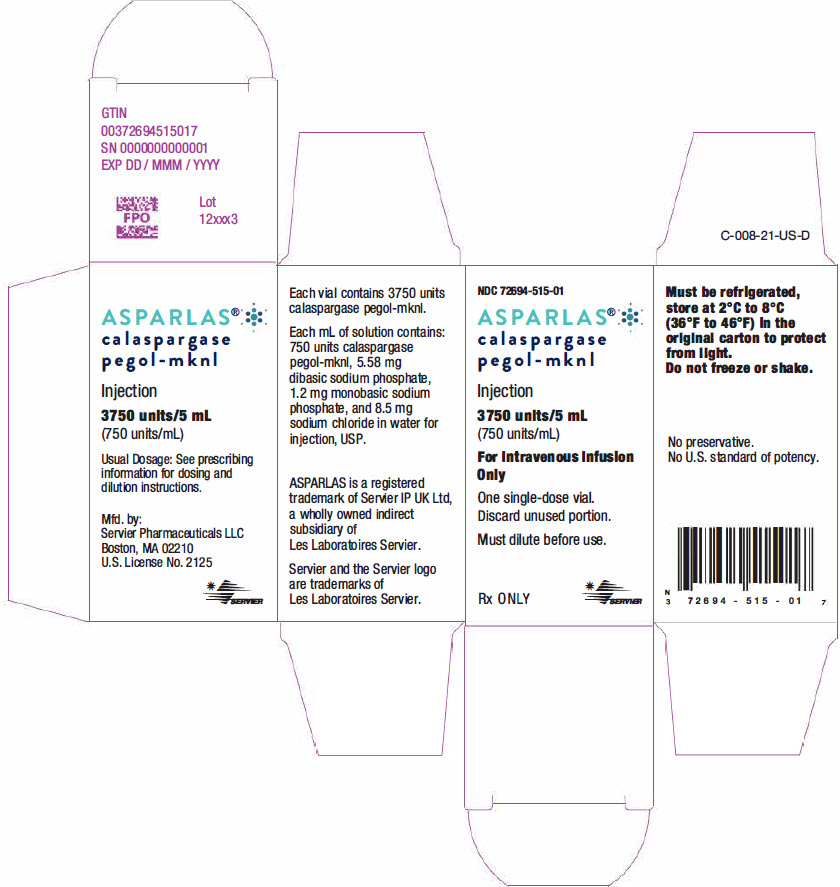
Label: ASPARLAS- calaspargase pegol injection, solution

- NDC Code(s): 72694-515-01
- Packager: Servier Pharmaceuticals LLC
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated December 6, 2023
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use ASPARLAS safely and effectively. See full prescribing information for ASPARLAS.
ASPARLAS® (calaspargase pegol-mknl) injection, for intravenous use
Initial U.S. Approval: 2018RECENT MAJOR CHANGES
Warnings and Precautions: Hepatotoxicity (5.5) 11/2023 INDICATIONS AND USAGE
ASPARLAS is an asparagine specific enzyme indicated as a component of a multi-agent chemotherapeutic regimen for the treatment of acute lymphoblastic leukemia in pediatric and young adult patients age 1 month to 21 years. (1.1)
DOSAGE AND ADMINISTRATION
DOSAGE FORMS AND STRENGTHS
Injection: 3,750 units/5 mL (750 units/mL) in a single-dose vial. (3)
CONTRAINDICATIONS
- History of serious hypersensitivity reactions to pegylated L-asparaginase. (4)
- History of serious thrombosis during L-asparaginase therapy. (4)
- History of serious pancreatitis related to previous L-asparaginase treatment. (4)
- History of serious hemorrhagic events during previous L-asparaginase therapy. (4)
- Severe hepatic impairment. (4)
WARNINGS AND PRECAUTIONS
- Hypersensitivity: Observe patients for one hour after administration. Discontinue ASPARLAS in patients with serious hypersensitivity reactions. (5.1)
- Pancreatitis: Discontinue ASPARLAS in patients with pancreatitis. Monitor blood glucose. (5.2)
- Thrombosis: Discontinue ASPARLAS for severe or life-threatening thrombosis. (5.3)
- Hemorrhage: Discontinue ASPARLAS for severe or life-threatening hemorrhage. Evaluate for etiology and treat. (5.4)
- Hepatotoxicity, including hepatic veno-occlusive disease (VOD): Monitor for toxicity through recovery from cycle. Discontinue ASPARLAS for severe liver toxicity. (5.5)
ADVERSE REACTIONS
The most common (incidence ≥10%) grade ≥3 adverse reactions were elevated transaminase, bilirubin increased, pancreatitis, and abnormal clotting studies. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Servier Pharmaceuticals LLC at 1-800-807-6124 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 11/2023
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Acute Lymphoblastic Leukemia
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Recommended Premedication
2.3 Recommended Monitoring and Dosage Modifications for Adverse Reactions
2.4 Preparation and Administration
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
5.2 Pancreatitis
5.3 Thrombosis
5.4 Hemorrhage
5.5 Hepatotoxicity, Including Hepatic Veno-Occlusive Disease
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Acute Lymphoblastic Leukemia
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
The recommended dose of ASPARLAS is 2,500 units/m2 given intravenously no more frequently than every 21 days.
