Label: BEOVU- brolucizumab injection, solution

- NDC Code(s): 0078-0827-60, 0078-0827-61, 0078-0827-98, 0078-0827-99
- Packager: Novartis Pharmaceuticals Corporation
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated November 3, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BEOVU safely and effectively. See full prescribing information for BEOVU.
BEOVU® (brolucizumab-dbll) injection, for intravitreal use
Initial U.S. Approval: 2019INDICATIONS AND USAGE
DOSAGE AND ADMINISTRATION
Neovascular (Wet) Age-Related Macular Degeneration (AMD)
- The recommended dose for BEOVU is 6 mg (0.05 mL of 120 mg/mL solution) monthly (approximately every 25-31 days) for the first three doses, followed by one dose of 6 mg (0.05 mL) every 8-12 weeks (2.2).
Diabetic Macular Edema (DME)
- The recommended dose for BEOVU is 6 mg (0.05 mL of 120 mg/mL solution) every six weeks (approximately every 39-45 days) for the first five doses, followed by one dose of 6 mg (0.05 mL of 120 mg/mL solution) every 8-12 weeks (2.3).
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- Endophthalmitis and retinal detachment may occur following intravitreal injections. Patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment without delay (5.1).
- Retinal vasculitis and/or retinal vascular occlusion, typically in the presence of intraocular inflammation, have been reported following BEOVU injections. Patients should be instructed to report any change in vision without delay (5.2).
- Increases in intraocular pressure (IOP) have been seen within 30 minutes of an intravitreal injection (5.3).
- There is a potential risk of arterial thromboembolic events (ATE) following intravitreal use of VEGF inhibitors (5.4).
ADVERSE REACTIONS
The most common adverse reactions reported in patients receiving BEOVU are vision blurred, cataract, conjunctival hemorrhage, eye pain, and vitreous floaters (6.1).
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 7/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Neovascular (Wet) Age-related Macular Degeneration (AMD)
1.2 Diabetic Macular Edema (DME)
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
2.2 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
2.3 Diabetic Macular Edema (DME)
2.4 Preparation for Administration – Pre-filled Syringe and Vial
2.5 Injection Procedure
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 Ocular or Periocular Infections
4.2 Active Intraocular Inflammation
4.3 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachment
5.2 Retinal Vasculitis and/or Retinal Vascular Occlusion
5.3 Increase in Intraocular Pressure
5.4 Thromboembolic Events
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Immunogenicity
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
14.2 Diabetic Macular Edema (DME)
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 General Dosing Information
For ophthalmic intravitreal injection. BEOVU must be administered by a qualified physician.
BEOVU is available packaged as follows [see How Supplied/Storage and Handling (16)]:
- Pre-filled Syringe
- Vial kit with injection components (vial, filter needle)
2.2 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
The recommended dose for BEOVU is 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection monthly (approximately every 25 to 31 days) for the first three doses, followed by 6 mg (0.05 mL) by intravitreal injection once every 8 to 12 weeks.
2.3 Diabetic Macular Edema (DME)
The recommended dose for BEOVU is 6 mg (0.05 mL of 120 mg/mL solution) administered by intravitreal injection every six weeks (approximately every 39-45 days) for the first five doses, followed by 6 mg (0.05 mL) by intravitreal injection once every 8-12 weeks.
2.4 Preparation for Administration – Pre-filled Syringe and Vial

Store BEOVU in the refrigerator between 2°C to 8°C (36°F to 46°F); do not freeze. Keep BEOVU in the outer carton to protect from light. 
Prior to use, the unopened glass vial or sealed blister pack of BEOVU may be kept at room temperature, 20°C to 25°C (68°F to 77°F) for up to 24 hours. After opening, proceed under aseptic conditions. BEOVU is a clear to slightly opalescent and colorless to slightly brownish-yellow solution. ![Preparation for Administration - BEOVU should be inspected visually upon removal from the refrigerator and prior to administration. If particulates, cloudiness, or discoloration are visible, the glass vial must not be used. The BEOVU kit includes the sterile glass vial and filter needle which are for single use only. Do not use if the packaging, vial and/or filter needle are damaged or expired [see How Supplied/Storage and Handling (16)].](/dailymed/image.cfm?name=rth258-04.jpg&setid=5d1dc1fa-a2d3-46ed-9e9a-c1a036590d3d)
BEOVU should be inspected visually upon removal from the refrigerator and prior to administration. If particulates, cloudiness, or discoloration are visible, the BEOVU must not be used. Use aseptic technique for preparation of the intravitreal injection.
Pre-filled Syringe
The BEOVU pre-filled glass syringe is sterile and for the treatment of a single eye. It should be inspected visually prior to administration. Do not use if the packaging, or pre-filled syringe are opened, damaged, or expired [see How Supplied/Storage and Handling (16)].
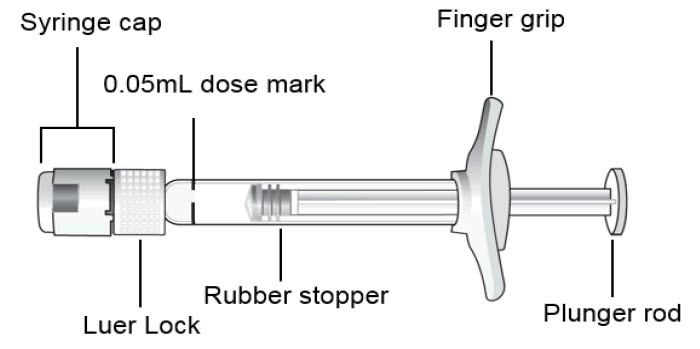
STEP 1: PREPARE
Peel the lid off the blister package and, using aseptic technique, remove the sterile syringe.STEP 2: SNAP OFF SYRINGE CAP
Snap off the syringe cap and dispose of it (see Figure 1).
