Label: IMURAN- azathioprine tablet


- NDC Code(s): 54766-590-10
- Packager: Sebela Pharmaceuticals Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated July 20, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
WARNING - MALIGNANCY
Chronic immunosuppression with IMURAN, a purine antimetabolite increases risk of malignancy in humans. Reports of malignancy include post-transplant lymphoma and hepatosplenic T-cell lymphoma (HSTCL) in patients with inflammatory bowel disease. Physicians using this drug should be very familiar with this risk as well as with the mutagenic potential to both men and women and with possible hematologic toxicities. Physicians should inform patients of the risk of malignancy with IMURAN. See WARNINGS.
-
DESCRIPTION:
IMURAN (azathioprine), an immunosuppressive antimetabolite, is available in tablet form for oral administration. Each scored tablet contains 50 mg azathioprine and the inactive ingredients lactose, magnesium stearate, potato starch, povidone, and stearic acid.
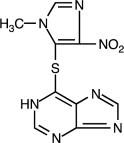
Azathioprine is chemically 6-[(1-methyl-4-nitro-1 H-imidazol-5-yl)thio]-1 H-purine. The structural formula of azathioprine is:
It is an imidazolyl derivative of 6-mercaptopurine and many of its biological effects are similar to those of the parent compound.
Azathioprine is insoluble in water, but may be dissolved with addition of one molar equivalent of alkali. Azathioprine is stable in solution at neutral or acid pH but hydrolysis to mercaptopurine occurs in excess sodium hydroxide (0.1N), especially on warming. Conversion to mercaptopurine also occurs in the presence of sulfhydryl compounds such as cysteine, glutathione, and hydrogen sulfide.
-
CLINICAL PHARMACOLOGY:
Azathioprine is well absorbed following oral administration. Maximum serum radioactivity occurs at 1 to 2 hours after oral 35S-azathioprine and decays with a half-life of 5 hours. This is not an estimate of the half-life of azathioprine itself, but is the decay rate for all 35S-containing metabolites of the drug. Because of extensive metabolism, only a fraction of the radioactivity is present as azathioprine. Usual doses produce blood levels of azathioprine, and of mercaptopurine derived from it, which are low (<1 mcg/mL). Blood levels are of little predictive value for therapy since the magnitude and duration of clinical effects correlate with thiopurine nucleotide levels in tissues rather than with plasma drug levels. Azathioprine and mercaptopurine are moderately bound to serum proteins (30%) and are partially dialyzable. See OVERDOSAGE.
Azathioprine is metabolized to 6-mercaptopurine (6-MP). Both compounds are rapidly eliminated from blood and are oxidized or methylated in erythrocytes and liver; no azathioprine or mercaptopurine is detectable in urine after 8 hours. Activation of 6-mercaptopurine occurs via hypoxanthine-guanine phosphoribosyltransferase (HGPRT) and a series of multi-enzymatic processes involving kinases to form 6-thioguanine nucleotides (6-TGNs) as major metabolites). The cytotoxicity of azathioprine is due, in part, to the incorporation of 6-TGN into DNA.
6-MP undergoes two major inactivation routes. One is thiol methylation, which is catalyzed by the enzyme thiopurine S-methyltransferase (TPMT), to form the inactive metabolite methyl-6-MP (6-MeMP). Another inactivation pathway is oxidation, which is catalyzed by xanthine oxidase (XO) to form 6-thiouric acid. The nucleotide diphosphatase (NUDT15) enzyme is involved in conversion of the 6-TGNs to inactive 6-TG monophosphates. TPMT activity correlates inversely with 6-TGN levels in erythrocytes and presumably other hematopoietic tissues, since these cells have negligible xanthine oxidase (involved in the other inactivation pathway) activities.
Genetic polymorphisms influence TPMT and NUDT15 activity. Several published studies indicate that patients with reduced TPMT or NUDT15 activity receiving usual doses of 6-MP or azathioprine, accumulate excessive cellular concentrations of active 6-TGNs, and are at higher risk for severe myelosuppression. Because of the risk of toxicity, patients with TPMT or NUDT15 deficiency require alternative therapy or dose modification (see DOSAGE and ADMINISTRATION).
