Label: MEKTOVI- binimetinib tablet, film coated


- NDC Code(s): 70255-010-02, 70255-010-03
- Packager: Array BioPharma Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated October 18, 2023
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MEKTOVI safely and effectively. See full prescribing information for MEKTOVI.
MEKTOVI® (binimetinib) tablets, for oral use
Initial U.S. Approval: 2018RECENT MAJOR CHANGES
INDICATIONS AND USAGE
MEKTOVI is a kinase inhibitor indicated:
- •
- in combination with encorafenib, for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test. (1.1, 2.1)
- •
- in combination with encorafenib, for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with a BRAF V600E mutation, as detected by an FDA-approved test. (1.2, 2.1)
DOSAGE AND ADMINISTRATION
Melanoma
- •
- Confirm the presence of BRAF V600E or V600K mutation in tumor specimens prior to the initiation of MEKTOVI. (2.1)
- •
- The recommended dose is 45 mg orally twice daily in combination with encorafenib. Take MEKTOVI with or without food. (2.2)
- •
- For patients with moderate or severe hepatic impairment the recommended dose is 30 mg orally twice daily. (2.4, 8.6)
NSCLC
DOSAGE FORMS AND STRENGTHS
Tablets: 15 mg. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- •
- New Primary Malignancies, Cutaneous and Non-cutaneous: Can occur when MEKTOVI is used in combination with encorafenib. Monitor patients for new malignancies prior to initiation of treatment, during treatment, and after discontinuation of treatment. (5.1)
- •
- Cardiomyopathy: Assess left ventricular ejection fraction (LVEF) before initiating treatment, after one month of treatment, then every 2 to 3 months thereafter. The safety of MEKTOVI has not been established in patients with LVEF below 50%. (5.2)
- •
- Venous Thromboembolism: Deep vein thrombosis and pulmonary embolism can occur. (5.3)
- •
- Ocular Toxicities: Serous retinopathy, retinal vein occlusion (RVO) and uveitis have occurred. Perform an ophthalmologic evaluation at regular intervals and for any visual disturbances. (5.4)
- •
- Interstitial Lung Disease (ILD): Assess new or progressive unexplained pulmonary symptoms or findings for possible ILD. (5.5)
- •
- Hepatotoxicity: Monitor liver function tests before and during treatment with MEKTOVI and encorafenib and as clinically indicated. (5.6)
- •
- Rhabdomyolysis: Monitor creatine phosphokinase and creatinine periodically and as clinically indicated. (5.7)
- •
- Hemorrhage: Major hemorrhagic events can occur in patients receiving MEKTOVI and encorafenib. (5.8)
- •
- Embryo-Fetal Toxicity: Can cause fetal harm. Advise females with reproductive potential of potential risk to the fetus and to use effective contraception. (5.9, 8.1, 8.3)
ADVERSE REACTIONS
Melanoma: Most common adverse reactions (≥25%) for MEKTOVI, in combination with encorafenib, are fatigue, nausea, diarrhea, vomiting, and abdominal pain. (6.1)
NSCLC: Most common adverse reactions (≥25%) for MEKTOVI, in combination with encorafenib, are fatigue, nausea, diarrhea, musculoskeletal pain, vomiting, abdominal pain, visual impairment, constipation, dyspnea, rash, and cough. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc.at 1-800-438-1985 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 10/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
1.2 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer (NSCLC)
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
2.2 Recommended Dosage and Administration
2.3 Dosage Modifications for Adverse Reactions
2.4 Dosage Modifications for Moderate or Severe Hepatic Impairment
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 New Primary Malignancies
5.2 Cardiomyopathy
5.3 Venous Thromboembolism
5.4 Ocular Toxicities
5.5 Interstitial Lung Disease
5.6 Hepatotoxicity
5.7 Rhabdomyolysis
5.8 Hemorrhage
5.9 Embryo-Fetal Toxicity
5.10 Risks Associated with Combination Treatment
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
14.2 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
MEKTOVI is indicated, in combination with encorafenib, for the treatment of patients with unresectable or metastatic melanoma with a BRAF V600E or V600K mutation, as detected by an FDA-approved test [see Dosage and Administration (2.1)].
1.2 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer (NSCLC)
MEKTOVI is indicated, in combination with encorafenib, for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) with a BRAF V600E mutation, as detected by an FDA-approved test. [see Dosage and Administration (2.1)].
-
2 DOSAGE AND ADMINISTRATION
2.1 Patient Selection
BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
Confirm the presence of a BRAF V600E or V600K mutation in tumor specimens prior to initiating MEKTOVI [see Clinical Studies (14)]. Information on FDA-approved tests for the detection of BRAF V600E and V600K mutations in melanoma is available at: http://www.fda.gov/CompanionDiagnostics.
BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer (NSCLC)
Confirm the presence of a BRAF V600E mutation in tumor or plasma specimens prior to initiating MEKTOVI [see Clinical Studies (14.2)]. If no mutation is detected in a plasma specimen, test tumor tissue. Information on FDA-approved tests for the detection of BRAF V600E mutations in NSCLC is available at: http://www.fda.gov/CompanionDiagnostics.
2.2 Recommended Dosage and Administration
The recommended dosage of MEKTOVI is 45 mg orally taken twice daily, approximately 12 hours apart, in combination with encorafenib until disease progression or unacceptable toxicity. Refer to the encorafenib prescribing information for recommended encorafenib dosing information.
MEKTOVI may be taken with or without food [see Clinical Pharmacology (12.3)]. Do not take a missed dose of MEKTOVI within 6 hours of the next dose of MEKTOVI.
Do not take an additional dose if vomiting occurs after MEKTOVI administration but continue with the next scheduled dose.
2.3 Dosage Modifications for Adverse Reactions
If encorafenib is permanently discontinued, discontinue MEKTOVI.
Dose reductions for adverse reactions associated with MEKTOVI are presented in Table 1.
Table 1: Recommended Dose Reductions for MEKTOVI for Adverse Reactions Action Recommended Dose First Dose Reduction
30 mg orally twice daily
Subsequent Modification
Permanently discontinue if unable to tolerate MEKTOVI 30 mg orally twice daily
Dosage modifications for adverse reactions associated with MEKTOVI are presented in Table 2.
Table 2: Recommended Dosage Modifications for MEKTOVI for Adverse Reactions Severity of Adverse Reaction* Dose Modification for MEKTOVI - *
- National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE) version 4.03.
- †
- Dose modification of MEKTOVI when administered with encorafenib is not recommended for the following adverse reactions: palmar- plantar erythrodysesthesia syndrome (PPES), non-cutaneous RAS mutation-positive malignancies, and QTc prolongation.
Cardiomyopathy [see Warnings and Precautions (5.2)]
- •
- Asymptomatic, absolute decrease in LVEF of greater than 10% from baseline that is also below lower limit of normal (LLN)
Withhold MEKTOVI for up to 4 weeks, evaluate LVEF every 2 weeks.
Resume MEKTOVI at a reduced dose if the following are present:- •
- LVEF is at or above the lower limit of normal and
- •
- Absolute decrease from baseline is 10% or less and
- •
- Patient is asymptomatic.
If the LVEF does not recover within 4 weeks permanently discontinue MEKTOVI.
- •
- Symptomatic congestive heart failure or absolute decrease in LVEF of greater than 20% from baseline that is also below LLN
Permanently discontinue MEKTOVI.
Venous Thromboembolism [see Warnings and Precautions (5.3)]
- •
- Uncomplicated deep venous thrombosis (DVT) or pulmonary embolism (PE)
Withhold MEKTOVI.
- •
- If improves to Grade 0-1, resume at a reduced dose.
- •
- If no improvement, permanently discontinue MEKTOVI.
