Label: DOBUTAMINE injection, solution, concentrate





- NDC Code(s): 51662-1337-1, 51662-1337-2, 51662-1337-3
- Packager: HF Acquisition Co LLC, DBA HealthFirst
- This is a repackaged label.
- Source NDC Code(s): 0409-2344
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated February 19, 2024
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED
-
DESCRIPTION
Dobutamine Injection, USP is a clear, practically colorless, sterile, nonpyrogenic solution of dobutamine hydrochloride for intravenous use only. Each milliliter contains 12.5 mg (41.5 µmol) dobutamine, as the hydrochloride and sodium metabisulfite, 0.2 mg added as antioxidant. May contain hydrochloric acid and/or sodium hydroxide for pH adjustment. pH is 3.3 (2.5 to 5.5).
Dobutamine Hydrochloride, USP is chemically designated (±)-4-[2-[[3-(ρ-hydroxyphenyl)-1-methylpropyl] amino]ethyl]-pyrocatechol hydrochloride.
It is a synthetic catecholamine.
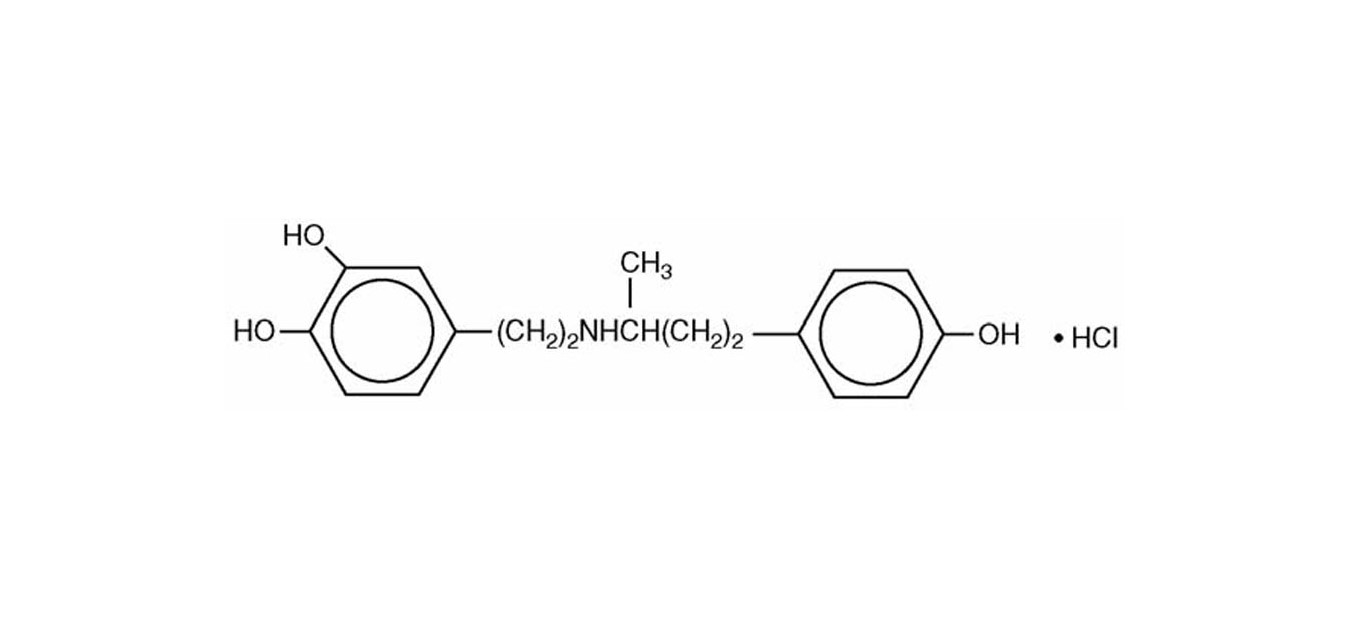
Molecular Weight: 337.85
Molecular Formula: C18H23NO3 • HCl
-
CLINICAL PHARMACOLOGY
Dobutamine hydrochloride is a direct-acting inotropic agent whose primary activity results from stimulation of the β receptors of the heart while producing comparatively mild chronotropic, hypertensive, arrhythmogenic, and vasodilative effects. It does not cause the release of endogenous norepinephrine, as does dopamine. In animal studies, dobutamine hydrochloride produces less increase in heart rate and less decrease in peripheral vascular resistance for a given inotropic effect than does isoproterenol.