Approximately 0.3% (1:300) of patients of European or African ancestry have two loss-of-function alleles of the TPMT gene and have little or no TPMT activity (homozygous deficient or poor metabolizers), and approximately 10% of patients have one loss-of-function TPMT allele leading to intermediate TPMT activity (heterozygous deficient or intermediate metabolizers). The TPMT*2, TPMT*3A, and TPMT*3C alleles account for about 95% of individuals with reduced levels of TPMT activity. NUDT15 deficiency is detected in <1% of patients of European or African ancestry. Among patients of East Asian ancestry (i.e., Chinese, Japanese, Vietnamese), 2% have two loss-of-function alleles of the NUDT15 gene, and approximately 21% have one loss-of-function allele. The p.R139C variant of NUDT15 (present on the *2 and *3 alleles) is the most commonly observed, but other less common loss-of-function NUDT15 alleles have been observed.
Inhibition of xanthine oxidase (XO) may cause increased plasma concentrations of azathioprine or its metabolites leading to toxicity (see PRECAUTIONS: Drug Interactions). Proportions of metabolites are different in individual patients, and this presumably accounts for variable magnitude and duration of drug effects. Renal clearance is probably not important in predicting biological effectiveness or toxicities, although dose reduction is practiced in patients with poor renal function.
Homograft Survival: The use of azathioprine for inhibition of renal homograft rejection is well established, the mechanism(s) for this action are somewhat obscure. The drug suppresses hypersensitivities of the cell-mediated type and causes variable alterations in antibody production. Suppression of T-cell effects, including ablation of T-cell suppression, is dependent on the temporal relationship to antigenic stimulus or engraftment. This agent has little effect on established graft rejections or secondary responses.
Alterations in specific immune responses or immunologic functions in transplant recipients are difficult to relate specifically to immunosuppression by azathioprine. These patients have subnormal responses to vaccines, low numbers of T-cells, and abnormal phagocytosis by peripheral blood cells, but their mitogenic responses, serum immunoglobulins, and secondary antibody responses are usually normal.
Immunoinflammatory Response: Azathioprine suppresses disease manifestations as well as underlying pathology in animal models of autoimmune disease. For example, the severity of adjuvant arthritis is reduced by azathioprine.
The mechanisms whereby azathioprine affects autoimmune diseases are not known. Azathioprine is immunosuppressive, delayed hypersensitivity and cellular cytotoxicity tests being suppressed to a greater degree than are antibody responses. In the rat model of adjuvant arthritis, azathioprine has been shown to inhibit the lymph node hyperplasia , which precedes the onset of the signs of the disease. Both the immunosuppressive and therapeutic effects in animal models are dose-related. Azathioprine is considered a slow-acting drug and effects may persist after the drug has been discontinued.
-
INDICATIONS AND USAGE:
IMURAN is indicated as an adjunct for the prevention of rejection in renal homotransplantation. It is also indicated for the management of active rheumatoid arthritis to reduce signs and symptoms.
Renal Homotransplantation: IMURAN is indicated as an adjunct for the prevention of rejection in renal homotransplantation. Experience with over 16,000 transplants shows a 5-year patient survival of 35% to 55%, but this is dependent on donor, match for HLA antigens, anti-donor or anti-B-cell alloantigen antibody, and other variables. The effect of IMURAN on these variables has not been tested in controlled trials.
Rheumatoid Arthritis: IMURAN is indicated for the treatment of active rheumatoid arthritis (RA) to reduce signs and symptoms. Aspirin, non-steroidal anti-inflammatory drugs and/or low dose glucocorticoids may be continued during treatment with IMURAN. The combined use of IMURAN with disease modifying anti-rheumatic drugs (DMARDs) has not been studied for either added benefit or unexpected adverse effects. The use of IMURAN with these agents cannot be recommended.
-
CONTRAINDICATIONS:
IMURAN should not be given to patients who have shown hypersensitivity to the drug. IMURAN should not be used for treating rheumatoid arthritis in pregnant women. Patients with rheumatoid arthritis previously treated with alkylating agents (cyclophosphamide, chlorambucil, melphalan, or others) may have a prohibitive risk of malignancy if treated with IMURAN.