Do not turn or twist the syringe cap.Figure 1:
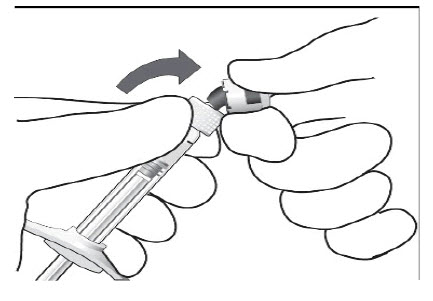
STEP 3: ATTACH INJECTION NEEDLE
Aseptically and firmly assemble a 30-gauge x ½ inch sterile injection needle (not included) onto the Luer lock syringe.STEP 4: DISLODGE AIR BUBBLES
To check for air bubbles, hold the syringe with the needle pointing up. If there are any air bubbles, gently tap the syringe with your finger until the bubbles rise to the top (see Figure 2).
Carefully remove the needle cap by pulling it straight off.Figure 2:
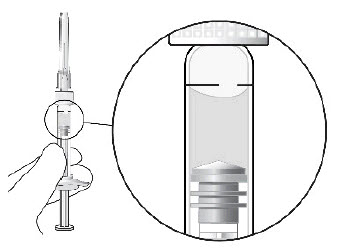
STEP 5: EXPEL AIR AND SET THE DOSE
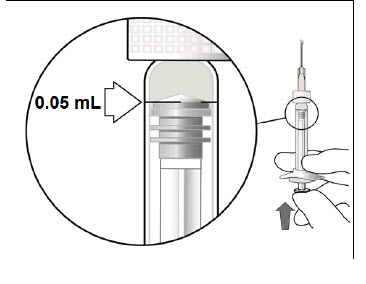
Hold the syringe at eye level and carefully push the plunger until the edge below the dome of the rubber stopper is aligned with the 0.05 mL dose mark (see Figure 3). This will expel the air and the excess liquid and set the dose to the 0.05 mL dose mark. The syringe is ready for the injection.Figure 3:
Vial
The BEOVU vial kit includes the sterile glass vial and filter needle which are sterile and for the treatment of a single eye. It should be inspected visually prior to administration. Do not use if the packaging, vial, and/or filter needle are damaged or expired [see How Supplied/Storage and Handling (16)].
STEP 1: Gather the supplies needed. - One BEOVU vial (included)
- One sterile 5-micron blunt filter needle (18-gauge x 1½ inch, 1.2 mm x 40 mm) (included)
- One sterile 30-gauge x ½ inch injection needle (not included)
- One sterile 1 mL syringe with a 0.05 mL dose mark (not included)
- Alcohol swab (not included)
STEP 2:
Inspect the solution. If particulates, cloudiness, or discoloration are visible, discard the vial and obtain a new vial.STEP 3:
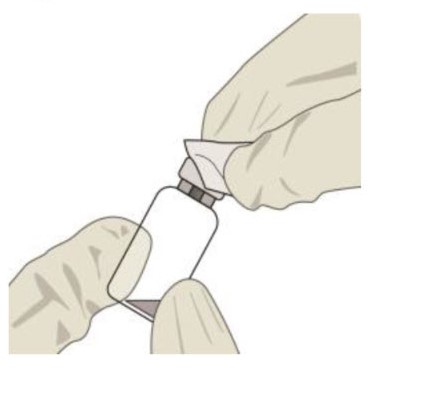
Remove the vial cap and clean the vial septum (e.g., with alcohol swab) (see Figure 4).Figure 4:
STEP 4:
Assemble the 5-micron filter needle (18-gauge x 1½ inch) onto a 1-mL syringe using aseptic technique.STEP 5:
Push the filter needle into the center of the vial septum until the needle touches the bottom of the vial.STEP 6:
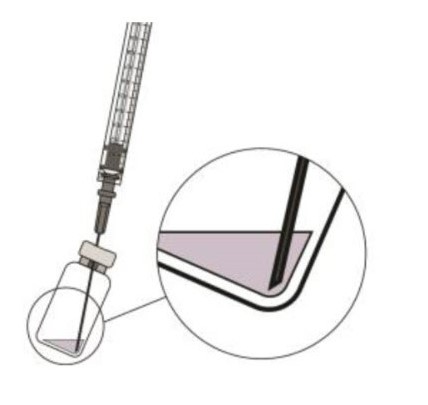
To withdraw the liquid, hold the vial slightly inclined and slowly withdraw all the liquid from the vial and filter needle (see Figure 5).
Ensure that the plunger rod is drawn sufficiently back when emptying the vial in order to completely empty the filter needle.Figure 5:
STEP 7:
Disconnect the filter needle from the syringe in an aseptic manner and dispose of it. The filter needle is not to be used for the intravitreal injection.STEP 8:
Aseptically and firmly assemble a 30-gauge x ½ inch injection needle onto the syringe.STEP 9:
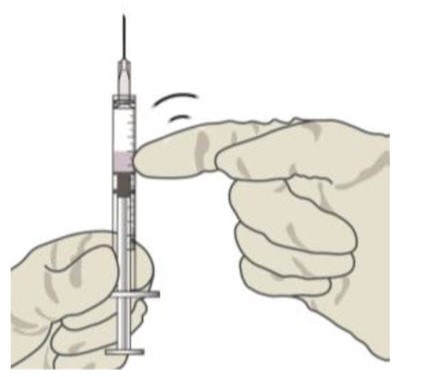
Check for air bubbles by holding the syringe with the needle pointing up. If there are any air bubbles, gently tap the syringe with your finger until the bubbles rise to the top (see Figure 6).Figure 6:
STEP 10:
Carefully expel the air from the syringe and adjust the dose to the 0.05 mL mark (see Figure 7).