- •
- Life threatening PE
Permanently discontinue MEKTOVI.
Serous Retinopathy [see Warnings and Precautions (5.4)]
- •
- Symptomatic serous retinopathy/Retinal pigment epithelial detachments
Withhold MEKTOVI for up to 10 days.
- •
- If improves and becomes asymptomatic, resume at same dose.
- •
- If not improved, resume at a lower dose level or permanently discontinue MEKTOVI.
Retinal Vein Occlusion (RVO) [see Warnings and Precautions (5.4)]
- •
- Any Grade
Permanently discontinue MEKTOVI.
Uveitis [see Warnings and Precautions (5.4)]
- •
- Grade 1-3
If Grade 1 or 2 does not respond to specific ocular therapy, or for Grade 3 uveitis, withhold MEKTOVI for up to 6 weeks.
- •
- If improved, resume at same or reduced dose.
- •
- If not improved, permanently discontinue MEKTOVI.
- •
- Grade 4
Permanently discontinue MEKTOVI.
Interstitial Lung Disease [see Warnings and Precautions (5.5)]
- •
- Grade 2
Withhold MEKTOVI for up to 4 weeks.
- •
- If improved to Grade 0-1, resume at a reduced dose.
- •
- If not resolved within 4 weeks, permanently discontinue MEKTOVI.
- •
- Grade 3 or Grade 4
Permanently discontinue MEKTOVI.
Hepatotoxicity [see Warnings and Precautions (5.6)]
- •
- Grade 2 AST or ALT increased
Maintain MEKTOVI dose.
- •
- If no improvement within 2 weeks, withhold MEKTOVI until improved to Grade 0-1 or to pretreatment/baseline levels and then resume at the same dose.
- •
- Grade 3 or 4 AST or ALT increased
Rhabdomyolysis or Creatine Phosphokinase (CPK) elevations [see Warnings and Precautions (5.7)]
- •
- Grade 4 asymptomatic CPK elevation or
- •
- Any Grade CPK elevation with symptoms or with renal impairment
Withhold MEKTOVI dose for up to 4 weeks.
- •
- If improved to Grade 0-1 resume at a reduced dose.
- •
- If not resolved within 4 weeks, permanently discontinue MEKTOVI.
Dermatologic [other than palmar plantar erythrodysesthesia syndrome (PPES)] [see Adverse Reactions (6.1)]
- •
- Grade 2
If no improvement within 2 weeks, withhold MEKTOVI until Grade 0-1. Resume at same dose if first occurrence or reduce dose if recurrent.
- •
- Grade 3
Withhold MEKTOVI until Grade 0-1. Resume at same dose if first occurrence or reduce dose if recurrent.
- •
- Grade 4
Permanently discontinue MEKTOVI.
Other Adverse Reactions (including Hemorrhage) [see Warnings and Precautions (5.8), Adverse Reactions (6.1)]†
- •
- Recurrent Grade 2 or
- •
- First occurrence of any Grade 3
Withhold MEKTOVI for up to 4 weeks.
- •
- If improves to Grade 0-1 or to pretreatment/baseline levels, resume at reduced dose.
- •
- If no improvement, permanently discontinue MEKTOVI.
- •
- First occurrence of any Grade 4
Permanently discontinue MEKTOVI, or
Withhold MEKTOVI for up to 4 weeks.- •
- If improves to Grade 0-1 or to pretreatment/baseline levels, then resume at a reduced dose.
- •
- If no improvement, permanently discontinue MEKTOVI.
- •
- Recurrent Grade 3
Consider permanently discontinuing MEKTOVI.
- •
- Recurrent Grade 4
Permanently discontinue MEKTOVI.
Refer to the encorafenib prescribing information for dose modifications for adverse reactions associated with encorafenib.
2.4 Dosage Modifications for Moderate or Severe Hepatic Impairment
For patients with moderate (total bilirubin greater than 1.5 and less than or equal to 3 × ULN and any AST) or severe (total bilirubin levels greater than 3 × ULN and any AST) hepatic impairment, the recommended dosage is 30 mg orally taken twice daily [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 New Primary Malignancies
New primary malignancies, cutaneous and non-cutaneous, can occur when MEKTOVI is used in combination with encorafenib.
In PHAROS, cutaneous squamous cell carcinoma and skin papilloma each occurred in 2% of patients who received MEKTOVI in combination with encorafenib.
Monitor patients for new malignancies prior to initiation of treatment, while on treatment, and after discontinuation of treatment [see Dosage and Administration (2.3)].
5.2 Cardiomyopathy
Cardiomyopathy, manifesting as left ventricular dysfunction associated with symptomatic or asymptomatic decreases in ejection fraction, has been reported in patients treated with MEKTOVI in combination with encorafenib. In COLUMBUS, evidence of cardiomyopathy (decrease in LVEF below the institutional LLN with an absolute decrease in LVEF ≥10% below baseline as detected by echocardiography or MUGA) occurred in 7% of patients receiving MEKTOVI plus encorafenib. Grade 3 left ventricular dysfunction occurred in 1.6% of patients. The median time to first occurrence of left ventricular dysfunction (any grade) in patients receiving MEKTOVI in combination with encorafenib was 3.6 months (range 0 to 21 months). Cardiomyopathy resolved in 87% of patients receiving MEKTOVI plus encorafenib.
In PHAROS, evidence of cardiomyopathy (decrease in LVEF below the institutional LLN with an absolute decrease in LVEF ≥10% below baseline as detected by echocardiography or MUGA) occurred in 11% of patients receiving MEKTOVI in combination with encorafenib. Grade 3 left ventricular dysfunction occurred in 1% of patients. Cardiomyopathy resolved in 82% of patients receiving MEKTOVI plus encorafenib.
Assess ejection fraction by echocardiogram or MUGA scan prior to initiating treatment, one month after initiating treatment, and then every 2 to 3 months during treatment. The safety of MEKTOVI in combination with encorafenib has not been established in patients with a baseline ejection fraction that is either below 50% or below the institutional lower limit of normal (LLN). Patients with cardiovascular risk factors should be monitored closely when treated with MEKTOVI.
Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.3 Venous Thromboembolism
In COLUMBUS, venous thromboembolism (VTE) occurred in 6% of patients receiving MEKTOVI in combination with encorafenib, including 3.1% of patients who developed pulmonary embolism. In PHAROS, VTE occurred in 7% of patients receiving MEKTOVI in combination with encorafenib, including 1% of patients who developed pulmonary embolism.
Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.4 Ocular Toxicities
Serous Retinopathy
In COLUMBUS, serous retinopathy occurred in 20% of patients treated with MEKTOVI in combination with encorafenib; 8% were retinal detachment and 6% were macular edema. Symptomatic serous retinopathy occurred in 8% of patients with no cases of blindness. No patient discontinued MEKTOVI due to serous retinopathy; 6% of patients required dose interruptions or dose reductions. The median time to onset of the first event of serous retinopathy (all grades) was 1.2 months (range 0 to 17.5 months).
In PHAROS, serous retinopathy (retinal detachment) occurred in 2% of patients with no cases of blindness treated with MEKTOVI in combination with encorafenib. No patient permanently discontinued MEKTOVI due to serous retinopathy; 1% of patients required dose interruptions.
Assess for visual symptoms at each visit. Perform an ophthalmologic examination at regular intervals, for new or worsening visual disturbances, and to follow new or persistent ophthalmologic findings. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
Retinal Vein Occlusion
RVO is a known class-related adverse reaction of MEK inhibitors and may occur in patients treated with MEKTOVI in combination with encorafenib. In patients with BRAF mutation-positive melanoma receiving MEKTOVI with encorafenib (n=690), 1 patient experienced RVO (0.1%).