-
WARNINGS:
Malignancy
Patients receiving immunosuppressants, including IMURAN, are at increased risk of developing lymphoma and other malignancies, particularly of the skin. Physicians should inform patients of the risk of malignancy with IMURAN. As usual for patients with increased risk for skin cancer, exposure to sunlight and ultraviolet light should be limited by wearing protective clothing and using a sunscreen with a high protection factor.
Post-transplant
Renal transplant patients are known to have an increased risk of malignancy, predominantly skin cancer and reticulum cell or lymphomatous tumors. The risk of post-transplant lymphomas may be increased in patients who receive aggressive treatment with immunosuppressive drugs, including IMURAN. Therefore, immunosuppressive drug therapy should be maintained at the lowest effective levels.
Rheumatoid Arthritis
Information is available on the risk of malignancy with the use of IMURAN in rheumatoid arthritis (see ADVERSE REACTIONS). It has not been possible to define the precise risk of malignancy due to IMURAN. The data suggest the risk may be elevated in patients with rheumatoid arthritis, though lower than for renal transplant patients. However, acute myelogenous leukemia as well as solid tumors have been reported in patients with rheumatoid arthritis who have received IMURAN.
Inflammatory Bowel Disease
Postmarketing cases of hepatosplenic T-cell lymphoma (HSTCL), a rare type of T-cell lymphoma, have been reported in patients treated with IMURAN. These cases have had a very aggressive disease course and have been fatal. The majority of reported cases have occurred in patients with Crohn's disease or ulcerative colitis and the majority were in adolescent and young adult males. Some of the patients were treated with IMURAN as monotherapy and some had received concomitant treatment with a TNFα blocker at or prior to diagnosis. The safety and efficacy of IMURAN for the treatment of Crohn's disease and ulcerative colitis have not been established.
Cytopenias
Severe leukopenia, thrombocytopenia, anemias including macrocytic anemia, and/or pancytopenia may occur in patients being treated with IMURAN. Severe bone marrow suppression may also occur. Hematologic toxicities are dose-related and may be more severe in renal transplant patients whose homograft is undergoing rejection. It is suggested that patients on IMURAN have complete blood counts, including platelet counts, weekly during the first month, twice monthly for the second and third months of treatment, then monthly or more frequently if dosage alterations or other therapy changes are necessary. Delayed hematologic suppression may occur. Prompt reduction in dosage or temporary withdrawal of the drug may be necessary if there is a rapid fall in or persistently low leukocyte count, or other evidence of bone marrow depression. Leukopenia does not correlate with therapeutic effect; therefore, the dose should not be increased intentionally to lower the white blood cell count.
TPMT or NUDT15 Deficiency
Patients with thiopurine S-methyl transferase (TPMT) or nucleotide diphosphatase (NUDT15) deficiency may be at an increased risk of severe and life-threatening myelotoxicity if receiving conventional doses of IMURAN (see CLINCIAL PHARMACOLOGY). Death associated with pancytopenia has been reported in patients with absent TPMT activity receiving azathioprine. In patients with severe myelosuppression, consider evaluation for TPMT and NUDT15 deficiency (see PRECAUTIONS: Laboratory Tests). Consider alternative therapy in patients with homozygous TPMT or NUDT15 deficiency and reduced dosages in patients with heterozygous deficiency (see DOSAGE AND ADMINISTRATION).
Serious infections
Patients receiving immunosuppressants, including Imuran, are at increased risk for bacterial, viral, fungal, protozoal, and opportunistic infections, including reactivation of latent infections. These infections may lead to serious, including fatal outcomes.
Progressive Multifocal Leukoencephalopathy
Cases of JC virus-associated infection resulting in progressive multifocal leukoencephalopathy (PML), sometimes fatal, have been reported in patients treated with immunosuppressants, including Imuran. Risk factors for PML include treatment with immunosuppressant therapies and impairment of immune function. Consider the diagnosis of PML in any patient presenting with new-onset neurological manifestations and consider consultation with a neurologist as clinically indicated. Consider reducing the amount of immunosuppression in patients who develop PML. In transplant patients, consider the risk that the reduced immunosuppression represents to the graft.