The syringe is ready for the injection.Figure 7:
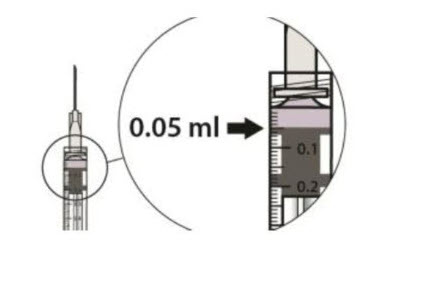
2.5 Injection Procedure
Ensure that the injection is given immediately after preparation of the dose.
The intravitreal injection procedure must be carried out under aseptic conditions, which includes the use of surgical hand disinfection, sterile gloves, a sterile drape and a sterile eyelid speculum (or equivalent), and the availability of sterile paracentesis equipment (if required). Adequate anesthesia and a broad-spectrum topical microbicide to disinfect the periocular skin, eyelid, and ocular surface should be administered prior to the injection.
Inject slowly until the rubber stopper reaches the end of the syringe to deliver the volume of 0.05 mL. Confirm delivery of the full dose by checking that the rubber stopper has reached the end of the syringe barrel.
Immediately following the intravitreal injection, patients should be monitored for elevation in intraocular pressure (IOP). Appropriate monitoring may consist of a check for perfusion of the optic nerve head or tonometry. If required, a sterile paracentesis needle should be available.
Following intravitreal injection, patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment (e.g., eye pain, redness of the eye, photophobia, blurring of vision) without delay [see Patient Counseling Information (17)].
Each vial or pre-filled syringe should only be used for the treatment of a single eye. If the contralateral eye requires treatment, a new vial or pre-filled syringe should be used and the sterile field, syringe, gloves, drapes, eyelid speculum, filter, and injection needles should be changed before BEOVU is administered to the other eye.
Any unused medicinal product or waste material should be disposed of in accordance with local regulations.
- 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Endophthalmitis and Retinal Detachment
Intravitreal injections, including those with BEOVU, have been associated with endophthalmitis and retinal detachment [see Contraindications (4.1) and Adverse Reactions (6.1)]. Proper aseptic injection techniques must always be used when administering BEOVU. Patients should be instructed to report any symptoms suggestive of endophthalmitis or retinal detachment without delay and should be managed appropriately [see Dosage and Administration (2.4) and Patient Counseling Information (17)].
5.2 Retinal Vasculitis and/or Retinal Vascular Occlusion
Retinal vasculitis and/or retinal vascular occlusion, typically in the presence of intraocular inflammation, have been reported with the use of BEOVU. These immune-mediated adverse events may occur following the first intravitreal injection. Discontinue treatment with BEOVU in patients who develop these events. Patients treated with BEOVU who experience intraocular inflammation may be at risk of developing retinal vasculitis and/or retinal vascular occlusion and should be closely monitored [see Contraindications (4.2) and Adverse Reactions (6.1, 6.2)]. Patients should be instructed to report any change in vision without delay.
5.3 Increase in Intraocular Pressure
Acute increases in intraocular pressure (IOP) have been seen within 30 minutes of intravitreal injection, including with BEOVU [see Adverse Reactions (6.1)]. Sustained IOP increases have also been reported. Both IOP and perfusion of the optic nerve head must be monitored and managed appropriately [see Dosage and Administration (2.4)].
5.4 Thromboembolic Events
Although there was a low rate of arterial thromboembolic events (ATEs) observed in the BEOVU clinical trials, there is a potential risk of ATEs following intravitreal use of VEGF inhibitors. Arterial thromboembolic events are defined as nonfatal stroke, nonfatal myocardial infarction, or vascular death (including deaths of unknown cause).
The ATE rate in the two controlled 96-week neovascular AMD studies (HAWK and HARRIER) during the first 96 weeks was 4.5% (33 of 730) in the pooled brolucizumab arms compared with 4.7% (34 of 729) in the pooled aflibercept arms [see Clinical Studies (14.1)].
-
6 ADVERSE REACTIONS
The following potentially serious adverse reactions are described elsewhere in the labeling:
- Hypersensitivity [see Contraindications (4.3)]
- Endophthalmitis and Retinal Detachment [see Warnings and Precautions (5.1)]
- Retinal Vasculitis and/or Retinal Vascular Occlusion [see Warnings and Precautions (5.2)]
- Increase in Intraocular Pressure [see Warnings and Precautions (5.3)]
- Thromboembolic Events [see Warnings and Precautions (5.4)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in one clinical trial of a drug cannot be directly compared with rates in the clinical trials of the same or another drug and may not reflect the rates observed in practice.
A total of 1,646 patients treated with brolucizumab constituted the safety population in four Phase 3 studies. Among these, 1,098 patients were treated with the recommended dose of 6 mg.
A total of 1,088 patients treated with brolucizumab, constituted the safety population in the two controlled neovascular AMD Phase 3 studies (HAWK and HARRIER) with a cumulative 96-week exposure to BEOVU, and 730 patients treated with the recommended dose of 6 mg [see Clinical Studies (14.1)]. A total of 558 patients treated with brolucizumab constituted the safety population in the two controlled DME Phase 3 studies (KESTREL and KITE) from baseline to week 52, including 368 patients treated with the recommended dose of 6 mg [see Clinical Studies (14.2)].
