs or symptoms associated with hypersensitivity reactions including flushing, chest tightness, tachycardia, and difficulty breathing [see Warnings and Precautions (5.9)].
Secondary Malignancies
Secondary hematological malignancies, including myelodysplastic syndrome, acute myeloid leukemia, and T-cell malignancies have occurred [see Boxed Warning, Warnings and Precautions (5.10), Adverse Reactions (6.1, 6.3)].
Advise patients of the need to:
- Have periodic monitoring of blood counts before and after CARVYKTI infusion [see Warnings and Precautions (5.6)] .
- Contact Janssen Biotech, Inc. at 1-800-526-7736 if they are diagnosed with a secondary malignancy [see Warnings and Precautions (5.10)] .
- Refrain from driving and engaging in hazardous occupations or activities, such as operating heavy or potentially dangerous machinery, for at least 8 weeks after treatment and in the event of any new onset of neurologic toxicities [see Warnings and Precautions (5.11)] .
- Tell their physician about their treatment with CARVYKTI before receiving a live virus vaccine [see Warnings and Precautions (5.8)] .
- SPL UNCLASSIFIED SECTION
-
MEDICATION GUIDE
MEDICATION GUIDE
CARVYKTI ®(car-vick-tee)
(ciltacabtagene autoleucel)Manufactured/Marketed by: Janssen Biotech, Inc., Horsham, PA 19044, USA. U.S. License Number 1864 Marketed by: Legend Biotech, Somerset, NJ 08873, USA. For patent information: www.janssenpatents.com. For more information, call 1-800-526-7736 or go to www.CARVYKTI.com. © Johnson & Johnson and its affiliates 2022–2024 This Medication guide has been approved by the U.S. Food and Drug Administration. Read this Medication Guide before you start your CARVYKTI treatment. The more you know about your treatment, the more active you can be in your care. Talk with your healthcare provider if you have questions about your health condition or treatment. Reading this Medication Guide does not take the place of talking with your healthcare provider about your treatment. What is the most important information I should know about CARVYKTI?
CARVYKTI may cause side effects that are severe or life-threatening and can lead to death. Call your healthcare provider or get emergency help right away if you get any of the following:- fever (100.4°F/38°C or higher)
- chills or shaking chills
- fast or irregular heartbeat
- difficulty breathing
- very low blood pressure
- dizziness/light headedness
- effects on your nervous system, some of which can occur days or weeks after you receive the infusion, and may initially be subtle such as:
- feeling confused, less alert, or disoriented, having difficulty speaking or slurred speech, having difficulty reading, writing, and understanding words, memory loss
- loss of coordination affecting movement and balance, slower movements, changes in handwriting
- personality changes including a reduced ability to express emotions, being less talkative, disinterest in activities, and reduced facial expression
- tingling, numbness, and pain of hands and feet, difficulty walking, leg and/or arm weakness, and difficulty breathing
- facial numbness, difficulty moving muscles of face and eyes
What is CARVYKTI? - CARVYKTI is a treatment used for adult patients who have cancer of the bone marrow called multiple myeloma. It is used when at least one other treatment has not worked or has stopped working.
- CARVYKTI is a medicine made from your own white blood cells, which have been changed (genetically modified) to recognize and attack your multiple myeloma cells.
Before you receive CARVYKTI tell your healthcare provider about all your medical conditions, including if you have: - Current or past neurologic problems (such as seizures, stroke, new or worsening memory loss)
- Lung or breathing problems
- Heart problems
- Liver problems
- Kidney problems
- A recent or active infection
- Low blood counts
How will I receive CARVYKTI? - CARVYKTI is made from your own white blood cells, so your blood will be collected by a process called 'leukapheresis' (loo-kah-fur-ee-sis). The procedure can take 3 to 6 hours and may need to be repeated.
- Your white blood cells are sent to a manufacturing center to make CARVYKTI. It takes about 4–5 weeks from the time your cells are received at the manufacturing site and are available to be shipped back to your healthcare provider, but the time may vary.
- While CARVYKTI is being made you may get other medicines to treat the multiple myeloma. This is so that your multiple myeloma does not get worse.
30 to 60 minutes before you are given CARVYKTI, you may be given other medicines. These may include:- medicines for an allergic reaction (antihistamines)
- medicines for fever (such as acetaminophen)
After getting CARVYKTI,you will be monitored at the certified healthcare facility where you received your treatment for at least 10 days after the infusion.