Effect on Sperm in Animals
IMURAN has been reported to cause temporary depression in spermatogenesis and reduction in sperm viability and sperm count in mice at doses 10 times the human therapeutic dose 1; a reduced percentage of fertile matings occurred when animals received 5 mg/kg. 2
IMURAN can cause fetal harm when administered to a pregnant woman. IMURAN should not be given during pregnancy without careful weighing of risk versus benefit. Whenever possible, use of IMURAN in pregnant patients should be avoided. This drug should not be used for treating rheumatoid arthritis in pregnant women. 3
IMURAN is teratogenic in rabbits and mice when given in doses equivalent to the human dose (5 mg/kg daily). Abnormalities included skeletal malformations and visceral anomalies. 2
Postmarketing cases of intrahepatic cholestasis of pregnancy (ICP) have been reported in women treated with azathioprine during pregnancy. ICP symptoms and evaluated bile acid levels improved following azathioprine discontinuation. Discontinue IMURAN if ICP develops in a pregnant woman.
Limited immunologic and other abnormalities have occurred in a few infants born of renal allograft recipients on IMURAN. In a detailed case report, 4 documented lymphopenia, diminished IgG and IgM levels, CMV infection, and a decreased thymic shadow were noted in an infant born to a mother receiving 150 mg azathioprine and 30 mg prednisone daily throughout pregnancy. At 10 weeks most features were normalized. DeWitte et al reported pancytopenia and severe immune deficiency in a preterm infant whose mother received 125 mg azathioprine and 12.5 mg prednisone daily. 5 There have been two published reports of abnormal physical findings. Williamson and Karp described an infant born with preaxial polydactyly whose mother received azathioprine 200 mg daily and prednisone 20 mg every other day during pregnancy. 6 Tallent et al described an infant with a large myelomeningocele in the upper lumbar region, bilateral dislocated hips, and bilateral talipes equinovarus. The father was on long-term azathioprine therapy. 7
Benefit versus risk must be weighed carefully before use of IMURAN in patients of reproductive potential. There are no adequate and well-controlled studies in pregnant women. If this drug is used during pregnancy or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing age should be advised to avoid becoming pregnant.
-
PRECAUTIONS:
General: A gastrointestinal hypersensitivity reaction characterized by severe nausea and vomiting has been reported. These symptoms may also be accompanied by diarrhea, rash, fever, malaise, myalgias, elevations in liver enzymes, and occasionally, hypotension. Symptoms of gastrointestinal toxicity most often develop within the first several weeks of therapy with IMURAN and are reversible upon discontinuation of the drug. The reaction can recur within hours after re-challenge with a single dose of IMURAN.
Information for Patients: Patients being started on IMURAN should be informed of the necessity of periodic blood counts while they are receiving the drug and should be encouraged to report any unusual bleeding or bruising to their physician. They should be informed of the danger of infection while receiving IMURAN and asked to report signs and symptoms of infection to their physician. Careful dosage instructions should be given to the patient, especially when IMURAN is being administered in the presence of impaired renal function or concomitantly with allopurinol (see Drug Interactions subsection and DOSAGE AND ADMINISTRATION). Patients should be advised of the potential risks of the use of IMURAN during pregnancy and during the nursing period. The increased risk of malignancy following therapy with IMURAN should be explained to the patient.
Laboratory Tests: Complete Blood Count (CBC) Monitoring: Patients on IMURAN should have complete blood counts, including platelet counts, weekly during the first month, twice monthly for the second and third months of treatment, then monthly or more frequently if dosage alterations or other therapy changes are necessary.
TPMT and NUDT15 Testing: Consider genotyping or phenotyping patients for TPMT deficiency and genotyping for NUDT15 deficiency in patients with severe myelosuppression. TPMT and NUDT15 testing cannot substitute for complete blood count (CBC) monitoring in patients receiving IMURAN. Accurate phenotyping (red blood cell TPMT activity) results are not possible in patients who have received recent blood transfusions (see CLINICAL PHARMACOLOGY, WARNINGS: Cytopenias, ADVERSE REACTIONS and DOSAGE AND ADMINISTRATION sections).