Table 1: Common Adverse Reactions (≥ 1%) in the AMD and DME Clinical Trials aIncluding vision blurred, visual acuity reduced, visual acuity reduced transiently, and visual impairment.
bIncluding anterior chamber cell, anterior chamber flare, anterior chamber inflammation, chorioretinitis, eye inflammation, iridocyclitis, iritis, retinal vasculitis, retinal vascular occlusion, uveitis, vitreous haze, vitritis.
cIncluding urticaria, rash, pruritus, erythema.
dIncluding blindness, blindness transient, amaurosis, and amaurosis fugax.BEOVU Active Control (aflibercept) Adverse Drug Reactions AMD
(N = 730)DME
(N = 368)AMD
(N = 729)DME
(N = 368)Vision blurreda 10% 2% 11% 4% Cataract 7% 4% 11% 5% Conjunctival hemorrhage 6% 6% 7% 7% Vitreous floaters 5% 3% 3% 2% Eye pain 5% 3% 6% 2% Intraocular inflammationb 4% 3% 1% 1% Intraocular pressure increased 4% 2% 5% 1% Retinal hemorrhage 4% 3% 1% Vitreous detachment 4% 2% 3% 1% Conjunctivitis 3% 2% 2% < 1% Retinal pigment epithelial tear 3% 1% Corneal abrasion 2% 1% 2% 2% Hypersensitivityc 2% 1% 1% 1% Punctate keratitis 1% 1% 2% Retinal tear 1% < 1% 1% < 1% Endophthalmitis 1% < 1% < 1% 1% Blindnessd 1% < 1% < 1% Retinal artery occlusion 1% 1% < 1% < 1% Retinal detachment 1% < 1% 1% Conjunctival hyperemia 1% < 1% 1% 1% Lacrimation increased 1% < 1% 1% < 1% Abnormal sensation in eye 1% < 1% 2% 1% Detachment of retinal pigment epithelium 1% < 1% Vitreous hemorrhage < 1% 1% < 1% 1% In clinical trials, scleritis and episcleritis were reported (incidence < 1%).
In a clinical study (MERLIN), patients with nAMD who received BEOVU every 4-week maintenance dosing experienced a higher incidence of intraocular inflammation (including retinal vasculitis) and retinal vascular occlusion than patients who received BEOVU every 8 or 12-week maintenance dosing in the clinical studies (HAWK and HARRIER). The interval between two BEOVU doses during maintenance treatment should not be less than 8 weeks.
6.2 Immunogenicity
As with all therapeutic proteins, there is a potential for an immune response in patients treated with BEOVU. The immunogenicity of BEOVU was evaluated in serum samples. The immunogenicity data reflect the percentage of patients whose test results were considered positive for antibodies to BEOVU in immunoassays. The detection of an immune response is highly dependent on the sensitivity and specificity of the assays used, sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to BEOVU with the incidence of antibodies to other products may be misleading.
Anti-brolucizumab antibodies were detected in the pre-treatment sample of 36% to 64% of treated naive patients. After initiation of dosing, anti-brolucizumab antibodies were detected in at least one serum sample in 53% to 76% of patients treated with BEOVU. Intraocular inflammation was observed in 6% of patients with anti-brolucizumab antibodies detected during dosing with BEOVU in clinical trials. Retinal vasculitis and/or retinal vascular occlusion, typically in the presence of intraocular inflammation, are immune-mediated adverse events related to exposure to BEOVU. This treatment-emergent antibody response may develop following the first intravitreal injection. Anti-brolucizumab antibodies were not associated with an impact on clinical efficacy.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no adequate and well-controlled studies of BEOVU administration in pregnant women. In an animal reproduction study, intravitreal administration of brolucizumab to pregnant monkeys once every 4 weeks in one eye from organogenesis to birth did not indicate any harmful effects with respect to pre- or postnatal development at 10-fold the maximum recommended human dose (MRHD) on a mg/kg basis (see Data).
Based on the anti-VEGF mechanism of action for brolucizumab [see Clinical Pharmacology (12.1)], treatment with BEOVU may pose a risk to human embryo-fetal development. BEOVU should be used during pregnancy only if the potential benefit outweighs the potential risk to the fetus.
All pregnancies have a background risk of birth defect, loss, and other adverse outcomes. The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Data
Animal Data
In an enhanced pre- and postnatal development (ePPND) study in pregnant cynomolgus monkeys, brolucizumab was administered to all animals by intravitreal (IVT) injection to one eye at doses of 3 or 6 mg once every 4 weeks until delivery. There was no impact of IVT administration of brolucizumab on embryo-fetal development, pregnancy or parturition; or on the survival, growth, or postnatal development of offspring at 6 mg/eye (10-fold the MRHD on a mg/kg basis).
VEGF inhibition has been shown to cause malformations, embryo-fetal resorption, and decreased fetal weight. VEGF also has been shown to affect follicular development, corpus luteum function, and fertility.
8.2 Lactation
Risk Summary
There is no information regarding the presence of brolucizumab in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production/excretion. Because many drugs are transferred in human milk and because of the potential for absorption and adverse reactions in the breastfed child, breastfeeding is not recommended during treatment and for at least one month after the last dose when stopping treatment with BEOVU.
8.3 Females and Males of Reproductive Potential
Contraception
Females
Females of reproductive potential should use highly effective contraception (methods that result in less than 1% pregnancy rates) during treatment with BEOVU and for at least one month after the last dose when stopping treatment with BEOVU.
Infertility
No studies on the effects of brolucizumab on fertility have been conducted. Based on its anti-VEGF mechanism of action, treatment with BEOVU may pose a risk to reproductive capacity.
8.5 Geriatric Use
In the two Phase 3 clinical studies in AMD, approximately 90% (978/1089) of patients randomized to treatment with BEOVU were ≥ 65 years of age and approximately 60% (648/1089) were ≥ 75 years of age. In the two Phase 3 clinical studies in DME, approximately 45% (164/368) of patients randomized to treatment with BEOVU were ≥ 65 years of age and approximately 10% (37/368) were ≥ 75 years of age. No significant differences in efficacy or safety were seen with increasing age in these studies. No dosage regimen adjustment is required in patients 65 years and above.
