Label: BREYANZI- lisocabtagene maraleucel kit

- NDC Code(s): 73153-900-01, 73153-901-08, 73153-902-04
- Packager: Juno Therapeutics, Inc.
- Category: CELLULAR THERAPY
- DEA Schedule: None
- Marketing Status: Biologic Licensing Application
Drug Label Information
Updated December 4, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BREYANZI safely and effectively. See full prescribing information for BREYANZI.
BREYANZI® (lisocabtagene maraleucel) suspension for intravenous infusion
Initial U.S. Approval: 2021WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, AND SECONDARY HEMATOLOGICAL MALIGNANCIES
See full prescribing information for complete boxed warning.
- •
- Cytokine Release Syndrome (CRS), including fatal or life-threatening reactions, occurred in patients receiving BREYANZI. Do not administer BREYANZI to patients with active infection or inflammatory disorders. Treat severe or life-threatening CRS with tocilizumab with or without corticosteroids. (2.2, 2.3, 5.1)
- •
- Neurologic toxicities, including fatal or life-threatening reactions, occurred in patients receiving BREYANZI, including concurrently with CRS, after CRS resolution, or in the absence of CRS. Monitor for neurologic events after treatment with BREYANZI. Provide supportive care and/or corticosteroids as needed. (2.2, 2.3, 5.2)
- •
- T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies, including BREYANZI. (5.7)
RECENT MAJOR CHANGES
INDICATIONS AND USAGE
BREYANZI is a CD19-directed genetically modified autologous T cell immunotherapy indicated for the treatment of:
- •
- adult patients with large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B, who have:
- •
- refractory disease to first-line chemoimmunotherapy or relapse within 12 months of first-line chemoimmunotherapy; or
- •
- refractory disease to first-line chemoimmunotherapy or relapse after first-line chemoimmunotherapy and are not eligible for hematopoietic stem cell transplantation (HSCT) due to comorbidities or age; or
- •
- relapsed or refractory disease after 2 or more lines of systemic therapy. (1.1)
Limitations of Use: BREYANZI is not indicated for the treatment of patients with primary central nervous system lymphoma. (1, 14)
- •
- adult patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) who have received at least 2 prior lines of therapy, including a Bruton tyrosine kinase (BTK) inhibitor and a B-cell lymphoma 2 (BCL-2) inhibitor. This indication is approved under accelerated approval based on response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s). (1.2)
- •
- adult patients with relapsed or refractory follicular lymphoma (FL) who have received 2 or more prior lines of systemic therapy. This indication is approved under accelerated approval based on response rate and duration of response. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s). (1.3)
- •
- adult patients with relapsed or refractory mantle cell lymphoma (MCL) who have received at least 2 prior lines of systemic therapy, including a Bruton tyrosine kinase (BTK) inhibitor. (1.4)
- •
- adult patients with relapsed or refractory marginal zone lymphoma (MZL) who have received at least 2 prior lines of systemic therapy. (1.5)
DOSAGE AND ADMINISTRATION
For autologous use only. For intravenous use only.