In patients with depressed cardiac function, both dobutamine hydrochloride and isoproterenol increase the cardiac output to a similar degree. In the case of dobutamine hydrochloride, this increase is usually not accompanied by marked increases in heart rate (although tachycardia is occasionally observed), and the cardiac stroke volume is usually increased. In contrast, isoproterenol increases the cardiac index primarily by increasing the heart rate while stroke volume changes little or declines.
Facilitation of atrioventricular conduction has been observed in human electrophysiologic studies and in patients with atrial fibrillation.
Systemic vascular resistance is usually decreased with administration of dobutamine hydrochloride. Occasionally, minimum vasoconstriction has been observed.
Most clinical experience with dobutamine hydrochloride is short-term − not more than several hours in duration. In the limited number of patients who were studied for 24, 48, and 72 hours, a persistent increase in cardiac output occurred in some, whereas output returned toward baseline values in others.
The onset of action of dobutamine is within 1 to 2 minutes; however, as much as 10 minutes may be required to obtain the peak effect of a particular infusion rate.
The plasma half-life of dobutamine hydrochloride in humans is 2 minutes. The principal routes of metabolism are methylation of the catechol and conjugation. In human urine, the major excretion products are the conjugates of dobutamine and 3-O-methyl dobutamine. The 3-O-methyl derivative of dobutamine is inactive.
Alteration of synaptic concentrations of catecholamines with either reserpine or tricyclic antidepressants does not alter the actions of dobutamine in animals, which indicates that the actions of dobutamine hydrochloride are not dependent on presynaptic mechanisms.
-
INDICATIONS & USAGE
Dobutamine Injection, USP is indicated when parenteral therapy is necessary for inotropic support in the short-term treatment of adults with cardiac decompensation due to depressed contractility resulting either from organic heart disease or from cardiac surgical procedures.
In patients who have atrial fibrillation with rapid ventricular response, a digitalis preparation should be used prior to institution of therapy with dobutamine hydrochloride.
- CONTRAINDICATIONS
-
WARNINGS
Increase in Heart Rate or Blood Pressure
Dobutamine hydrochloride may cause a marked increase in heart rate or blood pressure, especially systolic pressure. Approximately 10% of patients in clinical studies have had rate increases of 30 beats/minute or more, and about 7.5% have had a 50 mm Hg or greater increase in systolic pressure. Usually, reduction of dosage promptly reverses these effects. Because dobutamine hydrochloride facilitates atrioventricular conduction, patients with atrial fibrillation are at risk of developing rapid ventricular response. Patients with pre-existing hypertension appear to face an increased risk of developing an exaggerated pressor response.
Ectopic Activity
Dobutamine hydrochloride may precipitate or exacerbate ventricular ectopic activity, but it rarely has caused ventricular tachycardia.
Hypersensitivity
Reactions suggestive of hypersensitivity associated with administration of Dobutamine Injection, USP, including skin rash, fever, eosinophilia, and bronchospasm, have been reported occasionally.
Dobutamine Injection, USP contains sodium metabisulfite, a sulfite that may cause allergic-type reactions, including anaphylactic symptoms and life-threatening or less severe asthmatic episodes, in certain susceptible people. The overall prevalence of sulfite sensitivity in the general population is unknown and probably low. Sulfite sensitivity is seen more frequently in asthmatic than in nonasthmatic people.
