Label: TIGLUTIK- riluzole liquid

- NDC Code(s): 70726-0303-1, 70726-0303-2
- Packager: EDW PHARMA, INC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated February 8, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use TIGLUTIK® safely and effectively. See full prescribing information for TIGLUTIK.
TIGLUTIK (riluzole) oral suspension
Initial U.S. Approval: 1995INDICATIONS AND USAGE
TIGLUTIK is indicated for the treatment of amyotrophic lateral sclerosis (ALS) ( 1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Oral suspension: 50 mg/10 mL (5 mg/mL) in 300 mL multiple-dose bottle ( 3)
CONTRAINDICATIONS
Patients with a history of severe hypersensitivity reactions to riluzole or to any of its components ( 4)
WARNINGS AND PRECAUTIONS
- Hepatic injury: Use of TIGLUTIK is not recommended in patients with baseline elevations of serum aminotransferases greater than 5 times the upper limit of normal; discontinue TIGLUTIK if there is evidence of liver dysfunction ( 5.1)
- Neutropenia: Advise patients to report any febrile illness ( 5.2)
- Interstitial lung disease: Discontinue TIGLUTIK if interstitial lung disease develops ( 5.3)
ADVERSE REACTIONS
Most common adverse reactions (incidence greater than or equal to 5% and greater than placebo) were oral hypoesthesia, asthenia, nausea, decreased lung function, hypertension, and abdominal pain ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact ITF Pharma Inc. at 1-800-664-1490 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Strong to moderate CYP1A2 inhibitors: Co-administration may increase TIGLUTIK-associated adverse reactions ( 7.1)
- Strong to moderate CYP1A2 inducers: Co-administration may result in decreased efficacy ( 7.2)
- Hepatotoxic drugs: TIGLUTIK-treated patients that take other hepatotoxic drugs may be at increased risk for hepatotoxicity ( 7.3)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2020
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
2.2 Monitoring to Assess Safety
2.3 Important Administration Instructions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Injury
5.2 Neutropenia
5.3 Interstitial Lung Disease
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Agents that may Increase Riluzole Blood Concentrations
7.2 Agents that may Decrease Riluzole Plasma Concentrations
7.3 Hepatotoxic Drugs
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Japanese Patients
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Dosage Information
The recommended dosage for TIGLUTIK is 50 mg (10 mL) taken orally or via Percutaneous Endoscopic Gastrostomy tubes (PEG-tubes) twice daily, every 12 hours. TIGLUTIK should be taken at least 1 hour before or 2 hours after a meal [see Clinical Pharmacology (12.3)].
2.2 Monitoring to Assess Safety
Measure serum aminotransferases before and during treatment with TIGLUTIK [see Warnings and Precautions (5.1)].
2.3 Important Administration Instructions
Gently shake the TIGLUTIK bottle for at least 30 seconds before administration.Gently shake the TIGLUTIK bottle for at least 30 seconds before administration.
TIGLUTIK can be administered by mouth or via percutaneous endoscopic gastrostomy tubes (PEG-tubes). Both silicone and polyurethane PEG tubes can be used.
See the Instructions for Use for further administration details.See the Instructions for Use for further administration details.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
TIGLUTIK is contraindicated in patients with a history of severe hypersensitivity reactions to riluzole or to any of its components (anaphylaxis has occurred) [see Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Hepatic Injury
TIGLUTIK can cause liver injury. Cases of drug-induced liver injury, some of which were fatal, have been reported in patients taking riluzole. Asymptomatic elevations of hepatic transaminases have also been reported, and in some patients have recurred upon re-challenge with riluzole.
In clinical studies, the incidence of elevations in hepatic transaminases was greater in riluzole-treated patients than placebo-treated patients. The incidence of elevations of ALT above 5 times the upper limit of normal (ULN) was 2% in riluzole-treated patients. Maximum increases in ALT occurred within 3 months after starting riluzole. About 50% and 8% of riluzole-treated patients in pooled controlled efficacy studies (Studies 1 and 2) had at least one elevated ALT level above ULN and above 3 times ULN, respectively [see Clinical Studies (14)].