2.2 Recommended Premedication
Premedicate patients with acetaminophen, an H-1 receptor blocker (such as diphenhydramine), and an H-2 receptor blocker (such as famotidine) 30-60 minutes prior to administration of ASPARLAS to decrease the risk and severity of both infusion and hypersensitivity reactions [see Warnings and Precautions (5.1)].
2.3 Recommended Monitoring and Dosage Modifications for Adverse Reactions
Monitor patients at least weekly with bilirubin, transaminases, glucose, and clinical examinations until recovery from the cycle of therapy. If an adverse reaction should occur, modify treatment according to Table 1.
Table 1: Dosage Modifications Adverse Reaction Severity* Action - *
- Grade 1 is mild, grade 2 is moderate, grade 3 is severe, and grade 4 is life-threatening.
Infusion Reaction/ Hypersensitivity Reaction [see Warnings and Precautions (5.1)] Grade 1 - Reduce the infusion rate by 50%
Grade 2 - Interrupt the infusion of ASPARLAS
- Treat the symptoms
- When symptoms resolve, resume the infusion and reduce the infusion rate by 50%
Grade 3 to 4 - Discontinue ASPARLAS permanently
Pancreatitis [see Warnings and Precautions (5.2)] Grades 3 to 4 - Hold ASPARLAS for elevations in lipase or amylase >3 × upper limit of normal (ULN) until enzyme levels stabilize or are declining
- Discontinue ASPARLAS permanently if clinical pancreatitis is confirmed
Thrombosis [see Warnings and Precautions (5.3)] Uncomplicated deep vein thrombosis - Hold ASPARLAS
- Treat with appropriate antithrombotic therapy
- Upon resolution of symptoms consider resuming ASPARLAS, while continuing antithrombotic therapy
Severe or life-threatening thrombosis - Discontinue ASPARLAS permanently
- Treat with appropriate antithrombotic therapy
Hemorrhage [see Warnings and Precautions (5.4)] Grade 3 to 4 - Hold ASPARLAS
- Evaluate for coagulopathy and consider clotting factor replacement as needed
- Resume ASPARLAS with the next scheduled dose if bleeding is controlled
Hepatotoxicity [see Warnings and Precautions (5.5)] Total bilirubin more than 3 times to no more than 10 times the ULN - Hold ASPARLAS until total bilirubin is ≤1.5 times the ULN
Total bilirubin more than 10 times the ULN - Discontinue ASPARLAS and do not make up for missed doses
2.4 Preparation and Administration
ASPARLAS is a clear and colorless solution. Visually inspect parenteral drug products for particulate matter, cloudiness, or discoloration prior to administration. If any of these are present, discard the vial. Do not administer if ASPARLAS has been shaken or vigorously agitated, frozen, or stored at room temperature for more than 48 hours.
- Dilute ASPARLAS in 100 mL of 0.9% Sodium Chloride Injection, USP or 5% Dextrose Injection, USP using sterile/aseptic technique. Discard any unused portion left in a vial.
- After dilution, administer immediately into a running infusion of either 0.9% sodium chloride or 5% dextrose, respectively.