Drug Interactions: Use with xanthine oxidase (XO) inhibitors: One of the pathways for inactivation of azathioprine is inhibited by XO inhibitors (allopurinol or febuxostat). Patients receiving IMURAN and allopurinol concomitantly should have a dose reduction of IMURAN, to approximately 1/ 3 to 1/ 4 the usual dose. Concomitant use of IMURAN and febuxostat is not recommended. Inhibition of XO may cause increased plasma concentrations of azathioprine or its metabolite, 6-MP, leading to toxicity. It is recommended that a further dose reduction or alternative therapies be considered for patients with low or absent TPMT activity receiving IMURAN and xanthine oxidase inhibitors because both TPMT and XO inactivation pathways are affected (see CLINICAL PHARMACOLOGY, WARNINGS, PRECAUTIONS: Laboratory Tests and ADVERSE REACTIONS sections).
Use with Aminosalicylates: There is in vitro evidence that aminosalicylate derivatives (e.g., sulphasalazine, mesalazine, or olsalazine) inhibit the TPMT enzyme. Concomitant use of these agents with IMURAN should be done with caution.
Use with Other Agents Affecting Myelopoesis: Drugs which may affect leukocyte production, including co-trimoxazole, may lead to exaggerated leukopenia, especially in renal transplant recipients.
Use with Angiotensin-Converting Enzyme Inhibitors: The use of angiotensin-converting enzyme inhibitors to control hypertension in patients on azathioprine has been reported to induce anemia and severe leukopenia.
Use with Warfarin: IMURAN may inhibit the anticoagulant effect of warfarin.
Use with ribavirin: The use of ribavirin for hepatitis C in patients receiving azathioprine has been reported to induce severe pancytopenia and may increase the risk of azathioprine-related myelotoxicity. Inosine monophosphate dehydrogenase (IMDH) is required for one of the metabolic pathways of azathioprine. Ribavirin is known to inhibit IMDH, thereby leading to accumulation of an azathioprine metabolite, 6-methylthioionosine monophosphate (6-MTITP), which is associated with myelotoxicity (neutropenia, thrombocytopenia, and anemia). Patients receiving azathioprine with ribavirin should have complete blood counts, including platelet counts, monitored weekly for the first month, twice monthly for the second and third months of treatment, then monthly or more frequently if dosage or other therapy changes are necessary.
Nursing Mothers: The use of IMURAN in nursing mothers is not recommended. Azathioprine or its metabolites are transferred at low levels, both transplacentally and in breast milk. 8, 9, 10 Because of the potential for tumorigenicity shown for azathioprine, a decision should be made whether to discontinue nursing or discontinue the drug, taking into account the importance of the drug to the mother.
-
ADVERSE REACTIONS:
The principal and potentially serious toxic effects of IMURAN are hematologic and gastrointestinal. The risks of secondary infection and malignancy are also significant (see WARNINGS). The frequency and severity of adverse reactions depend on the dose and duration of IMURAN as well as on the patient's underlying disease or concomitant therapies. The incidence of hematologic toxicities and neoplasia encountered in groups of renal homograft recipients is significantly higher than that in studies employing IMURAN for rheumatoid arthritis. The relative incidences in clinical studies are summarized below:
* Data on the rate and risk of neoplasia among persons with rheumatoid arthritis treated with azathioprine are limited. The incidence of lymphoproliferative disease in patients with RA appears to be significantly higher than that in the general population. In one completed study, the rate of lymphoproliferative disease in RA patients receiving higher than recommended doses of azathioprine (5 mg/kg per day) was 1.8 cases per 1000 patient-years of follow-up, compared with 0.8 cases per 1000 patient-years of follow-up in those not receiving azathioprine. However, the proportion of the increased risk attributable to the azathioprine dosage or to other therapies (i.e., alkylating agents) received by patients treated with azathioprine cannot be determined.
Toxicity Renal Homograft Rheumatoid Arthritis Leukopenia (any degree) >50% 28% <2500 cells/mm 3 16% 5.3% Infections 20% <1% Neoplasia * Lymphoma 0.5% Others 2.8% Hematologic: Leukopenia and/or thrombocytopenia are dose-dependent and may occur late in the course of therapy with IMURAN. Dose reduction or temporary withdrawal may result in reversal of these toxicities. Infection may occur as a secondary manifestation of bone marrow suppression or leukopenia, but the incidence of infection in renal homotransplantation is 30 to 60 times that in rheumatoid arthritis. Anemias, including macrocytic anemia, and/or bleeding have been reported.
