Label: BUMETANIDE injection




- NDC Code(s): 0641-6007-01, 0641-6007-10, 0641-6008-01, 0641-6008-10
- Packager: Hikma Pharmaceuticals USA Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Abbreviated New Drug Application
Drug Label Information
Updated April 8, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
BOXED WARNING
(What is this?)
WARNING
Bumetanide Injection, USP is a potent diuretic which, if given in excessive amounts, can lead to a profound diuresis with water and electrolyte depletion. Therefore, careful medical supervision is required, and dose and dosage schedule have to be adjusted to the individual patient’s needs. (See DOSAGE AND ADMINISTRATION.)
-
DESCRIPTION
Bumetanide is a loop diuretic, available as 4 mL vials and 10 mL vials (0.25 mg/mL) for intravenous or intramuscular injection as a sterile solution.
Each mL contains bumetanide 0.25 mg, sodium chloride 8.5 mg and ammonium acetate 4 mg as buffers, edetate disodium 0.1 mg and benzyl alcohol 10 mg as preservative in Water for Injection. pH adjusted to 6.8 – 7.8 with sodium hydroxide.
Chemically, bumetanide is 3-(butylamino)-4-phenoxy-5-sulfamoylbenzoic acid. It is a practically white powder, slightly soluble in water, soluble in alkaline solutions, having the following structural formula:
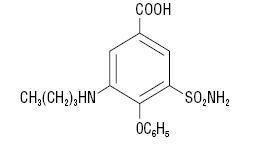
C17H20N2O5S Molecular weight: 364.42
-
CLINICAL PHARMACOLOGY
Bumetanide is a loop diuretic with a rapid onset and short duration of action. Pharmacological and clinical studies have shown that 1 mg bumetanide has a diuretic potency equivalent to approximately 40 mg furosemide. The major site of bumetanide action is the ascending limb of the loop of Henle.
The mode of action has been determined through various clearance studies in both humans and experimental animals. Bumetanide inhibits sodium reabsorption in the ascending limb of the loop of Henle, as shown by marked reduction of free-water clearance (CH2O) during hydration and tubular free-water reabsorption (TcH2O) during hydropenia. Reabsorption of chloride in the ascending limb is also blocked by bumetanide, and bumetanide is somewhat more chloruretic than natriuretic.
Potassium excretion is also increased by bumetanide, in a dose-related fashion.
Bumetanide may have an additional action in the proximal tubule. Since phosphate reabsorption takes place largely in the proximal tubule, phosphaturia during bumetanide-induced diuresis is indicative of this additional action. This is further supported by the reduction in the renal clearance of bumetanide by probenecid, associated with diminution in the natriuretic response. This proximal tubular activity does not seem to be related to an inhibition of carbonic anhydrase. Bumetanide does not appear to have a noticeable action on the distal tubule.
Bumetanide decreases uric acid excretion and increases serum uric acid. Diuresis starts within minutes following an intravenous injection and reaches maximum levels within 15 to 30 minutes.
Several pharmacokinetic studies have shown that bumetanide, administered orally or parenterally, is eliminated rapidly in humans, with a half-life of between 1 and 1 ½ hours. Plasma protein binding is in the range of 94% to 96%.
Oral administration of carbon-14 labeled bumetanide to human volunteers revealed that 81% of the administered radioactivity was excreted in the urine, 45% of it as unchanged drug. Urinary and biliary metabolites identified in this study were formed by oxidation of the N-butyl side chain. Biliary excretion of bumetanide amounted to only 2% of the administered dose.
Pediatric Pharmacology
Elimination of bumetanide appears to be considerably slower in neonatal patients compared with adults, possibly because of immature renal and hepatobiliary function in this population. Small pharmacokinetic studies of intravenous bumetanide in preterm and full-term neonates with respiratory disorders have reported an apparent half-life of approximately 6 hours with a range up to 15 hours and a serum clearance ranging from 0.2 to 11 mL/min/kg. In a population of neonates receiving bumetanide for volume overload, mean serum clearance rates were 2.17 mL/min/kg in patients less than 2 months of age and 3.8 mL/min/kg in patients aged 2 to 6 months. Mean serum half-life of bumetanide was 2.5 hours and 1.5 hours in patients aged less than 2 months and those aged 2 to 6 months, respectively. Elimination half-life decreased considerably during the first month of life, from a mean of approximately 6 hours at birth to approximately 2.4 hours at 1 month of age.