- Administer the dose over a period of 1 hour.
- Do not infuse other drugs through the same intravenous line during administration of ASPARLAS.
- The diluted solution may be stored for up to 4 hours at room temperature (15°C to 25°C [59°F to 77°F]) or refrigerated at 2°C to 8°C (36°F to 46°F) for up to 24 hours.
- Protect from light. Do not shake or freeze.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
ASPARLAS is contraindicated in patients with:
- History of serious hypersensitivity reactions, including anaphylaxis, to pegylated L-asparaginase therapy [see Warnings and Precautions (5.1)]
- History of serious pancreatitis during previous L-asparaginase therapy [see Warnings and Precautions (5.2)]
- History of serious thrombosis during previous L-asparaginase therapy [see Warnings and Precautions (5.3)]
- History of serious hemorrhagic events during previous L-asparaginase therapy [see Warnings and Precautions (5.4)]
- Severe hepatic impairment [see Warnings and Precautions (5.5)]
-
5 WARNINGS AND PRECAUTIONS
5.1 Hypersensitivity
Grade 3 and 4 hypersensitivity reactions, including anaphylaxis, have been reported in clinical trials with ASPARLAS with an incidence between 7 and 21% [see Contraindications (4), Adverse Reactions (6.1)]. Hypersensitivity reactions observed with other asparaginases include angioedema, lip swelling, eye swelling, erythema, blood pressure decreased, bronchospasm, dyspnea, pruritus, and rash [see Adverse Reactions (6.1)].
Premedicate patients 30-60 minutes prior to administration of ASPARLAS [see Dosage and Administration (2.2)]. Because of the risk of serious allergic reactions (e.g., life-threatening anaphylaxis), administer ASPARLAS in a clinical setting with resuscitation equipment and other agents necessary to treat anaphylaxis (e.g., epinephrine, oxygen, intravenous steroids, antihistamines) [see Dosage and Administration (2.4)] and observe patients for 1 hour after administration. Discontinue ASPARLAS in patients with serious hypersensitivity reactions.
5.2 Pancreatitis
Cases of pancreatitis have been reported in clinical trials with ASPARLAS with an incidence between 12 and 16% [see Adverse Reactions (6.1)]. Hemorrhagic or necrotizing pancreatitis have been reported with other asparaginases.
Inform patients of the signs and symptoms of pancreatitis, which, if left untreated, could be fatal. Assess serum amylase and/or lipase levels to identify early signs of pancreatic inflammation. Discontinue ASPARLAS if pancreatitis is suspected; if pancreatitis is confirmed, do not resume ASPARLAS [see Dosage and Administration (2.3)].
5.3 Thrombosis
Serious thrombotic events, including sagittal sinus thrombosis, have been reported in clinical trials with ASPARLAS with an incidence of 9 to 12%. Discontinue ASPARLAS in patients experiencing serious thrombotic events [see Dosage and Administration (2.3), Adverse Reactions (6.1)].
5.4 Hemorrhage
Hemorrhage associated with increased prothrombin time (PT), increased partial thromboplastin time (PTT), and hypofibrinogenemia have been reported in patients receiving ASPARLAS [see Adverse Reactions (6.1)]. Evaluate patients with signs and symptoms of hemorrhage with coagulation parameters including PT, PTT, fibrinogen. Consider appropriate replacement therapy in patients with severe or symptomatic coagulopathy [see Dosage and Administration (2.3)].
5.5 Hepatotoxicity, Including Hepatic Veno-Occlusive Disease
Hepatotoxicity, including severe, life-threatening, and potentially fatal cases of hepatic veno-occlusive disease (VOD), have been observed in patients treated with ASPARLAS in combination with standard chemotherapy, including during the induction phase of multiphase chemotherapy [see Adverse Reactions (6)]. Do not administer ASPARLAS to patients with severe hepatic impairment [see Contraindications (4)].