- •
- Do NOT use a leukodepleting filter. (2.2)
- •
- Administer a lymphodepleting regimen of fludarabine and cyclophosphamide before infusion of BREYANZI. (2.2)
- •
- Verify the patient’s identity prior to infusion. (2.2)
- •
- Premedicate with acetaminophen and an H1 antihistamine. (2.2)
- •
- Confirm availability of tocilizumab prior to infusion. (2.2, 5.1)
- •
- Dosing of BREYANZI is based on the number of chimeric antigen receptor (CAR)-positive viable T cells. (2.1)
- For LBCL:
- •
- after one line of therapy, the dose is 90 to 110 × 106 CAR-positive viable T cells. (2.1)
- •
- after two or more lines of therapy, the dose is 50 to 110 × 106 CAR-positive viable T cells. (2.1)
- For CLL/SLL, FL, MCL and MZL:
- •
- the dose is 90 to 110 × 106 CAR-positive viable T cells. (2.1)
DOSAGE FORMS AND STRENGTHS
- •
- BREYANZI is a cell suspension for infusion. (3)
- •
- A single dose of BREYANZI consists of 1:1 CAR-positive viable T cells of the CD8 and CD4 components, with each component supplied separately in one to four single-dose 5 mL vials. Each mL contains ≥ 1.5 × 106 to 70 × 106 CAR-positive viable T cells. (3)
CONTRAINDICATIONS
None. (4)
WARNINGS AND PRECAUTIONS
- •
- Hypersensitivity Reactions: Monitor for hypersensitivity reactions during infusion. (5.3)
- •
- Serious Infections: Monitor patients for signs and symptoms of infection; treat appropriately. (5.4)
- •
- Prolonged Cytopenias: Patients may exhibit Grade 3 or higher cytopenias for several weeks following BREYANZI infusion. Monitor complete blood counts. (5.5)
- •
- Hypogammaglobulinemia: Monitor and consider immunoglobulin replacement therapy. (5.6)
- •
- Secondary Malignancies: T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies, including BREYANZI. In the event that a secondary malignancy occurs after treatment with BREYANZI, contact Bristol-Myers Squibb at 1-888-805-4555. (5.7)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥ 30%) in:
- •
- LBCL are fever, CRS, fatigue, musculoskeletal pain, and nausea. The most common Grade 3-4 laboratory abnormalities include lymphocyte count decrease, neutrophil count decrease, platelet count decrease, and hemoglobin decrease. (6.1)
- •
- CLL/SLL are CRS, encephalopathy, fatigue, musculoskeletal pain, nausea, edema and diarrhea. The most common Grade 3-4 laboratory abnormalities include neutrophil count decrease, white blood cell decrease, hemoglobin decrease, platelet count decrease, and lymphocyte count decrease. (6.1)
- •
- FL are CRS. The most common Grade 3-4 laboratory abnormalities include lymphocyte count decreased, neutrophil count decreased, and white blood cell decreased. (6.1)
- •
- MCL are CRS, fatigue, musculoskeletal pain, and encephalopathy. The most common Grade 3-4 laboratory abnormalities include neutrophil count decrease, white blood cell decrease, and platelet count decrease. (6.1)
- •
- MZL are CRS. The most common Grade 3-4 laboratory abnormalities include lymphocyte count decreased, neutrophil count decreased, and white blood cell decreased. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 12/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, AND SECONDARY HEMATOLOGICAL MALIGNANCIES
1 INDICATIONS AND USAGE
1.1 Large B-cell Lymphoma (LBCL)
1.2 Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL)
1.3 Follicular Lymphoma (FL)
1.4 Mantle Cell Lymphoma (MCL)
1.5 Marginal Zone Lymphoma (MZL)
2 DOSAGE AND ADMINISTRATION
2.1 Dose
2.2 Administration
2.3 Management of Severe Adverse Reactions
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome
5.2 Neurologic Toxicities
5.3 Hypersensitivity Reactions
5.4 Serious Infections
5.5 Prolonged Cytopenias
5.6 Hypogammaglobulinemia
5.7 Secondary Malignancies
5.8 Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome (IEC-HS)
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Drug-Laboratory Test Interactions
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.6 Immunogenicity
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Large B-Cell Lymphoma
14.2 Relapsed or Refractory Chronic Lymphocytic Lymphoma or Small Lymphocytic Lymphoma
14.3 Relapsed or Refractory Follicular Lymphoma
14.4 Relapsed or Refractory Mantle Cell Lymphoma
14.5 Relapsed or Refractory Marginal Zone Lymphoma
15 REFERENCES
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: CYTOKINE RELEASE SYNDROME, NEUROLOGIC TOXICITIES, AND SECONDARY HEMATOLOGICAL MALIGNANCIES
- •
- Cytokine Release Syndrome (CRS), including fatal or life-threatening reactions, occurred in patients receiving BREYANZI. Do not administer BREYANZI to patients with active infection or inflammatory disorders. Treat severe or life-threatening CRS with tocilizumab with or without corticosteroids [see Dosage and Administration (2.2, 2.3) and Warnings and Precautions (5.1)].