The safety of MEKTOVI has not been established in patients with a history of RVO or current risk factors for RVO including uncontrolled glaucoma or a history of hyperviscosity or hypercoagulability syndromes.
Perform ophthalmologic evaluation for patient-reported acute vision loss or other visual disturbance within 24 hours. Permanently discontinue MEKTOVI in patients with documented RVO [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
Uveitis
Uveitis, including iritis and iridocyclitis, has been reported in patients treated with MEKTOVI in combination with encorafenib. In COLUMBUS, the incidence of uveitis among patients treated with MEKTOVI in combination with encorafenib was 4%. In PHAROS, uveitis occurred in 1% of patients receiving MEKTOVI in combination with encorafenib.
Assess for visual symptoms at each visit. Perform an ophthalmologic evaluation at regular intervals and for new or worsening visual disturbances, and to follow new or persistent ophthalmologic findings. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.5 Interstitial Lung Disease
In patients with BRAF mutation-positive melanoma receiving MEKTOVI with encorafenib (n=690), 2 patients (0.3%) developed interstitial lung disease (ILD), including pneumonitis. In PHAROS, 1 patient (1%) receiving MEKTOVI with encorafenib developed pneumonitis.
Assess new or progressive unexplained pulmonary symptoms or findings for possible ILD. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.6 Hepatotoxicity
Hepatotoxicity can occur when MEKTOVI is administered in combination with encorafenib. In COLUMBUS, the incidence of Grade 3 or 4 increases in liver function laboratory tests in patients receiving MEKTOVI in combination with encorafenib was 6% for alanine aminotransferase (ALT), 2.6% for aspartate aminotransferase (AST), and 0.5% for alkaline phosphatase. No patient experienced Grade 3 or 4 serum bilirubin elevation. In PHAROS, the incidence of Grade 3 or 4 increases in liver function laboratory tests in patients receiving MEKTOVI in combination with encorafenib was 10% for AST, 9% for ALT, and 3.2% for alkaline phosphatase.
Monitor liver laboratory tests before initiation of MEKTOVI, monthly during treatment, and as clinically indicated. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.7 Rhabdomyolysis
Rhabdomyolysis can occur when MEKTOVI is administered in combination with encorafenib. In COLUMBUS, elevation of laboratory values of serum CPK occurred in 58% of patients treated with MEKTOVI in combination with encorafenib. In patients with BRAF mutation-positive melanoma receiving MEKTOVI with encorafenib (n=690), rhabdomyolysis was reported in 1 patient (0.1%). In PHAROS, elevation of laboratory values of serum creatine kinase (CK) occurred in 41% of patients treated with MEKTOVI in combination with encorafenib. No patient experienced rhabdomyolysis.
Monitor CPK and creatinine levels prior to initiating MEKTOVI, periodically during treatment, and as clinically indicated. Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.8 Hemorrhage
Hemorrhage can occur when MEKTOVI is administered in combination with encorafenib. In COLUMBUS, hemorrhage occurred in 19% of patients receiving MEKTOVI in combination with encorafenib. Grade 3 or greater hemorrhage occurred in 3.2% of patients. The most frequent hemorrhagic events were gastrointestinal, including rectal hemorrhage (4.2%), hematochezia (3.1%), and hemorrhoidal hemorrhage (1%). Fatal intracranial hemorrhage in the setting of new or progressive brain metastases occurred in 1.6% of patients.
In PHAROS, hemorrhage occurred in 12% of patients receiving MEKTOVI in combination with encorafenib including fatal hemorrhage intracranial (1%); Grade 3 or 4 hemorrhage occurred in 4.1% of patients. The most frequent hemorrhagic events were anal hemorrhage and hemothorax (2% each).
Withhold, reduce dose, or permanently discontinue based on severity of adverse reaction [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.9 Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, MEKTOVI can cause fetal harm when administered to a pregnant woman. Binimetinib was embryotoxic and abortifacient when administered to rabbits during the period of organogenesis at doses greater than or equal to those resulting in exposures approximately 5 times the human exposure at the recommended clinical dose of 45 mg twice daily.
Advise pregnant women and females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with MEKTOVI and for 30 days after the last dose [see Use in Specific Populations (8.1, 8.3)].
-
6 ADVERSE REACTIONS
The following adverse reactions are described elsewhere in the labeling:
- •
- New Primary Malignancies [see Warnings and Precautions (5.1)]
- •
- Cardiomyopathy [see Warnings and Precautions (5.2)]
- •
- Venous Thromboembolism [see Warnings and Precautions (5.3)]
- •
- Ocular Toxicities [see Warnings and Precautions (5.4)]
- •
- Interstitial Lung Disease [see Warnings and Precautions (5.5)]
- •
- Hepatotoxicity [see Warnings and Precautions (5.6)]
- •
- Rhabdomyolysis [see Warnings and Precautions (5.7)]
- •
- Hemorrhage [see Warnings and Precautions (5.8)]
- •
- Embryo-Fetal Toxicity [see Warnings and Precautions (5.9)]
- •
- Risks Associated with Combination Treatment [see Warnings and Precautions (5.10)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described in WARNINGS AND PRECAUTIONS reflect exposure of 192 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma to MEKTOVI 45 mg twice daily in combination with encorafenib 450 mg once daily in a randomized open-label, active-controlled trial (COLUMBUS) [see Clinical Studies (14.1)] or, for rare events, exposure of 690 patients with BRAF V600 mutation-positive melanoma to MEKTOVI 45 mg twice daily in combination with encorafenib once daily across multiple clinical trials (NCT03915951, NCT01909453).
The pooled safety population described in the WARNINGS AND PRECAUTIONS also reflect exposure of 98 patients with BRAF V600E mutation-positive metastatic non-small cell lung cancer to MEKTOVI 45 mg twice daily and encorafenib 450 mg once daily until disease progression or unacceptable toxicity in PHAROS [see Clinical Studies (14.2)].
BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
The data described below reflect exposure of 192 patients with BRAF V600 mutation-positive unresectable or metastatic melanoma to MEKTOVI (45 mg twice daily) in combination with encorafenib (450 mg once daily) in COLUMBUS.
The COLUMBUS trial [see Clinical Studies (14.1)] excluded patients with a history of Gilbert's syndrome, abnormal left ventricular ejection fraction, prolonged QTc (>480 msec), uncontrolled hypertension, and history or current evidence of retinal vein occlusion. The median duration of exposure was 11.8 months for patients treated with MEKTOVI in combination with encorafenib and 6.2 months for patients treated with vemurafenib.
The most common (≥25%) adverse reactions in patients receiving MEKTOVI in combination with encorafenib were fatigue, nausea, diarrhea, vomiting, and abdominal pain.
Adverse reactions leading to dose interruptions of MEKTOVI occurred in 33% of patients receiving MEKTOVI in combination with encorafenib; the most common were left ventricular dysfunction (6%) and serous retinopathy (5%). Adverse reactions leading to dose reductions of MEKTOVI occurred in 19% of patients receiving MEKTOVI in combination with encorafenib; the most common were left ventricular dysfunction (3%), serous retinopathy (3%), and colitis (2%). Five percent (5%) of patients receiving MEKTOVI in combination with encorafenib experienced an adverse reaction that resulted in permanent discontinuation of MEKTOVI. The most common adverse reactions resulting in permanent discontinuation of MEKTOVI were hemorrhage in 2% and headache in 1% of patients.
Table 3 and Table 4 present adverse drug reactions and laboratory abnormalities, respectively, identified in COLUMBUS. The COLUMBUS trial was not designed to demonstrate a statistically significant difference in adverse reaction rates for MEKTOVI in combination with encorafenib, as compared to vemurafenib, for any specific adverse reaction listed in Table 3.