Evaluate bilirubin and transaminases prior to each dose of ASPARLAS and at least weekly, during cycles of treatment that include ASPARLAS, through 6 weeks after the last dose of ASPARLAS. Monitor frequently for signs and symptoms of hepatic VOD, which may include rapid weight gain, fluid retention with ascites, hepatomegaly (which may be painful), and rapid increase of bilirubin. For patients who develop abnormal liver tests after ASPARLAS, more frequent monitoring for liver test abnormalities and clinical signs and symptoms of VOD is recommended. In the event of serious liver toxicity, including VOD, discontinue treatment with ASPARLAS and provide supportive care [see Dosage and Administration (2.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described elsewhere in the labeling:
- Hypersensitivity [see Warnings and Precautions (5.1)]
- Pancreatitis [see Warnings and Precautions (5.2)]
- Thrombosis [see Warnings and Precautions (5.3)]
- Hemorrhage [see Warnings and Precautions (5.4)]
- Hepatotoxicity, including VOD [see Warnings and Precautions (5.5)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Study DFCI 11-001
The safety of ASPARLAS was investigated in Study DFCI 11-001, an open-label, randomized, active-controlled multicenter clinical trial that treated 237 children and adolescents with newly diagnosed ALL or lymphoblastic lymphoma, with ASPARLAS 2,500 U/m2 (n=118) or pegaspargase 2,500 U/m2 (n=119) as part of a Dana-Farber Cancer Institute (DFCI) ALL Consortium backbone therapy. The median age on enrollment was 5 years (range, 1-20 years). The majority of patients were male (62%) and white (70%). Most patients were considered standard risk (SR, 59%) and had B-cell lineage ALL (87%).
The median number of doses during the study was 11 doses for ASPARLAS (administered every three weeks) and 16 doses for pegaspargase (administered every two weeks). The median duration of exposure was 8 months for both ASPARLAS and pegaspargase.
There was 1 fatal adverse reaction (multi-organ failure in the setting of chronic pancreatitis associated with a pancreatic pseudocyst).
Table 2 summarizes the incidence of selected grades ≥3 adverse reactions that occurred in 2 or more patients receiving ASPARLAS. Because not all grade 1 and 2 adverse reactions were collected prospectively, only grade 3 and 4 adverse events are presented in Table 2.
Table 2: Selected Grades ≥3 Adverse Reactions in Patients Receiving ASPARLAS with Multi-Agent Chemotherapy (Study DFCI 11-001)* Adverse Reaction† ASPARLAS
2,500 U/m2
N=118Pegaspargase
2,500 U/m2
N=119Grades ≥3
n (%)‡Grades ≥3
n (%)‡- *
- ASPARLAS or pegaspargase were administered as a component of multi-agent chemotherapy regimens.
- †
- Grouped terms: Elevated transaminase: Alanine aminotransferase increased, Aspartate aminotransferase increased, Transaminases increased; Bilirubin increased: Bilirubin conjugated increased, Blood bilirubin increased; Pancreatitis: Amylase increased, Lipase increased, Pancreatic necrosis, Pancreatitis, Pancreatitis relapsing; Abnormal clotting studies: Activated partial thromboplastin time prolonged, Blood fibrinogen decreased; Diarrhea: Colitis, Diarrhea, Enterocolitis, Neutropenic colitis; Hypersensitivity: Anaphylactic reaction, Drug hypersensitivity, Hypersensitivity; Embolic and thrombotic events SMQ: Device related thrombosis, Disseminated intravascular coagulation, Embolism, Intracardiac thrombus, Intracranial venous sinus thrombosis, Pulmonary embolism, Superior sagittal sinus thrombosis, Thrombosis in device, Venous thrombosis, Venous thrombosis limb; Sepsis: Bacterial sepsis, Sepsis; Dyspnea: Hypoxia, Respiratory failure; Hemorrhages SMQ (excludes laboratory terms): Disseminated intravascular coagulation, Epistaxis, Hematoma, Hemorrhage intracranial, Melena, Esophageal ulcer hemorrhage, Small intestinal hemorrhage, Upper gastrointestinal hemorrhage; Fungal infection: Fungal infection, Hepatic infection fungal, Respiratory tract infection fungal, Splenic infection fungal, Systemic candida; Pneumonia: Lung infection, Pneumonia, Pneumonitis; Arrhythmia: Atrioventricular block complete, Sinus tachycardia, Ventricular arrhythmia; Cardiac failure: Ejection fraction decreased, Left ventricular dysfunction.