Monitor patients for signs and symptoms of hepatic injury, every month for the first 3 months of treatment, and periodically thereafter. The use of TIGLUTIK is not recommended if patients develop hepatic transaminases levels greater than 5 times the ULN. Discontinue TIGLUTIK if there is evidence of liver dysfunction (e.g., elevated bilirubin). Concomitant use with other hepatotoxic drugs may increase the risk for hepatotoxicity [see Drug Interactions (7.3)] .
-
6 ADVERSE REACTIONS
The following adverse reactions are described below and elsewhere in the labeling:
- Hepatic Injury [see Warnings and Precautions (5.1)]
- Neutropenia [see Warnings and Precautions (5.2)]
- Interstitial Lung Disease [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Controlled Clinical Trials of Riluzole Tablets
In the placebo-controlled clinical trials in patients with ALS (Study 1 and 2), a total of 313 patients received riluzole 50 mg twice daily [see Clinical Studies (14)]. The most common adverse reactions in riluzole-treated patients (in at least 5% of patients and more frequently than on placebo) were asthenia, nausea, decreased lung function, hypertension, and abdominal pain. The most common adverse reactions leading to discontinuation in the riluzole group were nausea, abdominal pain, constipation, and elevated ALT.
There was no difference in the rate of adverse reactions leading to discontinuation between females and males. However, the incidence of dizziness was higher in females (11%) than in males (4%). The adverse reaction profile was similar in older and younger patients. There are insufficient data to assess racial differences in the adverse reaction profile.
Table 1 lists adverse reactions that occurred in at least 2% of riluzole-treated patients (50 mg twice daily) in pooled Study 1 and 2, and at a higher rate than on placebo.
Table 1. Adverse Reactions in Pooled Placebo-Controlled Trials (Studies 1 and 2) in Patients with ALS Adverse Reaction Riluzole Tablets
50 mg twice daily
(N=313)
%Placebo
(N=320)
%Asthenia 19 12 Nausea 16 11 Decreased lung function 10 9 Hypertension 5 4 Abdominal pain 5 4 Vomiting 4 2 Arthralgia 4 3 Dizziness 4 3 Dry mouth 4 3 Insomnia 4 3 Pruritus 4 3 Tachycardia 3 1 Flatulence 3 2 Increased cough 3 2 Peripheral edema 3 2 Urinary Tract Infection 3 2 Circumoral paresthesia 2 0 Somnolence 2 1 Vertigo 2 1 Eczema 2 1 6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of riluzole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
- Acute hepatitis and icteric toxic hepatitis [see Warnings and Precautions (5.1)]
- Renal tubular impairment
- Pancreatitis
-
7 DRUG INTERACTIONS
7.1 Agents that may Increase Riluzole Blood Concentrations
CYP1A2 Inhibitors
Co-administration of riluzole (a CYP1A substrate) with CYP1A2 inhibitors was not evaluated in a clinical trial; however, in vitro findings suggest an increase in riluzole exposure is likely. The concomitant use of strong or moderate CYP1A2 inhibitors (e.g., ciprofloxacin, enoxacin, fluvoxamine, methoxsalen, mexiletine, oral contraceptives, thiabendazole, vemurafenib, zileuton) with TIGLUTIK may increase the risk of TIGLUTIK - associated adverse reactions [see Clinical Pharmacology (12.3)].
7.2 Agents that may Decrease Riluzole Plasma Concentrations
CYP1A2 Inducers
Co-administration of riluzole (a CYP1A substrate) with CYP1A2 inducers was not evaluated in a clinical trial; however, in vitro findings suggest a decrease in riluzole exposure is likely. Lower exposures may result in decreased efficacy [see Clinical Pharmacology (12.3)] .
7.3 Hepatotoxic Drugs
Clinical trials in ALS patients excluded patients on concomitant medications which were potentially hepatotoxic (e.g., allopurinol, methyldopa, sulfasalazine). TIGLUTIK-treated patients who take other hepatotoxic drugs may be at an increased risk for hepatotoxicity [see Warnings and Precautions (5.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no studies of riluzole in pregnant women, and case reports have been inadequate to inform the drug-associated risk. The background risk for major birth defects and miscarriage in patients with amyotrophic lateral sclerosis is unknown. In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
In studies in which riluzole was administered orally to pregnant animals, developmental toxicity (decreased embryofetal/offspring viability, growth, and functional development) was observed at clinically relevant doses [see Data] . Based on these results, women should be advised of a possible risk to the fetus associated with use of TIGLUTIK during pregnancy.