You should plan to stay close to the location where you received your treatment for at least 4 weeks. Your healthcare provider will check to see that your treatment is working and help you with any side effects that may occur. You may be hospitalized if you develop serious side effects until your side effects are under control and it is safe for you to leave the hospital.
Your healthcare provider will want to do blood tests to follow your progress. It is important that you have your blood tested. If you miss an appointment, call your healthcare provider as soon as possible to reschedule.What should I avoid after receiving CARVYKTI? - Do not drive, or operate heavy machinery, or do other activities that could be dangerous if you are not mentally alert, for at least 8 weeks after you get CARVYKTI. This is because the treatment can cause memory and coordination problems, sleepiness, confusion, dizziness, seizures, or other neurologic side effects as discussed by your healthcare provider.
- You must not be given certain vaccines called live vaccines for some time before and after CARVYKTI treatment. Talk to your healthcare provider if you need to have any vaccinations.
- Do not donate blood, organs, tissues, or cells for transplantation.
What are the possible or reasonably likely side effects of CARVYKTI?
The most common side effects of CARVYKTI include:- fever (100.4°F/38°C or higher), chills
- dizziness or light-headedness
- headache, muscle or joint pain, feeling very tired
- altered mental state, confusion
- infections
- low levels of antibodies (immunoglobulins) in the blood
- cough, being short of breath
- diarrhea, nausea, decreased appetite, constipation
- fast or irregular heartbeat
- problems with blood clotting
CARVYKTI can cause a very common side effect called cytokine release syndrome or CRS, which can be severe or fatal. Symptoms of CRS include fever, difficulty breathing, dizziness or lightheadedness, nausea, headache, fast heartbeat, low blood pressure, or fatigue. Tell your healthcare provider right away if you develop fever or any of these other symptoms after receiving CARVYKTI.
CARVYKTI can increase the risk of life-threatening infections including COVID-19 that may lead to death. Tell your healthcare provider right away if you develop fever, chills, or any signs or symptoms of an infection.
CARVYKTI can cause various neurologic side effects, some of which may be severe or fatal. Symptoms include but are not limited to confusion, disorientation, loss of consciousness, seizures, difficulty speaking, reading or writing, tremor, slower movements, changes in personality, depression, tingling and numbness of hands and feet, leg and arm weakness, and facial numbness.
CARVYKTI can lower one or more types of your blood cells (red blood cells, white blood cells, or platelets [cells that help blood to clot]), which may make you feel weak or tired or increase your risk of severe infection or bleeding that may lead to death. After treatment, your healthcare provider will test your blood to check for this. Tell your healthcare provider right away if you get a fever, chills, or any signs or symptoms of an infection, are feeling tired, or have bruising or bleeding.
CARVYKTI may increase your risk of getting cancers including certain types of blood cancers. Your healthcare provider should monitor you for this.
Having CARVYKTI in your blood may cause some commercial Human Immunodeficiency Virus (HIV) tests to incorrectly give you an HIV-positive result even though you may be HIV-negative.
These are not all the possible side effects of CARVYKTI. Call your healthcare provider if you have any side effects.
You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of CARVYKTI
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. If you would like more information about CARVYKTI, talk with your healthcare provider. You can ask your healthcare provider for information about CARVYKTI that is written for health professionals. For more information go to www.CARVYKTI.com or call 1-800-526-7736.What are the ingredients in CARVYKTI?
Active ingredient: ciltacabtagene autoleucel
Inactive ingredients:DMSORevised: April 2024
-
PRINCIPAL DISPLAY PANEL - 70 mL Bag Label
 ciltacabtagene autoleucel
ciltacabtagene autoleucel
CARVYKTI™
Suspension for Intravenous InfusionNDC 57894-111-01
Dose: One sterile bag for infusion.
Contents: A maximum of 1x10 8CAR-positive viable T cells in a 70 mL frozen suspension per
patient-specific infusion bag, containing 5% DMSO.FOR AUTOLOGOUS USE ONLY.
FOR INTRAVENOUS USE ONLY.Dosage: See Prescribing Information.
Storage: Store and transport in a vapor phase of
liquid nitrogen ≤ -120°C (-184°F). Thaw before using.DO NOT re-freeze or refrigerate once thawed.