In preterm neonates, mean serum concentrations following a single 0.05 mg/kg dose ranged from 126 mcg/L at 1 hour to 57 mcg/L at 8 hours. In another study, mean serum concentrations following a single 0.05 mg/kg dose were 338 ng/mL at 30 minutes and 176 ng/mL after 4 hours. A single dose of 0.1 mg/kg produced mean serum levels of 314 ng/mL at 1 hour, and 196 ng/mL at 6 hours. Mean volume of distribution in neonates has been reported to range from 0.26 L/kg to 0.38 L/kg.
The degree of protein binding of bumetanide in cord sera from healthy neonates was approximately 97%, suggesting the potential for bilirubin displacement. A study using pooled sera from critically ill neonates found that bumetanide at concentrations of 0.5 to 50 mcg/mL, but not 0.25 mcg/mL, caused a linear increase in unbound bilirubin concentrations.
In 56 infants aged 4 days to 6 months, bumetanide doses ranging from 0.005 mg/kg to 0.1 mg/kg were studied for pharmacodynamic effect. Peak bumetanide excretion rates increased linearly with increasing doses of drug. Maximal diuretic effect was observed at a bumetanide excretion rate of about 7 mcg/kg/hr, corresponding to doses of 0.035 to 0.040 mg/kg. Higher doses produced a higher bumetanide excretion rate but no increase in diuretic effect. Urine flow rate peaked during the first hour after drug administration in 80% of patients and by 3 hours in all patients.
Geriatric Pharmacology
In a group of ten geriatric subjects between the ages of 65 and 73 years, total bumetanide clearance was significantly lower (1.8 ± 0.3 mL/min/kg) compared with younger subjects (2.9 ± 0.2 mL/min/kg) after a single oral bumetanide 0.5 mg dose. Maximum plasma concentrations were higher in geriatric subjects (16.9 ± 1.8 ng/mL) compared with younger subjects (10.3 ± 1.5 ng/mL). Urine flow rate and total excretion of sodium and potassium were increased less in the geriatric subjects compared with younger subjects, although potassium excretion and fractional sodium excretion were similar between the two age groups. Nonrenal clearance, bioavailability, and volume of distribution were not significantly different between the two groups.
-
INDICATIONS AND USAGE
Bumetanide Injection is indicated for the treatment of edema associated with congestive heart failure, hepatic and renal disease, including the nephrotic syndrome.
Almost equal diuretic response occurs after oral and parenteral administration of bumetanide. Therefore, if impaired gastrointestinal absorption is suspected or oral administration is not practical, bumetanide should be given by the intramuscular or intravenous route.
Successful treatment with bumetanide following instances of allergic reactions to furosemide suggests a lack of cross-sensitivity.
-
CONTRAINDICATIONS
Bumetanide is contraindicated in anuria. Although bumetanide can be used to induce diuresis in renal insufficiency, any marked increase in blood urea nitrogen or creatinine, or the development of oliguria during therapy of patients with progressive renal disease, is an indication for discontinuation of treatment with bumetanide. Bumetanide is also contraindicated in patients in hepatic coma or in states of severe electrolyte depletion until the condition is improved or corrected. Bumetanide is contraindicated in patients hypersensitive to this drug.
-
WARNINGS
Volume and Electrolyte Depletion
The dose of bumetanide should be adjusted to the patient’s need. Excessive doses or too frequent administration can lead to profound water loss, electrolyte depletion, dehydration, reduction in blood volume and circulatory collapse with the possibility of vascular thrombosis and embolism, particularly in elderly patients.
Hypokalemia
Hypokalemia can occur as a consequence of bumetanide administration. Prevention of hypokalemia requires particular attention in the following conditions: patients receiving digitalis and diuretics for congestive heart failure, hepatic cirrhosis and ascites, states of aldosterone excess with normal renal function, potassium-losing nephropathy, certain diarrheal states, or other states where hypokalemia is thought to represent particular added risks to the patient, i.e., history of ventricular arrhythmias.
In patients with hepatic cirrhosis and ascites, sudden alterations of electrolyte balance may precipitate hepatic encephalopathy and coma. Treatment in such patients is best initiated in the hospital with small doses and careful monitoring of the patient’s clinical status and electrolyte balance. Supplemental potassium and/or spironolactone may prevent hypokalemia and metabolic alkalosis in these patients.