- •
- Neurologic toxicities, including fatal or life-threatening reactions, occurred in patients receiving BREYANZI, including concurrently with CRS, after CRS resolution, or in the absence of CRS. Monitor for neurologic events after treatment with BREYANZI. Provide supportive care and/or corticosteroids as needed [see Dosage and Administration (2.2, 2.3) and Warnings and Precautions (5.2)].
- •
- T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies, including BREYANZI [see Warnings and Precautions (5.7)].
-
1 INDICATIONS AND USAGE
1.1 Large B-cell Lymphoma (LBCL)
BREYANZI is indicated for the treatment of adult patients with large B-cell lymphoma (LBCL), including diffuse large B-cell lymphoma (DLBCL) not otherwise specified (including DLBCL arising from indolent lymphoma), high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma, and follicular lymphoma grade 3B who have:
- •
- refractory disease to first-line chemoimmunotherapy or relapse within 12 months of first-line chemoimmunotherapy; or
- •
- refractory disease to first-line chemoimmunotherapy or relapse after first-line chemoimmunotherapy and are not eligible for hematopoietic stem cell transplantation (HSCT) due to comorbidities or age; or
- •
- relapsed or refractory disease after 2 or more lines of systemic therapy.
Limitations of Use: BREYANZI is not indicated for the treatment of patients with primary central nervous system (CNS) lymphoma [see Clinical Studies (14.1)].
1.2 Chronic Lymphocytic Leukemia (CLL) or Small Lymphocytic Lymphoma (SLL)
BREYANZI is indicated for the treatment of adult patients with relapsed or refractory chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) who have received at least 2 prior lines of therapy including, a Bruton tyrosine kinase (BTK) inhibitor and a B-cell lymphoma 2 (BCL-2) inhibitor.
This indication is approved under accelerated approval based on response rate and duration of response [see Clinical Studies (14.2)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
1.3 Follicular Lymphoma (FL)
BREYANZI is indicated for the treatment of adult patients with relapsed or refractory follicular lymphoma (FL) who have received 2 or more prior lines of systemic therapy.
This indication is approved under accelerated approval based on response rate and duration of response [see Clinical Studies (14.3)]. Continued approval for this indication may be contingent upon verification and description of clinical benefit in confirmatory trial(s).
-
2 DOSAGE AND ADMINISTRATION
2.1 Dose
For autologous use only. For intravenous use only.
See the respective Certificate of Release for Infusion (RFI Certificate) for each component, for the actual cell counts and volumes to be infused [see Dosage and Administration (2.2) and Dosage Forms and Strengths (3)].
A single dose of BREYANZI contains CAR-positive viable T cells (consisting of 1:1 CAR-positive viable T cells of the CD8 and CD4 components), with each component supplied separately in one to four single-dose vials. See Table 1 for dose range per indication.
Table 1: Dose Range Abbreviations: LBCL = large B-cell lymphoma; CLL = chronic lymphocytic leukemia; SLL = small lymphocytic lymphoma; FL = follicular lymphoma; MCL = mantle cell lymphoma; MZL = marginal zone lymphoma. Indication
BREYANZI dose range
LBCL after two or more lines of therapy (1.1)
50 to 110 × 106 CAR-positive viable T cells
LBCL after one line of therapy (1.1)
90 to 110 × 106 CAR-positive viable T cells
CLL or SLL (1.2)
90 to 110 × 106 CAR-positive viable T cells
FL (1.3)
90 to 110 × 106 CAR-positive viable T cells
MCL (1.4)
90 to 110 × 106 CAR-positive viable T cells
MZL (1.5)
90 to 110 × 106 CAR-positive viable T cells
2.2 Administration
BREYANZI is for autologous use only. The patient’s identity must match the patient identifiers on the BREYANZI cartons, vials, and syringe labels. Do not infuse BREYANZI if the information on the patient-specific labels does not match the intended patient.
Preparing the Patient for BREYANZI
Confirm the availability of BREYANZI before starting lymphodepleting chemotherapy.