DO NOT irradiate.
DO NOT use a leukodepleting filter.CULTURED, GENETICALLY MODIFIED.
NO U.S. STANDARD OF POTENCY.
NOT EVALUATED FOR INFECTIOUS SUBSTANCES.NO PRESERVATIVE
Attention: Dispense the enclosed Medication
Guide to each patient.Rx only
One Sterile Bag for Infusion
Mfg./Mktd. by: Janssen Biotech, Inc., Horsham,
PA 19044, USA; U.S. License No. 1864Mktd. by: Legend Biotech, Somerset, NJ 08873, USA
Upon receipt: Match the identity of
the patient with the patient
identifiers on the cassette and
infusion bag.BAG ID: COI [followed by Bag Number]
LOT: XXXXXXXX
EXP: YYYY-MMM-DDORDER ID:
PATIENT
NAME:DOB: YYYY-MMM-DD
MEDICAL RECORD NO.:
DIN:
© 2022 Janssen
10579200LEGEND
BIOTECHjanssen
-
PRINCIPAL DISPLAY PANEL - 30 mL Bag Label
 ciltacabtagene autoleucel
ciltacabtagene autoleucel
CARVYKTI™
Suspension for Intravenous InfusionNDC 57894-111-02
Dose: One sterile bag for infusion.
Contents: A maximum of 1x10 8CAR-positive viable T cells in a 30 mL frozen suspension per
patient-specific infusion bag, containing 5% DMSO.FOR AUTOLOGOUS USE ONLY.
FOR INTRAVENOUS USE ONLY.Dosage: See Prescribing Information.
Storage: Store and transport in a vapor phase of
liquid nitrogen ≤ -120°C (-184°F). Thaw before using.DO NOT re-freeze or refrigerate once thawed.
DO NOT irradiate.
DO NOT use a leukodepleting filter.CULTURED, GENETICALLY MODIFIED.
NO U.S. STANDARD OF POTENCY.
NOT EVALUATED FOR INFECTIOUS SUBSTANCES.NO PRESERVATIVE
Attention: Dispense the enclosed Medication
Guide to each patient.Rx only
One Sterile Bag for Infusion
Mfg./Mktd. by: Janssen Biotech, Inc., Horsham,
PA 19044, USA; U.S. License No. 1864Mktd. by: Legend Biotech, Somerset, NJ 08873, USA
Upon receipt: Match the identity of
the patient with the patient
identifiers on the cassette and
infusion bag.BAG ID: COI [followed by Bag Number]
LOT: XXXXXXXX
EXP: YYYY-MMM-DDORDER ID:
PATIENT
NAME:DOB: YYYY-MMM-DD
MEDICAL RECORD NO.:
DIN:
© 2022 Janssen
10580600LEGEND
BIOTECHjanssen
-
INGREDIENTS AND APPEARANCE
CARVYKTI
ciltacabtagene autoleucel injection, suspensionProduct Information Product Type CELLULAR THERAPY Item Code (Source) NDC:57894-111 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CILTACABTAGENE AUTOLEUCEL (UNII: 0L1F17908Q) (CILTACABTAGENE AUTOLEUCEL - UNII:0L1F17908Q) CILTACABTAGENE AUTOLEUCEL 100000000 Inactive Ingredients Ingredient Name Strength DIMETHYL SULFOXIDE (UNII: YOW8V9698H) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:57894-111-01 1 in 1 BAG; Type 0: Not a Combination Product 2 NDC:57894-111-02 1 in 1 BAG; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125746 02/28/2022 Labeler - Janssen Biotech, Inc (099091753) Establishment Name Address ID/FEI Business Operations Janssen Biotech, Inc. 038978363 analysis(57894-111) Establishment Name Address ID/FEI Business Operations BioReliance Corporation 147227730 analysis(57894-111) Establishment Name Address ID/FEI Business Operations Janssen Biologics B.V. 409612918 analysis(57894-111) Establishment Name Address ID/FEI Business Operations Janssen Vaccines, Zweigniederlassung der Cilag GmbH International 480244564 manufacture(57894-111) , api manufacture(57894-111) Establishment Name Address ID/FEI Business Operations Janssen Pharmaceuticals, Inc. 868441320 analysis(57894-111) , manufacture(57894-111)