Patients with low or absent TPMT or NUDT15 activity are at increased risk for severe, life-threatening myelosuppression from IMURAN (see CLINICAL PHARMACOLOGY, WARNINGS: Cytopenias and PRECAUTIONS: Laboratory Tests, DOSAGE AND ADMINISTRATION).
Gastrointestinal: Nausea and vomiting may occur within the first few months of therapy with IMURAN, and occurred in approximately 12% of 676 rheumatoid arthritis patients. The frequency of gastric disturbance often can be reduced by administration of the drug in divided doses and/or after meals. However, in some patients, nausea and vomiting may be severe and may be accompanied by symptoms such as diarrhea, fever, malaise, and myalgias (see PRECAUTIONS). Vomiting with abdominal pain may occur rarely with a hypersensitivity pancreatitis. Hepatotoxicity manifest by elevation of serum alkaline phosphatase, bilirubin, and/or serum transaminases is known to occur following azathioprine use, primarily in allograft recipients. Hepatotoxicity has been uncommon (less than 1%) in rheumatoid arthritis patients. Hepatotoxicity following transplantation most often occurs within 6 months of transplantation and is generally reversible after interruption of IMURAN. A rare, but life-threatening hepatic veno-occlusive disease associated with chronic administration of azathioprine has been described in transplant patients and in one patient receiving IMURAN for panuveitis. 11, 12, 13 Periodic measurement of serum transaminases, alkaline phosphatase, and bilirubin is indicated for early detection of hepatotoxicity. If hepatic veno-occlusive disease is clinically suspected, IMURAN should be permanently withdrawn.
Others: Additional side effects of low frequency have been reported. These include skin rashes, alopecia, fever, arthralgias, diarrhea, steatorrhea, negative nitrogen balance, reversible interstitial pneumonitis, hepatosplenic T-cell lymphoma (see Warnings – Malignancy), and Sweet's Syndrome (acute febrile neutrophilic dermatosis).
Postmarketing Experience
The following adverse reactions have been identified during postapproval use of IMURAN. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a casual relationship to drug exposure.- intrahepatic cholestasis of pregnancy (see WARNINGS, Pregnancy).
-
OVERDOSAGE:
The oral LD 50s for single doses of IMURAN in mice and rats are 2500 mg/kg and 400 mg/kg, respectively. Very large doses of this antimetabolite may lead to marrow hypoplasia, bleeding, infection, and death. About 30% of IMURAN is bound to serum proteins, but approximately 45% is removed during an 8-hour hemodialysis. 14 A single case has been reported of a renal transplant patient who ingested a single dose of 7500 mg IMURAN. The immediate toxic reactions were nausea, vomiting, and diarrhea, followed by mild leukopenia and mild abnormalities in liver function. The white blood cell count, SGOT, and bilirubin returned to normal 6 days after the overdose.
-
DOSAGE AND ADMINISTRATION:
TPMT TESTING CANNOT SUBSTITUTE FOR COMPLETE BLOOD COUNT (CBC) MONITORING IN PATIENTS RECEIVING IMURAN. TPMT genotyping or phenotyping can be used to identify patients with absent or reduced TPMT activity. Patients with low or absent TPMT activity are at an increased risk of developing severe, life-threatening myelotoxicity from IMURAN if conventional doses are given. Physicians may consider alternative therapies for patients who have low or absent TPMT activity (homozygous for non-functional alleles). IMURAN should be administered with caution to patients having one non-functional allele (heterozygous) who are at risk for reduced TPMT activity that may lead to toxicity if conventional doses are given. Dosage reduction is recommended in patients with reduced TPMT activity. Early drug discontinuation may be considered in patients with abnormal CBC results that do not respond to dose reduction.
