Label: MYOBLOC- rimabotulinumtoxinb injection, solution



- NDC Code(s): 10454-710-10, 10454-711-10, 10454-712-10
- Packager: Solstice Neurosciences, LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated October 22, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use MYOBLOC ® safely and effectively. See full prescribing information for MYOBLOC ®.
MYOBLOC ® (rimabotulinumtoxinB) injection, for intramuscular or intraglandular use
Initial U.S. Approval: 2000WARNING: DISTANT SPREAD OF TOXIN EFFECT
See full prescribing information for complete boxed warning.
The effects of MYOBLOC and all botulinum toxin products may spread from the area of injection to produce symptoms consistent with botulinum toxin effects. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults, particularly in those patients who have an underlying condition that would predispose them to these symptoms ( 5.1).
INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
- Cervical Dystonia: for patients with demonstrated tolerance of botulinum toxin injection, recommended total dosage is 2,500 Units to 5,000 Units divided among effected muscles ( 2.2)
- Chronic Sialorrhea: recommended dosage is 1,500 Units to 3,500 Units; 500 Units to 1,500 Units per parotid gland and 250 Units per submandibular gland; no more frequent than every 12 weeks ( 2.3)
DOSAGE FORMS AND STRENGTHS
Injection: 2,500 Units/0.5 mL; 5,000 Units/mL; or 10,000 Units/2 mL (5,000 Units/mL) in a single-dose vial ( 3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Spread of toxin effects; swallowing and breathing difficulties can lead to death. Seek immediate medical attention if respiratory, speech or swallowing difficulties occur ( 5.1, 5.4)
- MYOBLOC potency units cannot be compared to or converted into units of other botulinum toxins ( 5.2)
- Patients with neuromuscular disorders should be monitored closely for swallowing/breathing difficulty ( 5.4)
ADVERSE REACTIONS
The most common adverse reactions (>5% of MYOBLOC-treated patients at any dose and >5% more common than placebo) include:
- Cervical Dystonia: dry mouth, dysphagia, injection site pain, and headache ( 6.1)
- Chronic Sialorrhea: dry mouth and dysphagia ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Solstice Neurosciences at 1-888-461-2255 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2021
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: DISTANT SPREAD OF TOXIN EFFECT
1 INDICATIONS AND USAGE
1.1 Cervical Dystonia
1.2 Chronic Sialorrhea
2 DOSAGE AND ADMINISTRATION
2.1 Instructions for Safe Use
2.2 Dosing for Cervical Dystonia
2.3 Dosing for Chronic Sialorrhea
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Spread of Toxin Effect
5.2 Lack of Interchangeability between Botulinum Toxin Products
5.3 Hypersensitivity Reactions
5.4 Dysphagia and Breathing Difficulties
5.5 Human Albumin and Transmission of Viral Diseases
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
6.3 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Aminoglycosides and Other Agents Interfering with Neuromuscular Transmission
7.2 Anticholinergic Drugs
7.3 Other Botulinum Neurotoxin Products
7.4 Muscle Relaxants
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Cervical Dystonia
14.2 Chronic Sialorrhea
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: DISTANT SPREAD OF TOXIN EFFECT
Postmarketing reports indicate that the effects of MYOBLOC and all botulinum toxin products may spread from the area of injection to produce symptoms consistent with botulinum toxin effects. These may include asthenia, generalized muscle weakness, diplopia, blurred vision, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence, and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death. The risk of symptoms is probably greatest in children treated for spasticity but symptoms can also occur in adults treated for spasticity and other conditions, particularly in those patients who have underlying conditions that would predispose them to these symptoms. In unapproved uses, including spasticity in children and adults, and in approved indications, cases of spread of effect have occurred at doses comparable to those used to treat cervical dystonia and at lower doses [see Warnings and Precautions (5.1)].
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Instructions for Safe Use
The potency units of MYOBLOC are specific to the preparation and assay method utilized. They are not interchangeable with other preparations of botulinum toxin products and cannot be compared to or converted into units of any other botulinum toxin products [see Warnings and Precautions (5.2), Description (11)] .
Each single-dose vial should only be used during one session and only for one patient. Discard any remaining solution in the vial.
MYOBLOC is ready to use; no reconstitution required.
MYOBLOC may be diluted with 0.9% Sodium Chloride Injection. Once diluted, the product must be used within 4 hours as the formulation does not contain a preservative.
2.2 Dosing for Cervical Dystonia
The recommended initial dosage of MYOBLOC for cervical dystonia patients with a prior history of tolerating botulinum toxin injections is 2,500 Units to 5,000 Units divided among affected muscles [see Clinical Studies (14.1)] . Patients without a prior history of tolerating botulinum toxin injections should receive a lower initial dosage [see Adverse Reactions (6.1)] . Subsequent dosing should be determined by the patient's individual response. MYOBLOC should be administered by physicians familiar with and experienced in the assessment and management of patients with cervical dystonia.
The duration of effect in patients responding to MYOBLOC treatment for cervical dystonia has been observed in studies to be between 12 and 16 weeks at doses of 5,000 Units or 10,000 Units [see Clinical Studies (14.1)] .
2.3 Dosing for Chronic Sialorrhea
Dosing Information
The recommended dosage of MYOBLOC for chronic sialorrhea is 1,500 Units to 3,500 Units, divided among the parotid and submandibular glands (Table 1). Patient response to treatment should be considered when determining subsequent MYOBLOC dosage [see Clinical Studies (14.2)] . The typical duration of effect of each treatment is up to 3 months; however, the effect may vary in individual patients. The frequency of MYOBLOC repeat treatments should be determined by clinical response but should generally be no more frequent than every 12 weeks.
Table 1: Dosing by Gland for Chronic Sialorrhea in Adults Gland Recommended Dosage Parotid 500 Units to 1,500 Units per gland Submandibular 250 Units per gland Administration Information
A suitable sterile needle (e.g., 30-gauge, 0.5 inch) should be used for intra-salivary gland administration.
Figure 1: Glands for Injection in Chronic Sialorrhea
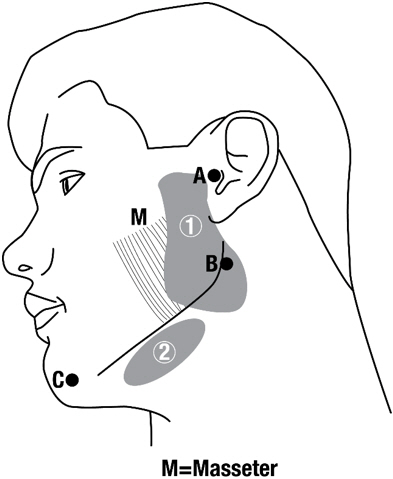
Guidelines for locating salivary glands using anatomical landmarks (Figure 1):
- 1. To inject the parotid gland, bisect the distance between the tip of the tragus (Site A) and the angle of the mandible (Site B). Inject one finger breadth anterior to this site (Injection Site 1).