Table 3: Adverse Reactions Occurring in ≥10% of Patients Receiving MEKTOVI in Combination with Encorafenib in COLUMBUS* Adverse Reaction MEKTOVI with
encorafenib
N=192Vemurafenib
N=186All
Grades
(%)Grades
3 and 4†
(%)All
Grades
(%)Grades
3 and 4†
(%)General Disorders and Administration Site Conditions
Fatigue‡
43
3
46
6
Pyrexia‡
18
4
30
0
Peripheral edema‡
13
1
15
1
Gastrointestinal Disorders
Nausea
41
2
34
2
Diarrhea
36
3
34
2
Vomiting‡
30
2
16
1
Abdominal pain‡
28
4
16
1
Constipation
22
0
6
1
Skin and Subcutaneous Tissue Disorders
Rash‡
22
1
53
13
Nervous System Disorders
Dizziness‡
15
3
4
0
Visual Disorders
Visual impairment‡
20
0
4
0
Serous retinopathy/RPED‡
20
3
2
0
Vascular Disorders
Hemorrhage‡
19
3
9
2
Hypertension‡
11
6
11
3
Other clinically important adverse reactions occurring in <10% of patients who received MEKTOVI in combination with encorafenib were:
Gastrointestinal disorders: Colitis
Skin and subcutaneous tissue disorders: Panniculitis
Immune system disorders: Drug hypersensitivity
Table 4: Laboratory Abnormalities Occurring in ≥10% (All grades) of Patients Receiving MEKTOVI in Combination with Encorafenib in COLUMBUS* Laboratory Abnormality MEKTOVI
with encorafenib
N=192Vemurafenib
N=186All
Grades
(%)Grades
3 and 4
(%)All
Grades
(%)Grades
3 and 4
(%)- *
- Grades per National Cancer Institute CTCAE v4.03.
Hematology
Anemia
36
3.6
34
2.2
Leukopenia
13
0
10
0.5
Lymphopenia
13
2.1
30
7
Neutropenia
13
3.1
4.8
0.5
Chemistry
Increased Creatinine
93
3.6
92
1.1
Increased Creatine Phosphokinase
58
5
3.8
0
Increased Gamma Glutamyl Transferase
45
11
34
4.8
Increased ALT
29
6
27
2.2
Increased AST
27
2.6
24
1.6
Increased Alkaline Phosphatase
21
0.5
35
2.2
Hyponatremia
18
3.6
15
0.5
BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer (NSCLC)
The safety of MEKTOVI in combination with encorafenib is described in 98 patients with BRAF V600E mutation-positive metastatic NSCLC who received MEKTOVI (45 mg twice daily) in combination with encorafenib (450 mg once daily) in an open-label, single-arm trial (PHAROS).
The PHAROS trial [see Clinical Studies (14.2)] excluded patients with abnormal LVEF, prolonged QTc (>480 ms), uncontrolled hypertension, and history or current evidence of retinal vein occlusion. The median duration of treatment for MEKTOVI and encorafenib was 8.4 and 9.2 months respectively.
The most common (≥25%) adverse reactions in patients receiving MEKTOVI were fatigue, nausea, diarrhea, musculoskeletal pain, vomiting, abdominal pain, visual impairment, constipation, dyspnea, rash, and cough.
Adverse reactions leading to dose interruptions of MEKTOVI occurred in 62% of patients receiving MEKTOVI; the most common (≥5%) were diarrhea (17%); nausea (15%); fatigue (9%); AST increased (7%); ALT increased, anemia, musculoskeletal pain, vomiting (6% each); and acute kidney injury, hemorrhage, and LV dysfunction/cardiomyopathy (5% each). Adverse reactions leading to dose reductions of MEKTOVI occurred in 33% of patients receiving MEKTOVI; the most common (≥5%) were diarrhea (8%), nausea (6%), and AST increased (5%). A total of 17% of patients receiving MEKTOVI experienced an adverse reaction that resulted in permanent discontinuation of MEKTOVI; the most common (≥2%) were diarrhea (3.1%); musculoskeletal pain, LV dysfunction/cardiomyopathy, fatigue, nausea, rash, visual impairment, and vomiting (2% each). None of the other adverse reactions leading to permanent discontinuation of MEKTOVI occurred in more than 1 patient.
Serious adverse reactions occurred in 38% of patients who received MEKTOVI in combination with encorafenib. Serious adverse reactions in ≥2% of patients included hemorrhage (6%); diarrhea (4.1%); anemia, dyspnea, pneumonia (3.1% each); arrhythmia, device related infection, edema, myocardial infarction, and pleural effusion (2% each). Fatal adverse reactions occurred in 2% of patients who received MEKTOVI (45 mg twice-daily) in combination with encorafenib, including intracranial hemorrhage and myocardial infarction (1% each).
Table 5 and Table 6 present adverse drug reactions and laboratory abnormalities, respectively, identified in PHAROS.
Table 5: Adverse Reactions Occurring in ≥10% of Patients Receiving MEKTOVI in Combination with Encorafenib in PHAROS* - *
- Grades per National Cancer Institute CTCAE v4.03.
- †
- One Grade 5 adverse reaction of hemorrhage occurred.
- ‡
- Fatigue includes fatigue, asthenia.
- §
- Edema includes edema peripheral, generalized edema, swelling, localized edema, face edema.
- ¶
- Diarrhea includes diarrhea, colitis.
- #
- Abdominal pain includes abdominal pain, abdominal pain upper, abdominal discomfort, epigastric discomfort.
- Þ
- Visual impairment includes vision blurred, visual impairment, vitreous floaters, photophobia, visual acuity reduced, photopsia.
- ß
- Musculoskeletal pain includes back pain, arthralgia, pain in extremity, myalgia, musculoskeletal chest pain, non-cardiac chest pain, neck pain.
- à
- Rash includes rash, rash macular, rash maculo-papular, rash papular, rash pustular,dermatitis acneiform, palmar-plantar erythrodysesthesia syndrome, eczema, skin exfoliation.
- è
- Pruritis includes pruritus, pruritus genital.
- ð
- Dyspnea includes dyspnea, dyspnea exertional.
- ø
- Cough includes cough, productive cough.
- ý
- Dizziness includes dizziness, balance disorder.
- £
- Hemorrhage includes anal hemorrhage, hemothorax, gastrointestinal hemorrhage, hematochezia, hematuria, hemoptysis, hemorrhage intracranial, hyphema, small intestinal hemorrhage, upper gastrointestinal hemorrhage, vaginal hemorrhage.
- ¥
- Left ventricular dysfunction/cardiomyopathy includes ejection fraction decreased, cardiac failure, cardiac failure congestive.