-
11 DESCRIPTION
Brolucizumab-dbll is a recombinant human vascular endothelial growth factor inhibitor. Brolucizumab-dbll is a humanized monoclonal single-chain Fv (scFv) antibody fragment. Brolucizumab-dbll has a molecular weight of ~26 kilodaltons and is produced in Escherichia coli cells by recombinant DNA technology.
BEOVU (brolucizumab-dbll) injection is a sterile, clear to slightly opalescent, colorless to slightly brownish-yellow solution in a single-dose pre-filled syringe or a single-dose vial for intravitreal administration. Each single-dose pre-filled syringe and vial is designed to deliver 0.05 mL of solution containing 6 mg brolucizumab-dbll, polysorbate 80 (0.02%), sodium citrate (10 mM), sucrose (5.8%), and Water for Injection, USP and with a pH of approximately 7.2. This product does not contain an antimicrobial preservative.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Brolucizumab is a human VEGF inhibitor. Brolucizumab binds to the three major isoforms of VEGF-A (e.g., VEGF110, VEGF121, and VEGF165), thereby preventing interaction with receptors VEGFR-1 and VEGFR-2. By inhibiting VEGF-A, brolucizumab suppresses endothelial cell proliferation, neovascularization, and vascular permeability.
12.2 Pharmacodynamics
Leakage of blood and fluid from choroidal neovascularization (CNV) may cause retinal thickening or edema. Increased retinal thickness and accumulation of intraretinal fluid (IRF)/subretinal fluid (SRF), assessed by optical coherence tomography (OCT), is associated with nAMD and DME. Reductions in central retinal subfield thickness (CST) were observed across all treatment arms.
12.3 Pharmacokinetics
Following a single intravitreal dose of BEOVU to 25 AMD patients, the mean (range) serum Cmax of free brolucizumab (unbound to VEGF-A) was 49 ng/mL (9 to 548 ng/mL) and was attained at 24 hours post-dose. Brolucizumab concentrations were near or less than 0.5 ng/mL (lower limit of assay quantitation) at approximately 4 weeks after repeat dose administration and no accumulation in serum was observed in most patients.
Elimination
The estimated mean (± standard deviation) systemic half-life of brolucizumab is 4.4 days (± 2.0 days) after a single intravitreal dose.
Metabolism
Metabolism of brolucizumab has not been fully characterized. However, free brolucizumab is expected to undergo metabolism via proteolysis.
Excretion
The excretion of brolucizumab has not been fully characterized. However, free brolucizumab is expected to undergo target-mediated disposition and/or passive renal excretion.
Specific Populations
Following repeat intravitreal dose administration of BEOVU in the HAWK and HARRIER clinical studies, no differences in the systemic pharmacokinetics of brolucizumab were observed based on age (50 years and above), sex, or mild to moderate renal impairment (glomerular filtration rate (GFR) = 30 to 70 mL/min, estimated using the Modification of Diet in Renal Disease (MDRD) equation). The effect of severe renal impairment or any degree of hepatic impairment on the pharmacokinetics of BEOVU is unknown. As significant increases in serum brolucizumab exposures are not expected with intravitreal route of administration, no dosage adjustment is needed based on renal or hepatic impairment status.
Drug Interaction Studies
No studies evaluating the drug interaction potential of BEOVU have been conducted.
- 13 NONCLINICAL TOXICOLOGY
-
14 CLINICAL STUDIES
14.1 Neovascular (Wet) Age-Related Macular Degeneration (AMD)
The safety and efficacy of BEOVU were assessed in two randomized, multi-center, double-masked, active-controlled studies (HAWK - NCT02307682 and HARRIER - NCT02434328) in patients with neovascular AMD. A total of 1,817 patients were treated in these studies for two years (1,088 on brolucizumab and 729 on control). Patient ages ranged from 50 to 97 years with a mean of 76 years.
In HAWK, patients were randomized in a 1:1:1 ratio to the following dosing regimens:
1) brolucizumab 3 mg administered every 8 or 12 weeks after the first 3 monthly doses,
2) brolucizumab 6 mg administered every 8 or 12 weeks after the first 3 monthly doses,
3) aflibercept 2 mg administered every 8 weeks after the first 3 monthly doses.
In HARRIER, patients were randomized in a 1:1 ratio to the following dosing regimens:
1) brolucizumab 6 mg administered every 8 or 12 weeks after the first 3 monthly doses,
2) aflibercept 2 mg administered every 8 weeks after the first 3 monthly doses.
In both studies, after three initial monthly doses (Week 0, 4, and 8), treating physicians decided whether to treat each individual patient on an every 8-week or 12-week dosing interval guided by visual and anatomical measures of disease activity, although the utility of these measures has not been established. Patients on 12-week dosing intervals could be changed based on the same measures to an 8-week schedule after subsequent treatment visits. Any patient placed on an 8-week schedule, remained on the 8-week dosing interval until the end of the study. Protocol-specified visits in the initial three months occurred every 28 ± 3 days followed by every 28 ± 7 days for the remainder of the studies. Baseline anatomical measures may have contributed to the regimen selection because the majority of patients on the 12-week dosing schedule at the end of the trial had less baseline macular edema and/or smaller baseline lesions.