Ototoxicity
In cats, dogs and guinea pigs, bumetanide has been shown to produce ototoxicity. In these test animals bumetanide was 5 to 6 times more potent than furosemide and, since the diuretic potency of bumetanide is about 40 to 60 times furosemide, it is anticipated that blood levels necessary to produce ototoxicity will rarely be achieved. The potential exists, however, and must be considered a risk of intravenous therapy, especially at high doses, repeated frequently in the face of renal excretory function impairment. Potentiation of aminoglycoside ototoxicity has not been tested for bumetanide. Like other members of this class of diuretics, bumetanide probably shares this risk.
-
PRECAUTIONS
General
Serum potassium should be measured periodically and potassium supplements or potassium-sparing diuretics added if necessary. Periodic determinations of other electrolytes are advised in patients treated with high doses or for prolonged periods, particularly in those on low salt diets.
Hyperuricemia may occur; it has been asymptomatic in cases reported to date. Reversible elevations of the BUN and creatinine may also occur, especially in association with dehydration and particularly in patients with renal insufficiency. Bumetanide may increase urinary calcium excretion with resultant hypocalcemia.
Diuretics have been shown to increase the urinary excretion of magnesium; this may result in hypomagnesemia.
Laboratory Tests
Studies in normal subjects receiving bumetanide revealed no adverse effects on glucose tolerance, plasma insulin, glucagon and growth hormone levels, but the possibility of an effect on glucose metabolism exists. Periodic determinations of blood sugar should be done, particularly in patients with diabetes or suspected latent diabetes.
Patients under treatment should be observed regularly for possible occurrence of blood dyscrasias, liver damage or idiosyncratic reactions, which have been reported occasionally in foreign marketing experience. The relationship of these occurrences to bumetanide use is not certain.
Drug Interactions
Drugs With Ototoxic Potential
(see WARNINGS )
Especially in the presence of impaired renal function, the use of parenterally administered bumetanide in patients to whom aminoglycoside antibiotics are also being given should be avoided, except in life-threatening conditions.
Drugs With Nephrotoxic Potential
There has been no experience with the concurrent use of bumetanide with drugs known to have a nephrotoxic potential. Therefore, the simultaneous administration of these drugs should be avoided.
Lithium
Lithium should generally not be given with diuretics (such as bumetanide) because they reduce its renal clearance and add a high risk of lithium toxicity.
Probenecid
Pretreatment with probenecid reduces both the natriuresis and hyperreninemia produced by bumetanide. This antagonistic effect of probenecid on bumetanide natriuresis is not due to a direct action on sodium excretion but is probably secondary to its inhibitory effect on renal tubular secretion of bumetanide. Thus, probenecid should not be administered concurrently with bumetanide.
Indomethacin
Indomethacin blunts the increases in urine volume and sodium excretion seen during bumetanide treatment and inhibits the bumetanide-induced increase in plasma renin activity. Concurrent therapy with bumetanide is thus not recommended.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Bumetanide was devoid of mutagenic activity in various strains of Salmonella typhimurium when tested in the presence or absence of an in vitro metabolic activation system. An 18-month study showed an increase in mammary adenomas of questionable significance in female rats receiving oral doses of 60 mg/kg/day (2000 times a 2 mg human dose). A repeat study at the same doses failed to duplicate this finding.
Reproduction studies were performed to evaluate general reproductive performance and fertility in rats at oral dose levels of 10, 30, 60 or 100 mg/kg/day. The pregnancy rate was slightly decreased in the treated animals; however, the differences were small and not statistically significant.
Pregnancy
Teratogenic Effects
Pregnancy Category C
Bumetanide is neither teratogenic nor embryocidal in mice when given in doses up to 3400 times the maximum human therapeutic dose.
Bumetanide has been shown to be nonteratogenic, but it has a slight embryocidal effect in rats when given in doses of 3400 times the maximum human therapeutic dose and in rabbits at doses of 3.4 times the maximum human therapeutic dose. In one study, moderate growth retardation and increased incidence of delayed ossification of sternebrae were observed in rats at oral doses of 100 mg/kg/day, 3400 times the maximum human therapeutic dose. These effects were associated with maternal weight reductions noted during dosing. No such adverse effects were observed at 30 mg/kg/day (1000 times the maximum human therapeutic dose). No fetotoxicity was observed at 1000 to 2000 times the human therapeutic dose.