- 2. To inject the submandibular gland, bisect the distance between the angle of the mandible (Site B) and the tip of the chin (Site C). Inject one finger breadth medial to the inferior surface of the point of bisection (Injection Site 2).
In clinical studies, MYOBLOC was injected using anatomical landmarks to localize the salivary glands, or using ultrasound guidance for gland location. Both methods produced similar reduction in the unstimulated salivary flow rate.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
MYOBLOC is contraindicated in patients with:
- A known hypersensitivity to any botulinum toxin product or to any of the components in the formulation [see Warnings and Precautions (5.3), Description (11)]
- Infection at the proposed injection site(s)
-
5 WARNINGS AND PRECAUTIONS
5.1 Spread of Toxin Effect
Postmarketing safety data from MYOBLOC and other approved botulinum toxins suggest that botulinum toxin effects may, in some cases, be observed beyond the site of local injection. The symptoms are consistent with the mechanism of action of botulinum toxin and may include asthenia, generalized muscle weakness, diplopia, blurred vision, ptosis, dysphagia, dysphonia, dysarthria, urinary incontinence, and breathing difficulties. These symptoms have been reported hours to weeks after injection. Swallowing and breathing difficulties can be life threatening and there have been reports of death related to spread of toxin effects. The risk of symptoms is probably greatest in children treated for spasticity, but symptoms can also occur in adults treated for spasticity and other conditions, and particularly in those patients who have underlying conditions that would predispose them to these symptoms. In unapproved uses, including spasticity in children and adults, and in approved indications, symptoms consistent with spread of toxin effect have been reported at doses comparable to or lower than doses used to treat cervical dystonia.
5.2 Lack of Interchangeability between Botulinum Toxin Products
The potency units of MYOBLOC are specific to the preparation and biological activity assay method utilized. Due to differences in the aspects of this assay such as the vehicle, dilution scheme, and laboratory protocols for various potency assays, potency units are not interchangeable with other preparations of botulinum toxin products and, therefore, units of biological activity of MYOBLOC cannot be compared to or converted into units of any other botulinum toxin products assessed with any other specific assay method [see Description (11)] .
5.3 Hypersensitivity Reactions
Serious hypersensitivity reactions have been reported with botulinum toxin products. Angioedema, urticaria, and rash have occurred with MYOBLOC treatment [see Adverse Reactions (6.3)]. Hypersensitivity reactions can also include anaphylaxis, serum sickness, soft tissue edema, and dyspnea. If serious and/or immediate hypersensitivity reactions occur, discontinue further injection of MYOBLOC and institute appropriate medical therapy immediately. The use of MYOBLOC in patients with a known hypersensitivity to any botulinum neurotoxin or to any of the excipients (human albumin, sucrose), could lead to a life-threatening allergic reaction [see Contraindications (4), Adverse Reactions (6.3)].
5.4 Dysphagia and Breathing Difficulties
Treatment with MYOBLOC and other botulinum toxin products can result in swallowing or breathing difficulties. Patients with pre-existing swallowing or breathing difficulties may be more susceptible to these complications. In most cases, this is a consequence of weakening of muscles in the area of injection that are involved in breathing or swallowing. When distant effects occur, additional respiratory muscles may be involved.
Deaths as a complication of severe dysphagia have been reported after treatment with botulinum toxin. Dysphagia may persist for several months and require use of a feeding tube to maintain adequate nutrition and hydration. Aspiration may result from severe dysphagia and is a particular risk when treating patients in whom swallowing or respiratory function is already compromised.
Treatment of cervical dystonia with botulinum toxins may weaken neck muscles that serve as accessory muscles of ventilation. This may result in a critical loss of breathing capacity in patients with respiratory disorders who may have become dependent upon these accessory muscles. There have been postmarketing reports of serious breathing difficulties, including respiratory failure, in cervical dystonia patients. Patients treated with botulinum toxin may require immediate medical attention should they develop problems with swallowing, speech or respiratory disorders. These reactions can occur within hours to weeks after injection with botulinum toxin [see Adverse Reactions (6.1)] .
Individuals with peripheral motor neuropathic diseases, amyotrophic lateral sclerosis, or neuromuscular junctional disorders (e.g., myasthenia gravis or Lambert-Eaton syndrome) should be monitored particularly closely when given botulinum toxin. Patients with neuromuscular disorders may be at increased risk of clinically significant effects including severe dysphagia and respiratory compromise from typical doses of MYOBLOC [see Adverse Reactions (6.1)] .
5.5 Human Albumin and Transmission of Viral Diseases
This product contains albumin, a derivative of human blood. Based on effective donor screening and product manufacturing processes, it carries an extremely remote risk for transmission of viral diseases and variant Creutzfeldt-Jakob disease (vCJD). There is a theoretical risk for transmission of Creutzfeldt-Jakob disease (CJD), but if that risk actually exists, the risk of transmission would be considered extremely remote. No cases of transmission of viral diseases, CJD, or vCJD have ever been identified for licensed albumin or albumin contained in other licensed products.
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions to MYOBLOC are discussed in greater detail in other sections of the labeling:
- Spread of Toxin Effect [see Warnings and Precautions (5.1)]
- Lack of Interchangeability Between Botulinum Toxin Products [see Warnings and Precautions (5.2)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.3)]
- Dysphagia and Breathing Difficulties [see Warnings and Precautions (5.4)]
- Human Albumin and Transmission of Viral Diseases [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in clinical trials of another drug and may not reflect the rates observed in practice.
Cervical Dystonia
In the treatment of cervical dystonia, MYOBLOC was studied in both placebo-controlled single treatment studies and open-label repeated treatment studies; most treatment sessions and patients were in the uncontrolled studies. The data described below reflect exposure to MYOBLOC at varying dosages in 570 subjects, including more than 300 patients with 4 or more treatment sessions. Most treatment sessions were at dosages of 12,500 Units or less.
Adverse reactions occurring in at least 5% of patients exposed to MYOBLOC treatment in pooled placebo-controlled clinical trials are shown in Table 2. The mean age of the population in these studies was 55 years, and approximately 66% were female. Most of the patients studied were Caucasian, and all had moderate to severe symptoms of cervical dystonia.