Data
Animal Data
Oral administration of riluzole (3, 9, or 27 mg/kg/day) to pregnant rats during the period of organogenesis resulted in decreases in fetal growth (body weight and length) at the high dose. The mid dose, a no-effect dose for embryofetal developmental toxicity, is approximately equal to the recommended human daily dose (RHDD, 100 mg) on a mg/m2 basis. When riluzole was administered orally (3, 10, or 60 mg/kg/day) to pregnant rabbits during the period of organogenesis, embryofetal mortality was increased at the high dose and fetal body weight was decreased and morphological variations increased at all but the lowest dose tested. The no-effect dose (3 mg/kg/day) for embryofetal developmental toxicity is less than the RHDD on a mg/m2 basis. Maternal toxicity was observed at the highest dose tested in rat and rabbit.
When riluzole was orally administered (3, 8, or 15 mg/kg/day) to male and female rats prior to and during mating and to female rats throughout gestation and lactation, increased embryofetal mortality and decreased postnatal offspring viability, growth, and functional development were observed at the high dose. The mid dose, a no-effect dose for pre- and postnatal developmental toxicity, is approximately equal to the RHDD on a mg/m2 basis.
8.2 Lactation
Risk Summary
There are no data on the presence of riluzole in human milk, the effects on the breastfed infant, or the effects on milk production. Riluzole or its metabolites have been detected in milk of lactating rat. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for TIGLUTIK and any potential adverse effects on the breastfed infant from TIGLUTIK or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
In rats, oral administration of riluzole resulted in decreased fertility indices and increases in embryolethality [see Nonclinical Toxicology (13.1)].
8.5 Geriatric Use
In clinical studies of riluzole, 30% of patients were 65 years and over. No overall differences in safety or effectiveness were observed between these patients and younger patients, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
Patients with mild [Child-Pugh's (CP) score A] or moderate (CP score B) hepatic impairment had increases in AUC, compared to patients with normal hepatic function. Thus, patients with mild or moderate hepatic impairment may be at increased risk of adverse reactions. The impact of severe hepatic impairment on riluzole exposure is unknown.
Use of TIGLUTIK is not recommended in patients with baseline elevations of serum aminotransferases greater than 5 times upper limit of normal or evidence of liver dysfunction (e.g., elevated bilirubin) [see Clinical Pharmacology (12.3)].
8.7 Japanese Patients
Japanese patients are more likely to have higher riluzole concentrations. Consequently, the risk of adverse reactions may be greater in Japanese patients [see Clinical Pharmacology (12.3)].
-
10 OVERDOSAGE
Reported symptoms of overdose following ingestion of riluzole ranging from 1.5 to 3 grams (30 to 60 times the recommended dose) included acute toxic encephalopathy, coma, drowsiness, memory loss, and methemoglobinemia.
No specific antidote for the treatment of TIGLUTIK overdose is available. For current information on the management of poisoning or overdosage, contact a certified poison control center.
-
11 DESCRIPTION
Riluzole is a member of the benzothiazole class. The chemical designation for riluzole is 2-amino-6-(trifluoromethoxy)benzothiazole. Its molecular formula is C 8H 5F 3N 2OS, and its molecular weight is 234.2. The chemical structure is:
Riluzole is a white to slightly yellow powder that is very soluble in dimethylformamide, dimethylsulfoxide, and methanol; freely soluble in dichloromethane; sparingly soluble in 0.1 N HCl; and very slightly soluble in water and in 0.1 N NaOH.
TIGLUTIK (50 mg/10mL) oral suspension is a slightly brown, opaque, homogeneous suspension containing 50 mg of riluzole per 10 mL of suspension.
TIGLUTIK also contains the following inactive ingredients: magnesium aluminum silicate, noncrystallizing sorbitol solution, polyoxyl 20 cetostearyl ether, purified water, saccharin sodium, simethicone emulsion, sodium lauryl sulfate, and xanthan gum.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
The mechanism by which riluzole exerts its therapeutic effects in patients with ALS is unknown.
12.3 Pharmacokinetics
A pharmacokinetic study in healthy adult subjects who received riluzole suspension 50 mg under fasting conditions demonstrated similar pharmacokinetics following intragastric administration via feeding tubes and oral administration.
