Label: BICALUTAMIDE tablet, film coated

- NDC Code(s): 60429-177-01, 60429-177-05, 60429-177-30
- Packager: Golden State Medical Supply, Inc.
- This is a repackaged label.
- Source NDC Code(s): 16729-023
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated February 22, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
BICALUTAMIDE TABLETS. These highlights do not include all the information needed to use BICALUTAMIDE TABLETS safely and effectively. See full prescribing information for BICALUTAMIDE TABLETS.
BICALUTAMIDE TABLETS, for oral use
Initial U.S. Approval: 1995INDICATIONS AND USAGE
-
Bicalutamide tablets 50 mg is an androgen receptor inhibitor indicated for use in combination therapy with a luteinizing hormone-releasing hormone (LHRH) analog for the treatment of Stage D 2metastatic carcinoma of the prostate. ( 1)
-
Bicalutamide tablets 150 mg daily is not approved for use alone or with other treatments. ( 1)
DOSAGE AND ADMINISTRATION
The recommended dose for bicalutamide therapy in combination with an LHRH analog is one 50 mg tablet once daily (morning or evening). ( 2)
DOSAGE FORMS AND STRENGTHS
50 mg tablets ( 3)
WARNINGS AND PRECAUTIONS
- Severe hepatic injury and fatal hepatic failure have been observed. Monitor serum transaminase levels prior to starting treatment with bicalutamide, at regular intervals for the first four months of treatment and periodically thereafter, and for symptoms or signs suggestive of hepatic dysfunction. Use bicalutamide with caution in patients with hepatic impairment. ( 5.1)
- Hemorrhage with Concomitant Use of Coumarin Anticoagulant. Closely monitor the Prothrombin Time (PT) and International Normalized Ratio (INR), and adjust the anticoagulant dose as needed. ( 5.2)
- Gynecomastia and breast pain have been reported during treatment with bicalutamide 150 mg when used as a single agent. ( 5.3)
- Bicalutamide is used in combination with an LHRH agonist. LHRH agonists have been shown to cause a reduction in glucose tolerance in males. Consideration should be given to monitoring blood glucose in patients receiving bicalutamide in combination with LHRH agonists. ( 5.4)
- Monitoring Prostate Specific Antigen (PSA) is recommended. Evaluate for clinical progression if PSA increases. ( 5.5)
ADVERSE REACTIONS
Adverse reactions that occurred in more than 10% of patients receiving bicalutamide plus an LHRH-A were: hot flashes, pain (including general, back, pelvic and abdominal), asthenia, constipation, infection, nausea, peripheral edema, dyspnea, diarrhea, hematuria, nocturia and anemia. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Accord Healthcare Inc. at 1-866-941-7875 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch
DRUG INTERACTIONS
USE IN SPECIFIC POPULATIONS
See 17 for PATIENT COUNSELING INFORMATION and FDA-approved patient labeling.
Revised: 10/2023
-
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1. INDICATIONS AND USAGE
2. DOSAGE AND ADMINISTRATION
2.1. Recommended Dose and Schedule
2.2. Dosage Adjustment in Renal Impairment
2.3. Dosage Adjustment in Hepatic Impairment
3. DOSAGE FORMS AND STRENGTHS
4. CONTRAINDICATIONS
5. WARNINGS AND PRECAUTIONS
5.1. Hepatitis
5.2. Hemorrhage with Concomitant Use of Coumarin Anticoagulant
5.3. Gynecomastia and Breast Pain
5.4. Glucose Tolerance
5.5. Laboratory Tests
6. ADVERSE REACTIONS
6.1. Clinical Trials Experience
6.2. Postmarketing Experience
7. DRUG INTERACTIONS
8. USE IN SPECIFIC POPULATIONS
8.1. Pregnancy
8.2. Lactation
8.3. Females and Males of Reproductive Potential
8.4. Pediatric Use
8.5. Geriatric Use
8.6. Hepatic Impairment
8.7. Renal Impairment
10. OVERDOSAGE
11. DESCRIPTION
12. CLINICAL PHARMACOLOGY
12.1. Mechanism of Action
12.3. Pharmacokinetics
13. NONCLINICAL TOXICOLOGY
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
14. CLINICAL STUDIES
14.1. Bicalutamide Tablets 50 mg Daily in Combination with an LHRH-A
14.2 Safety Data from Clinical Studies using Bicalutamide Tablets 150 mg
16. HOW SUPPLIED/STORAGE AND HANDLING
16.1. Storage and Handling
17. PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1. INDICATIONS AND USAGE
Bicalutamide tablets 50 mg daily is indicatedfor use in combination therapy with a luteinizing hormone-releasing hormone (LHRH) analog for the treatment of Stage D 2metastatic carcinoma of the prostate.
Bicalutamide tablets 150 mg daily is not approvedfor use alone or with other treatments [see Clinical Studies (14.2)].
-
2. DOSAGE AND ADMINISTRATION
2.1. Recommended Dose and Schedule
The recommended dose for bicalutamide tablets therapy in combination with an LHRH analog is one 50 mg tablet once daily (morning or evening), with or without food. It is recommended that bicalutamide tablets be taken at the same time each day. Treatment with bicalutamide tablets should be started at the same time as treatment with an LHRH analog. If a dose of bicalutamide is missed, take the next dose at the scheduled time. Do not take the missed dose and do not double the next dose.
2.2. Dosage Adjustment in Renal Impairment
No dosage adjustment is necessary for patients with renal impairment [see Use in Specific Populations (8.7)] .
2.3. Dosage Adjustment in Hepatic Impairment
No dosage adjustment is necessary for patients with mild to moderate hepatic impairment. In patients with severe liver impairment (n=4), although there was a 76% increase in the half-life (5.9 and 10.4 days for normal and impaired patients, respectively) of the active enantiomer of bicalutamide, no dosage adjustment is necessary [see Use in Specific Populations (8.6)] .