Adverse Reaction
MEKTOVI
with encorafenib
N=98
All
Grades
(%)
Grade 3 and 4†
(%)
General Disorders and Administration Site Conditions
Fatigue‡
61
8
Edema§
23
1
Pyrexia
22
0
Gastrointestinal Disorders
Nausea
58
3.1
Diarrhea¶
52
7
Vomiting
37
1
Abdominal pain#
32
1
Constipation
27
0
Eye Disorders
Visual impairmentÞ
29
2
Musculoskeletal and Connective Tissue Disorders
Musculoskeletal painß
48
4.1
Skin and Subcutaneous Tissue Disorders
Rashà
27
3.1
Pruritisè
16
0
Dry skin
13
0
Alopecia
12
0
Respiratory, Thoracic and Mediastinal Disorders
Dyspneað
27
8
Coughø
26
0
Nervous System Disorders
Dizzinessý
17
1
Headache
11
0
Metabolism and Nutrition Disorders
Decreased appetite
14
1
Vascular Disorders
12
4.1
Hypertension
10
5
Cardiac Disorders
Left ventricular dysfunction/cardiomyopathy¥
11
1
Investigations
Weight increased
11
1
Psychiatric Disorders
Insomnia
10
0
Other clinically important adverse reactions occurring in <10% of patients who received MEKTOVI in combination with encorafenib were:
Nervous system disorders: Peripheral neuropathy, Dysgeusia, Facial paresis
Gastrointestinal disorders: Pancreatitis
Skin and subcutaneous tissue disorders: Hyperkeratosis, Erythema
Immune system disorders: Drug hypersensitivity
Table 6: Laboratory Abnormalities Occurring in ≥10% (All Grades) of Patients Receiving MEKTOVI with Encorafenib in PHAROS* Laboratory Abnormality†
MEKTOVI with encorafenib
All
Grades
(%)
Grades
3 and 4
(%)
Hematology
Anemia
47
11
Lymphopenia
24
6
Thrombocytopenia
20
1.1
Leukopenia
12
0
Neutropenia
12
1.1
Chemistry
Increased creatinine
91
3.2
Hyperglycemia
48
6
Increased creatine kinase
41
3.3
Lipase increased
40
14
Increased ALT
34
9
Hypoalbuminemia
32
0
Increased AST
31
10
Increased alkaline phosphatase
31
3.2
Hyperkalemia
31
2.1
Hyponatremia
26
11
Serum amylase increased
22
1.1
Hypocalcemia
12
2.1
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on findings from animal reproduction studies and its mechanism of action [see Clinical Pharmacology (12.1)], MEKTOVI can cause fetal harm when administered to a pregnant woman. There are no available clinical data on the use of MEKTOVI during pregnancy. In animal reproduction studies, oral administration of binimetinib during the period of organogenesis was embryotoxic and an abortifacient in rabbits at doses greater than or equal to those resulting in exposures approximately 5 times the human exposure at the clinical dose of 45 mg twice daily (see Data). Advise pregnant women and females of reproductive potential of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Animal Data
In reproductive toxicity studies, administration of binimetinib to rats during the period of organogenesis resulted in maternal toxicity, decreased fetal weights and increased variations in ossification at doses ≥30 mg/kg/day (approximately 37 times the human exposure based on AUC at the recommended clinical dose of 45 mg twice daily). In pregnant rabbits, administration of binimetinib during the period of organogenesis resulted in maternal toxicity, decreased fetal body weights, an increase in malformations, and increased post-implantation loss, including total loss of pregnancy at doses ≥10 mg/kg/day (approximately 5 times the human exposure based on AUC at the recommended clinical dose of 45 mg twice daily). There was a significant increase in fetal ventricular septal defects and pulmonary trunk alterations at 20 mg/kg/day of binimetinib (less than 8 times the human exposure at the recommended clinical dose of 45 mg twice daily).
8.2 Lactation
Risk Summary
There are no data on the presence of binimetinib or its active metabolite in human milk, or the effects of binimetinib on the breastfed child, or the effects on milk production. Because of the potential for serious adverse reactions in a breastfed child, advise women not to breastfeed during treatment with MEKTOVI and for 3 days after the last dose.
8.3 Females and Males of Reproductive Potential
Based on animal data, MEKTOVI can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating MEKTOVI [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of MEKTOVI have not been established in pediatric patients.
8.5 Geriatric Use
Of the 690 patients with BRAF mutation-positive melanoma who received MEKTOVI in combination with encorafenib across multiple clinical trials, 20% were aged 65 to 74 years and 8% were aged 75 years and older [see Clinical Pharmacology (12.3)].
Of the 98 patients with BRAF V600E mutation-positive metastatic NSCLC who received MEKTOVI in combination with encorafenib, 62 (63.2%) were 65 years of age and over and 20 (20.4%) were 75 years and over [see Clinical Studies (14.2)].
No overall differences in the safety or effectiveness of MEKTOVI plus encorafenib were observed in older patients as compared to younger patients.
8.6 Hepatic Impairment
Binimetinib concentrations may increase in patients with moderate or severe hepatic impairment. Dose adjustment for MEKTOVI is not recommended in patients with mild hepatic impairment (total bilirubin >1 and ≤1.5 × ULN and any AST or total bilirubin ≤ ULN and AST > ULN). Reduce the dose of MEKTOVI for patients with moderate (total bilirubin >1.5 and ≤3 × ULN and any AST) or severe (total bilirubin levels >3 × ULN and any AST) hepatic impairment [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
Binimetinib is a kinase inhibitor. The chemical name is 5-[(4-bromo-2-fluorophenyl)amino]-4-fluoro-N-(2-hydroxyethoxy)-1-methyl-1H-benzimidazole-6-carboxamide. The molecular formula is C17H15BrF2N4O3 and the molecular weight is 441.2 daltons. The chemical structure of binimetinib is shown below:
Binimetinib is a white to slightly yellow powder. In aqueous media, binimetinib is slightly soluble at pH 1, very slightly soluble at pH 2, and practically insoluble at pH 4.5 and higher.
MEKTOVI (binimetinib) tablets for oral use contain 15 mg of binimetinib with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, magnesium stearate (vegetable source), and colloidal silicon dioxide. The coating contains polyvinyl alcohol, polyethylene glycol, titanium dioxide, talc, ferric oxide yellow, and ferrosoferric oxide.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Binimetinib is a reversible inhibitor of mitogen-activated extracellular signal regulated kinase 1 (MEK1) and MEK2 activity. MEK proteins are upstream regulators of the extracellular signal-related kinase (ERK) pathway. In vitro, binimetinib inhibited extracellular signal-related kinase (ERK) phosphorylation in cell-free assays as well as viability and MEK-dependent phosphorylation of BRAF-mutant human melanoma cell lines. Binimetinib also inhibited in vivo ERK phosphorylation and tumor growth in BRAF-mutant murine xenograft models.
Binimetinib and encorafenib target two different kinases in the RAS/RAF/MEK/ERK pathway. Compared to either drug alone, coadministration of encorafenib and binimetinib resulted in greater anti-proliferative activity in vitro in BRAF mutation-positive cell lines and greater anti-tumor activity with respect to tumor growth inhibition in BRAF V600E mutant human melanoma xenograft studies in mice. Additionally, the combination of binimetinib and encorafenib delayed the emergence of resistance in BRAF V600E mutant human melanoma xenografts in mice compared to either drug alone. In a BRAF V600E mutant NSCLC patient-derived xenograft model in mice, coadministration of encorafenib and binimetinib resulted in greater anti-tumor activity compared to binimetinib alone, with respect to tumor growth inhibition. Increased tumor growth delay after dosing cessation was also observed with the coadministration compared to either drug alone.
12.3 Pharmacokinetics
The pharmacokinetics of binimetinib was studied in healthy subjects and patients with solid tumors. After twice-daily dosing, the accumulation is 1.5-fold and the coefficient of variation (CV%) of the area under the concentration-time curve (AUC) is <40% at steady state. The systemic exposure of binimetinib is approximately dose proportional.
Absorption
After oral administration, at least 50% of the binimetinib dose was absorbed with a median time to maximum concentration (Tmax) of 1.6 hours.
Distribution
Binimetinib is 97% bound to human plasma proteins and the blood-to-plasma ratio is 0.72. The geometric mean (CV%) of apparent volume of distribution of binimetinib is 92 L (45%).
Elimination
The mean (CV%) terminal half-life (t1/2) of binimetinib is 3.5 hours (28.5%) and apparent clearance (CL/F) is 20.2 L/h (24%).
Metabolism
The primary metabolic pathway is glucuronidation with UGT1A1 contributing up to 61% of the binimetinib metabolism. Other pathways of binimetinib metabolism include N-dealkylation, amide hydrolysis, and loss of ethane-diol from the side chain. The active metabolite M3 produced by CYP1A2 and CYP2C19 represents 8.6% of the binimetinib exposure. Following a single oral dose of 45 mg radiolabeled binimetinib, approximately 60% of the circulating radioactivity AUC in plasma was attributable to binimetinib.