-
PRECAUTIONS
General
During the administration of Dobutamine Injection, USP, as with any adrenergic agent, ECG and blood pressure should be continuously monitored. In addition, pulmonary wedge pressure and cardiac output should be monitored whenever possible to aid in the safe and effective infusion of dobutamine hydrochloride.
Hypovolemia should be corrected with suitable volume expanders before treatment with dobutamine hydrochloride is instituted.
No improvement may be observed in the presence of marked mechanical obstruction, such as severe valvular aortic stenosis.
Usage Following Acute Myocardial Infarction − Clinical experience with dobutamine hydrochloride following myocardial infarction has been insufficient to establish the safety of the drug for this use. There is concern that any agent that increases contractile force and heart rate may increase the size of an infarction by intensifying ischemia, but it is not known whether dobutamine hydrochloride does so.
Laboratory Tests − Dobutamine, like other β2-agonists, can produce a mild reduction in serum potassium concentration, rarely to hypokalemic levels. Accordingly, consideration should be given to monitoring serum potassium.
Drug Interactions − Animal studies indicate that dobutamine may be ineffective if the patient has recently received a β-blocking drug. In such a case, the peripheral vascular resistance may increase.
Preliminary studies indicate that the concomitant use of dobutamine and nitroprusside results in a higher cardiac output and, usually, a lower pulmonary wedge pressure than when either drug is used alone.
There was no evidence of drug interactions in clinical studies in which dobutamine was administered concurrently with other drugs, including digitalis preparations, furosemide, spironolactone, lidocaine, nitroglycerin, isosorbide dinitrate, morphine, atropine, heparin, protamine, potassium chloride, folic acid, and acetaminophen.
Carcinogenesis, Mutagenesis, Impairment of Fertility − Studies to evaluate the carcinogenic or mutagenic potential of dobutamine hydrochloride, or its potential to affect fertility, have not been conducted.
Pregnancy – Teratogenic Effects − Reproduction studies performed in rats at doses up to the normal human dose (10 mcg/kg/min for 24 h, total daily dose of 14.4 mg/kg), and in rabbits at doses up to twice the normal human dose, have revealed no evidence of harm to the fetus due to dobutamine hydrochloride. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Labor and Delivery − The effect of dobutamine hydrochloride on labor and delivery is unknown.
Nursing Mothers − It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk, caution should be exercised when dobutamine hydrochloride is administered to a nursing woman. If a mother requires dobutamine hydrochloride treatment, breastfeeding should be discontinued for the duration of treatment.
Pediatric Use − The safety and effectiveness of Dobutamine Injection, USP for use in pediatric patients have not been studied.
-
ADVERSE REACTIONS
Increased Heart Rate, Blood Pressure, and Ventricular Ectopic Activity − A 10 to 20 mm increase in systolic blood pressure and an increase in heart rate of 5 to 15 beats/minute have been noted in most patients (see WARNINGS regarding exaggerated chronotropic and pressor effects). Approximately 5% of patients have had increased premature ventricular beats during infusions. These effects are dose related.
Hypotension − Precipitous decreases in blood pressure have occasionally been described in association with dobutamine therapy. Decreasing the dose or discontinuing the infusion typically results in rapid return of blood pressure to baseline values. In rare cases, however, intervention may be required and reversibility may not be immediate.
Reactions at Sites of Intravenous Infusion − Phlebitis has occasionally been reported. Local inflammatory changes have been described following inadvertent infiltration. Isolated cases of cutaneous necrosis (destruction of skin tissue) have been reported.
Miscellaneous Uncommon Effects − The following adverse effects have been reported in 1% to 3% of patients: nausea, headache, anginal pain, nonspecific chest pain, palpitations, and shortness of breath. Isolated cases of thrombocytopenia have been reported.
Administration of dobutamine hydrochloride, like other catecholamines, can produce a mild reduction in serum potassium concentration, rarely to hypokalemic levels (see PRECAUTIONS).
Longer-Term Safety − Infusions of up to 72 hours have revealed no adverse effects other than those seen with shorter infusions.