- ‡
- Grading is based on the Common Terminology Criteria for Adverse Events (CTCAE) v4.0.
Elevated transaminase 61 (52) 79 (66) Bilirubin increased 24 (20) 30 (25) Pancreatitis 21 (18) 29 (24) Abnormal clotting studies 17 (14) 25 (21) Diarrhea 10 (9) 6 (5) Hypersensitivity 9 (8) 8 (7) Embolic and thrombotic events 9 (8) 10 (8) Sepsis 6 (5) 7 (6) Dyspnea 5 (4) 1 (1) Hemorrhages 5 (4) 5 (4) Fungal infection 4 (3) 3 (3) Pneumonia 4 (3) 8 (7) Arrhythmia 2 (2) 1 (1) Cardiac failure 2 (2) 1 (1) In the subgroup of patients with B-cell lineage ALL, the complete remission rate in the ASPARLAS arm was 98% (95/97), compared to 99% in the pegaspargase arm; the Kaplan-Meier estimates of overall survival of the treatment arms were comparable.
Study AALL07P4
The safety of ASPARLAS was also evaluated in Study AALL07P4, an open-label, randomized, active-controlled, multicenter clinical trial that treated patients with newly diagnosed high-risk B-precursor ALL using ASPARLAS 2,500 U/m2 (n=43) or 2,100 U/m2 (n=68), or pegaspargase 2,500 U/m2 (n=52), as a component of an augmented Berlin-Frankfurt-Münster (BFM) therapy regimen. The median age was 11 years (range, 1-26 years); the median duration of exposure was 7 months for both ASPARLAS and pegaspargase. In this study, the induction mortality of patients treated with ASPARLAS was 2.8% (3 out of 111); there were no induction deaths among 52 patients treated with pegaspargase.
Immunogenicity: Anti-Drug Antibody-Associated Adverse Reactions
In Study DFCI 11-001, hypersensitivity reactions occurred in 80% of ASPARLAS-treated patients with new or an increased titer of anti-drug antibodies (ADA) and in 6% of those without ADA [see Clinical Pharmacology (12.6)]. Two patients with ADA experienced anaphylaxis [see Warnings and Precautions (5.1)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of ASPARLAS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hepatic: Veno-occlusive disease
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Based on published literature studies with L-asparaginase in pregnant animals, ASPARLAS can cause fetal harm when administered to a pregnant woman. There are no available data on ASPARLAS use in pregnant women to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction studies, intravenous administration of calaspargase pegol-mknl to pregnant rats during organogenesis at doses 0.2 to 1 times the maximum recommended human doses did not result in adverse developmental outcomes. Published literature studies in pregnant rabbits, however, suggest asparagine depletion may cause harm to the animal offspring (see Data). Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In an embryo-fetal development study, calaspargase pegol-mknl was administered intravenously at doses of 75, 150, and 300 U/kg (0.2, 0.6 and 1 times the maximum recommended human dose, respectively, based on AUC) to pregnant rats during the period of organogenesis. Maternal toxicity of decreased body weight and food consumption was seen at all dose levels resulting in reductions in gravid uterine and placental weights, and slight reductions in fetal body weights. No evidence of structural abnormalities or embryo-fetal mortality were observed in this study at any of the doses tested. Published literature studies in which pregnant rabbits were administered L-asparaginase suggested harm to the animal offspring.
8.2 Lactation
Risk Summary
There are no data on the presence of calaspargase pegol-mknl in human milk, the effects on the breastfed child, or the effects on milk production. Because of the potential for adverse reactions in the breastfed child, advise women not to breastfeed during treatment with ASPARLAS and for 3 months after the last dose.
8.3 Females and Males of Reproductive Potential
ASPARLAS can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
8.4 Pediatric Use
The safety and effectiveness of ASPARLAS in the treatment of ALL have been established in pediatric patients 1 month to <17 years (no data for the age group <1 month old). Use of ASPARLAS in these age groups is supported by evidence from an adequate and well-controlled trial with additional safety from a second trial. The trials included 208 children with ALL or lymphoblastic lymphoma treated with ASPARLAS; there were 19 infants (1 month to <2 years old), 128 children (2 years to <12 years old), and 61 adolescents (12 years to <17 years old). There were no clinically meaningful differences in safety or nadir serum asparaginase activity across age groups [see Adverse Reactions (6.1), Clinical Studies (14)].