Specific Populations
Age (20 to 94 years), sex, or body weight do not have a clinically important effect on the systemic exposure of binimetinib. The effect of race or ethnicity on the pharmacokinetics of binimetinib is unknown.
Hepatic Impairment: No clinically meaningful changes in binimetinib exposure (AUC and Cmax) were observed in subjects with mild hepatic impairment (total bilirubin >1 and ≤1.5 × ULN and any AST or total bilirubin ≤ ULN and AST > ULN) as compared to subjects with normal liver function (total bilirubin ≤ ULN and AST ≤ ULN). A 2-fold increase in AUC was observed in subjects with moderate (total bilirubin >1.5 and ≤3 × ULN and any AST) or severe (total bilirubin levels >3 × ULN and any AST) hepatic impairment [see Dosage and Administration (2.4)].
Drug Interaction Studies
Clinical Studies
Effect of UGT1A1 Inducers or Inhibitors on Binimetinib: UGT1A1 genotype and smoking (UGT1A1 inducer) do not have a clinically important effect on binimetinib exposure. Simulations predict similar Cmax of binimetinib 45 mg in the presence or absence of atazanavir 400 mg (UGT1A1 inhibitor).
No differences in binimetinib exposure have been observed when MEKTOVI is coadministered with encorafenib.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with binimetinib have not been conducted. Binimetinib was not genotoxic in studies evaluating reverse mutations in bacteria, chromosomal aberrations in mammalian cells, or micronuclei in bone marrow of rats.
No dedicated fertility studies have been conducted with binimetinib in animals. In general toxicology studies in rats and monkeys, there were no remarkable findings in male or female reproductive organs.
-
14 CLINICAL STUDIES
14.1 BRAF V600E or V600K Mutation-Positive Unresectable or Metastatic Melanoma
MEKTOVI in combination with encorafenib was evaluated in a randomized, active-controlled, open-label, multicenter trial (COLUMBUS; NCT01909453). Eligible patients were required to have BRAF V600E or V600K mutation-positive unresectable or metastatic melanoma, as detected using the bioMerieux THxID™BRAF assay. Patients were permitted to have received immunotherapy in the adjuvant setting and one prior line of immunotherapy for unresectable locally advanced or metastatic disease. Prior use of BRAF inhibitors or MEK inhibitors was prohibited. Randomization was stratified by American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c), Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1), and prior immunotherapy for unresectable or metastatic disease (yes versus no).
Patients were randomized (1:1:1) to receive MEKTOVI 45 mg twice daily in combination with encorafenib 450 mg once daily (MEKTOVI in combination with encorafenib), encorafenib 300 mg once daily, or vemurafenib 960 mg twice daily. Treatment continued until disease progression or unacceptable toxicity. Only the results of the approved dosing (MEKTOVI 45 mg in combination with encorafenib 450 mg) are described below.
The major efficacy outcome measure was progression-free survival (PFS), as assessed by a blinded independent central review, to compare MEKTOVI in combination with encorafenib with vemurafenib. Additional efficacy measures included overall survival (OS), as well as objective response rate (ORR) and duration of response (DoR) which were assessed by central review.
A total of 577 patients were randomized, 192 to the MEKTOVI in combination with encorafenib arm, 194 to the encorafenib arm, and 191 to the vemurafenib arm. Of the 383 patients randomized to either the MEKTOVI in combination with encorafenib or the vemurafenib arms, the median age was 56 years (20 to 89 years), 59% were male, 91% were White, and 72% had baseline ECOG performance status of 0. Ninety-five percent (95%) had metastatic disease, 65% were Stage IVM1c, and 4% received prior CTLA-4, PD-1, or PD-L1 directed antibodies. Twenty-eight percent (28%) had elevated baseline serum lactate dehydrogenase (LDH), 45% had ≥3 organs with tumor involvement at baseline, and 3% had brain metastases. Based on centralized testing, 100% of patients’ tumors tested positive for BRAF mutations; BRAF V600E (88%), BRAF V600K (11%), or both (<1%).
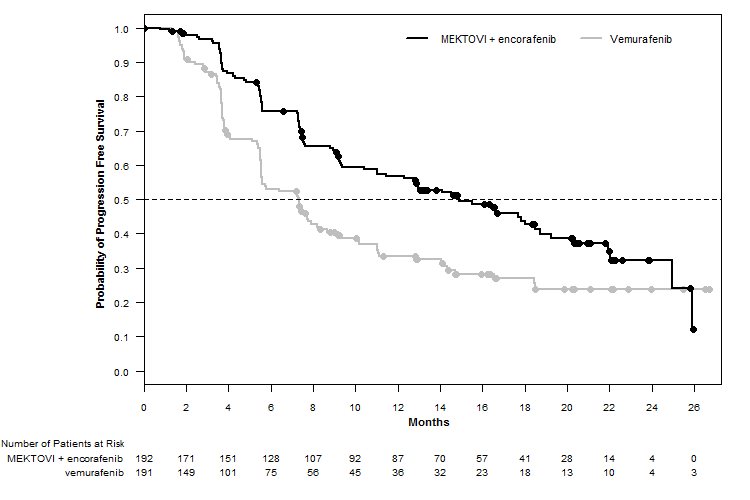
MEKTOVI in combination with encorafenib demonstrated a statistically significant improvement in PFS compared to vemurafenib. Efficacy results are summarized in Table 7 and Figure 1.
Table 7: Efficacy Results for COLUMBUS CI = Confidence interval; CR = Complete response; DoR = Duration of response; HR = Hazard ratio; NE = Not estimable; ORR = Overall response rate; OS = Overall survival; PFS = Progression-free survival; PR = Partial response. - *
- Estimated with Cox proportional hazard model adjusted by the following stratification factors: American Joint Committee on Cancer (AJCC) Stage (IIIB, IIIC, IVM1a or IVM1b, versus IVM1c) and Eastern Cooperative Oncology Group (ECOG) performance status (0 versus 1).
- †
- Log-rank test adjusted by the same stratification factors.
- ‡
- Based on a cutoff date 17.6 months after the date of PFS analysis.
MEKTOVI
with encorafenib
N=192
Vemurafenib
N=191
Progression-Free Survival
Number of events (%)
98 (51)
106 (55)
Progressive disease
88 (46)
104 (54)
Death
10 (5)
2 (1)
Median PFS, months (95% CI)
14.9 (11, 18.5)
7.3 (5.6, 8.2)
HR (95% CI)*
0.54 (0.41, 0.71)
P value†
<0.0001
Overall Survival‡
Number of events (%)
105 (55)
127 (67)
Median OS, months (95% CI)
33.6 (22.4, 39.2)
16.9 (14.0, 24.5)
HR (95% CI) *
0.61 (0.47, 0.79)
Overall Response Rate
ORR (95% CI)
63% (56%, 70%)
40% (33%, 48%)
CR
8%
6%
PR
55%
35%
Duration of Response
Median DoR, months (95% CI)
16.6 (12.2, 20.4)
12.3 (6.9, 16.9)
Figure 1: Kaplan-Meier Curves for Progression-Free Survival in COLUMBUS
14.2 BRAF V600E Mutation-Positive Metastatic Non-Small Cell Lung Cancer
MEKTOVI in combination with encorafenib was evaluated in an open-label, multicenter, single-arm study in patients with BRAF V600E mutation-positive metastatic non-small cell lung cancer (NSCLC) (PHAROS; NCT03915951). Eligible patients had a diagnosis of histologically-confirmed metastatic NSCLC with BRAF V600E mutation that was treatment-naïve or had been previously treated with 1 prior line of systemic therapy in the metastatic setting (platinum-based chemotherapy and/or anti-PD-1/PD-L1 therapies), age 18 years or older, Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0 or 1, and measurable disease as defined by Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Prior use of BRAF inhibitors or MEK inhibitors was not allowed.