The most common adverse reactions (greater than 5% of MYOBLOC-treated patients at any dosage and at least 5% more common than placebo) in studies of cervical dystonia (Studies 1, 2, and 4) were dry mouth, dysphagia, injection site pain, and headache. Dry mouth and dysphagia were the adverse reactions most frequently resulting in discontinuation of treatment. There was an increased incidence of dysphagia with increased dose in the sternocleidomastoid muscle. The incidence of dry mouth showed some dose-related increase with doses injected into the splenius capitis, trapezius, and sternocleidomastoid muscles.
In the cervical dystonia program, only nine patients without a prior history of tolerating injections of type A botulinum toxin have been studied. Adverse reaction rates have not been adequately evaluated in these patients and may be higher than those described in Table 2.
Table 2: Adverse Reactions in at Least 5% of MYOBLOC-Treated Patients and Greater than Placebo, Following Single Treatment Session in Controlled Cervical Dystonia Studies (Studies 1, 2, and 4) Adverse Reaction MYOBLOC 2,500 Units
(N=31)
%MYOBLOC 5,000 Units
(N=67)
%MYOBLOC 10,000 Units
(N=106)
%Placebo
(N=104)
%Dry Mouth 3 12 34 3 Dysphagia 16 10 25 3 Injection Site Pain 16 12 15 9 Pain 6 6 13 10 Headache 10 16 11 8 Dyspepsia 3 0 10 5 Flu Syndrome 6 9 8 4 Arthralgia 0 1 7 5 Back Pain 3 4 7 3 Cough Increased 3 6 7 3 Asthenia 3 0 6 4 Dizziness 3 3 6 2 In the overall clinical trial experience with MYOBLOC in cervical dystonia (570 patients, including the uncontrolled studies), most cases of dry mouth or dysphagia were reported as mild or moderate in severity. Severe dysphagia was reported by 3% of patients [see Warnings and Precautions (5.4)]. Severe dry mouth was reported by 6% of patients. Dysphagia and dry mouth were the most frequent adverse reactions reported as a reason for discontinuation from repeated treatment studies. These adverse reactions led to discontinuation from further treatments with MYOBLOC in some patients even when not reported as severe.
The following additional adverse events were reported in 2% or greater of patients participating in any of the clinical studies in cervical dystonia (by body system):
Body as a Whole: chest pain, chills, hernia, malaise, abscess, cyst, viral infection; Respiratory: dyspnea, pneumonia; Nervous System: migraine; anxiety, hyperesthesia, vertigo, vasodilation; Digestive System: gastrointestinal disorder; Skin and Appendages: pruritis; Urogenital System: urinary tract infection, cystitis; Special Senses: amblyopia, abnormal vision; Metabolic and Nutritional Disorders: edema; Hemic and Lymphatic System: ecchymosis.
Chronic Sialorrhea
In the double-blind placebo-controlled studies (Study 1 and Study 2), 166 patients were treated with a single treatment of MYOBLOC (1,500 Units; 2,500 Units; or 3,500 Units) and 75 patients received placebo. The mean age of patients treated with MYOBLOC in these studies was 65 years; 83% of the patients were male; and 95% were White. Four MYOBLOC-treated patients and three patients on placebo discontinued because of an adverse event. One patient discontinued because of dry mouth (3,500 Unit dose). The adverse reactions that occurred in at least 5% of MYOBLOC-treated patients and were more frequent than placebo are shown in Table 3.
Table 3: Adverse Reactions in At Least 5% of MYOBLOC-Treated Patients and Greater than Placebo in Pooled Chronic Sialorrhea Studies (Studies 1 and 2) Adverse Reaction MYOBLOC 1,500 Units
(N=14) *
%MYOBLOC 2,500 Units
(N=75) †
%MYOBLOC 3,500 Units
(N=77) †
%Placebo
(N=75)
%Dry mouth 14 36 39 7 Dental caries 0 7 5 3 Dysphagia 0 9 4 3 6.2 Immunogenicity
As with all therapeutic proteins, there is potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies in the studies described below with the incidence of antibodies in other studies or to other rimabotulinumtoxinB products may be misleading.
Cervical Dystonia
A two-stage assay was used to test for immunogenicity and neutralizing activity induced by treatment with MYOBLOC. In order to account for varying lengths of follow-up, life-table analysis methods were used to estimate the rates of development of immune responses and neutralizing activity. During the repeated treatment studies, 446 subjects were followed with periodic ELISA based evaluations for development of antibody responses against MYOBLOC. Only patients who showed a positive ELISA assay were subsequently tested for the presence of neutralizing activity against MYOBLOC in the mouse neutralization assay (MNA). 12% of patients had positive ELISA assays at baseline. Patients began to develop new ELISA responses after a single treatment session with MYOBLOC. By six months after initiating treatment, estimates for ELISA positive rate were 20%, which continued to rise to 36% at one year and 50% positive ELISA status at 18 months. Serum neutralizing activity was primarily not seen in patients until after 6 months. Estimated rates of development were 10% at one year and 18% at 18 months in the overall group of patients, based on analysis of samples from ELISA positive individuals. The effect of conversion to ELISA or MNA positive status on efficacy was not evaluated in these studies, and the clinical significance of development of antibodies has not been determined.
The data reflect the percentage of patients whose test results were considered positive for antibodies to MYOBLOC in both an in vitro and in vivo assay. The results of these antibody tests are highly dependent on the sensitivity and specificity of the assays. Additionally, the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to MYOBLOC with the incidence of antibodies to other products may be misleading.
6.3 Postmarketing Experience
The following adverse reactions have been reported during postmarketing use of MYOBLOC: angioedema, urticaria, rash, constipation, dry eye, and accommodation disorder. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
-
7 DRUG INTERACTIONS
7.1 Aminoglycosides and Other Agents Interfering with Neuromuscular Transmission
Co-administration of MYOBLOC and aminoglycosides or other agents interfering with neuromuscular transmission (e.g., curare-like compounds) should only be performed with caution as the effect of the toxin may be potentiated.
7.2 Anticholinergic Drugs
Use of anticholinergic drugs after administration of MYOBLOC may potentiate systemic anticholinergic effects.
7.3 Other Botulinum Neurotoxin Products
The effect of administering different botulinum toxin products at the same time or within several months of each other is unknown. Excessive neuromuscular weakness may be exacerbated by administration of another botulinum toxin prior to the resolution of the effects of a previously administered botulinum toxin.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate data on the developmental risks associated with the use of MYOBLOC in pregnant women. No developmental toxicity was observed in pregnant rats administered MYOBLOC by intramuscular injection during gestation and lactation, at doses producing maternal toxicity.