-
11 DESCRIPTION
Calaspargase pegol-mknl contains an asparagine specific enzyme derived from Escherichia coli, as a conjugate of L-asparaginase (L-asparagine amidohydrolase) and monomethoxypolyethylene glycol (mPEG) with a succinimidyl carbonate (SC) linker. The SC linker is a chemically stable carbamate bond between the mPEG moiety and the lysine groups of L-asparaginase.
L-asparaginase is a tetrameric enzyme that is produced endogenously by E. coli and consists of identical 34.5 kDa subunits. Approximately 31 to 39 molecules of SC-PEG are linked to L-asparaginase; the molecular weight of each SC-PEG molecule is about 5 kDa. The activity of ASPARLAS is expressed in units (U).
ASPARLAS injection is supplied as a clear, colorless, preservative-free, isotonic sterile solution in phosphate-buffered saline, pH 7.3 that requires dilution prior to intravenous infusion. Each vial of ASPARLAS contains 3,750 units in 5 mL of solution. Each milliliter contains 750 units of calaspargase pegol-mknl; dibasic sodium phosphate, USP (5.58 mg); monobasic sodium phosphate, USP (1.20 mg); and sodium chloride, USP (8.50 mg) in water for injection, USP.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
L-asparaginase is an enzyme that catalyzes the conversion of the amino acid L-asparagine into aspartic acid and ammonia. The pharmacological effect of ASPARLAS is thought to be based on the killing of leukemic cells due to depletion of plasma asparagine. Leukemic cells with low expression of asparagine synthetase have a reduced ability to synthesize asparagine, and therefore depend on an exogenous source of asparagine for survival.
12.2 Pharmacodynamics
Calaspargase pegol-mknl pharmacodynamic (PD) response was assessed through measurement of plasma and cerebrospinal fluid (CSF) asparagine concentrations via an LC-MS/MS assay.
Asparagine concentrations in plasma (N=41) were maintained below the assay limit of quantification for more than 18 days following a single dose of ASPARLAS 2,500 U/m2 during the induction phase. Mean CSF asparagine concentrations decreased from a pretreatment concentration of 0.8 µg/mL (N=10) to 0.2 µg/mL on Day 4 (N=37) and remained decreased at 0.2 µg/mL (N=35) 25 days after the administration of a single dose of ASPARLAS 2,500 U/m2 in the induction phase.
12.3 Pharmacokinetics
Calaspargase pegol-mknl pharmacokinetics (PK) were assessed through measurement of plasma asparaginase activity via a coupled enzymatic assay.
The plasma asparaginase activity pharmacokinetics were characterized in 43 patients (1 to 26 years) with newly diagnosed high risk B-precursor ALL treated with a multidrug backbone therapy. Table 3 summarizes the plasma asparaginase activity pharmacokinetic parameters after a single dose of ASPARLAS 2,500 U/m2 in the induction phase.
Table 3: Plasma Asparaginase Activity Pharmacokinetic Parameters After a Single Dose of ASPARLAS 2,500 U/m2 in Patients with ALL in Study AALL07P4 Parameter Arithmetic Mean (%CV)
N=43General Cmax (U/mL) 1.62 (23.0) AUC0-25day (day∙U/mL) 16.9 (23.2)* AUC0∞ (day∙U/mL)† 25.5 (30.4)* Absorption Tmax (h)† 1.17 (1.05, 5.47)‡ Distribution Vss (L) 2.96 (84.3)* Elimination t1/2 (day)§ 16.1 (51.9)* Clearance (L/day) 0.147 (76.1)* 12.6 Immunogenicity
As with all therapeutic proteins, there is a potential for immunogenicity. The detection of antibody formation is highly dependent on the sensitivity and specificity of the assay. Additionally, the observed incidence of antibody (including neutralizing antibody) positivity in an assay may be influenced by several factors, including assay methodology, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to ASPARLAS in the studies described below with the incidence of antibodies in other studies or to other asparaginase products may be misleading.