Pretreatment
Administer the lymphodepleting chemotherapy regimen before infusion of BREYANZI: fludarabine 30 mg/m2/day intravenously (IV), and cyclophosphamide 300 mg/m2/day IV for 3 days. See the prescribing information for fludarabine and cyclophosphamide for information on dose adjustment in renal impairment.
Infuse BREYANZI 2 to 7 days after completion of lymphodepleting chemotherapy.
Delay the infusion of BREYANZI if the patient has unresolved serious adverse events from preceding chemotherapies, active uncontrolled infection, or active graft-versus-host disease (GVHD).
Premedication
To minimize the risk of infusion reactions, premedicate the patient with acetaminophen (650 mg orally) and diphenhydramine (25-50 mg, IV or orally), or another H1-antihistamine, 30 to 60 minutes prior to treatment with BREYANZI.
Avoid prophylactic use of systemic corticosteroids, as they may interfere with the activity of BREYANZI.
Receipt of BREYANZI
- •
- BREYANZI is shipped directly to the cell-associated lab or clinical pharmacy associated with the infusion center in the vapor phase of a liquid nitrogen shipper.
- •
- Confirm the patient’s identity with the patient identifiers on the shipper.
- •
- If the patient is not expected to be ready for administration before the shipper expires and the infusion site is qualified for onsite storage, transfer BREYANZI to onsite vapor phase of liquid nitrogen storage prior to preparation.
- •
- If the patient is not expected to be ready for administration before the shipper expires and the infusion site is not qualified for onsite storage, contact Bristol-Myers Squibb at 1-888-805-4555 to arrange for return shipment.
Preparing BREYANZI
Before thawing the vials
- •
- Confirm the patient’s identity with the patient identifiers on the RFI Certificate.
- •
- Read the RFI Certificate (affixed inside the shipper) for information on the number of syringes you will need to administer the CD8 and CD4 components (syringe labels are provided with the RFI Certificate). There is a separate RFI Certificate for each cell component.
- •
- Confirm tocilizumab and emergency equipment are available prior to infusion and during the recovery period.
- •
- Confirm the infusion time in advance and adjust the start time of BREYANZI thaw such that it will be available for infusion when the patient is ready.
Thawing the vials
- 1.
- Confirm the patient’s identity with the patient identifiers on the outer carton and on the syringe labels.
Once the vials of CAR-positive viable T cells (CD8 component and CD4 component) are removed from frozen storage, the thaw must be carried to completion and the cells administered within 2 hours. - 2.
- Remove the CD8 component carton and CD4 component carton from the outer carton.
- 3.
- Confirm the patient’s identity with the patient identifiers on the inner carton.
- 4.
- Open each inner carton and visually inspect the vial(s) for damage. If the vials are damaged, contact Bristol-Myers Squibb at 1-888-805-4555.
- 5.
- Confirm the patient’s identity with the patient identifiers on the vials.
- 6.
- Carefully remove the vials from the cartons, place vials on a protective barrier pad, and thaw at room temperature until there is no visible ice in the vials. Thaw all of the vials at the same time. Keep the CD8 and CD4 components separate.
Dose preparation
- •
- Prepare BREYANZI using sterile technique.
- •
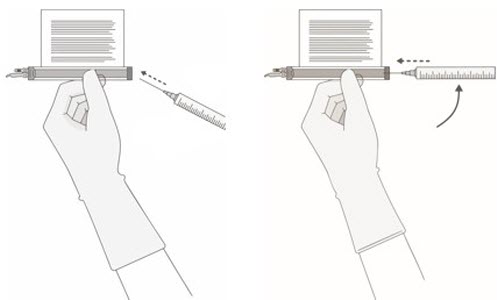
- Based on the concentration of CAR-positive viable T cells for each component, more than one vial of each of the CD8 and CD4 components may be required to complete a dose. A separate syringe should be prepared for each CD8 or CD4 component vial received.