In the U.S. general population, the background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
When MYOBLOC was administered by intramuscular injection to pregnant rats (0, 300, 1000, or 3000 Units/kg/day) or rabbits (0, 0.03, 0.1, 0.3, or 1.0 Units/kg/day) throughout gestation, no adverse effects on embryofetal development were observed. The highest dose tested in rat, which was associated with maternal toxicity, was 36 times the maximum recommended human dose (MRHD) for cervical dystonia (5000 Units) on a body weight (Units/kg) basis. The highest dose tested in rabbit was substantially less than the MRHD for cervical dystonia on a Units/kg basis; maternal toxicity was observed at all but the lowest dose tested.
8.2 Lactation
Risk Summary
There are no data on the presence of MYOBLOC in human milk, the effects on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for MYOBLOC and any potential adverse effects on the breastfed infant from MYOBLOC or from the underlying maternal condition.
8.5 Geriatric Use
Cervical Dystonia
In the controlled studies for MYOBLOC in patients with cervical dystonia, 152 (75%) were under the age of 65, and 52 (26%) were 65 years of age or older [see Clinical Studies (14.1)] . For these age groups, the most frequently reported adverse reactions occurred at similar rates in both age groups. Efficacy results did not suggest any large differences between these age groups.
Very few patients age 75 or older were enrolled; therefore, no conclusions regarding the safety and efficacy of MYOBLOC within this age group can be determined.
Chronic Sialorrhea
Of the 166 MYOBLOC-treated patients in the placebo-controlled studies for treatment of chronic sialorrhea [see Clinical Studies (14.2)] , 105 (63%) were 65 years of age or older, and 43 (26%) were 75 years of age or older. No overall differences in safety or effectiveness were observed between patients over 65 years of age and younger patients, but greater sensitivity of some older patients cannot be ruled out.
-
10 OVERDOSAGE
Excessive doses of MYOBLOC may be expected to produce neuromuscular weakness with a variety of symptoms. Respiratory support may be required where excessive doses cause paralysis of respiratory muscles. In the event of overdose, the patient should be medically monitored for symptoms of excessive muscle weakness or muscle paralysis [see Warnings and Precautions (5.1, 5.4)] . Symptomatic treatment may be necessary.
Symptoms of overdose are likely not to be present immediately following injection. Should accidental injection or oral ingestion occur, the person should be medically supervised for several weeks for signs and symptoms of excessive muscle weakness or muscle paralysis.
In the event of overdose, antitoxin raised against botulinum toxin is available from the Centers for Disease Control and Prevention (CDC) in Atlanta, GA. However, the antitoxin will not reverse any botulinum toxin-induced effects already apparent by the time of antitoxin administration. In the event of suspected or actual cases of botulinum toxin poisoning, please contact your local or state Health Department to process a request for antitoxin through the CDC. If you do not receive a response within 30 minutes, please contact the CDC directly at 770-488-7100. More information can be obtained at http://cdc.gov/ncidod/srp/drugs/drug-service.html.
-
11 DESCRIPTION
RimabotulinumtoxinB is an acetylcholine release inhibitor. RimabotulinumtoxinB is a 700 kDA botulinum toxin type B complex produced from fermentation of the bacterium Clostridium botulinum type B (Bean strain) and exists in noncovalent association with hemagglutinin and nonhemagglutinin proteins as a neurotoxin complex. The neurotoxin complex is recovered from the fermentation process and purified through a series of precipitation and chromatography steps.
MYOBLOC (rimabotulinumtoxinB) injection is a sterile, preservative-free, clear and colorless to light-yellow solution in a single-dose vial for intramuscular or intraglandular use. Each vial contains 2,500 Units/0.5 mL; 5,000 units/mL; or 10,000 Units/2 mL of rimabotulinumtoxinB at a concentration of 5,000 Units/mL at approximately pH 5.6.
Each 2,500 Units/0.5 mL vial of MYOBLOC contains 2,500 Units rimabotulinumtoxinB, 0.235 mg albumin human, 2.9 mg sodium chloride and 1.35 mg sodium succinate.
Each 5,000 Units/mL vial of MYOBLOC contains 5,000 Units rimabotulinumtoxinB, 0.47 mg albumin human, 5.8 mg sodium chloride and 2.7 mg sodium succinate.
Each 10,000 Units/2 mL vial of MYOBLOC contains 10,000 Units rimabotulinumtoxinB, 0.94 mg albumin human, 11.6 mg sodium chloride and 5.4 mg sodium succinate.
One unit of MYOBLOC corresponds to the calculated median lethal intraperitoneal dose (LD50) in mice. The method for performing the assay is specific to Solstice Neurosciences' manufacture of MYOBLOC. Due to differences in specific details such as the vehicle, dilution scheme and laboratory protocols for various mouse LD50 assays, Units of biological activity of MYOBLOC cannot be compared to or converted into Units of any other botulinum toxin or any toxin assessed with any other specific assay method. Therefore, differences in species sensitivities to different botulinum neurotoxin serotypes preclude extrapolation of animal dose-activity relationships to human dose estimates. The specific activity of MYOBLOC ranges between 70 to 130 Units/ng.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
MYOBLOC blocks cholinergic transmission at the neuromuscular and salivary neuroglandular junction by inhibiting the release of acetylcholine from peripheral cholinergic nerved terminals. This inhibition occurs according to the following sequence: neurotoxin binding to cholinergic nerve terminals, internalization of the neurotoxin into the nerve terminal, translocation of the light-chain part of the molecule into the cytosol of the nerve terminal, and enzymatic cleavage of synaptic Vesicle Associated Membrane Protein (VAMP, also known as synaptobrevin), a presynaptic target protein essential for the release of acetylcholine. In both muscles and glands, impulse transmission is re-established by the formation of new nerve endings.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Impairment of Fertility
Intramuscular administration of MYOBLOC (0, 300, 1000, or 3000 Units/kg/day) to male and female rats prior to and throughout the mating period and continuing in females to gestation day 6 resulted in decreases in implantation sites and viable fetuses at the high dose of 3000 Units/kg/day, which was associated with maternal toxicity. The no-effect dose for reproductive toxicity (1000 Units/kg/day) is 12 times the maximum recommended human dose for cervical dystonia (5000 Units) on a body weight (Units/kg) basis.