- 3. DOSAGE FORMS AND STRENGTHS
-
4. CONTRAINDICATIONS
Bicalutamide is contraindicated in:
- Hypersensitivity
Bicalutamide is contraindicated in any patient who has shown a hypersensitivity reaction to the drug or any of the tablet’s components. Hypersensitivity reactions including angioneurotic edema and urticaria have been reported.
- Women
Bicalutamide has no indication for women, and should not be used in this population.
- Pregnancy
Bicalutamide can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
-
5. WARNINGS AND PRECAUTIONS
5.1. Hepatitis
Cases of death or hospitalization due to severe liver injury (hepatic failure) have been reported postmarketing in association with the use of bicalutamide. Hepatotoxicity in these reports generally occurred within the first three to four months of treatment. Hepatitis or marked increases in liver enzymes leading to drug discontinuation occurred in approximately 1% of bicalutamide patients in controlled clinical trials.
Serum transaminase levels should be measured prior to starting treatment with bicalutamide, at regular intervals for the first four months of treatment, and periodically thereafter. If clinical symptoms or signs suggestive of liver dysfunction occur (e.g., nausea, vomiting, abdominal pain, fatigue, anorexia, “flu-like” symptoms, dark urine, jaundice, or right upper quadrant tenderness), the serum transaminases, in particular the serum ALT, should be measured immediately. If at any time a patient has jaundice, or their ALT rises above two times the upper limit of normal, bicalutamide should be immediately discontinued with close follow-up of liver function.
5.2. Hemorrhage with Concomitant Use of Coumarin Anticoagulant
In the postmarketing setting, there have been reports of excessive prolongation of the prothrombin time (PT) and International Normalized Ratio (INR) days to weeks after the introduction of bicalutamide in patients who were previously stable on coumarin anticoagulants. Some patients had serious bleeding including intracranial, retroperitoneal, and gastrointestinal requiring blood transfusion and/or administration of vitamin K. Closely monitor the PT/INR, and adjust the anticoagulant dose as needed [see Drug Interactions (7)and Adverse Reactions (6.2)].
5.3. Gynecomastia and Breast Pain
In clinical trials with bicalutamide 150 mg as a single agent for prostate cancer, gynecomastia and breast pain have been reported in up to 38% and 39% of patients, respectively.
5.4. Glucose Tolerance
A reduction in glucose tolerance has been observed in males receiving LHRH agonists. This may manifest as diabetes or loss of glycemic control in those with pre-existing diabetes. Consideration should therefore be given to monitoring blood glucose in patients receiving bicalutamide in combination with LHRH agonists.
5.5. Laboratory Tests
Regular assessments of serum Prostate Specific Antigen (PSA) may be helpful in monitoring the patient’s response. If PSA levels rise during bicalutamide therapy, the patient should be evaluated for clinical progression. For patients who have objective progression of disease together with an elevated PSA, a treatment-free period of antiandrogen, while continuing the LHRH analog, may be considered.
-
6. ADVERSE REACTIONS
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
6.1. Clinical Trials Experience
In patients with advanced prostate cancer treated with bicalutamide in combination with an LHRH analog, the most frequent adverse reaction was hot flashes (53%).
In the multi-center, double-blind, controlled clinical trial comparing bicalutamide 50 mg once daily with flutamide 250 mg three times a day, each in combination with an LHRH analog, the following adverse reactions with an incidence of 5% or greater, regardless of causality, have been reported.
Table 1. Incidence of Adverse Reactions (≥ 5% in Either Treatment Group) Regardless of Causality Body System Adverse Reaction Treatment Group Number of Patients (%) Bicalutamide Plus LHRH Analog (n=401) Flutamide Plus LHRH Analog (n=407) Body as a Whole Pain (General) 142 (35) 127 (31) Back Pain 102 (25) 105 (26) Asthenia 89 (22) 87 (21) Pelvic Pain 85 (21) 70 (17) Infection
71(18)
57 (14)
Abdominal Pain
46 (11)
46 (11)
Chest Pain
34 (8)
34 (8)
Headache
29 (7)
27 (7)
Flu Syndrome
28 (7)
30 (7)
Cardiovascular
Hot Flashes
211 (53)
217 (53)
Hypertension
34 (8)
29 (7)
Digestive
Constipation
87 (22)
69 (17)
Nausea
62 (15)
58 (14)
Diarrhea
49 (12)
107 (26)
Increased Liver Enzyme Test
30 (7)
46 (11)
Dyspepsia
30 (7)
23 (6)
Flatulence
26 (6)
22 (5)
Anorexia
25 (6)
29 (7)
Vomiting
24 (6)
32 (8)
Hemic and Lymphatic
Anemia
45 (11)
53 (13)
Metabolic and Nutritional
Peripheral Edema
53 (13)
42 (10)
Weight Loss
30 (7)
39 (10)
Hyperglycemia
26 (6)
27 (7)
Alkaline Phosphatase Increased
22 (5)
24 (6)
Weight Gain
22 (5)
18 (4)
Musculoskeletal
Bone Pain
37 (9)
43 (11)
Myasthenia
27 (7)
19 (5)
Arthritis
21 (5)
29 (7)
Pathological Fracture
17 (4)
32 (8)
Nervous System
Dizziness
41 (10)
35 (9)
Paresthesia
31 (8)
40 (10)
Insomnia
27 (7)
39 (10)
Anxiety
20 (5)
9 (2)
Depression
16 (4)
33 (8)
Respiratory System
Dyspnea
51 (13)
32 (8)
Cough Increased
33 (8)
24 (6)
Pharyngitis
32 (8)
23 (6)
Bronchitis
24 (6)
22 (3)
Pneumonia
18 (4)
19 (5)
Rhinitis
15 (4)
22 (5)
Skin and Appendages
Rash
35 (9)
30 (7)
Sweating
25 (6)
20 (5)
Urogenital
Nocturia
49 (12)
55 (14)
Hematuria
48 (12)
26 (6)
Urinary Tract Infection
35 (9)
36 (9)
Gynecomastia
36 (9)
30 (7)
Impotence
27 (7)
35 (9)
Breast Pain
23 (6)
15 (4)
Urinary Frequency
23 (6)
29 (7)
Urinary Retention
20 (5)
14 (3)
Urinary Impaired
19 (5)
15 (4)
Urinary Incontinence
15 (4)
32 (8)
Other adverse reactions (greater than or equal to 2%, but less than 5%) reported in the bicalutamide-LHRH analog treatment group are listed below by body system and are in order of decreasing frequency within each body system regardless of causality.