In rabbits, a dose-related decrease in litter size and an increase in resorption rate were noted at oral doses of 0.1 and 0.3 mg/kg/day (3.4 and 10 times the maximum human therapeutic dose). A slightly increased incidence of delayed ossification of sternebrae occurred at 0.3 mg/kg/day; however, no such adverse effects were observed at the dose of 0.03 mg/kg/day. The sensitivity of the rabbit to bumetanide parallels the marked pharmacologic and toxicologic effects of the drug in this species.
Bumetanide was not teratogenic in the hamster at an oral dose of 0.5 mg/kg/day (17 times the maximum human therapeutic dose). Bumetanide was not teratogenic when given intravenously to mice and rats at doses up to 140 times the maximum human therapeutic dose.
There are no adequate and well-controlled studies in pregnant women. A small investigational experience in the United States and marketing experience in other countries to date have not indicated any evidence of adverse effects on the fetus, but these data do not rule out the possibility of harmful effects. Bumetanide should be given to a pregnant woman only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
It is not known whether this drug is excreted in human milk. As a general rule, nursing should not be undertaken while the patient is on bumetanide since it may be excreted in human milk.
Pediatric Use
Safety and effectiveness in pediatric patients below the age of 18 have not been established.
In vitro studies using pooled sera from critically ill neonates have shown bumetanide to be a potent displacer of bilirubin (see CLINICAL PHARMACOLOGY, Pediatric Pharmacology). The administration of bumetanide could present a particular concern if given to critically ill or jaundiced neonates at risk for kernicterus.
Geriatric Use
Clinical studies of bumetanide did not include sufficient numbers of subjects aged 65 and over to determine whether they responded differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
This drug is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function. Because elderly patients are more likely to have decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal function.
-
ADVERSE REACTIONS
The most frequent clinical adverse reactions considered probably or possibly related to bumetanide are muscle cramps (seen in 1.1% of treated patients), dizziness (1.1%), hypotension (0.8%), headache (0.6%), nausea (0.6%), and encephalopathy (in patients with preexisting liver disease) (0.6%). One or more of these adverse reactions have been reported in approximately 4.1% of bumetanide-treated patients.
Less frequent clinical adverse reactions to bumetanide are impaired hearing (0.5%), pruritus (0.4%), electrocardiogram changes (0.4%), weakness (0.2%), hives (0.2%), abdominal pain (0.2%), arthritic pain (0.2%), musculoskeletal pain (0.2%), rash (0.2%) and vomiting (0.2%). One or more of these adverse reactions have been reported in approximately 2.9% of bumetanide-treated patients.
Other clinical adverse reactions, which have each occurred in approximately 0.1% of patients, are vertigo, chest pain, ear discomfort, fatigue, dehydration, sweating, hyperventilation, dry mouth, upset stomach, renal failure, asterixis, itching, nipple tenderness, diarrhea, premature ejaculation and difficulty maintaining an erection.
Laboratory abnormalities reported have included hyperuricemia (in 18.4% of patients tested), hypochloremia (14.9%), hypokalemia (14.7%), azotemia (10.6%), hyponatremia (9.2%), increased serum creatinine (7.4%), hyperglycemia (6.6%), and variations in phosphorus (4.5%), CO2 content (4.3%), bicarbonate (3.1%) and calcium (2.4%). Although manifestations of the pharmacologic action of bumetanide, these conditions may become more pronounced by intensive therapy.
Also reported have been thrombocytopenia (0.2%) and deviations in hemoglobin (0.8%), prothrombin time (0.8%), hematocrit (0.6%), WBC (0.3%) and differential counts (0.1%). There have been rare spontaneous reports of thrombocytopenia from postmarketing experience.
Diuresis induced by bumetanide may also rarely be accompanied by changes in LDH (1.0%), total serum bilirubin (0.8%), serum proteins (0.7%), SGOT (0.6%), SGPT (0.5%), alkaline phosphatase (0.4%), cholesterol (0.4%) and creatinine clearance (0.3%). Increases in urinary glucose (0.7%) and urinary protein (0.3%) have also been seen.