-
14 CLINICAL STUDIES
14.1 Cervical Dystonia
Two Phase 3, randomized, multi-center, double-blind, placebo-controlled studies of the treatment of cervical dystonia were conducted (Study 1 and Study 2). Both studies enrolled only adult patients who had a history of receiving botulinum toxin type A in an open-label manner, with a perceived good response and tolerable adverse effects. Study 1 enrolled patients who were perceived as having an acceptable response to type A toxin, while Study 2 enrolled only patients who had secondarily lost responsiveness to type A toxin. Other eligibility criteria common to both studies were that all patients had moderate or greater severity of cervical dystonia with at least 2 muscles involved, no neck contractures or other causes of decreased neck range of motion, and no history of any other neuromuscular disorder. Patients in Study 1 were randomized to receive placebo, MYOBLOC 5,000 Units or MYOBLOC 10,000 Units. Patients in Study 2 were randomized to receive placebo or 10,000 Units of MYOBLOC. The study agent was administered to subjects in a single treatment session by investigators who selected 2 to 4 muscles per subject from the following: splenius capitis, sternocleidomastoid, levator scapulae, trapezius, semispinalis capitis, and scalene muscles. The total dose was divided between the selected muscles, and from 1 to 5 injections were made per muscle. There were 109 patients enrolled into Study 1, and 77 into Study 2. Patient evaluations continued for 16 weeks post injection.
The primary efficacy outcome variable for both studies was the Toronto Western Spasmodic Torticollis Rating Scale (TWSTRS)-Total Score (scale range of possible scores is 0–87) at Week 4. TWSTRS is comprised of three sub-scales which examine 1) Severity—the severity of the patient's abnormal head position; 2) Pain—the severity and duration of pain due to the dystonia; and 3) Disability— the effects of the abnormal head position and pain on a patient's activities. The secondary endpoints were the Patient Global and Physician Global Assessments of change at Week 4. Both Global Assessments used a 100-point visual-analog scale (VAS). The Patient Global Assessment allows patients to indicate how they feel at the time of their evaluation compared to the pre-injection baseline. Likewise, the Physician Global Assessment indicates the physician's assessment of a patient's change from baseline to Week 4. Scores of 50 indicate no change, 0 much worse, and 100 much better. Results of comparisons of the primary and secondary efficacy variables are summarized in Table 4.
Table 4: Efficacy Results From Two Phase 3 MYOBLOC Studies in Cervical Dystonia STUDY 1 STUDY 2 Assessments * Placebo
(N=36)MYOBLOC 5,000 Units
(N=36)MYOBLOC 10,000 Units
(N=37)Placebo
(N=38)MYOBLOC 10,000 Units
(N=39)- *
- 95% Cl are for the differences between the active and placebo groups. The P values are for the comparison of active dose and placebo. For TWSTRS-Total and TWSTRS-subscale scores, P values are from ANCOVA for each variable with center and treatment in the model and the baseline value of the variable included as a covariate. For the Patient Global and Physician Global Assessments, P values are from ANOVA for each variable with center and treatment in the model.
TWSTRS Total Mean at Baseline 43.6 46.4 46.9 51.2 52.8 Change from Baseline -4.3 -9.3 -11.7 -2.0 -11.1 95% Confidence Interval (-8.9, -1.2) (-11.1, -3.3) (-12.2, -5.2) P value 0.012 0.0004 0.0001 Patient Global Mean at Week Four 43.6 60.6 64.6 39.5 60.2 95% Confidence Interval (7.0, 26.9) (11.3, 31.1) (11.2, 29.1) P value 0.001 0.0001 0.0001 Physician Global Mean at Week Four 52.0 65.3 64.2 47.9 60.6 95% Confidence Interval (5.5, 21.3) (3.9, 19.7) (7.4, 18.1) P value 0.001 0.004 0.0001 TWSTRS-Subscales – Severity Mean at Baseline 18.4 20.2 20.2 22.1 22.6 Change from Baseline -2.3 -3.2 -4.8 -1.2 -3.7 95% Confidence Interval (-2.5, 0.6) (-4.0, -1.0) (-3.9, -1.0) P value 0.22 0.002 0.001 – Pain Mean at Baseline 10.9 11.8 12.4 12.2 11.9 Change from Baseline -0.5 -3.6 -4.2 -0.2 -3.6 95% Confidence Interval (-4.7, -1.1) (-5.1, -1.4) (-5.0, -2.1) P value 0.002 0.0008 0.0001 – Disability Mean at Baseline 14.3 14.4 14.4 16.9 18.3 Change from Baseline -1.6 -2.5 -2.7 0.8 -3.8 95% Confidence Interval (-2.7, 0.7) (-2.8, 0.6) (-4.1, -1.0) P value 0.26 0.19 0.002 There were no statistically significant differences in results between the 5,000 Unit and 10,000 Unit doses in Study 1. Exploratory analyses of these two studies suggested that the majority of patients who showed a beneficial response by Week 4 had returned to their baseline status between Weeks 12 to 16 post injection. Although there was a MYOBLOC-associated decrease in pain, there remained many patients who experienced an increase in dystonia-related neck pain irrespective of treatment group [ See Adverse Reactions, (6.1)] . TWSTRS Total Score at Week 4 and Patient Global Assessment among subgroups by gender or age showed consistent treatment-associated effects across these subgroups [see Use in Specific Populations (8.5)]. There were too few non-Caucasian patients enrolled to draw any conclusions regarding relative efficacy in racial subsets.
MYOBLOC was studied in two Phase 2 dose-ranging studies, Studies 3 and 4, which preceded the Phase 3 studies. Studies 3 and 4 had a study design similar to the Phase 3 studies, including eligibility criteria. Study 3 enrolled 85 patients randomized to placebo, MYOBLOC 400 Units, MYOBLOC 1,200 Units, or MYOBLOC 2,400 Units (21 or 22 patients per group). Study 4 enrolled 122 patients randomized to placebo, MYOBLOC 2,500 Units, MYOBLOC 5,000 Units, or MYOBLOC 10,000 Units (30 or 31 patients per group). These studies demonstrated efficacy on the TWSTRS-Total, baseline to Week 4, at doses of 2,400 Units; 2,500 Units; 5,000 Units; and 10,000 Units. Study 3 showed mean improvement from baseline on the Week 4 TWSTRS for placebo and 2,400 Units of 2.0 and 8.5 points respectively (from baselines of 42.0 and 42.4 points). Study 4 showed mean improvement from baseline to Week 4 for placebo, MYOBLOC 2,500 Units, MYOBLOC 5,000 Units, and MYOBLOC 10,000 Units of 3.3, 11.6, 12.5, and 16.4 points, respectively (from baseline of 45.5, 45.6, 45.2, and 47.5 points). Study 3 also showed less response for doses below 2,400 Units.