Body as a Whole:Neoplasm; Neck Pain; Fever; Chills; Sepsis; Hernia; Cyst
Cardiovascular:Angina Pectoris; Congestive Heart Failure; Myocardial Infarct; Heart Arrest; Coronary Artery Disorder; Syncope
Digestive:Melena; Rectal Hemorrhage; Dry Mouth; Dysphagia; Gastrointestinal Disorder; Periodontal Abscess; Gastrointestinal Carcinoma
Metabolic and Nutritional:Edema; BUN Increased; Creatinine Increased; Dehydration; Gout; Hypercholesteremia
Musculoskeletal:Myalgia; Leg Cramps
Nervous:Hypertonia; Confusion; Somnolence; Libido Decreased; Neuropathy; Nervousness
Respiratory:Lung Disorder; Asthma; Epistaxis; Sinusitis
Skin and Appendages:Dry Skin; Alopecia; Pruritus; Herpes Zoster; Skin Carcinoma; Skin Disorder
Special Senses:Cataract Specified
Urogenital:Dysuria; Urinary Urgency; Hydronephrosis; Urinary Tract Disorder
Abnormal Laboratory Test Values:
Laboratory abnormalities including: elevated AST, ALT, bilirubin, BUN, and creatinine; and decreased hemoglobin and white cell count, have been reported in both bicalutamide-LHRH analog treated and flutamide-LHRH analog treated patients.
6.2. Postmarketing Experience
The following adverse reactions have been identified during post-approval use of bicalutamide. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Respiratory disorders:Interstitial lung disease (some fatal) including interstitial pneumonitis and pulmonary fibrosis, most often at doses greater than 50 mg.
Hemorrhage:Increased PT/INR due to interaction between coumarin anticoagulants and bicalutamide. Serious bleeding reported. [see Warnings and Precautions (5.2)]
Skin and subcutaneous tissue disorders:Photosensitivity
-
7. DRUG INTERACTIONS
Clinical studies have not shown any drug interactions between bicalutamide and LHRH analogs (goserelin or leuprolide). There is no evidence that bicalutamide induces hepatic enzymes.
In vitrostudies have shown that R-bicalutamide is an inhibitor of CYP 3A4 with lesser inhibitory effects on CYP 2C9, 2C19 and 2D6 activity. Clinical studies have shown that with co-administration of bicalutamide, mean midazolam (a CYP 3A4 substrate) levels may be increased 1.5-fold (for C max) and 1.9-fold (for AUC). Hence, caution should be exercised when bicalutamide is co-administered with CYP 3A4 substrates.
In vitroprotein-binding studies have shown that bicalutamide can displace coumarin anticoagulants from binding sites. PT/INR should be closely monitored in patients concomitantly receiving coumarin anticoagulants and bicalutamide. Adjustment of the anticoagulant dose may be necessary. [see Warnings and Precautions (5.2)and Adverse reaction (6.2)]
-
8. USE IN SPECIFIC POPULATIONS
8.1. Pregnancy
Risk Summary
Bicalutamide is contraindicated for use in pregnant women because it can cause fetal harm. Bicalutamide is not indicated for use in females. There are no human data on the use of bicalutamide in pregnant women. In animal reproduction studies, oral administration of bicalutamide to pregnant rats during organogenesis caused abnormal development of reproductive organs in male fetuses at exposures approximately 0.7 to 2 times the human exposure at the recommended dose (see Data).
Data
Animal Data
In an embryo-fetal development study in pregnant rats dosed during the period of organogenesis from gestation days 6 to15, male fetuses had reduced anogenital distance at doses of 10 mg/kg/day and above (approximately 0.7 to 2 times the human exposure at the recommended dose).
In a pre- and post-natal development study, female rats were dosed from gestation day 7 to 16 and allowed to litter and rear their offspring to weaning. Male offspring of rats receiving doses of 10 mg/kg/day (approximately 0.7 times the human exposure at the recommended dose) and above, were observed to have reduced anogenital distance.
In a peri- and post-natal development study, female rats were dosed from gestation day 16 to lactation day 22 and allowed to litter and rear their offspring to weaning. Survival and weights of offspring during lactation were reduced for litters from maternal rats receiving doses of 250 mg/kg/day (approximately 2 times the human exposure at the recommended dose). Male offspring of rats receiving doses of 10 mg/kg/day (approximately 0.7 times the human exposure at the recommended dose) and above, were observed to have reduced anogenital distance, smaller secondary sex organs, cryptorchidism and hypospadias resulting in an inability to mate and impregnate their female partners. Female offspring of rats receiving doses of 10 mg/kg/day (approximately 0.7 times the human exposure at the recommended dose) and above had reduced pregnancy rates.
8.2. Lactation
Risk Summary
Bicalutamide is not indicated for use in pregnant women. There is no information available on the presence of bicalutamide in human milk, or on the effects on the breastfed infant or on milk production. Bicalutamide has been detected in rat milk.
8.3. Females and Males of Reproductive Potential
Contraception
Males
Antiandrogen therapy may cause morphological changes in spermatozoa [see Nonclinical Toxicology (13.1)]. Based on findings in animal reproduction studies and its mechanism of action, advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 130 days after the final dose of bicalutamide [see Use in Specific Populations (8.1)and Clinical Pharmacology (12.1)].
Infertility
Males
Based on animal studies, bicalutamide can lead to inhibition of spermatogenesis and may impair fertility in males of reproductive potential. The long-term effects of bicalutamide tablets on male fertility have not been studied. [see Nonclinical Toxicology (13.1)] .