-
OVERDOSAGE
Overdosage can lead to acute profound water loss, volume and electrolyte depletion, dehydration, reduction of blood volume and circulatory collapse with a possibility of vascular thrombosis and embolism. Electrolyte depletion may be manifested by weakness, dizziness, mental confusion, anorexia, lethargy, vomiting and cramps. Treatment consists of replacement of fluid and electrolyte losses by careful monitoring of the urine and electrolyte output and serum electrolyte levels.
-
DOSAGE AND ADMINISTRATION
Dosage should be individualized with careful monitoring of patient response.
Parenteral Administration
Bumetanide Injection may be administered parenterally (IV or IM) to patients in whom gastrointestinal absorption may be impaired or in whom oral administration is not practical.
Parenteral treatment should be terminated and oral treatment instituted as soon as possible.
The usual initial dose is 0.5 to 1 mg intravenously or intramuscularly. Intravenous administration should be given over a period of 1 to 2 minutes. If the response to an initial dose is deemed insufficient, a second or third dose may be given at intervals of 2 to 3 hours, but should not exceed a daily dosage of 10 mg.
Miscibility and Parenteral Solutions
The compatibility tests of bumetanide injection with 5% Dextrose Injection in Water, 0.9% Sodium Chloride Injection, and Lactated Ringer’s Injection in both glass and plasticized PVC (Viaflex) containers have shown no significant absorption effect with either containers, nor a measurable loss of potency due to degradation of the drug. However, solutions should be freshly prepared and used within 24 hours.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
-
HOW SUPPLIED
Bumetanide Injection, USP, 0.25 mg/mL is a sterile, clear, colorless to slightly yellow solution supplied in amber vials as follows:
4 mL Single Dose Vial packaged in 10s (NDC 0641-6008-10)
10 mL Multiple Dose Vial packaged in 10s (NDC 0641-6007-10)
This product, including the packaging components, is free of latex.
Storage
Store at 20° to 25°C (68° to 77°F), excursions permitted to 15° to 30° C (59° to 86°F) [See USP Controlled Room Temperature]. Protect from light.
To report SUSPECTED ADVERSE REACTIONS, contact Hikma Pharmaceuticals USA Inc. at 1-877-845-0689, or the FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
For Product Inquiry call 1-877-845-0689.
Manufactured by:
Hikma Pharmaceuticals USA Inc.
Berkeley Heights, NJ 07922Revised December 2019
462-485-02
- PRINCIPAL DISPLAY PANEL
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BUMETANIDE
bumetanide injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0641-6007 Route of Administration INTRAMUSCULAR, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BUMETANIDE (UNII: 0Y2S3XUQ5H) (BUMETANIDE - UNII:0Y2S3XUQ5H) BUMETANIDE 0.25 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 8.5 mg in 1 mL AMMONIUM ACETATE (UNII: RRE756S6Q2) 4 mg in 1 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 0.1 mg in 1 mL BENZYL ALCOHOL (UNII: LKG8494WBH) 10 mg in 1 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0641-6007-10 10 in 1 CARTON 04/30/2008 1 NDC:0641-6007-01 10 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA079196 04/30/2008 BUMETANIDE
bumetanide injectionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0641-6008 Route of Administration INTRAMUSCULAR, INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BUMETANIDE (UNII: 0Y2S3XUQ5H) (BUMETANIDE - UNII:0Y2S3XUQ5H) BUMETANIDE 0.25 mg in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM CHLORIDE (UNII: 451W47IQ8X) 8.5 mg in 1 mL AMMONIUM ACETATE (UNII: RRE756S6Q2) 4 mg in 1 mL EDETATE DISODIUM (UNII: 7FLD91C86K) 0.1 mg in 1 mL BENZYL ALCOHOL (UNII: LKG8494WBH) 10 mg in 1 mL WATER (UNII: 059QF0KO0R) SODIUM HYDROXIDE (UNII: 55X04QC32I) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0641-6008-10 10 in 1 CARTON 04/30/2008 1 NDC:0641-6008-01 4 mL in 1 VIAL; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date ANDA ANDA079196 04/30/2008 Labeler - Hikma Pharmaceuticals USA Inc. (946499746) Establishment Name Address ID/FEI Business Operations Hikma Pharmaceuticals USA Inc. 946499746 analysis(0641-6008, 0641-6007) , label(0641-6008, 0641-6007) , manufacture(0641-6008, 0641-6007)