Study 5 was an open-label, intrapatient dose-escalation study of 3 treatment sessions where each patient with cervical dystonia sequentially received 10,000 Units; 12,500 Units; and 15,000 Units of MYOBLOC, at periods of 12 to 16 weeks between treatment sessions irrespective of their response to their previous dose. This study enrolled 145 patients, of whom 125 received all three treatments. Although this was an open-label design where investigators and patients knew the dose at each treatment session, there were similar mean improvements on the TWSTRS-Total, from baseline to Week 4, for all three doses.
In the MYOBLOC-treated patients (n=112) of the Phase 3 studies, 19% had 2 muscles injected, 48% had 3 muscles injected, and 33% had 4 muscles injected. Table 5 indicates the frequency of use for each of the permitted muscles, and the fraction of the total dose of the treatment injected into each muscle, for those patients in whom the muscle was injected.
Table 5: Study 1 and Study 2 Combined Data Fraction of Total Dose Injected into Involved Muscles in Patients with Cervical Dystonia Muscle Injected Percent Frequency Injected * Fraction of Total Dose Injected by Percentiles 25th 50th 75th - *
- Percent frequency of patients in whom each muscle was injected
Splenius Capitis 88 0.30 0.40 0.50 Sternocleidomastoid 80 0.20 0.25 0.30 Semispinalis Capitis 52 0.30 0.36 0.50 Levator Scapulae 46 0.13 0.20 0.20 Trapezius 38 0.20 0.25 0.35 Scalene Complex 13 0.20 0.25 0.30 14.2 Chronic Sialorrhea
Study 1
Study 1 (NCT01994109) was a multicenter, randomized, double-blind, placebo-controlled study of a single treatment of chronic sialorrhea (with 13-week follow-up), followed by an open-label treatment period. 187 adult patients with chronic, troublesome sialorrhea for at least 3 months were randomized to receive treatment with MYOBLOC 2,500 Units, MYOBLOC 3,500 Units, or placebo. Patients had chronic sialorrhea associated with Parkinson's disease (n=122), amyotrophic lateral sclerosis(ALS) (n=12), stroke (n=13), and other causes (n=40). Patients with a history of aspiration or severe dysphagia in the last 6 months and ALS patients with a forced vital capacity of less than 20% of predicted were excluded from the study. A single treatment was administered, consisting of bilateral injections of MYOBLOC into the parotid (1,000 Units or 1,500 Units per gland) and submandibular (250 Units per gland) salivary glands or volume matched placebo. A total of 114 patients received 4 consecutive treatments with 3,500 Units of MYOBLOC every 11 to 15 weeks.
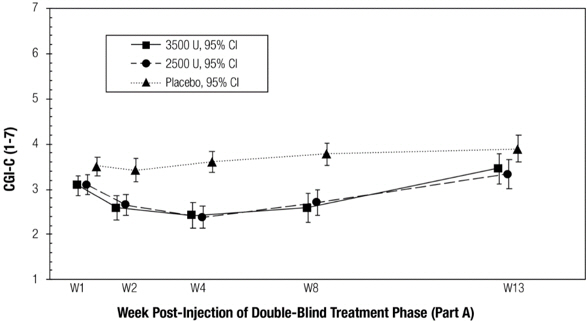
The co-primary efficacy endpoints for Study 1 were the change from baseline in Unstimulated Salivary Flow Rate (USFR) and the Clinical Global Impression of Change (CGI-C) assessed 4 weeks after treatment in the double-blind part of the study. The CGI-C is a seven-point Likert scale with scores ranging from "1=very much improved" to "7=very much worse". The change from baseline (i.e., decrease) in USFR at Week 4 was significantly greater for patients treated with MYOBLOC than in patients on placebo (Table 6). Similarly, CGI-C scores at Week 4 were significantly lower (i.e., better) in patients treated with MYOBLOC than in patients on placebo (Table 7). Chronic sialorrhea was 'much improved' or 'very much improved', according to CGI-C scores at Week 4 post injection, in patients treated with MYOBLOC 2,500 Units (60%) and MYOBLOC 3,500 Units (53%) than in patients on placebo (12%).
Figure 2 and Figure 3 show change in the USFR and CGI-C, respectively over the 13-week double-blind part of Study 1. The change from baseline to Week 4 on the USFR and the CGI-C was similar for MYOBLOC 2,500 Units and 3,500 Units, but there was a trend for more prolonged effect in patients treated with MYOBLOC 3,500 Units (Figure 2). CGI-C scores over the double-blind period were similar for both dose groups (Figure 3).
Table 6: Mean USFR Change from Baseline (g/min) at Week 4 in Study 1 Visit MYOBLOC 2,500 Units
(N=63)MYOBLOC 3,500 Units
(N=64)Placebo
(N=57)- *
- Primary endpoint, p< 0.0001
Week 4 -0.37 * -0.36 * -0.07 Table 7: CGI-C Score at Week 4 in Study 1 Visit MYOBLOC 2,500 Units
(N=63)MYOBLOC 3,500 Units
(N=64)Placebo
(N=57)- *
- Co-primary endpoint, p< 0.0001
Week 4 2.38 * 2.45 * 3.59 Figure 2: Mean Unstimulated Salivary Flow Rate over Time in Study 1
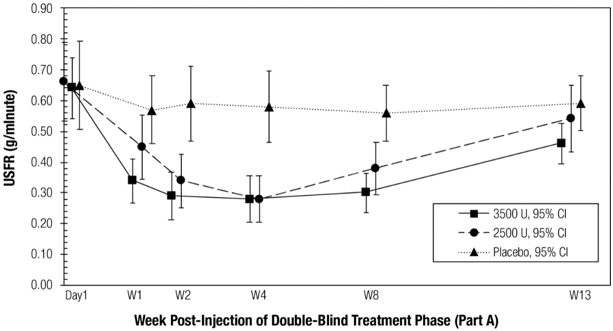
Figure 3: Mean Clinical Global Impression of Change Score over Time in Study 1
Study 2
Study 2 (NCT00515437) was a multicenter, double-blind, placebo-controlled, sequential dose-escalation study of MYOBLOC 1,500 Units; 2,500 units; or 3,500 Units versus matching placebo for the treatment of troublesome chronic sialorrhea in patients with Parkinson's disease. Patients were randomized to receive a single treatment with MYOBLOC 1,500 Units (n=14); MYOBLOC 2,500 Units (n=12); or MYOBLOC 3,500 Units (n=13). Each group also included 5 patients who received placebo (n=15). Patients were followed for up to 20 weeks after injection. The mean age of patients in the study was 71 years. In the study, 89% of patients were male, and 96% White.