8.4. Pediatric Use
The safety and effectiveness of bicalutamide in pediatric patients have not been established.
Bicalutamide orodispersible tablet was studied in combination with anastrozole orodispersible tablet in an open-label, non-comparative, multi-center study that assessed the efficacy and safety of this combination regimen over 12 months in the treatment of gonadotropin-independent precocious puberty in boys with familial male-limited precocious puberty, also known as testotoxicosis. Patients were enrolled in the study if they had a baseline age ≥ 2 years and a diagnosis of testotoxicosis based on clinical features of progressive precocious puberty, symmetrical testicular enlargement, advanced bone age, pubertal levels of serum testosterone, prepubertal pattern of gonadotropin secretion following a GnRH stimulation test, and absence of other clinical and biochemical causes of testosterone excess. Thirteen out of the 14 patients enrolled completed 12 months of combination treatment (one patient was lost to follow-up). If central precocious puberty (CPP) developed, an LHRH analog was to be added. Four patients were diagnosed with CPP during the 12-month study and received LHRH analog treatment and 2 additional patients were diagnosed at the end of the 12 months and received treatment subsequently. Mean ± SD characteristics at baseline were as follows: chronological age: 3.9±1.9 years; bone age 8.8±2.5; bone age/chronological age ratio: 2.06 ± 0.51; growth rate (cm/yr): 10.81 ± 4.22; growth rate standard deviation score (SDS): 0.41 ± 1.36.
The starting bicalutamide dose was 12.5 mg. Bicalutamide was titrated in each patient until steady-state R-bicalutamide (the active isomer of bicalutamide) trough plasma concentration reached 5 to 15 mcg/mL, which is the range of therapeutic concentrations achieved in adults with prostate cancer following the administration of the currently approved bicalutamide dose of 50 mg. The starting daily dose of anastrozole was 0.5 mg. Anastrozole was independently titrated in each patient until it reached at steady-state a serum estradiol concentration of <10 pmol/L (2.7 pg/mL). The following ascending doses were used for bicalutamide: 12.5 mg, 25 mg, 50 mg, and 100 mg. For anastrozole there were two ascending doses: 0.5 mg and 1 mg. At the end of the titration phase, 1 patient was on 12.5 mg bicalutamide, 8 patients were on 50 mg bicalutamide, and 4 patients were on 100 mg bicalutamide; 10 patients were on 0.5 mg anastrozole and 3 patients were on 1 mg anastrozole. In the majority of patients, steady-state trough concentrations of R-bicalutamide appeared to be attained by Day 21 with once daily dosing. Steady-state trough plasma anastrozole concentrations appeared to be attained by Day 8.
The primary efficacy analysis of the study was to assess the change in growth rate after 12 months of treatment, relative to the growth rate during the ≥6 months prior to entering the study. Pre-study growth rates were obtained retrospectively. There was no statistical evidence that the growth rate was reduced during treatment. During bicalutamide /anastrozole treatment the mean growth rate (cm/yr) decreased by 1.6 cm/year, 95% CI (-4.7 to 1.5) p=0.28; the mean growth rate SDS decreased by 0.1 SD, 95% CI (–1.2 to 1.0) p=0.88. Table 2 shows descriptive data for growth rates for the overall population and for subgroups defined by history of previous treatment for testotoxicosis with ketoconazole, spironolactone, anastrozole or other aromatase inhibitors.
Table 2. Growth Rates - *
- Change compared to pre-study growth rate.
- †
- PT = Previous treatment for testotoxicosis with ketoconazole, spironolactone, anastrozole or other aromatase inhibitors.
- ‡
- Median calculated as midpoint of 3rd and 4th ranked observations.
- §
- NPT = no previous treatment for testotoxicosis with ketoconazole, spironolactone, anastrozole, or other aromatase inhibitors.
Endpoint
Analysis population
Pre-study Mean
Change from pre-study to 12 months
% patients with growth reduction * Mean
Median
(Min, Max)
Growth rate (cm/yr)
All treated (n=13)
10.8
-1.6
-2.8
(-7.4, 8.4)
9/13 (69%)
PT †(n=6)
10.3
-0.2
-2.6 ‡
(-7.2, 8.4)
4/6 (67%)
NPT §(n=7)
11.2
-2.8
-2.8
(-7.4, 1.1)
5/7 (71%)
Growth rate (SD units)
All treated (n=13)
0.4
-0.1
-0.4
(-2.7, 3.5)
9/13 (69%)
PT †(n=6)
-0.1
+0.7
-0.2 ‡
(-1.6, 3.5)
4/6 (67%)
NPT §(n=7)
0.8
-0.7
-0.4
(-2.7, 0.5)
5/7 (71%)
Total testosterone concentrations increased by a mean of 5 mmol/L over the 12 months of treatment from a baseline mean of 10 mmol/L. Estradiol concentrations were at or below the level of quantification (9.81 pmol/L) for 11 of 12 patients after 12 months of treatment. Six of the 12 patients started treatment at an estradiol concentration below the level of quantification.
There were no deaths, serious adverse events, or discontinuations due to adverse events during the study. Of the 14 patients exposed to study treatment, 13 (92.9%) experienced at least one adverse event. The most frequently reported (>3 patients) adverse events were gynecomastia (7/14, 50%), central precocious puberty (6/14, 43%), vomiting (5/14, 36%), headache (3/14, 21%), pyrexia (3/14, 21%), and upper respiratory tract infection (3/14, 21%). Adverse reactions considered possibly related to bicalutamide by investigators included gynecomastia (6/14, 43%), central precocious puberty (2/14, 14%), breast tenderness (2/14, 14%), breast pain (1/14, 7%), asthenia (1/14, 7%), increased alanine aminotransferase [ALT] (1/14, 7%), increased aspartate aminotransferase [AST] (1/14, 7%), and musculoskeletal chest pain (1/14, 7%). Headache was the only adverse reaction considered possibly related to anastrozole by investigators. For the patient who developed elevated ALT and AST, the elevation was <3X ULN, and returned to normal without stopping treatment; there was no concomitant elevation in total bilirubin.