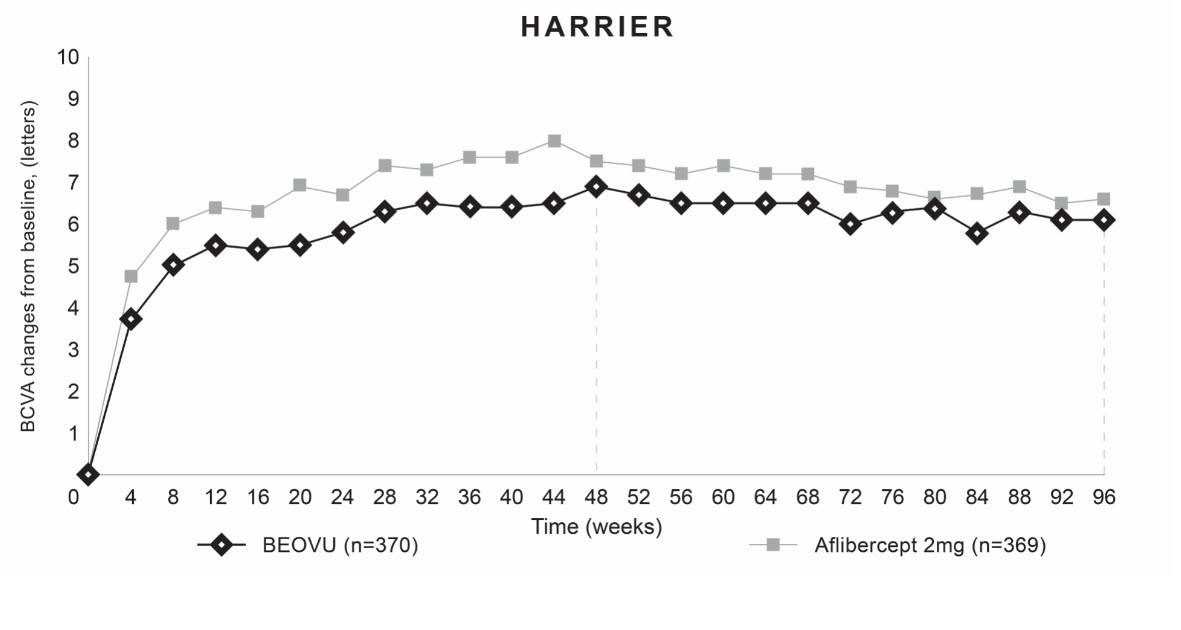
Both studies demonstrated efficacy in the primary endpoint defined as the change from baseline in Best Corrected Visual Acuity (BCVA) at Week 48, measured by the Early Treatment Diabetic Retinopathy Study (ETDRS) Letter Score. In both studies, BEOVU treated patients had a similar mean change from baseline in BCVA as the patients treated with aflibercept 2 mg (fixed every 8 weeks). Detailed results of both studies are shown in Table 2 and Figures 8 and 9 below.
Table 2: Efficacy Outcomes at Week 48 and 96 in Phase 3 HAWK and HARRIER Studies Abbreviations: BCVA, Best Corrected Visual Acuity; missing data are imputed using last observation carried forward (LOCF) method; ETDRS, Early Treatment Diabetic Retinopathy Study; SE, standard error. HAWK HARRIER Efficacy outcome At week BEOVU
(n = 360)Aflibercept
2 mg
(n = 360)Difference
(95% CI)
brolucizumab – afliberceptBEOVU
(n = 370)Aflibercept
2 mg
(n = 369)Difference
(95% CI)
brolucizumab – afliberceptMean (SD) BCVA at baseline 60.8 (13.7) 60.0 (13.9) 61.5 (12.6) 60.8 (12.9) Mean (SE) change from baseline in BCVA (measured by ETDRS letters score) 48
966.6
(0.71)
5.9
(0.78)6.8
(0.71)
5.3
(0.78)-0.2
(-2.1, 1.8)
+0.5
(-1.6, 2.7)6.9
(0.61)
6.1
(0.73)7.6
(0.61)
6.6
(0.73)-0.7
(-2.4, 1.0)
-0.4
(-2.5, 1.6)Proportion of patients who gained visual acuity (%) (≥ 15 letters of BCVA) 48
9633.6
34.2
25.4
27
8.2
(2.2, 15.0)
7.2
(1.4, 13.8)29.3
29.1
29.9
31.5
-0.6
(-7.1, 5.8)
-2.4
(-8.8, 4.1)Proportion of patients who lost visual acuity (%) (≥ 15 letters of BCVA) 48
966.4
8.1
5.5
7.4
0.9
(-2.7, 4.3)
0.7
(-3.6, 4.6)3.8
7.1
4.8
7.5
-1.0
(-3.9, 2.2)
-0.4
(-3.8, 3.3)Figure 8: Mean Change in Visual Acuity From Baseline to Week 96 in HAWK
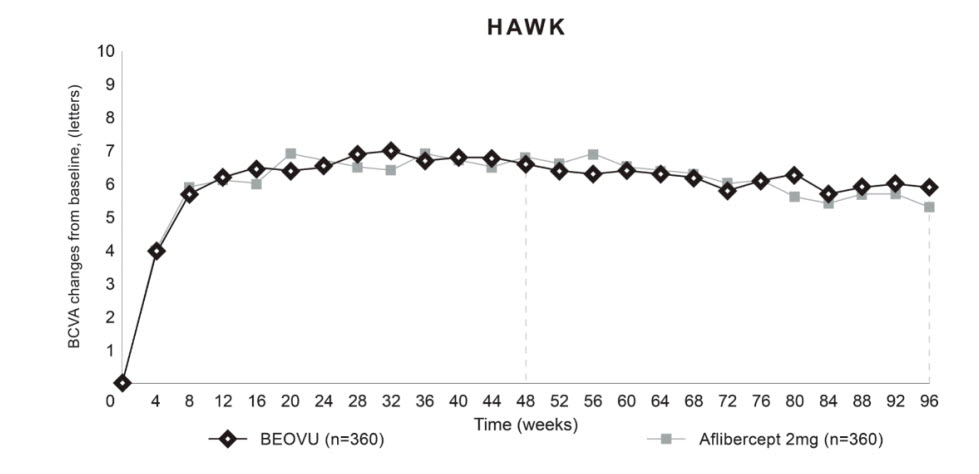
Figure 9: Mean Change in Visual Acuity From Baseline to Week 96 in HARRIER
Through Week 48, 56% (HAWK) and 51% (HARRIER) of patients remained on BEOVU every 12 weeks. The proportion of patients who were maintained on every 12-week dosing through Week 96 was 45% and 39% in HAWK and HARRIER, respectively. The probability of remaining on every 12-week dosing from Week 20 to Week 48 was 85% and 82%, and from Week 48 to Week 96 was 82% and 75% in HAWK and HARRIER, respectively.