The change from baseline in the unstimulated salivary flow rate (USFR) and the Clinical Global Impression of Change (CGI-C) was assessed 4 weeks after treatment. There was a significant reduction in the USFR for all three dosage groups of MYOBLOC, compared with patients on placebo (Table 8). Similarly, the CGI-C scores were significantly lower in all three MYOBLOC dosage groups than in patients on placebo (Table 9). The mean change from baseline to Week 4 on the USFR was similar in all three MYOBLOC dosage groups.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
MYOBLOC (rimabotulinumtoxinB) injection is supplied as a clear and colorless to light-yellow solution in single-dose glass vials.
MYOBLOC is available in three presentations (Table 10).
Table 10: MYOBLOC Presentations Strength per Vial Single-Vial Carton 2,500 Units/0.5 mL NDC 10454-710-10 5,000 Units/mL NDC 10454-711-10 10,000 Units/2 mL NDC 10454-712-10 No U.S. Standard of Potency
16.2 Storage and Handling
Store under refrigeration at 2°C to 8°C (36°F- 46°F) in the original carton to protect from light.
DO NOT FREEZE. DO NOT SHAKE.
All vials of expired MYOBLOC and equipment used in the administration of MYOBLOC should be carefully discarded according to standard medical waste practices.
Do not use after the expiration date stamped on the vial.
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Swallowing, Speaking, or Breathing Difficulties or Other Unusual Symptoms
Advise patients to inform their healthcare provider if they develop any unusual symptoms, including difficulty with swallowing, speaking or breathing, or if any existing symptom worsens [see Box Warning and Warnings and Precautions (5.1, 5.4)]. Inform patients of the risk of aspiration.
-
SPL UNCLASSIFIED SECTION
Manufactured By:
Solstice Neurosciences, LLC, Rockville, MD 20852
U.S. License No. 1718
"MYOBLOC" and the "MYOBLOC Logo" are registered trademarks of Solstice Neurosciences, Inc.
Protected by U.S. Patent No.
6,290,961; 6,632,433; 6,872,397; 6,887,476© 2019 Solstice Neurosciences, LLC
64183
RA-MYO-V5
-
MEDICATION GUIDE
MEDICATION GUIDE
MYOBLOC ® (My-o-block)
(rimabotulinumtoxinB)
injection, for intramuscular or intraglandular useThis Medication Guide has been approved by the U.S. Food and Drug Administration. Rev: 3/2021 What is the most important information I should know about MYOBLOC? MYOBLOC may cause serious side effects that can be life threatening. Call your doctor or get medical help right away if you have any of these problems after treatment with MYOBLOC: -
Problems swallowing, speaking, or breathing. These problems can happen hours to weeks after an injection of MYOBLOC if the muscles that you use to breathe and swallow become weak after the injection. Death can happen as a complication if you have severe problems with swallowing or breathing after treatment with MYOBLOC.
- People with certain breathing problems may need to use muscles in their neck to help them breathe. These people may be at greater risk for serious breathing problems with MYOBLOC.
- Swallowing problems may last for several months. People who cannot swallow well may need a feeding tube to receive food and water. If swallowing problems are severe, food or liquids may go into your lungs. People who already have swallowing or breathing problems before receiving MYOBLOC have the highest risk of getting these problems.
- Spread of toxin effects. In some cases, the effect of botulinum toxin may affect areas of the body away from the injection site and cause symptoms of a serious condition called botulism. The symptoms of botulism include:
- loss of strength and muscle weakness all over the body
- double vision
- blurred vision and drooping eyelids
- hoarseness or change or loss of voice (dysphonia)
- trouble saying words clearly (dysarthria)
- loss of bladder control
- trouble breathing
- trouble swallowing (dysphagia)
These symptoms can happen hours to weeks after you receive an injection of MYOBLOC. These problems could make it unsafe for you to drive a car or do other dangerous activities. See " What should I avoid while receiving MYOBLOC?" What is MYOBLOC? MYOBLOC is a prescription medicine used in adults that is injected into: - muscles and used to treat the abnormal head position and neck pain that happens with cervical dystonia (CD).
- glands that make saliva and is used to treat long-lasting (chronic) drooling (sialorrhea).
It is not known whether MYOBLOC is safe or effective in children. Do not receive MYOBLOC if you: - are allergic to MYOBLOC or any of the ingredients in MYOBLOC. See the end of this Medication Guide for a list of ingredients in MYOBLOC.
- had an allergic reaction to any other botulinum toxin product such as BOTOX® , BOTOX® Cosmetic (onabotulinumtoxinA), DYSPORT® (abobotulinumtoxinA), or XEOMIN® (incobotulinumtoxinA).
- have a skin infection at the planned injection site.
Before receiving MYOBLOC, tell your doctor about all your medical conditions, including if you have: - a disease that affects your muscles and nerves (such as amyotrophic lateral sclerosis [ALS or Lou Gehrig's disease], myasthenia gravis or Lambert-Eaton syndrome). See " What is the most important information I should know about MYOBLOC?"
- had any side effect from any botulinum toxin product in the past.
- a breathing problem, such as asthma or emphysema.
- a history of swallowing problems or inhaling food or fluid into your lungs (aspiration).
- bleeding problems.
- drooping eyelids.
- plans to have surgery.
- had surgery on your face.
Tell your doctor if you: - are pregnant or plan to become pregnant. It is not known if MYOBLOC can harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if MYOBLOC passes into breast milk.
Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. Talk to your doctor before you take any new medicine after you receive MYOBLOC. Using MYOBLOC with certain other medicines may cause serious side effects. Do not start any new medicines until you have told your doctor that you have received MYOBLOC in the past. Especially tell your doctor if you: - have received any other botulinum toxin product in the last 4 months.
- have received injections of botulinum toxin such as BOTOX®, BOTOX® Cosmetic (onabotulinumtoxinA), DYSPORT® (abobotulinumtoxinA), or XEOMIN® (incobotulinumtoxinA) in the past. Be sure your doctor knows exactly which product you received.
- have recently received an antibiotic by injection.
- take muscle relaxants.
- take an allergy or cold medicine.
- take a sleep medicine.
Ask your doctor if you are not sure if your medicine is one that is listed above. Know the medicines you take. Keep a list of your medicines with you to show your doctor and pharmacist each time you get a new medicine. How should I receive MYOBLOC? - MYOBLOC is a shot (injection) that your doctor will give you.
- MYOBLOC is injected into your affected muscles or glands.
- Your doctor may give you another dose of MYOBLOC after 12 weeks or longer, if it is needed.
- Your doctor may change your dose of MYOBLOC, until you and your doctor find the best dose for you.