Patients received MEKTOVI 45 mg orally twice daily and encorafenib 450 mg once daily until disease progression or unacceptable toxicity. The major efficacy outcome measures were objective response rate (ORR) per RECIST v1.1 and duration of response (DoR) as assessed by independent review committee (IRC).
In the efficacy population, BRAF V600E mutation status was determined by prospective local testing using tumor tissue (78%) or blood (22%) specimens. Of the 98 patients with BRAF V600E mutation, 6 patients were enrolled into the trial based on testing of their tumor tissue specimens with the FoundationOne CDx tissue test. Of the remaining 92 patients enrolled based on local testing, 68 patients had their tumor tissue specimens retrospectively confirmed as having BRAF V600E positive status by the FoundationOne CDx tissue test. The remaining patients had either BRAF V600E negative status (n=5) or had unevaluable results (n=19) by the FoundationOne CDx tissue test. In addition, plasma samples from 81 out of 98 patients were retrospectively tested using the FoundationOne Liquid CDx assay. Of the 81 patients, 48 were confirmed positive for BRAF V600E, while 33 patients were BRAF V600E mutation negative by FoundationOne Liquid CDx assay. The remaining 17 samples had unevaluable results with FoundationOne Liquid CDx assay.
The efficacy population included 59 treatment-naïve patients and 39 previously-treated patients. Among these 98 patients, the median age was 70 years (range: 47 to 86); 53% female; 88% White, 7% Asian, 3% Black or African American, and 1% American Indian or Alaska Native; 99% were not Hispanic or Latino; 13% were current smokers and 57% were former smokers; 73% had ECOG PS of 1; and 97% had adenocarcinoma. All patients had metastatic disease and 8% had brain metastases at baseline.
Efficacy results for patients with BRAF V600E mutation-positive metastatic NSCLC are summarized in Table 8.
Table 8: Efficacy Results for PHAROS CI = Confidence interval; CR = Complete response; DoR = Duration of response; N = Number of patients; NE = Not estimable; ORR = Objective response rate; PR = Partial response. - *
- Assessed by Independent Central Review (ICR).
MEKTOVI with encorafenib
Efficacy Parameter
Treatment naïve
(N=59)
Previously treated
(N=39)
Objective Response Rate*
ORR (95% CI)
75% (62, 85)
46% (30, 63)
CR
15%
10%
PR
59%
36%
Duration of Response*
N=44
N=18
Median DoR, months
(95% CI)NE (23.1, NE)
16.7 (7.4, NE)
% with DoR ≥6 months
75%
67%
% with DoR ≥12 months
59%
33%
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Inform patients of the following:
New Primary Malignancies
Advise patients that MEKTOVI administered with encorafenib can result in the development of new primary cutaneous and non-cutaneous malignancies. Advise patients to contact their healthcare provider immediately for any new lesions, changes to existing lesions on their skin, or other signs and symptoms of malignancies [see Warnings and Precautions (5.1)].
Cardiomyopathy
Advise patients to report any symptoms of heart failure to their healthcare provider [see Warnings and Precautions (5.2)].
Venous Thromboembolism
Advise patients to contact their healthcare provider if they experience symptoms of venous thrombosis or pulmonary embolism. Advise patients to seek medical attention for sudden onset of difficulty breathing, leg pain, or swelling [see Warnings and Precautions (5.3)].
Ocular Toxicities
Advise patients to contact their healthcare provider as soon as possible if they experience any changes in their vision [see Warnings and Precautions (5.4)].
Interstitial Lung Disease
Advise patients to contact their healthcare provider if they experience any new or worsening respiratory symptoms including cough or dyspnea [see Warnings and Precautions (5.5)].
Hepatotoxicity
Advise patients that serial testing of serum liver tests (ALT, AST, bilirubin) is recommended during treatment with MEKTOVI. Instruct patients to report symptoms of liver dysfunction including jaundice, dark urine, nausea, vomiting, loss of appetite, fatigue, bruising, or bleeding [see Warnings and Precautions (5.6)].
Rhabdomyolysis
Advise patients to contact their healthcare provider as soon as possible if they experience unusual or new onset weakness, myalgia, or darkened urine [see Warnings and Precautions (5.7)].
Hemorrhage
Advise patients to notify their healthcare provider if they experience symptoms suggestive of hemorrhage, such as unusual bleeding [see Warnings and Precautions (5.8)].
Females and Males of Reproductive Potential
Embryo-Fetal Toxicity: Advise females with reproductive potential of the potential risk to a fetus. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, during treatment with MEKTOVI [see Warnings and Precautions (5.9), Use in Specific Populations (8.1)]. Advise females of reproductive potential to use effective contraception during treatment with MEKTOVI and for 30 days after the last dose.
Lactation: Advise women not to breastfeed during treatment with MEKTOVI and for 3 days after the last dose [see Use in Specific Populations (8.2)].
-
SPL UNCLASSIFIED SECTION
Distributed by:
Array BioPharma Inc., a wholly owned subsidiary of Pfizer Inc.
3200 Walnut Street
Boulder, CO 80301© 2023 Array BioPharma Inc. All rights reserved.
MEKTOVI® is a registered trademark of Array BioPharma Inc. in the United States and various other countries.
Patented. See www.arraybiopharma.com/patentsLAB-1426-2.0
-
Medication Guide
MEDICATION GUIDE
MEKTOVI® (mek-TOE-vee)
(binimetinib)
tablets
Important Information: If your healthcare provider prescribes MEKTOVI with encorafenib, please read the Medication Guide that comes with encorafenib.
What is the most important information I should know about MEKTOVI when taken in combination with encorafenib?
MEKTOVI when taken in combination with encorafenib may cause serious side effects, including:
- •
- Risk of new skin cancers. MEKTOVI, when used with encorafenib, may cause skin cancers called cutaneous squamous cell carcinoma or basal cell carcinoma.
Talk to your healthcare provider about your risk for these cancers.
Check your skin and tell your healthcare provider right away about any skin changes, including a:
-
- o
- new wart
- o
- skin sore or reddish bump that bleeds or does not heal
- o
- change in size or color of a mole
- Your healthcare provider should check your skin before treatment with MEKTOVI, when taken in combination with encorafenib, every 2 months during treatment, and for up to 6 months after you stop treatment to look for any new skin cancers.
- Your healthcare provider should also check for cancers that may not occur on the skin. Tell your healthcare provider about any new symptoms that develop during treatment with MEKTOVI when taken in combination with encorafenib.
- See "What are the possible side effects of MEKTOVI?"for more information about side effects.
What is MEKTOVI?
MEKTOVI is a prescription medicine used:
- •
- in combination with a medicine called encorafenib to treat people with a type of skin cancer called melanoma:
- o
- that has spread to other parts of the body or cannot be removed by surgery, and
- o
- that has a certain type of abnormal “BRAF” gene
- •
- in combination with a medicine called encorafenib to treat adults with a type of lung cancer called non-small cell lung cancer (NSCLC):
- o
- that has spread to other parts of the body, and
- o
- that has a certain type of abnormal “BRAF” gene
Your healthcare provider will perform a test to make sure that MEKTOVI is right for you.
It is not known if MEKTOVI is safe and effective in children.
Before taking MEKTOVI, tell your healthcare provider about all of your medical conditions, including if you:
- •
- have heart problems
- •
- have had blood clots
- •
- have eye problems
- •
- have lung or breathing problems
- •
- have liver or kidney problems
- •
- have any muscle problems
- •
- have bleeding problems
- •
- have high blood pressure (hypertension)
- •
- are pregnant or plan to become pregnant. MEKTOVI can harm your unborn baby.