Treatment effects in evaluable subgroups (e.g., age, gender, race, baseline visual acuity) in each study were generally consistent with the results in the overall populations.
14.2 Diabetic Macular Edema (DME)
The safety and efficacy of BEOVU were assessed in two randomized, multi-center, double-masked, active controlled studies (KESTREL – NCT03481634 and KITE - NCT03481660) in patients with DME. A total of 926 patients were treated in these studies for 1 year (558 on brolucizumab and 368 on aflibercept 2 mg). Patient ages ranged from 23 to 87 years with a mean of 63 years.
In KESTREL, patients were randomized in a 1:1:1 ratio to the following dosing regimens:
1) brolucizumab 6 mg administered once every 6 weeks for first 5 doses, followed by brolucizumab 6 mg every 8 or 12 weeks.
2) brolucizumab 3 mg administered once every 6 weeks for first 5 doses, followed by brolucizumab 3 mg every 8 or 12 weeks.
3) aflibercept 2 mg administered once every 4 weeks for first 5 doses, followed by aflibercept 2 mg every 8 weeks.
In KITE, patients were randomized in a 1:1 ratio to the following dosing regimens:
1) brolucizumab 6 mg administered once every 6 weeks for first 5 doses, followed by brolucizumab 6 mg every 8 or 12 weeks.
2) aflibercept 2 mg administered once every 4 weeks for first 5 doses, followed by aflibercept 2 mg every 8 weeks.
In both studies, after the first five doses (Weeks 0, 6, 12, 18, and 24), brolucizumab patients were treated every 12 weeks, with the option of adjusting to an every 8-week dosing interval based on disease activity. Disease activity was assessed by changes in visual acuity and/or anatomical parameters, including CST and/or presence of IRF/SRF although the utility of the specific action parameters used has not been established. Disease activity was assessed by a physician during the first 12-week interval (at Weeks 32 and 36) and at each subsequent scheduled 12-week treatment visit. Patients who showed disease activity at any of these visits were adjusted to an every 8 week treatment interval. The comparator aflibercept was administered every 8 weeks after the first 5 monthly doses.
The primary efficacy endpoint for both studies was the change from baseline to Week 52 in Best Corrected Visual Acuity (BCVA) as measured by the Early Treatment Diabetic Retinopathy Study (ETDRS) Letter Score with the primary objective being to demonstrate non-inferiority of BEOVU vs. aflibercept 2 mg. In both studies, BEOVU was non-inferior to aflibercept 2 mg for the change in BCVA from baseline to Week 52 and the change from baseline over the period Week 40 through Week 52.
After 5 initial q6w loading doses, the patients in the BEOVU arm could have received between the minimum of 2 and maximum of 3 additional injections through Week 52. At Week 52, the median number of injections given over 12 months was 7 in patients treated with BEOVU.
Through Week 52, 55% (KESTREL) and 50% (KITE) of patients remained on BEOVU every 12 weeks. The probability of remaining on every 12-week dosing from Week 36 to Week 52 was 88% and 95% in KESTREL and KITE, respectively.
Results of both studies are shown in Table 3 and Figures 10 and 11 below.
Table 3: Efficacy Outcomes at Week 52 in Phase 3 - KESTREL and KITE Studies BCVA: Best Corrected Visual Acuity; BCVA assessments after start of alternative DME treatment in the study eye were censored and replaced by the last value prior to start of this alternative treatment. KESTREL KITE Efficacy outcome At Week BEOVU
(n = 189)aflibercept
2 mg
(n = 187)Difference
(95% CI)
BEOVU – afliberceptBEOVU
(n = 179)aflibercept
2 mg
(n = 181)Difference
(95% CI)
BEOVU – afliberceptChange from baseline in BCVA (measured by ETDRS letters score) – LS mean (SE) 52