Table 2 displays the pharmacokinetic parameters of riluzole.
Table 2. Pharmacokinetics of Riluzole * Absorption Bioavailability (oral) Approximately 60% Dose Proportionality Linear over a dose range of 25 mg to 100 mg every 12 hours (1/2 to 2 times the recommended dosage) Food effect † AUC ↓ 9% and Cmax ↓ 55% (high fat meal) Time to peak plasma concentration (median) † 0.8 hours Distribution Plasma Protein Binding 96% (Mainly to albumin and lipoproteins) Elimination Elimination half-life - 12 hours (CV=35%)
- The high individual variability in the clearance of riluzole is potentially attributable to variability of CYP1A2. The clinical implications are not known.
Accumulation Approximately 2-fold Metabolism Fraction metabolized (% dose) At least 88% Primary metabolic pathway(s) [in vitro] - Oxidation: CYP1A2
- Direct and sequential glucoronidation: UGT-HP4
Active Metabolites Some metabolites appear pharmacologically active in vitro, but the clinical implications are not known. Excretion Primary elimination pathways (% dose) - Feces: 5%
- Urine: 90% (2% unchanged riluzole)
Specific Populations
Hepatic Impairment
Compared with healthy volunteers, the AUC of riluzole was approximately 1.7-fold greater in patients with mild chronic hepatic impairment (CP score A), and approximately 3-fold greater in patients with moderate chronic hepatic impairment (CP score B). The pharmacokinetics of riluzole have not been studied in patients with severe hepatic impairment (CP score C) [see Use in Specific Populations (8.6)].
Race
The clearance of riluzole was 50% lower in male Japanese subjects than in Caucasian subjects, after normalizing for body weight [see Use in Specific Populations (8.7)].
Smokers
The clearance of riluzole in tobacco smokers was 20% greater than in nonsmokers. Geriatric Patients and Patients with Moderate to Severe Renal ImpairmentAge 65 years or older and moderate to severe renal impairment do not have a meaningful effect on the pharmacokinetics of riluzole. The pharmacokinetics of riluzole in patients undergoing hemodialysis are unknown.
Drug Interaction Studies
Drugs Highly Bound to Plasma Proteins
Riluzole and warfarin are highly bound to plasma proteins. In vitro, riluzole did not show any displacement of warfarin from plasma proteins. Riluzole binding to plasma proteins was unaffected by warfarin, digoxin, imipramine and quinine at high therapeutic concentrations in vitro.
- 12 hours (CV=35%)
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Riluzole was not carcinogenic in mice or rats when administered for 2 years at daily oral doses up to 20 and 10 mg/kg/day, respectively, which are approximately equal to the recommended human daily dose (RHDD, 100 mg) on a mg/m2 basis.
Mutagenesis
Riluzole was negative in in vitro(bacterial reverse mutation (Ames), mouse lymphoma tk, chromosomal aberration assay in human lymphocytes), and in in vivo(rat cytogenetic and mouse micronucleus) assays.
N-hydroxyriluzole, the major active metabolite of riluzole, was positive for clastogenicity in the in vitro mouse lymphoma tk assay and in the in vitro micronucleus assay using the same mouse lymphoma cell line. N-hydroxyriluzole was negative in the HPRT gene mutation assay, the Ames assay (with and without rat or hamster S9), the in vitro chromosomal aberration assay in human lymphocytes, and the in vivo mouse micronucleus assay.
Impairment of Fertility
When riluzole (3, 8, or 15 mg/kg) was administered orally to male and female rats prior to and during mating and continuing in females throughout gestation and lactation, fertility indices were decreased and embryolethality was increased at the high dose. This dose was also associated with maternal toxicity. The mid dose, a no-effect dose for effects on fertility and early embryonic development, is approximately equal to the RHDD on a mg/m2 basis.
-
14 CLINICAL STUDIES
The efficacy of TIGLUTIK is based upon bioavailability studies comparing oral riluzole tablets to TIGLUTIK oral suspension [see Clinical Pharmacology (12.3)].
The efficacy of riluzole was demonstrated in two studies (Study 1 and 2) that evaluated 50 mg riluzole oral tablets twice daily in patients with amyotrophic lateral sclerosis (ALS). Both studies included patients with either familial or sporadic ALS, disease duration of less than 5 years, and baseline forced vital capacity greater than or equal to 60% of normal.