What should I avoid while receiving MYOBLOC? MYOBLOC may cause loss of strength or general muscle weakness or vision problems within hours to weeks of receiving MYOBLOC. If this happens, do not drive a car, operate machinery or do other dangerous activities. See " What is the most important information I should know about MYOBLOC?" What are the possible side effects of MYOBLOC? MYOBLOC can cause serious side effects including: See " What is the most important information I should know about MYOBLOC?" - allergic reactions. Symptoms of an allergic reaction to MYOBLOC may include: itching, rash, redness, swelling, wheezing, trouble breathing, or dizziness or feeling faint. Tell your doctor or get medical help right away if you get wheezing, or trouble breathing, or if you get dizzy or faint.
The most common side effects of MYOBLOC in people with cervical dystonia include: - dry mouth
- injection site discomfort or pain
- trouble swallowing (dysphagia)
- headache
The most common side effects of MYOBLOC in people with sialorrhea include dry mouth and trouble swallowing (dysphagia). These are not all the possible side effects of MYOBLOC. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to Solstice Neurosciences at 1-888-461-2255. General information about the safe and effective use of MYOBLOC. Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. You can ask your doctor or pharmacist for information about MYOBLOC that is written for healthcare professionals. What are the ingredients in MYOBLOC? Active ingredient: botulinum toxin type B Inactive ingredients: albumin human, sodium chloride and sodium succinate. Manufactured by: Solstice Neurosciences, LLC, Rockville MD 20852
U.S. License No. 1718
SNI-MYO424A-0410
RA-MYO-MGV2
"MYOBLOC" and the "MYOBLOC Logo" are registered trademarks of Solstice Neurosciences, LLC
All trademarks are the property of their respective owners.For more information about MYOBLOC call 1-888-461-2255 or go to www.MYOBLOC.com. -
Problems swallowing, speaking, or breathing. These problems can happen hours to weeks after an injection of MYOBLOC if the muscles that you use to breathe and swallow become weak after the injection. Death can happen as a complication if you have severe problems with swallowing or breathing after treatment with MYOBLOC.
-
PRINCIPAL DISPLAY PANEL - 0.5 mL Vial Carton
NDC 10454-710-10
Rx Only
rimabotulinumtoxinB
MYOBLOC ®
InjectionFOR INTRAMUSCULAR AND
INTRAGLANDULAR USE2,500 Units/0.5 mL
WARNING: Dosing units of botulinum toxins are
not interchangeable between commercial products.Single-dose Vial
Discard Unused Portion
Dispense the accompanying Medication Guide to each patient.
SOLSTICE ®
NEUROSCIENCESUS WorldMeds ®

-
PRINCIPAL DISPLAY PANEL - 1 mL Vial Carton
NDC 10454-711-10
Rx Only
rimabotulinumtoxinB
MYOBLOC ®
InjectionFOR INTRAMUSCULAR AND
INTRAGLANDULAR USE5,000 Units/1 mL
WARNING: Dosing units of botulinum toxins are
not interchangeable between commercial products.Single-dose Vial
Discard Unused Portion
Dispense the accompanying Medication Guide to each patient.
SOLSTICE ®
NEUROSCIENCESUS WorldMeds ®

-
PRINCIPAL DISPLAY PANEL - 2 mL Vial Carton
NDC 10454-712-10
Rx Only
rimabotulinumtoxinB
MYOBLOC ®
InjectionFOR INTRAMUSCULAR AND
INTRAGLANDULAR USE10,000 Units/2 mL
(5,000 Units/mL)WARNING: Dosing units of botulinum toxins are
not interchangeable between commercial products.Single-dose Vial
Discard Unused Portion
Dispense the accompanying Medication Guide to each patient.
SOLSTICE ®
NEUROSCIENCESUS WorldMeds ®

-
INGREDIENTS AND APPEARANCE
MYOBLOC
rimabotulinumtoxinb injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:10454-710 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIMABOTULINUMTOXINB (UNII: 0Y70779M1F) (RIMABOTULINUMTOXINB - UNII:0Y70779M1F) RIMABOTULINUMTOXINB 2500 [USP'U] in 0.5 mL Inactive Ingredients Ingredient Name Strength SODIUM SUCCINATE HEXAHYDRATE (UNII: U16QOD6C4E) SODIUM CHLORIDE (UNII: 451W47IQ8X) ALBUMIN HUMAN (UNII: ZIF514RVZR) N-ACETYL-DL-TRYPTOPHAN SODIUM (UNII: 3EN9H0M2FX) SODIUM CAPRYLATE (UNII: 9XTM81VK2B) HYDROCHLORIC ACID (UNII: QTT17582CB) Product Characteristics Color yellow (colorless to light yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:10454-710-10 1 in 1 CARTON 01/01/2020 1 0.5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103846 12/08/2000 MYOBLOC
rimabotulinumtoxinb injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:10454-711 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIMABOTULINUMTOXINB (UNII: 0Y70779M1F) (RIMABOTULINUMTOXINB - UNII:0Y70779M1F) RIMABOTULINUMTOXINB 5000 [USP'U] in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM SUCCINATE HEXAHYDRATE (UNII: U16QOD6C4E) SODIUM CHLORIDE (UNII: 451W47IQ8X) ALBUMIN HUMAN (UNII: ZIF514RVZR) N-ACETYL-DL-TRYPTOPHAN SODIUM (UNII: 3EN9H0M2FX) SODIUM CAPRYLATE (UNII: 9XTM81VK2B) HYDROCHLORIC ACID (UNII: QTT17582CB) Product Characteristics Color yellow (colorless to light yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:10454-711-10 1 in 1 CARTON 01/01/2020 1 1 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103846 12/08/2000 MYOBLOC
rimabotulinumtoxinb injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:10454-712 Route of Administration INTRAMUSCULAR Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength RIMABOTULINUMTOXINB (UNII: 0Y70779M1F) (RIMABOTULINUMTOXINB - UNII:0Y70779M1F) RIMABOTULINUMTOXINB 10000 [USP'U] in 2 mL Inactive Ingredients Ingredient Name Strength SODIUM SUCCINATE HEXAHYDRATE (UNII: U16QOD6C4E) SODIUM CHLORIDE (UNII: 451W47IQ8X) ALBUMIN HUMAN (UNII: ZIF514RVZR) N-ACETYL-DL-TRYPTOPHAN SODIUM (UNII: 3EN9H0M2FX) SODIUM CAPRYLATE (UNII: 9XTM81VK2B) HYDROCHLORIC ACID (UNII: QTT17582CB) Product Characteristics Color yellow (colorless to light yellow) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:10454-712-10 1 in 1 CARTON 01/01/2020 1 2 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA103846 12/08/2000 Labeler - Solstice Neurosciences, LLC (780448184) Registrant - MDD US Operations, LLC (087875626)