Note: The volume to be drawn up and infused may differ for each component as indicated on the RFI Certificate. Do NOT draw up excess volume into the syringe. - •
- Each vial contains 5 mL with a total extractable volume of 4.6 mL of CD8 or CD4 component T cells. The RFI Certificate for each component indicates the volume (mL) of cells to be drawn up into each syringe. Use the smallest Luer-lock tip syringe necessary (1, 3, or 5 mL) to draw up the specified volume from each vial. A 5 mL syringe should not be used for volumes less than 3 mL.
- 7.
-
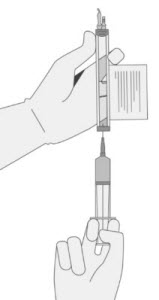
Prepare the syringe(s) of the CD8 component first. Affix the CD8 syringe labels to the syringe(s) prior to pulling the required volume into the syringe(s).
Note:It is important to confirm that the volume drawn up for each component matches the volume specified in the respective RFI Certificate. Do NOT draw up excess volume into the syringe.
Withdrawal of the required volume of cells from each vial into a separate syringe should be carried out using the following instructions:
- 8.
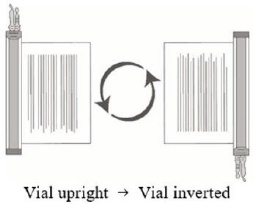
- Hold the thawed vial(s) upright and gently invert the vial(s) 5 times to mix the cell product. If any clumping is apparent, continue to invert the vial(s) until clumps have dispersed and cells appear to be evenly resuspended.
- 9.
- Visually inspect the thawed vial(s) for damage or leaks. Do not use if the vial is damaged or if the clumps do not disperse; contact Bristol-Myers Squibb at 1-888-805-4555. The liquid in the vials should be slightly opaque to opaque, colorless to yellow or brownish-yellow.
- 10.
- Remove the polyaluminum cover (if present) from the bottom of the vial and swab the septum with an alcohol wipe. Allow to air dry before proceeding.
Note: The absence of the polyaluminum cover does not impact the sterility of the vial.
- 11.
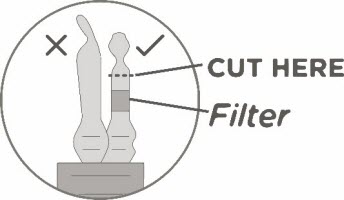
- Keeping the vial(s) upright, cut the seal on the tubing line on the top of the vial immediately above the filter to open the air vent on the vial.
Note: Be careful to select the correct tubing line with the filter. Cut ONLY the tubing with a filter.
- 12.
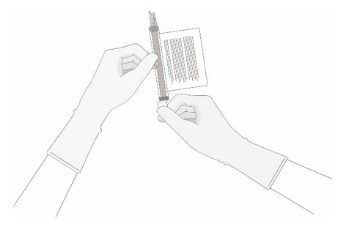
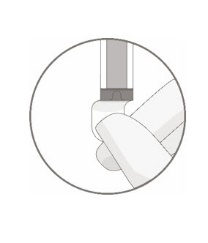
- Hold a 20-gauge, 1-1 ½ inch needle, with the opening of the needle tip away from the retrieval port septum.
- a.
- Insert the needle into the septum at a 45°- 60° angle to puncture the retrieval port septum.
- b.
- Increase the angle of the needle gradually as the needle enters the vial.
- 13.
- WITHOUT drawing air into the syringe, slowly withdraw the target volume (as specified in the RFI Certificate). Carefully inspect the syringe for signs of debris prior to proceeding. If there is debris, contact Bristol-Myers Squibb at 1-888-805-4555.
- 14.
- Verify that the volume of CD8/CD4 component matches the volume specified for the relevant component in the RFI Certificate.
Once the volume is verified, remove the syringe/needle from the vial, carefully detach the needle from the syringe and cap the syringe.
- 15.
- Continue to keep the vial horizontal and return it to the carton to avoid leaking from the vial.
- 16.
- Dispose of any unused portion of BREYANZI (according to local biosafety guidelines).
- 17.
- Repeat the process steps 7-16 for the CD4 Component.
- 18.
- Transport the labeled CD8 and CD4 syringes to the bedside by placing with protective barrier pad inside an insulated room temperature container.
BREYANZI Administration
- •
- Do NOT use a leukodepleting filter.