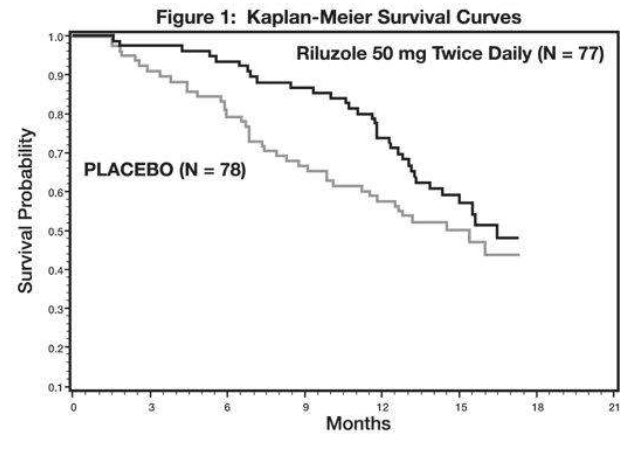
Study 1 was a randomized, double-blind, placebo-controlled clinical study that enrolled 155 patients with ALS. Patients were randomized to receive riluzole 50 mg twice daily (n=77) or placebo (n=78) and were followed for at least 13 months (up to a maximum duration of 18 months). The clinical outcome measure was time to tracheostomy or death.
The time to tracheostomy or death was longer for patients receiving riluzole compared to placebo. There was an early increase in survival in patients receiving riluzole compared to placebo. Figure 1 displays the survival curves for time to death or tracheostomy. The vertical axis represents the proportion of individuals alive without tracheostomy at various times following treatment initiation (horizontal axis). Although these survival curves were not statistically significantly different when evaluated by the analysis specified in the study protocol (Logrank test p=0.12), the difference was found to be significant by another appropriate analysis (Wilcoxon test p=0.05). As seen in Figure 1, the study showed an early increase in survival in patients given riluzole. Among the patients in whom the endpoint of tracheostomy or death was reached during the study, the difference in median survival between the riluzole 50 mg twice daily and placebo groups was approximately 90 days.
Figure 1. Time to Tracheostomy or Death in ALS Patients in Study 1 (Kaplan-Meier Curves)
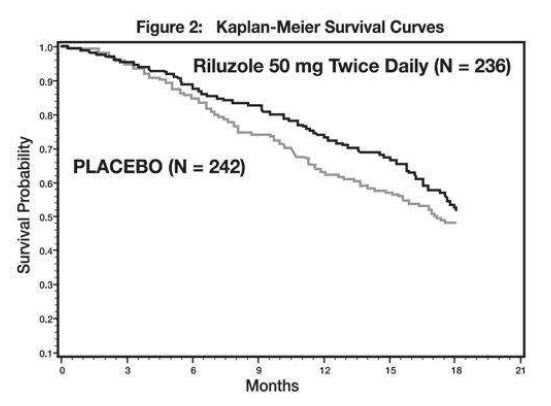
Study 2 was a randomized, double-blind, placebo-controlled clinical study that enrolled 959 patients with ALS. Patients were randomized to riluzole 50 mg twice daily (n=236) or placebo (n=242) and were followed for at least 12 months (up to a maximum duration of 18 months). The clinical outcome measure was time to tracheostomy or death.
The time to tracheostomy or death was longer for patients receiving riluzole compared to placebo. Figure 2 displays the survival curves for time to death or tracheostomy for patients randomized to either riluzole 100 mg per day or placebo. Although these survival curves were not statistically significantly different when evaluated by the analysis specified in the study protocol (Logrank test p=0.076), the difference was found to be significant by another appropriate analysis (Wilcoxon test p=0.05). Not displayed in Figure 2 are the results of riluzole 50 mg per day (one-half of the recommended daily dose), which could not be statistically distinguished from placebo, or the results of riluzole 200 mg per day (two times the recommended daily dose), which were not distinguishable from the 100 mg per day results. Among the patients in whom the endpoint of tracheostomy or death was reached during the study, the difference in median survival between riluzole and placebo was approximately 60 days.
Although riluzole improved survival in both studies, measures of muscle strength and neurological function did not show a benefit.