Renal Homotransplantation: The dose of IMURAN required to prevent rejection and minimize toxicity will vary with individual patients; this necessitates careful management. The initial dose is usually 3 to 5 mg/kg daily, beginning at the time of transplant. IMURAN is usually given as a single daily dose on the day of, and in a minority of cases 1 to 3 days before, transplantation. Dose reduction to maintenance levels of 1 to 3 mg/kg daily is usually possible. The dose of IMURAN should not be increased to toxic levels because of threatened rejection. Discontinuation may be necessary for severe hematologic or other toxicity, even if rejection of the homograft may be a consequence of drug withdrawal.
Rheumatoid Arthritis: IMURAN is usually given on a daily basis. The initial dose should be approximately 1.0 mg/kg (50 to 100 mg) given as a single dose or on a twice-daily schedule. The dose may be increased, beginning at 6 to 8 weeks and thereafter by steps at 4-week intervals, if there are no serious toxicities and if initial response is unsatisfactory. Dose increments should be 0.5 mg/kg daily, up to a maximum dose of 2.5 mg/kg per day. Therapeutic response occurs after several weeks of treatment, usually 6 to 8; an adequate trial should be a minimum of 12 weeks. Patients not improved after 12 weeks can be considered refractory. IMURAN may be continued long-term in patients with clinical response, but patients should be monitored carefully, and gradual dosage reduction should be attempted to reduce risk of toxicities.
Maintenance therapy should be at the lowest effective dose, and the dose given can be lowered decrementally with changes of 0.5 mg/kg or approximately 25 mg daily every 4 weeks while other therapy is kept constant. The optimum duration of maintenance IMURAN has not been determined. IMURAN can be discontinued abruptly, but delayed effects are possible.
Patients with TPMT and/or NUDT15 Deficiency
Consider testing for TPMT and NUDT15 deficiency in patients who experience severe bone marrow toxicities. Early drug discontinuation may be considered in patients with abnormal CBC results that do not respond to dose reduction (see CLINICAL PHARMACOLOGY, WARNINGS: Cytopenias, and PRECAUTIONS: Laboratory Tests).
Homozygous deficiency in either TPMT or NUDT15
Because of the risk of increased toxicity, consider alternative therapies for patients who are known to have TPMT or NUDT15 deficiency (see CLINICAL PHARMACOLOGY, WARNINGS: Cytopenias, and PRECAUTIONS: Laboratory Tests).
Heterozygous deficiency in TPMT and/or NUDT15
Because of the risk of increased toxicity, dosage reduction is recommended in patients known to have heterozygous deficiency of TPMT or NUDT15. Patients who are heterozygous for both TPMT and NUDT15 deficiency may require more substantial dosage reductions (see CLINICAL PHARMACOLOGY, WARNINGS: Cytopenias, and PRECAUTIONS: Laboratory Tests).
Use in Renal Dysfunction: Relatively oliguric patients, especially those with tubular necrosis in the immediate postcadaveric transplant period, may have delayed clearance of IMURAN or its metabolites, may be particularly sensitive to this drug, and are usually given lower doses.
Procedures for proper handling and disposal of this immunosuppressive antimetabolite drug should be considered. Several guidelines on this subject have been published. 15-21 There is no general agreement that all of the procedures recommended in the guidelines are necessary or appropriate.
- HOW SUPPLIED:
-
REFERENCES:
- Clark JM. The mutagenicity of azathioprine in mice, Drosophila melanogaster, and Neurospora crassa. Mutat Res. 1975; 28:87-99.
- Data on file, Sebela Ireland Ltd.
- Tagatz GE, Simmons RL. Pregnancy after renal transplantation. Ann Intern Med. 1975; 82:113-114. Editorial Notes.
- Cote' CJ, Meuwissen HJ, Pickering RJ. Effects on the neonate of prednisone and azathioprine administered to the mother during pregnancy . J Pediatr. 1974; 85:324-328.
- DeWitte DB, Buick MK, Cyran SE, et al. Neonatal pancytopenia and severe combined immunodeficiency associated with antenatal administration of azathioprine and prednisone. J Pediatr. 1984; 105:625-628.
- Williamson RA, Karp LE. Azathioprine teratogenicity: review of the literature and case report. Obstet Gynecol. 1981; 58:247-250.
- Tallent MB, Simmons RL, Najarian JS. Birth defects in child of male recipient of kidney transplant. JAMA. 1970; 211: 1854-1855.