8.5. Geriatric Use
In two studies in patients given 50 or 150 mg daily, no significant relationship between age and steady-state levels of total bicalutamide or the active R-enantiomer has been shown.
8.6. Hepatic Impairment
Bicalutamide should be used with caution in patients with moderate-to-severe hepatic impairment. Bicalutamide is extensively metabolized by the liver. Limited data in subjects with severe hepatic impairment suggest that excretion of bicalutamide may be delayed and could lead to further accumulation. Periodic liver function tests should be considered for hepatic-impaired patients on long-term therapy [see Warnings and Precautions (5.1)].
No clinically significant difference in the pharmacokinetics of either enantiomer of bicalutamide was noted in patients with mild-to-moderate hepatic disease as compared to healthy controls. However, the half-life of the R-enantiomer was increased approximately 76% (5.9 and 10.4 days for normal and impaired patients, respectively) in patients with severe liver disease (n=4).
-
10. OVERDOSAGE
Long-term clinical trials have been conducted with dosages up to 200 mg of bicalutamide daily and these dosages have been well tolerated. A single dose of bicalutamide that results in symptoms of an overdose considered to be life threatening has not been established.
There is no specific antidote; treatment of an overdose should be symptomatic.
In the management of an overdose with bicalutamide, vomiting may be induced if the patient is alert. It should be remembered that, in this patient population, multiple drugs may have been taken. Dialysis is not likely to be helpful since bicalutamide is highly protein bound and is extensively metabolized. General supportive care, including frequent monitoring of vital signs and close observation of the patient, is indicated.
-
11. DESCRIPTION
Bicalutamide tablets, USP contain 50 mg of bicalutamide, a non-steroidal androgen receptor inhibitor with no other known endocrine activity. The chemical name is propanamide, N [4 cyano-3-(trifluoromethyl)phenyl]-3-[(4-fluorophenyl)sulfonyl]-2-hydroxy-2-methyl-,(+-). The structural and empirical formulas are:
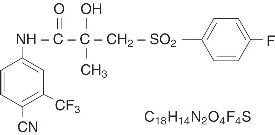
Bicalutamide has a molecular weight of 430.37. The pKa is approximately 12. Bicalutamide is a fine white to off-white powder which is practically insoluble in water at 37°C (5 mg per 1000 mL), slightly soluble in chloroform and absolute ethanol, sparingly soluble in methanol, and soluble in acetone and tetrahydrofuran.
Bicalutamide is a racemate with its antiandrogenic activity being almost exclusively exhibited by the R-enantiomer of bicalutamide; the S-enantiomer is essentially inactive.
The inactive ingredients of bicalutamide tablets, USP are lactose monohydrate, magnesium stearate, hypromellose E5, polyethylene glycol 400, povidone K 30, sodium starch glycolate, and titanium dioxide.
Bicalutamide tablets, USP 50 mg meets USP Dissolution Test 2.
-
12. CLINICAL PHARMACOLOGY
12.1. Mechanism of Action
Bicalutamide is a non-steroidal androgen receptor inhibitor. It competitively inhibits the action of androgens by binding to cytosol androgen receptors in the target tissue. Prostatic carcinoma is known to be androgen sensitive and responds to treatment that counteracts the effect of androgen and/or removes the source of androgen.
When bicalutamide is combined with LHRH analog therapy, the suppression of serum testosterone induced by the LHRH analog is not affected. However, in clinical trials with bicalutamide as a single agent for prostate cancer, rises in serum testosterone and estradiol have been noted.
In a subset of patients who have been treated with bicalutamide and an LHRH agonist, and who discontinue bicalutamide therapy due to progressive advanced prostate cancer, a reduction in Prostate Specific Antigen (PSA) and/or clinical improvement (antiandrogen withdrawal phenomenon) may be observed.
12.3. Pharmacokinetics
Absorption
Bicalutamide is well-absorbed following oral administration, although the absolute bioavailability is unknown. Co-administration of bicalutamide with food has no clinically significant effect on rate or extent of absorption.
Distribution
Bicalutamide is highly protein-bound (96%) [see Drug Interactions (7)] .
Metabolism/Elimination
Bicalutamide undergoes stereospecific metabolism. The S (inactive) isomer is metabolized primarily by glucuronidation. The R (active) isomer also undergoes glucuronidation but is predominantly oxidized to an inactive metabolite followed by glucuronidation. Both the parent and metabolite glucuronides are eliminated in the urine and feces. The S-enantiomer is rapidly cleared relative to the R-enantiomer, with the R-enantiomer accounting for about 99% of total steady-state plasma levels.
Pharmacokinetics of the active enantiomer of bicalutamide in normal males and patients with prostate cancer are presented in Table 3.
Table 3. Pharmacokinetics of Bicalutamide Active Enantiomer
Parameter Mean Standard Deviation Normal Males (n=30)
Apparent Oral Clearance (L/hr)
0.320
0.103
Single Dose Peak Concentration (µg/mL)
0.768
0.178
Single Dose Time to Peak Concentration (hours)
31.3
14.6
Half-life (days)
5.8
2.29
Patients with Prostate Cancer (n=40)
C ss(µg/mL)
8.939
3.504
-
13. NONCLINICAL TOXICOLOGY
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year oral carcinogenicity studies were conducted in both male and female rats and mice at doses of 5, 15, or 75 mg/kg/day of bicalutamide. A variety of tumor target organ effects were identified and were attributed to the antiandrogenicity of bicalutamide, namely, testicular benign interstitial (Leydig) cell tumors in male rats at all dose levels (the steady-state plasma concentration with the 5 mg/kg/day dose is approximately 0.7 times the human exposure at the recommended dose) and uterine adenocarcinoma in female rats at 75 mg/kg/day (approximately 1.5 times the human exposure at the recommended dose). There is no evidence of Leydig cell hyperplasia in patients; uterine tumors are not relevant to the indicated patient population
A small increase in the incidence of hepatocellular carcinoma in male mice given 75 mg/kg/day of bicalutamide (approximately 4 times the human exposure at the recommended dose) and an increased incidence of benign thyroid follicular cell adenomas in rats given 5 mg/kg/day (approximately 0.7 times the human exposure at the recommended dose) and above were recorded. These neoplastic changes were progressions of non-neoplastic changes related to hepatic enzyme induction observed in animal toxicity studies. Enzyme induction has not been observed following bicalutamide administration in man. There were no tumorigenic effects suggestive of genotoxic carcinogenesis.