40-529.2
(0.57)
9.0
(0.53)10.5
(0.57)
10.5
(0.53)-1.3
(-2.9, 0.3)
-1.5
(-3.0, 0.0)10.6
(0.66)
10.3
(0.62)9.4
(0.66)
9.4
(0.62)1.2
(-0.6, 3.1)
0.9
(-0.9, 2.6)Figure 10: Mean Change in Visual Acuity From Baseline to Week 52 in KESTREL
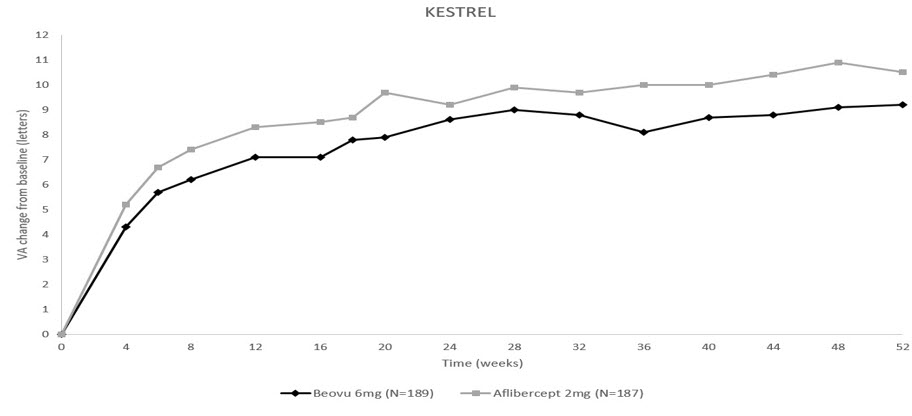
Figure 11: Mean Change in Visual Acuity From Baseline to Week 52 in KITE
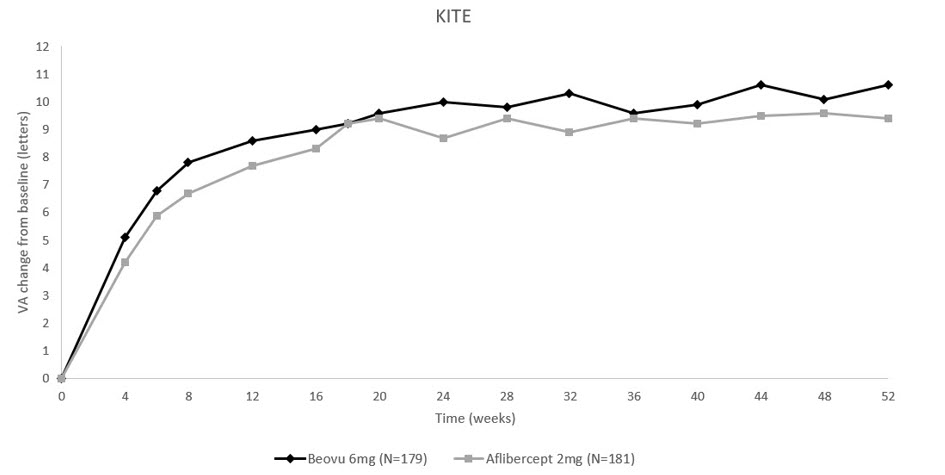
Treatment effects in evaluable subgroups (i.e., age, gender, baseline HbA1c, baseline visual acuity, baseline central subfield thickness, DME lesion type, duration of DME since diagnosis, retinal fluid status) in each study were generally consistent with the results in the overall population.
In both studies, BEOVU demonstrated a significant reduction from baseline in CST starting at Week 4 and continuing up to Week 52.
-
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
BEOVU (brolucizumab-dbll) injection is supplied as a clear to slightly opalescent and colorless to slightly brownish-yellow solution in a single-dose pre-filled syringe and a single-dose vial.
Each pre-filled syringe or vial is for the treatment of a single eye. BEOVU is supplied in the following presentations [see Dosage and Administration (2.3)].
NDC NUMBER CARTON TYPE CARTON CONTENTS 0078-0827-60 Pre-filled Syringe one sealed blister pack containing one BEOVU 6 mg/0.05 mL single-dose pre-filled syringe
one Prescribing Information0078-0827-61 Vial Kit with Injection Components one BEOVU 6 mg/0.05 mL single-dose vial
one 18-gauge x 1½ inch, 1.2 mm x 40 mm, 5-micron, filter needle for withdrawal of the contents
one Prescribing Information -
17 PATIENT COUNSELING INFORMATION
Advise patients that in the days following BEOVU administration, patients are at risk of developing endophthalmitis, retinal detachment, retinal vasculitis and/or retinal vascular occlusion. If the eye becomes red, sensitive to light, painful, or if a patient develops any change in vision, instruct the patient to seek immediate care from an ophthalmologist [see Warnings and Precautions (5.1, 5.2)].
Patients may experience temporary visual disturbances after an intravitreal injection with BEOVU and the associated eye examination [see Adverse Reactions (6.1)]. Advise patients not to drive or use machinery until visual function has recovered sufficiently.
Manufactured by:
Novartis Pharmaceuticals Corporation
East Hanover, New Jersey 07936
U.S. License Number: 1244© Novartis
T2024-60
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
BEOVU
brolucizumab injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0078-0827 Route of Administration INTRAVITREAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength BROLUCIZUMAB (UNII: XSZ53G39H5) (BROLUCIZUMAB - UNII:XSZ53G39H5) BROLUCIZUMAB 6 mg in 0.05 mL Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 13.3 mg in 0.05 mL SODIUM CITRATE, UNSPECIFIED FORM (UNII: 1Q73Q2JULR) 0.59 mg in 0.05 mL POLYSORBATE 80 (UNII: 6OZP39ZG8H) 0.05 mg in 0.05 mL SODIUM HYDROXIDE (UNII: 55X04QC32I) NITROGEN (UNII: N762921K75) WATER (UNII: 059QF0KO0R) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0078-0827-61 1 in 1 CARTON 10/07/2019 02/28/2023 1 0.05 mL in 1 VIAL, SINGLE-DOSE; Type 1: Convenience Kit of Co-Package 2 NDC:0078-0827-99 1 in 1 CARTON 10/07/2019 02/28/2023 2 0.05 mL in 1 VIAL, SINGLE-DOSE; Type 1: Convenience Kit of Co-Package 3 NDC:0078-0827-60 1 in 1 CARTON 03/09/2022 3 0.05 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) 4 NDC:0078-0827-98 1 in 1 CARTON 03/09/2022 4 0.05 mL in 1 SYRINGE, GLASS; Type 3: Prefilled Biologic Delivery Device/System (syringe, patch, etc.) Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA761125 10/07/2019 Labeler - Novartis Pharmaceuticals Corporation (002147023)