-
OVERDOSAGE
Overdoses of dobutamine have been reported rarely. The following is provided to serve as a guide if such an overdose is encountered.
Signs and Symptoms − Toxicity from dobutamine is usually due to excessive cardiac β-receptor stimulation. The duration of action of dobutamine is generally short (T1/2 = 2 minutes) because it is rapidly metabolized by catechol-O-methyltransferase. The symptoms of toxicity may include anorexia, nausea, vomiting, tremor, anxiety, palpitations, headache, shortness of breath, and anginal and nonspecific chest pain. The positive inotropic and chronotropic effects of dobutamine on the myocardium may cause hypertension, tachyarrhythmias, myocardial ischemia, and ventricular fibrillation. Hypotension may result from vasodilation.
Treatment − To obtain up-to-date information about the treatment of overdose, a good resource is your certified Regional Poison Control Center. Telephone numbers of certified poison control centers are listed in the Physicians' Desk Reference (PDR). In managing overdosage, consider the possibility of multiple drug overdoses, interaction among drugs, and unusual drug kinetics in your patient.
The initial actions to be taken in a dobutamine overdose are discontinuing administration, establishing an airway, and ensuring oxygenation and ventilation. Resuscitative measures should be initiated promptly. Severe ventricular tachyarrhythmias may be successfully treated with propranolol or lidocaine. Hypertension usually responds to a reduction in dose or discontinuation of therapy.
Protect the patient's airway and support ventilation and perfusion. If needed, meticulously monitor and maintain, within acceptable limits, the patient's vital signs, blood gases, serum electrolytes, etc. If the product is ingested, unpredictable absorption may occur from the mouth and the gastrointestinal tract. Absorption of drugs from the gastrointestinal tract may be decreased by giving activated charcoal, which, in many cases, is more effective than emesis or lavage; consider charcoal instead of or in addition to gastric emptying. Repeated doses of charcoal over time may hasten elimination of some drugs that have been absorbed. Safeguard the patient's airway when employing gastric emptying or charcoal.
Forced diuresis, peritoneal dialysis, hemodialysis, or charcoal hemo-perfusion have not been established as beneficial for an overdose of dobutamine.
-
DOSAGE & ADMINISTRATION
Note − Do not add Dobutamine Injection, USP to 5% Sodium Bicarbonate Injection or to any other strongly alkaline solution. Because of potential physical incompatibilities, it is recommended that dobutamine hydrochloride not be mixed with other drugs in the same solution. Dobutamine hydrochloride should not be used in conjunction with other agents or diluents containing both sodium bisulfite and ethanol.
Preparation and Stability − At the time of administration, Dobutamine Injection, USP must be further diluted in an intravenous container to at least a 50 mL solution using one of the following intravenous solutions as a diluent: 5% Dextrose Injection, USP; 5% Dextrose and 0.45% Sodium Chloride Injection, USP; 5% Dextrose and 0.9% Sodium Chloride Injection, USP; 10% Dextrose Injection, USP; Isolyte® M with 5% Dextrose Injection; Lactated Ringer's Injection; 5% Dextrose in Lactated Ringer's Injection; Normosol®-M in D5-W; 20% Osmitrol® in Water for Injection; 0.9% Sodium Chloride Injection, USP; or Sodium Lactate Injection, USP. Intravenous solutions should be used within 24 hours.