Figure 2. Time to Tracheostomy or Death in ALS Patients in Study 2 (Kaplan-Meier Curves)
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
TIGLUTIK (50 mg/10 mL) oral suspension is supplied in amber glass bottles closed with child-resistant tamper evident screw caps. Each bottle contains 300 mL of oral suspension and is intended for multi-dose use, NDC 70726-0303-2.
TIGLUTIK is supplied in a carton, NDC 70726-0303-1, containing:
- Two bottles, each containing 300 mL oral suspension
- Two 10 mL oral syringes
- Two syringe bottle adapters
- Two syringe tip caps
- Prescribing Information, including Instructions for Use
16.2 Storage and Handling
Store at 20-25°C (68-77°F), excursions permitted to 15-30°C (59-86°F) [see USP Controlled Room Temperature], and protect from bright light. Do not freeze. Store upright.
Use within 15 days after initially opening of each bottle. Discard any unused TIGLUTIK remaining after 15 days of first opening of the bottle.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Instructions for Use).
Administration Instructions
Instruct patients to discard any unused TIGLUTIK after 15 days of opening the bottle. If the patient requires administration of TIGLUTIK via PEG tube, refer the patient/caregiver to the Instructions for Use for steps on how to take/give TIGLUTIK.
Hepatic Injury
Advise patients that TIGLUTIK can cause liver injury, which can be fatal. Inform patients of the clinical signs or symptoms suggestive of hepatic dysfunction (e.g., unexplained nausea, vomiting, abdominal pain, fatigue, anorexia, or jaundice and/or dark urine) and to contact a healthcare provider promptly if these signs or symptoms occur [see Warnings and Precautions (5.1)].
Neutropenia
Advise patients that TIGLUTIK can cause neutropenia, and to report to their healthcare provider if they have a fever [see Warnings and Precautions (5.2)].
Interstitial Lung Disease
Advise patients that TIGLUTIK can cause interstitial lung disease, and to report to their healthcare provider if they have respiratory symptoms (e.g., dry cough and difficult or labored breathing) [see Warnings and Precautions (5.3)].
Pregnancy
Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during TIGLUTIK therapy [see Use in Specific Populations (8.1)].
Lactation
Advise patients to notify their healthcare provider if they are breastfeeding or intend to breastfeed during TIGLUTIK therapy [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
-
PRINCIPAL DISPLAY PANEL - 300 mL Bottle Carton
NDC 70726-0303-1
R xonly
Tiglutik ®
riluzole
oral suspension
50 mg/10 mL (5 mg/mL)For Oral Administration
Shake gently before useContents of this package:
- Two bottles, each containing 300 mL
- Two 10 mL oral syringes
- Two syringe bottle adapters
- Two syringe tip caps
- One Prescribing Information, including
Instructions for Use
This product is a liquid suspension and is
supplied with syringes for oral administration.
Before use, please read the enclosed
Prescribing Information.600 mL (two bottles/300 mL each)
Not To Be Dispensed Separately
-
INGREDIENTS AND APPEARANCE
TIGLUTIK
riluzole liquidProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70726-0303 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RILUZOLE (UNII: 7LJ087RS6F) (RILUZOLE - UNII:7LJ087RS6F) RILUZOLE 50 mg in 10 mL Inactive Ingredients Ingredient Name Strength SORBITOL (UNII: 506T60A25R) POLYOXYL 20 CETOSTEARYL ETHER (UNII: YRC528SWUY) MAGNESIUM ALUMINUM SILICATE (UNII: 6M3P64V0NC) WATER (UNII: 059QF0KO0R) SACCHARIN SODIUM (UNII: SB8ZUX40TY) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) DIMETHICONE (UNII: 92RU3N3Y1O) SODIUM LAURYL SULFATE (UNII: 368GB5141J) XANTHAN GUM (UNII: TTV12P4NEE) Product Characteristics Color brown (slightly brown, opaque) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70726-0303-1 2 in 1 CARTON 09/21/2018 1 NDC:70726-0303-2 300 mL in 1 BOTTLE, PLASTIC; Type 1: Convenience Kit of Co-Package Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209080 09/21/2018 Labeler - EDW PHARMA, INC (080260470) Registrant - EDW PHARMA, INC (080260470) Establishment Name Address ID/FEI Business Operations Aphena Pharma Solutions 829739833 manufacture(70726-0303)