Immunogenicity was assessed using enzyme linked immunosorbent assays (ELISA) in Study DFCI 11-001. Of 98 evaluable patients treated with ASPARLAS, 15 (15%) patients developed new or an increased titer of anti-drug antibodies (ADA) during treatment; 14 of these 15 patients were positive for anti-PEG antibodies. The presence of ADA correlated with the occurrence of hypersensitivity reactions [see Adverse Reactions (6.1)]. There is insufficient information to determine whether the development of antibodies is associated with altered pharmacokinetics (i.e., loss of asparaginase activity).
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Acute Lymphoblastic Leukemia
The determination of efficacy was based on a demonstration of the achievement and maintenance of nadir serum asparaginase activity (NSAA) above the level of 0.1 U/mL using ASPARLAS 2500 U/m2 intravenously every 3 weeks. The pharmacokinetics of ASPARLAS were studied when used in combination with multiagent chemotherapy in 124 patients with B-cell lineage acute lymphoblastic leukemia (ALL). Among these patients, the median age was 11.5 years (range, 1-26); 62 (50%) were male, 102 (82%) white, 6 (5%) Asian, 5 (4%) Black or African American, 2 (2%) Native Hawaiian or Pacific Islander and 9 (7%) other or unknown. The results showed that 123 (99%, 95% CI: 96%-100%) of the 124 patients maintained NSAA >0.1 U/mL at weeks 6, 12, 18, 24, and 30.
- 16 HOW SUPPLIED/STORAGE AND HANDLING
-
17 PATIENT COUNSELING INFORMATION
Hypersensitivity
Inform patients on the possibility of serious allergic reactions, including anaphylaxis. Instruct the patient on the symptoms of allergic reactions and to seek medical advice immediately if they experience such symptoms [see Warnings and Precautions (5.1)].
Pancreatitis
Instruct patients on the signs and symptoms of pancreatitis and to seek immediate medical attention if they experience severe abdominal pain [see Warnings and Precautions (5.2)].
Instruct patients on the risk of hyperglycemia and glucose intolerance. Advise patients to seek medical advice if they experience excessive thirst or any increase in the volume or frequency of urination [see Dosage and Administration (2.3)].
Thrombosis
Instruct patients on the risk of thrombosis and to seek medical advice immediately if they experience severe headache, arm or leg swelling, shortness of breath, or chest pain [see Warnings and Precautions (5.3)].
Hemorrhage
Advise patients to report any unusual bleeding or bruising to their healthcare provider [see Warnings and Precautions (5.4)].
Hepatotoxicity , including Veno-Occlusive Liver Disease (VOD)
Inform patients that liver problems, including severe, life-threatening, or fatal VOD and abnormalities in liver tests, may develop during ASPARLAS treatment. Advise patients to contact their healthcare provider immediately if they experience jaundice, rapid weight gain, abdominal swelling, or right upper abdominal pain or tenderness [see Warnings and Precautions (5.5)].
Pregnancy
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to inform their healthcare provider of a known or suspected pregnancy [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with ASPARLAS and for 3 months after the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise women not to breastfeed during treatment with ASPARLAS and for at least 3 months after the last dose [see Use in Specific Populations (8.2)].
- SPL UNCLASSIFIED SECTION
- PRINCIPAL DISPLAY PANEL - 5 mL Vial Carton
-
INGREDIENTS AND APPEARANCE
ASPARLAS
calaspargase pegol injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:72694-515 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength CALASPARGASE PEGOL (UNII: T9FVH03HMZ) (CALASPARGASE PEGOL - UNII:T9FVH03HMZ) CALASPARGASE PEGOL 750 U in 1 mL Inactive Ingredients Ingredient Name Strength SODIUM PHOSPHATE, MONOBASIC, DIHYDRATE (UNII: 5QWK665956) SODIUM PHOSPHATE, DIBASIC, UNSPECIFIED FORM (UNII: GR686LBA74) SODIUM CHLORIDE (UNII: 451W47IQ8X) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:72694-515-01 1 in 1 CARTON 09/27/2019 1 5 mL in 1 VIAL, SINGLE-USE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761102 09/27/2019 Labeler - Servier Pharmaceuticals LLC (116608503)