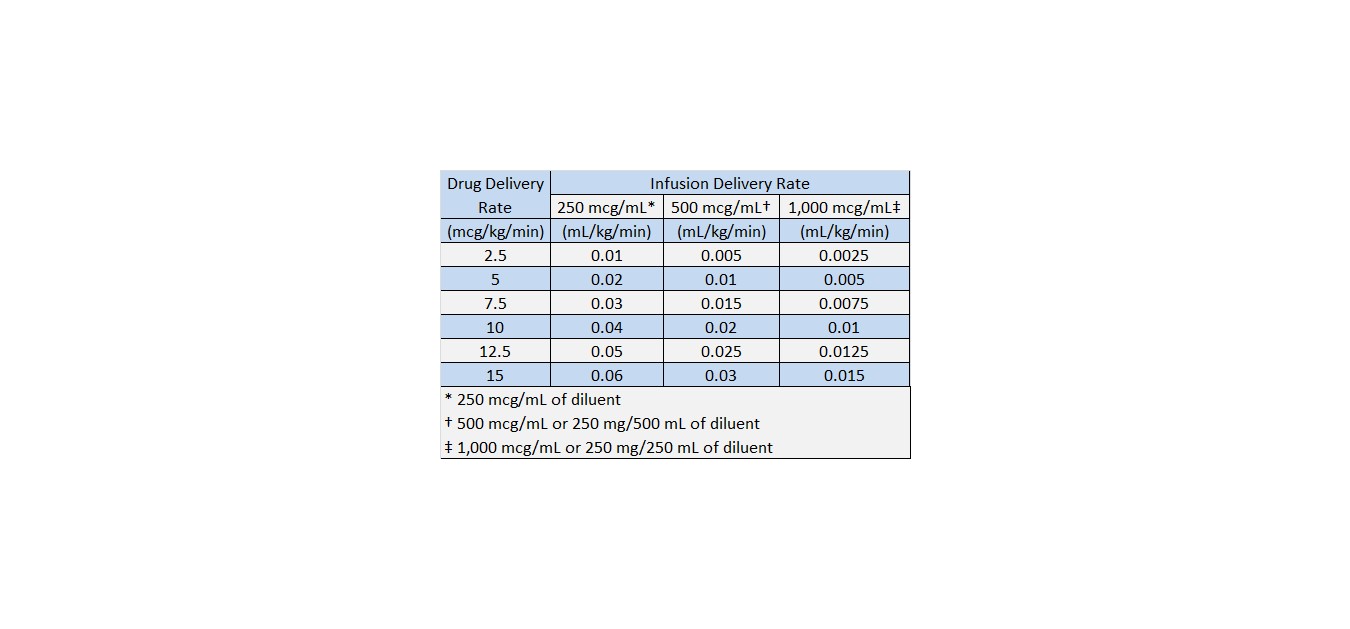
Recommended Dosage − The rate of infusion needed to increase cardiac output usually ranged from 2.5 to 15 mcg/kg/min (see Table 1). On rare occasions, infusion rates up to 40 mcg/kg/min have been required to obtain the desired effect.
Table 1 Dobutamine Infusion Rate (mL/kg/min) for Concentrations of 250, 500, and 1,000 mcg/mL
Rates of infusion in mL/h for Dobutamine concentrations of 500 mcg/mL, 1,000 mcg/mL, and 2,000 mcg/mL are given in Table 2.