A comprehensive battery of both in vitroand in vivogenotoxicity tests (yeast gene conversion, Ames, E. coli, CHO/HGPRT, human lymphocyte cytogenetic, mouse micronucleus, and rat bone marrow cytogenetic tests) has demonstrated that bicalutamide does not have genotoxic activity.
In repeat-dose toxicology studies, atrophy of seminiferous tubules of the testes has been observed for all species examined, which is a predicted class effect with antiandrogens. In the 6- and 12-month rat study, testicular atrophy was seen at approximately 2 times the human exposure at the recommended dose. In the 12- month dog study, the incidence of testicular atrophy was seen at approximately 7 times the human exposure at the recommended dose. In male rats administered 250 mg/kg/day (approximately 2 times human exposure at the recommended dose), the precoital interval and time to successful mating were increased in the first pairing, but no effects on fertility following successful mating were seen. These effects were reversed by 7 weeks after the end of an 11-week period of dosing.
Female rats dosed at 1, 10 and 250 mg/kg/day (less than to 2 times the human exposure at the recommended dose) had increased estrous cycle irregularity but there was no effect on fertility.
In a peri- and post-natal development study, female offspring of rats receiving doses of 10 mg/kg/day (approximately 0.7 times the human exposure at the recommended clinical dose) and above had reduced pregnancy rates. Administration of bicalutamide to pregnant females resulted in feminization of the male offspring leading to hypospadias at doses of 10 mg/kg/day (approximately 0.7 times the human exposure at the recommended dose) and above. Affected male offspring were also impotent.
-
14. CLINICAL STUDIES
14.1. Bicalutamide Tablets 50 mg Daily in Combination with an LHRH-A
In a multi-center, double-blind, controlled clinical trial, 813 patients with previously untreated advanced prostate cancer were randomized to receive bicalutamide 50 mg once daily (404 patients) or flutamide 250 mg (409 patients) three times a day, each in combination with LHRH analogs (either goserelin acetate implant or leuprolide acetate depot).
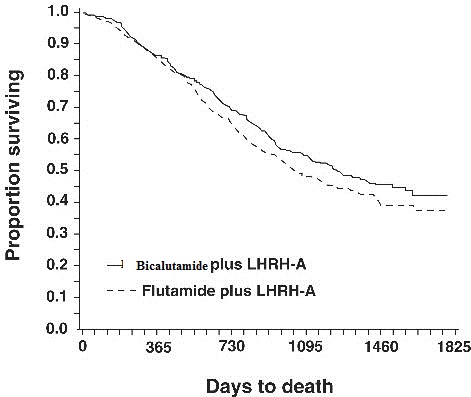
In an analysis conducted after a median follow-up of 160 weeks was reached, 213 (52.7%) patients treated with bicalutamide-LHRH analog therapy and 235 (57.5%) patients treated with flutamide-LHRH analog therapy had died. There was no significant difference in survival between treatment groups (see Figure 1). The hazard ratio for time to death (survival) was 0.87 (95% confidence interval 0.72 to 1.05).
Figure 1 - The Kaplan-Meier probability of death for both antiandrogen treatment groups.
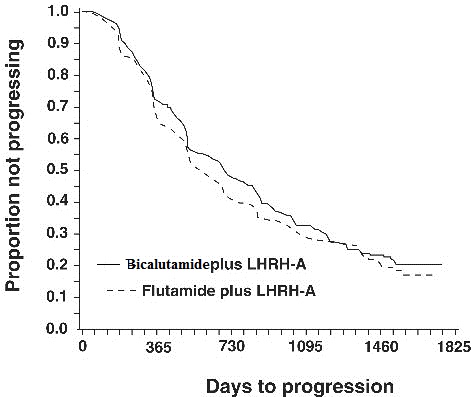
There was no significant difference in time to objective tumor progression between treatment groups (see Figure 2). Objective tumor progression was defined as the appearance of any bone metastases or the worsening of any existing bone metastases on bone scan attributable to metastatic disease, or an increase by 25% or more of any existing measurable extraskeletal metastases. The hazard ratio for time to progression of bicalutamide plus LHRH analog to that of flutamide plus LHRH analog was 0.93 (95% confidence interval, 0.79 to 1.10).
Figure 2 - Kaplan-Meier curve for time to progression for both antiandrogen treatment groups.
Quality of life was assessed with self-administered patient questionnaires on pain, social functioning, emotional well-being, vitality, activity limitation, bed disability, overall health, physical capacity, general symptoms, and treatment related symptoms. Assessment of the Quality of Life questionnaires did not indicate consistent significant differences between the two treatment groups.
14.2 Safety Data from Clinical Studies using Bicalutamide Tablets 150 mg
Bicalutamide tablet 150 mg is not approved for use either alone or with other treatments.
Two identical multi-center, randomized, open-label trials comparing bicalutamide 150 mg daily monotherapy to castration were conducted in patients that had locally advanced (T3-4, NX, M0) or metastatic (M1) prostate cancer.
Monotherapy — M1 Group
Bicalutamide 150 mg daily is not approved for use in patients with M1 cancer of the prostate. Based on an interim analysis of the two trials for survival, the Data Safety Monitoring Board recommended that bicalutamide treatment be discontinued in the M1 patients because the risk of death was 25% (HR 1.25, 95% CI 0.87 to 1.81) and 31% (HR 1.31, 95% CI 0.97 to 1.77) higher in the bicalutamide treated group compared to that in the castrated group, respectively.