- •
- Confirm tocilizumab and emergency equipment are available prior to infusion and during the recovery period.
- •
- Confirm the patient’s identity matches the patient identifiers on the syringe label.
- •
- Once BREYANZI has been drawn into syringes, proceed with administration as soon as possible. The total time from removal from frozen storage to patient administration should not exceed 2 hours as indicated by the time entered on the syringe label.
19. Use intravenous normal saline to flush all the infusion tubing prior to and after each CD8 or CD4 component administration.
20. Administer the entire volume of the CD8 component intravenously at an infusion rate of approximately 0.5 mL/minute, using the closest port or Y-arm.
Note: The time for infusion will vary but will usually be less than 15 minutes for each component.21. If more than one syringe is required for a full cell dose of the CD8 component, administer the volume in each syringe consecutively without any time between administering the contents of the syringes (unless there is a clinical reason (e.g., infusion reaction) to hold the dose).
22. After the CD8 component has been administered, flush the tubing with normal saline, using enough volume to clear the tubing and the length of the IV catheter.
23. Administer the CD4 component second, immediately after administration of the CD8 component is complete, using steps 1-4, as described for the CD8 component. Following administration of the CD4 component, flush the tubing with normal saline, using enough volume to clear the tubing and the length of the IV catheter.
BREYANZI contains human blood cells that are genetically modified with replication-incompetent, self-inactivating lentiviral vector. Follow universal precautions and local biosafety guidelines applicable for the handling and disposal, to avoid potential transmission of infectious diseases.
Monitoring
- •
- Monitor patients daily for at least 7 days following BREYANZI infusion for signs and symptoms of CRS and neurologic toxicities.
- •
- Instruct patients to remain within proximity of a healthcare facility for at least 2 weeks following infusion.
- •
- Advise patients to avoid driving for at least 2 weeks following infusion.
2.3 Management of Severe Adverse Reactions
Cytokine Release Syndrome
Identify cytokine release syndrome (CRS) based on clinical presentation [see Warnings and Precautions (5.1)]. Evaluate for and treat other causes of fever, hypoxia, and hypotension. If CRS is suspected, manage according to the recommendations in Table 2. Physicians may also consider management per current practice guidelines.
Patients who experience Grade 2 or higher CRS (e.g., hypotension not responsive to fluids, or hypoxia requiring supplemental oxygenation) should be monitored with continuous cardiac telemetry and pulse oximetry. For patients experiencing severe CRS, consider performing an echocardiogram to assess cardiac function. For severe or life-threatening CRS, consider intensive-care supportive therapy.
If concurrent neurologic toxicity is suspected during CRS, administer:
- •
- Corticosteroids according to the more aggressive intervention based on the CRS and neurologic toxicity grades in Tables 2 and 3
- •
- Tocilizumab according to the CRS grade in Table 2
- •
- Antiseizure medication according to the neurologic toxicity in Table 3
Table 2: CRS Grading and Management Guidance a Lee criteria for grading CRS (Lee et al, 2014). b If corticosteroids are initiated, continue corticosteroids for at least 3 doses or until complete resolution of symptoms, and consider corticosteroid taper. CRS Gradea
Tocilizumab
Corticosteroidsb
Grade 1
Fever
If less than 72 hours after infusion, consider tocilizumab 8 mg/kg IV over 1 hour (not to exceed 800 mg).
If 72 hours or more after infusion, treat symptomatically.
If less than 72 hours after infusion, consider dexamethasone 10 mg IV every 24 hours.
If 72 hours or more after infusion, treat symptomatically.
Grade 2
Symptoms require and respond to moderate intervention.
Oxygen requirement less than 40% FiO2, or hypotension responsive to fluids or low dose of one vasopressor, or Grade 2 organ toxicity.
Administer tocilizumab 8 mg/kg IV over 1 hour (not to exceed 800 mg).
Repeat tocilizumab every 8 hours as needed if not responsive to intravenous fluids or increasing supplemental oxygen.
Limit to a maximum of 3 doses in a 24-hour period; maximum total of 4 doses.If less than 72 hours after infusion, administer dexamethasone 10 mg IV every 12‑24 hours.