Table 2
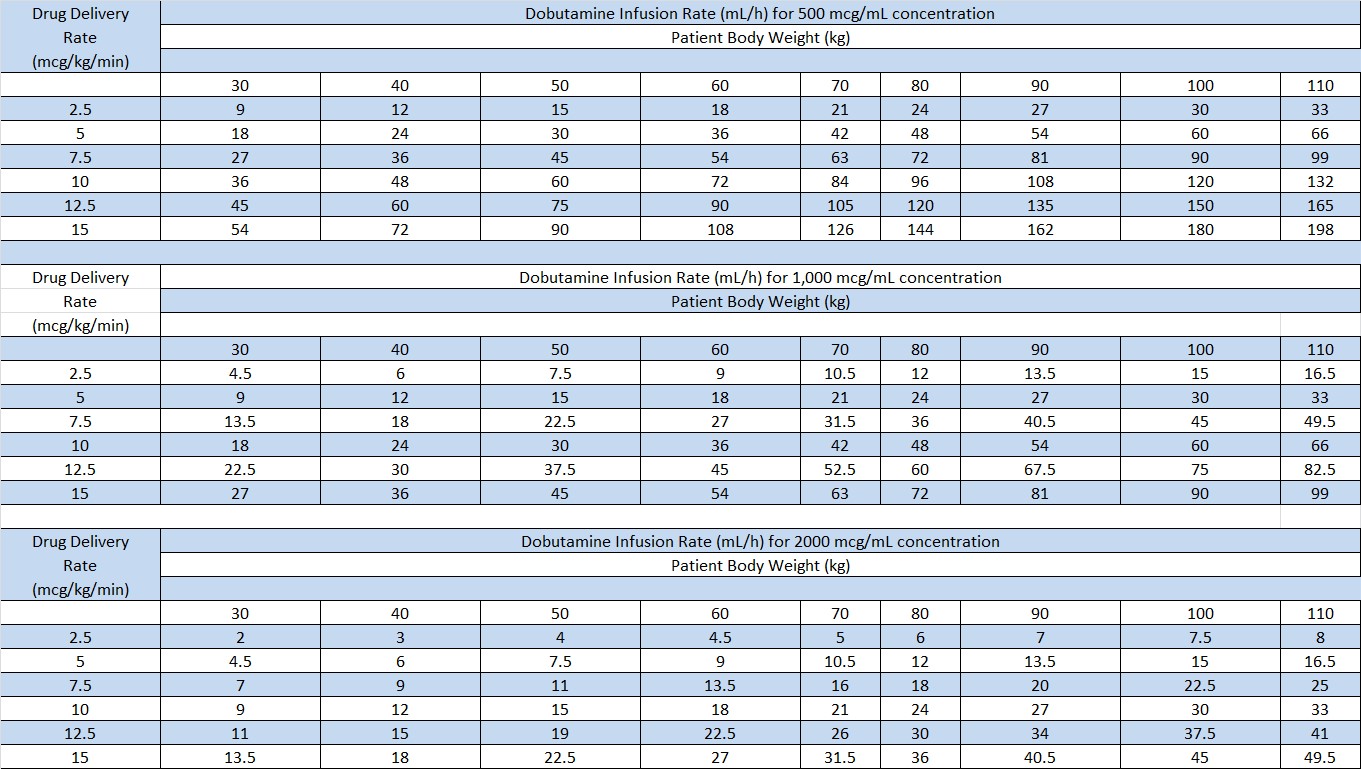
The rate of administration and the duration of therapy should be adjusted according to the patient's response as determined by heart rate, presence of ectopic activity, blood pressure, urine flow, and, whenever possible, measurement of central venous or pulmonary wedge pressure and cardiac output.
Concentrations of up to 5,000 mcg/mL have been administered to humans (250 mg/50 mL). The final volume administered should be determined by the fluid requirements of the patient.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
-
HOW SUPPLIED/STORAGE & HANDLING
DOBUTAMINE INJECTION, USP is supplied in the following dosage forms.