Locally Advanced (T 3-4, NX, M0) Group
Bicalutamide 150 mg daily is not approved for use in patients with locally advanced (T 3-4, NX, M0) cancer of the prostate. Following discontinuation of all M1 patients, the trials continued with the T3-4, NX, M0 patients until study completion. In the larger trial (N=352), the risk of death was 25% (HR 1.25, 95% CI 0.92 to 1.71) higher in the bicalutamide group and in the smaller trial (N=140), the risk of death was 36% (HR 0.64, 95% CI, 0.39 to 1.03) lower in the bicalutamide group.
In addition to the above two studies, there are three other ongoing clinical studies that provide additional safety information for bicalutamide 150 mg, a dose that is not approved for use. These are three multi-center, randomized, double-blind, parallel group trials comparing bicalutamide 150 mg daily monotherapy (adjuvant to previous therapy or under watchful waiting) with placebo, for death or time to disease progression, in a population of 8113 patients with localized or locally advanced prostate cancer.
Bicalutamide 150 mg daily is not approved for use as therapy for patients with localized prostate cancer who are candidates for watchful waiting. Data from a planned subgroup analysis of two of these trials in 1627 patients with localized prostate cancer who were under watchful waiting, revealed a trend toward decreased survival in the bicalutamide arm after a median follow-up of 7.4 years. There were 294 (37.7%) deaths in the bicalutamide treated patients versus 279 (32.9%) deaths in the placebo-treated patients (localized watchful waiting group) for a hazard ratio of 1.16 (95% CI 0.99 to 1.37).
-
16. HOW SUPPLIED/STORAGE AND HANDLING
Bicalutamide tablets, USP 50 mg are white to off-white, round, biconvex, film-coated tablets debossed "B 50" on one side and plain on other side and supplied in bottles of 30 tablets with a child-resistant closure (NDC 60429-177-30), bottles of 100 tablets with a child-resistant closure (NDC 60429-177-01) and bottles of 500 tablets with a child-resistant closure (NDC 60429-177-05)
-
17. PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Dose and Schedule:Inform patients that therapy with bicalutamide and the LHRH analog should be started at the same time and that they should not interrupt or stop taking these medications without consulting their healthcare provider [see Dosage and Administration (2.1)].
Hepatitis:Inform patients that bicalutamide can cause hepatitis, which may result in hepatic failure and death. Advise patients that liver function tests should be monitored regularly during treatment and to report signs and symptoms of hepatitis [see Warnings and Precautions (5.1)].
Hemorrhage with Concomitant Use of Coumarin Anticoagulant:Inform patients that serious bleeding has occurred with reported increased anticoagulant effects while taking bicalutamide. Advise patients to notify their healthcare provider of any bleeding or spontaneous bruising while on bicalutamide and taking anticoagulants [see Warnings and Precautions (5.2)and Adverse reaction (6.2)] .
Glucose Tolerance:Inform patients that diabetes or loss of glycemic control in patients with pre-existing diabetes has been reported during treatment with LHRH agonists. Consideration should therefore be given to monitoring blood glucose in patients receiving bicalutamide in combination with LHRH agonists [see Warnings and Precautions (5.4)].
Somnolence:During treatment with bicalutamide, somnolence has been reported. Advise patients who experience this symptom to observe caution when driving or operating machines [see Adverse Reactions (6.1)].
Photosensitivity:Inform patients that cases of photosensitivity have been reported during treatment with bicalutamide and that they should avoid direct exposure to excessive sunlight or UV-light exposure. Consideration should be given to the use of sunscreen [see Adverse Reactions (6.2)].
Contraception and fertility:Advise male patients with female partners of reproductive potential to use effective contraception during treatment and for 130 days after the last dose of bicalutamide therapy. Advise male patients that bicalutamide may impair fertility [see Use in Specific Populations (8.3)].
Manufactured For:
Accord Healthcare, Inc.,
8041 Arco Corporate Drive,
Suite 200,
Raleigh, NC 27617,
USA.Manufactured By:
Intas Pharmaceuticals Limited,
Plot No. : 457, 458,
Village - Matoda,
Bavla Road, Ta.:Sanand,
Dist.: Ahmedabad : 382 210.
India.10 0857 4 6025824
Issued October 2023
Marketed by:
GSMS, Inc.
Camarillo, CA 93012 USA
-
PATIENT PACKAGE INSERT
PATIENT INFORMATION
Bicalutamide Tablets
[bye-kah-LEW-tuh-mide]
What are bicalutamide tablets?
Bicalutamide is a prescription medicine called an androgen receptor inhibitor, used in combination with luteinizing hormone-releasing hormone (LHRH) medicines to treat Stage D 2metastatic prostate cancer.
Bicalutamide tablets 150 mg daily is not approved for use alone or with other treatments.
It is not known if bicalutamide tablets are safe and effective in children.
Do not take bicalutamide tablets if you are:
- allergic to bicalutamide or any of the ingredients in bicalutamide tablets. See the end of this Patient Information leaflet for a complete list of ingredients in bicalutamide tablets. Get medical help right away if you develop any of the following symptoms of an allergic reaction:
- itching
- hives(raised bumps)
- swelling of the face, lips or tongue
- trouble breathing or swallowing
- female. Bicalutamide tablets is not for use by women
- pregnant or may become pregnant. Bicalutamide tablets may harm your unborn baby.
Before taking bicalutamide tablets, tell your healthcare provider about all your medical conditions, including if you:
- have liver problems.
- take a medicine to thin your blood. Ask your healthcare provider or pharmacist if you are not sure if your medicine is a blood thinner.
- have diabetes.
- have a female partner who can become pregnant. Males who have a female partner who can become pregnant should use effective birth control during treatment with bicalutamide tablets and for 130 days after the final dose. Talk to your healthcare provider if you have any questions about birth control.
Tell your healthcare provider about all the medicines you take,including prescription and over-the-counter medicines, vitamins, and herbal supplements. Bicalutamide tablets may affect the way other medicines work and other medicines may affect how bicalutamide tablets works, causing side effects.