- o
- Females who are able to become pregnant should use effective birth control (contraception) during treatment with MEKTOVI and for at least 30 days after the last dose of MEKTOVI.
- o
- Talk to your healthcare provider about birth control methods that may be right for you during this time.
- o
- Your healthcare provider will do a pregnancy test before you start taking MEKTOVI. Tell your healthcare provider right away if you become pregnant or think you might be pregnant during treatment with MEKTOVI.
- •
- are breastfeeding or plan to breastfeed. It is not known if MEKTOVI passes into your breast milk. Do not breastfeed during treatment with MEKTOVI and for 3 days after the last dose of MEKTOVI. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take MEKTOVI?
- •
- Take MEKTOVI exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking MEKTOVI unless your healthcare provider tells you to.
- •
- Take MEKTOVI in combination with encorafenib by mouth 2 times a day, about 12 hours apart.
- •
- MEKTOVI may be taken with or without food.
- •
- If you miss a dose of MEKTOVI, take it as soon as you remember. If it is within 6 hours of your next scheduled dose, take your next dose at your regular time. Do not make up for the missed dose.
- •
- Do not take an extra dose if you vomit after taking your scheduled dose. Take your next dose at your regular time.
- •
- If you stop treatment with encorafenib, talk to your healthcare provider about whether your MEKTOVI treatment may need to be stopped.
What are the possible side effects of MEKTOVI?
MEKTOVI may cause serious side effects, including:
- •
-
Heart problems, including heart failure. MEKTOVI, when taken with encorafenib, can cause heart problems. Your healthcare provider should check your heart function before and during treatment with MEKTOVI. Call your healthcare provider right away if you have any of the following signs and symptoms of a heart problem:
- o
- feeling like your heart is pounding or racing
- o
- shortness of breath
- o
- swelling of your ankles and feet
- o
- feeling lightheaded
- •
-
Blood clots. MEKTOVI, when taken with encorafenib, can cause blood clots in your arms or legs, which can travel to your lungs and can lead to death. Get medical help right away if you have the following symptoms:
- o
- chest pain
- o
- sudden shortness of breath or trouble breathing
- o
- pain in your legs with or without swelling
- o
- swelling in your arms and legs
- o
- a cool pale arm or leg
- •
-
Eye problems. MEKTOVI, when taken with encorafenib, can cause eye problems. Your healthcare provider should perform an eye exam regularly during treatment with MEKTOVI. Tell your healthcare provider right away if you develop any new or worsening symptoms of eye problems, including:
- o
- blurred vision, loss of vision, or other vision changes
- o
- see colored dots
- o
- see halos (blurred outline around objects)
- o
- eye pain, swelling, or redness
- •
-
Lung or breathing problems. MEKTOVI, when taken with encorafenib, can cause lung or breathing problems. Tell your healthcare provider if you have any new or worsening symptoms of lung or breathing problems, including:
- o
- shortness of breath
- o
- cough
- •
- Liver problems. MEKTOVI, when taken with encorafenib, can cause liver problems. Your healthcare provider should perform blood tests to check your liver function before and during treatment with MEKTOVI. Tell your healthcare provider if you have any of the following signs and symptoms of a liver problem:
- o
- yellowing of your skin or your eyes
- o
- dark or brown (tea-colored) urine
- o
- nausea or vomiting
- o
- loss of appetite
- o
- tiredness
- o
- bruising
- o
- bleeding
- •
-
Muscle problems (rhabdomyolysis). MEKTOVI, when taken with encorafenib, can cause muscle problems that can be severe. Treatment with MEKTOVI may increase the level of an enzyme in your blood called creatine phosphokinase (CPK) and can be a sign of muscle damage. Your healthcare provider should perform a blood test to check your levels of CPK before and during treatment. Tell your healthcare provider right away if you develop any of these symptoms:
- o
- weakness
- o
- muscle aches or pain
- o
- dark, reddish urine
- •
-
Bleeding problems. MEKTOVI, when taken with encorafenib, can cause serious bleeding problems, including in your brain or stomach, that can lead to death. Tell your healthcare provider and get medical help right away if you develop any signs of bleeding, including:
- o
- headaches, dizziness, or feeling weak
- o
- cough up blood or blood clots
- o
- vomit blood or your vomit looks like “coffee grounds”
- o
- red or black stool that look like tar
Your healthcare provider may change your dose, temporarily stop, or permanently stop treatment with MEKTOVI if you have certain side effects.
The most common side effects of MEKTOVI when taken with encorafenib for melanoma include:
- •
- fatigue
- •
- nausea
- •
- diarrhea
- •
- vomiting
- •
- stomach-area (abdominal) pain
The most common side effects of MEKTOVI when taken with encorafenib for NSCLC include:
- •
- fatigue
- •
- nausea
- •
- diarrhea
- •
- muscle or joint pain
- •
- vomiting
- •
- stomach-area (abdominal) pain
- •
- blurred vision, loss of vision, or other vision changes
- •
- constipation
- •
- shortness of breath
- •
- rash
- •
- cough
These are not all of the possible side effects of MEKTOVI.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
You may also report side effects to Pfizer Inc. at 1-800-438-1985.
How should I store MEKTOVI?
- •
- Store MEKTOVI at room temperature between 68°F to 77°F (20°C to 25°C).
Keep MEKTOVI and all medicines out of the reach of children.
General information about the safe and effective use of MEKTOVI.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use MEKTOVI for a condition for which it was not prescribed. Do not give MEKTOVI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about MEKTOVI that is written for health professionals.
What are the ingredients in MEKTOVI?
Active ingredient: binimetinib
Inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, magnesium stearate (vegetable source), and colloidal silicon dioxide
Tablet coating: polyvinyl alcohol, polyethylene glycol, titanium dioxide, talc, ferric oxide yellow, ferrosoferric oxide
Distributed by: Array BioPharma Inc., a wholly owned subsidiary of Pfizer Inc., Boulder, Colorado 80301.
MEKTOVI® is a registered trademark of Array BioPharma Inc. in the United States and various other countries.
For more information, go to www.BRAFTOVIMEKTOVI.com or call 1-844-792-7729.
©2020 Array BioPharma Inc. All rights reserved.
LAB-1427-2.0
This Medication Guide has been approved by the U.S. Food and Drug Administration.
Revised: 10/2023 - PRINCIPAL DISPLAY PANEL - 15 mg Tablet Bottle Label
- PRINCIPAL DISPLAY PANEL - 15 mg Tablet Bottle Carton
-
INGREDIENTS AND APPEARANCE
MEKTOVI
binimetinib tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:70255-010 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BINIMETINIB (UNII: 181R97MR71) (BINIMETINIB - UNII:181R97MR71) BINIMETINIB 15 mg Inactive Ingredients Ingredient Name Strength LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) CROSCARMELLOSE SODIUM (UNII: M28OL1HH48) MAGNESIUM STEARATE (UNII: 70097M6I30) SILICON DIOXIDE (UNII: ETJ7Z6XBU4) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) POLYETHYLENE GLYCOL 3350 (UNII: G2M7P15E5P) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TALC (UNII: 7SEV7J4R1U) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) FERROSOFERRIC OXIDE (UNII: XM0M87F357) Product Characteristics Color YELLOW Score no score Shape OVAL Size 12mm Flavor Imprint Code A;15 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:70255-010-02 1 in 1 CARTON 06/27/2018 1 180 in 1 BOTTLE; Type 0: Not a Combination Product 2 NDC:70255-010-03 90 in 1 BOTTLE; Type 0: Not a Combination Product 06/27/2018 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA210498 06/27/2018 Labeler - Array BioPharma Inc. (004047838) Registrant - Pfizer Inc (113480771)