If 72 hours or more after infusion, consider dexamethasone 10 mg IV every 12‑24 hours.
If no improvement within 24 hours or rapid progression, repeat tocilizumab and escalate dose and frequency of dexamethasone (10‑20 mg IV every 6 to 12 hours).
If no improvement or continued rapid progression, maximize dexamethasone, switch to high-dose methylprednisolone 2 mg/kg if needed. After 2 doses of tocilizumab, consider alternative immunosuppressants. Do not exceed 3 doses tocilizumab in 24 hours, or 4 doses in total.
Grade 3
Symptoms require and respond to aggressive intervention.
Oxygen requirement greater than or equal to 40% FiO2, or hypotension requiring high-dose or multiple vasopressors, or Grade 3 organ toxicity, or Grade 4 transaminitis.
Per Grade 2.
Administer dexamethasone 10 mg IV every 12 hours.
If no improvement within 24 hours or rapid progression of CRS, repeat tocilizumab and escalate dose and frequency of dexamethasone (10-20 mg IV every 6 to 12 hours).
If no improvement or continued rapid progression, maximize dexamethasone, switch to high-dose methylprednisolone 2 mg/kg if needed. After 2 doses of tocilizumab, consider alternative immunosuppressants. Do not exceed 3 doses tocilizumab in 24 hours, or 4 doses in total.
Grade 4
Life-threatening symptoms.
Requirements for ventilator support or continuous veno‑venous hemodialysis (CVVHD) or Grade 4 organ toxicity (excluding transaminitis).
Per Grade 2.
Administer dexamethasone 20 mg IV every 6 hours.
If no improvement within 24 hours or rapid progression of CRS, escalate tocilizumab and corticosteroid use. If no improvement or continued rapid progression, maximize dexamethasone, switch to high-dose methylprednisolone 2 mg/kg if needed. After 2 doses of tocilizumab, consider alternative immunosuppressants. Do not exceed 3 doses tocilizumab in 24 hours, or 4 doses in total.
Neurologic Toxicity
Monitor patients for signs and symptoms of neurologic toxicities (Table 3). Rule out other causes of neurologic symptoms. Provide intensive care supportive therapy for severe or life-threatening neurologic toxicities. If neurologic toxicity is suspected, manage according to the recommendations in Table 3. Physicians may also consider management per current practice guidelines.
If concurrent CRS is suspected during neurologic toxicity, administer:
- •
- Corticosteroids according to the more aggressive intervention based on the CRS and neurologic toxicity grades in Tables 2 and 3
- •
- Tocilizumab according to the CRS grade in Table 2
- •
- Antiseizure medication according to the neurologic toxicity in Table 3
Table 3: Neurologic Toxicity (NT) Grading and Management Guidance NT Gradea Corticosteroids and Antiseizure Medication a NCI CTCAE criteria for grading neurologic toxicities, version 4.03. Grade 1
Start non-sedating, antiseizure medicines (e.g., levetiracetam) for seizure prophylaxis.
If 72 hours or more after infusion, observe.
If less than 72 hours after infusion, consider dexamethasone 10 mg IV every 12 to 24 hours for 2 to 3 days.Grade 2
Start non-sedating, antiseizure medicines (e.g., levetiracetam) for seizure prophylaxis.
Dexamethasone 10 mg IV every 12 hours for 2-3 days, or longer for persistent symptoms. Consider taper for a total steroid exposure of greater than 3 days.
If no improvement after 24 hours or worsening of neurologic toxicity, increase the dose and/or frequency of dexamethasone up to a maximum of 20 mg IV every 6 hours.
If no improvement after another 24 hours, rapidly progressing symptoms, or life-threatening complications arise, give methylprednisolone (2 mg/kg loading dose, followed by 2 mg/kg divided 4 times a day; taper within 7 days).Grade 3
Start non-sedating, antiseizure medicines (e.g., levetiracetam) for seizure prophylaxis.
Dexamethasone 10 to 20 mg IV every 8 to 12 hours. Steroids are not recommended for isolated Grade 3 headaches.