Know the medicines you take. Keep a list of your medicines with you to show your healthcare providers when you get a new medicine.
How should I take bicalutamide tablets?
- Take bicalutamide tablets exactly as your healthcare provider tells you to take it.
- Do not stop taking bicalutamide tablets unless your healthcare provider tells you to.
- Bicalutamide tablets can be taken either in the morning or in the evening, but you should take it at the same time every day.
- Your treatment with bicalutamide tablets should start at the same time as your treatment with the LHRH medicine.
- If you miss a bicalutamide tablets dose do not take the missed dose, take the next dose at your next scheduled time. Do not take 2 doses at the same time.
- Bicalutamide tablets can be taken with or without food.
- If you take too much bicalutamide tablets, call your healthcare provider or go to the nearest hospital emergency room right away
What should I avoid during treatment with bicalutamide tablets?
- Do not drive, operate machinery, or do other dangerous activities until you know how bicalutamide tablets affect you. Bicalutamide tablets can make you sleepy.
- Avoid sunlight, sunlamps and tanning beds and consider using sunscreen during treatment with bicalutamide tablets. Some people have had skin sensitivity to sunlight during treatment with bicalutamide tablets.
What are the possible side effects of bicalutamide tablets?
Bicalutamide tablets may cause serious side effects, including.
-
Liver problems.Severe liver problems including liver failure that may need to be treated in a hospital or that may lead to death have happened in people who take bicalutamide tablets Your healthcare provider should do blood tests to check your liver function before and during treatment with bicalutamide tablets. Tell your healthcare provider right away if you develop any of these symptoms of liver problems during treatment:
- yellowing of the skin and eyes (jaundice)
- dark urine
- right upper stomach pain
- nausea
- vomiting
- tiredness
- loss of appetite
- chills
- fever
- Bleeding problems.Serious bleeding problems have happened in people who take bicalutamide tablets in combination with a blood thinner medicine (coumarin anticoagulants). Bleeding problems have happened days to weeks after starting bicalutamide tablets treatment. If you take a blood thinner medicine during treatment with bicalutamide tablets, tell your healthcare provider if you develop any bleeding or unexplained bruising.
- Breast enlargement (gynecomastia) and breast pain.
- Blood sugar problems.Poor blood sugar control can happen in people who take bicalutamide tablets in combination with LHRH medicines.
Your healthcare provider may do blood tests during treatment with bicalutamide tablets to check for side effects.
Your prostate cancer may get worse during treatment with bicalutamide tablets in combination with LHRH medicines. Regular monitoring of your prostate cancer with your healthcare provider is important to determine if your disease is worse.
Tell your healthcare provider if you have trouble breathing with or without a cough or fever. Some people taking bicalutamide tablets get an inflammation in the lungs called interstitial lung disease.
The most common side effects of bicalutamide tablets include:
- hot flashes (short periods of feeling warm and sweating)
- body pain (including back, pelvis, stomach)
- feeling weak
- constipation
- infection
- nausea
- swelling in your arms, ankles, legs or feet
- shortness of breath (dyspnea)
- dizziness
- diarrhea
- blood in your urine
- frequent urination at night
- a decrease in red blood cells (anemia)
Bicalutamide tablets may have an effect on male fertility which could be reversible. Talk to your healthcare provider if this is a concern for you.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of bicalutamide tablets. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store bicalutamide tablets?
Store bicalutamide tablets at room temperature between 68°F to 77°F (20°C to 25°C).
Keep bicalutamide tablets and all medicines out of the reach of children.
General information about the safe and effective use of bicalutamide tablets.
Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use bicalutamide tablets for a condition for which it was not prescribed. Do not give bicalutamide tablets to other people, even if they have the same symptoms that you have. It may harm them.
This Patient Information leaflet summarizes the most important information about bicalutamide tablets. If you would like more information about bicalutamide tablets, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information about bicalutamide tablets that is written for health professionals.
For more information, go to www.accordhealthcare.usor call Accord Healthcare at 1-866-941-7875.
What are the ingredients in bicalutamide tablets?
Active ingredient: bicalutamide.
Inactive ingredients: lactose monohydrate, magnesium stearate, hypromellose E5, polyethylene glycol 400, povidone K30, sodium starch glycolate, and titanium dioxide.
Manufactured For:
Accord Healthcare Inc.
8041 Arco Corporate Drive,
Suite 200,
Raleigh, NC 27617,
USA.Manufactured By:
Intas Pharmaceuticals Limited
Plot No.: 457,458,
Village-Matoda,
Bavla Road, Ta. - Sanand,
Dist.-Ahmedabad 382 210,
India.
10 0857 4 6025824
Issued October 2023Marketed by:
GSMS, Inc.
Camarillo, CA 93012 USA
- allergic to bicalutamide or any of the ingredients in bicalutamide tablets. See the end of this Patient Information leaflet for a complete list of ingredients in bicalutamide tablets. Get medical help right away if you develop any of the following symptoms of an allergic reaction:
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BICALUTAMIDE
bicalutamide tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:60429-177(NDC:16729-023) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BICALUTAMIDE (UNII: A0Z3NAU9DP) (BICALUTAMIDE - UNII:A0Z3NAU9DP) BICALUTAMIDE 50 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE 2910 (5 MPA.S) (UNII: R75537T0T4) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) POVIDONE K30 (UNII: U725QWY32X) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color white (WHITE TO OFF WHITE) Score no score Shape ROUND Size 7mm Flavor Imprint Code B50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:60429-177-30 30 in 1 BOTTLE; Type 0: Not a Combination Product 05/08/2014 2 NDC:60429-177-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 05/08/2014 3 NDC:60429-177-05 500 in 1 BOTTLE; Type 0: Not a Combination Product 05/08/2014 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA078917 07/06/2009 Labeler - Golden State Medical Supply, Inc. (603184490) Establishment Name Address ID/FEI Business Operations Golden State Medical Supply, Inc. 603184490 relabel(60429-177) , repack(60429-177)