- Data on file, Sebela Ireland Ltd.
- Saarikoski S, Seppälä M. Immunosuppression during pregnancy: transmission of azathioprine and its metabolites from the mother to the fetus. Am J Obstet Gynecol. 1973; 115:1100-1106.
- Coulam CB, Moyer TP, Jiang NS, et al. Breast-feeding after renal transplantation. Transplant Proc. 1982; 14: 605-609.
- Read AE, Wiesner RH, LaBrecque DR, et al. Hepatic veno-occlusive disease associated with renal transplantation and azathioprine therapy. Ann Intern Med. 1986; 104:651-655.
- Katzka DA, Saul SH, Jorkasky D, et al. Azathioprine and hepatic veno-occlusive disease in renal transplant patients. Gastroenterology. 1986; 90:446-454.
- Weitz H, Gokel JM, Loeshke K, et al. Veno-occlusive disease of the liver in patients receiving immunosuppressive therapy. Virchows Arch A Pathol Anat Histol. 1982; 395:245-256.
- Schusziarra V, Ziekursch V, Schlamp R, et al. Pharmacokinetics of azathioprine under haemodialysis. Int J Clin Pharmacol Biopharm. 1976; 14:298-302.
- Recommendations for the safe handling of parenteral antineoplastic drugs. Washington, DC: Division of Safety; Clinical Center Pharmacy Department and Cancer Nursing Services, National Institute of Health; 1992. US Dept of Health and Human Services. Public Health Service Publication NIH 92-2621.
- AMA Council on Scientific Affairs. Guidelines for handling parenteral antineoplastics. JAMA. 1985; 253:1590-1592.
- National Study Commission on Cytotoxic Exposure. Recommendations for handling cytotoxic agents. 1987. Available from Louis P. Jeffrey, Chairman, National Study Commission on Cytotoxic Exposure. Massachusetts College of Pharmacy and Allied Health Sciences, 179 Longwood Avenue, Boston, MA 02115.
- Clinical Oncological Society of Australia. Guidelines and recommendations for safe handling of antineoplastic agents. Med J Aust. 1983; 1:426-428.
- Jones RB, Frank R, Mass T. Safe handling of chemotherapeutic agents: a report from The Mount Sinai Medical Center. CA Cancer J for Clinicians. 1983; 33:258-263.
- American Society of Hospital Pharmacists. ASHP technical assistance bulletin on handling cytotoxic and hazardous drugs. Am J Hosp Pharm. 1990; 47:1033-1049.
- Yodaiken RE, Bennett D. OSHA Work-Practice guidelines for personnel dealing with cytotoxic (antineoplastic) drugs. Am J Hosp Pharm, 1996; 43:1193-1204.
IMURAN is a registered trademark of Sebela International Ltd.
Distributed by Sebela Pharmaceuticals Inc.
645 Hembree Parkway, Suite I
Roswell, GA 30076
www.sebelapharma.com
Toll Free 1-844-732-3521
©2024 Sebela Pharmaceuticals Inc. All rights reserved.
Revised 07/2024PI59010001
Sebela Pharmaceuticals Inc.
- Imuran 50mg - 100 count Carton
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
IMURAN
azathioprine tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:54766-590 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength AZATHIOPRINE (UNII: MRK240IY2L) (AZATHIOPRINE - UNII:MRK240IY2L) AZATHIOPRINE 50 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) LACTOSE (UNII: J2B2A4N98G) STARCH, POTATO (UNII: 8I089SAH3T) POVIDONE (UNII: FZ989GH94E) STEARIC ACID (UNII: 4ELV7Z65AP) Product Characteristics Color yellow (YELLOW) Score 2 pieces Shape DOUBLE CIRCLE (DOUBLE CIRCLE) Size 11mm Flavor Imprint Code IMURAN;50 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:54766-590-10 100 in 1 BOTTLE; Type 0: Not a Combination Product 12/01/2017 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA016324 12/01/2017 Labeler - Sebela Pharmaceuticals Inc. (079104574) Establishment Name Address ID/FEI Business Operations Pharmaceutics International Inc. 878265586 manufacture(54766-590)

 100 Tablets
100 Tablets