NDC 51662-1337-1
DOBUTAMINE INJECTION, USP 250mg PER 20mL VIALNDC 51662-1337-2 DOBUTAMINE INJECTION, USP 250mg PER 20mL VIAL in a Pouch
NDC 51662-1337-3 Case of 10, DOBUTAMINE INJECTION, USP 250mg PER 20mL VIAL in a Pouch
HF Acquisition Co LLC, DBA HealthFirst
Mukilteo, WA 98275Also supplied in the following manufacture supplied dosage forms
Dobutamine Injection, USP is supplied in 20 mL single-dose glass vials containing 250 mg dobutamine, as the hydrochloride as follows:
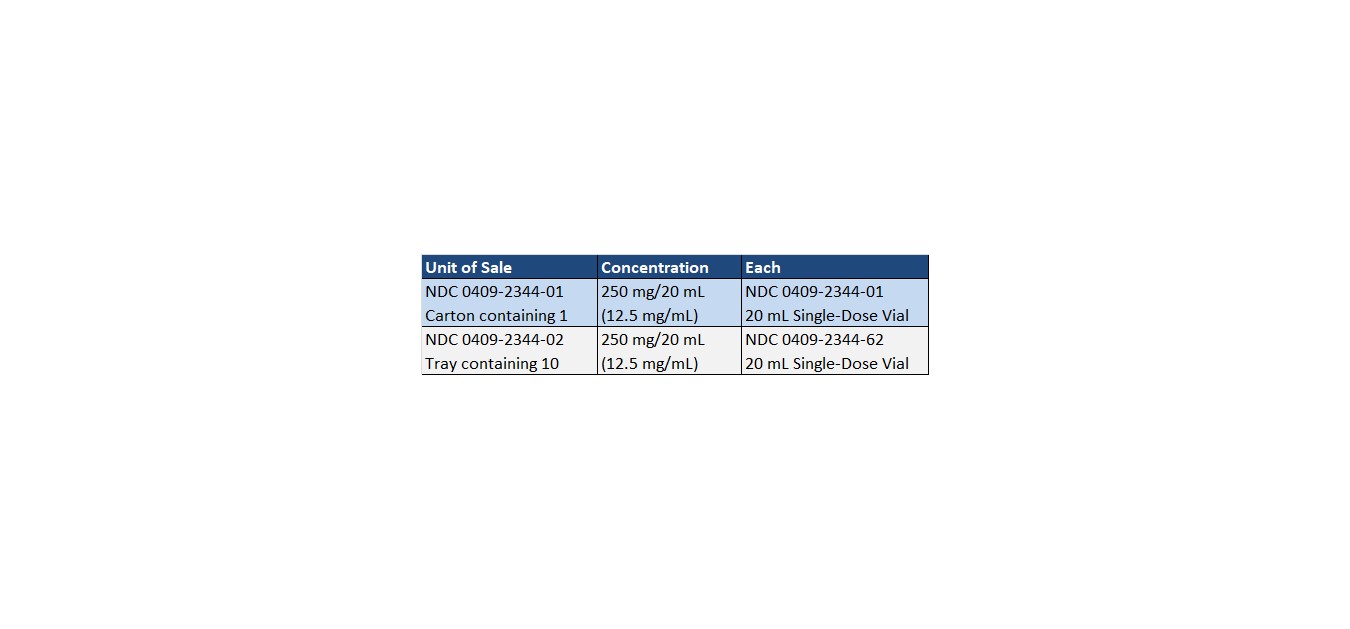
Store at 20 to 25°C (68° to 77°F). [See USP Controlled Room Temperature.]
- SPL UNCLASSIFIED
- PRINCIPAL DISPLAY PANEL, VIAL LABEL
- PRINCIPAL DISPLAY PANEL, SERIALIZED LABEL
- PRINCIPAL DISPLAY PANEL 51662-1337-3 CASE
- PRINCIPAL DISPLAY PANEL 51662-1337-2 POUCH
-
INGREDIENTS AND APPEARANCE
DOBUTAMINE
dobutamine injection, solution, concentrateProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:51662-1337(NDC:0409-2344) Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength DOBUTAMINE HYDROCHLORIDE (UNII: 0WR771DJXV) (DOBUTAMINE - UNII:3S12J47372) DOBUTAMINE 12.5 mg in 1 mL Inactive Ingredients Ingredient Name Strength HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM METABISULFITE (UNII: 4VON5FNS3C) 0.2 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:51662-1337-1 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product 11/26/2018 2 NDC:51662-1337-3 10 in 1 CASE 06/12/2022 2 NDC:51662-1337-2 1 in 1 POUCH 2 20 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA074086 11/26/2018 Labeler - HF Acquisition Co LLC, DBA HealthFirst (045657305) Registrant - HF Acquisition Co LLC, DBA HealthFirst (045657305) Establishment Name Address ID/FEI Business Operations HF Acquisition Co LLC, DBA HealthFirst 045657305 relabel(51662-1337)



 RFID LABEL
RFID LABEL