If no improvement after 24 hours or worsening of neurologic toxicity, escalate to methylprednisolone (dose and frequency as per Grade 2).
If cerebral edema is suspected, consider hyperventilation and hyperosmolar therapy. Give high-dose methylprednisolone (1-2 g, repeat every 24 hours if needed; taper as clinically indicated) and cyclophosphamide 1.5 g/m2.Grade 4
Start non-sedating, antiseizure medicines (e.g., levetiracetam) for seizure prophylaxis.
Dexamethasone 20 mg IV every 6 hours.
If no improvement after 24 hours or worsening of neurologic toxicity, escalate to methylprednisolone (dose and frequency as per Grade 2).
If cerebral edema is suspected, consider hyperventilation and hyperosmolar therapy. Give high-dose methylprednisolone (1-2 g, repeat every 24 hours if needed; taper as clinically indicated), and cyclophosphamide 1.5 g/m2. -
3 DOSAGE FORMS AND STRENGTHS
BREYANZI is a cell suspension for infusion.
A single dose of BREYANZI contains CAR-positive viable T cells that consist of CD8 and CD4 components, with each component supplied separately in single-dose vials [see Dosage and Administration (2.1)].
More than one vial of each of the CD8 component and/or CD4 component may be needed to achieve the dose of BREYANZI.
Each vial contains between 6.9 × 106 and 322 × 106 CAR-positive viable T cells in 4.6 mL cell suspension (between 1.5 × 106 and 70 x 106 CAR-positive viable T cells/mL).
The infusion volume is calculated based on the concentration of cryopreserved drug product CAR-positive viable T cells. The volume may differ for each component infused. See the RFI Certificate for details [see How Supplied/Storage and Handling (16)].
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Cytokine Release Syndrome
Cytokine release syndrome (CRS), including fatal or life-threatening reactions, occurred following treatment with BREYANZI.
In clinical trials of BREYANZI, which included a total of 769 patients with non-Hodgkin lymphoma (NHL) exposed to BREYANZI, CRS occurred in 56% of patients, including ≥ Grade 3 CRS (Lee grading system1) in 3.4% of patients. The median time to onset was 5 days (range: 1 to 63 days). CRS resolved in 99% of patients with a median duration of 5 days (range: 1 to 37 days). One patient had fatal CRS and 5 patients had ongoing CRS at the time of death. The most common manifestations of CRS (≥ 10%) included fever, hypotension, chills, tachycardia, hypoxia, and headache.
Serious events that may be associated with CRS include cardiac arrhythmias (including atrial fibrillation and ventricular tachycardia), cardiac arrest, cardiac failure, diffuse alveolar damage, renal insufficiency, capillary leak syndrome, hypotension, hypoxia, and hemophagocytic lymphohistiocytosis/macrophage activation syndrome (HLH/MAS) [see Adverse Reactions (6.1)].
Ensure that 2 doses of tocilizumab are available prior to infusion of BREYANZI.
Monitor patients daily for at least 7 days following BREYANZI infusion for signs and symptoms of CRS. Continue to monitor patients for signs or symptoms of CRS for at least 2 weeks after infusion. At the first sign of CRS, institute treatment with supportive care, tocilizumab, or tocilizumab and corticosteroids as indicated [see Dosage and Administration (2.2, 2.3)].
Counsel patients to seek immediate medical attention should signs or symptoms of CRS occur at any time [see Patient Counseling Information (17)].
5.2 Neurologic Toxicities
Neurologic toxicities that were fatal or life-threatening, including immune effector cell-associated neurotoxicity syndrome (ICANS), occurred following treatment with BREYANZI. Serious events including cerebral edema and seizures occurred with BREYANZI. Fatal and serious cases of leukoencephalopathy, some attributable to fludarabine, also occurred.
In clinical trials of BREYANZI, CAR T cell-associated neurologic toxicities occurred in 32% of patients, including ≥ Grade 3 cases in 10% of patients. The median time to onset of neurotoxicity was 8 days (range: 1 to 63 days). Neurologic toxicities resolved in 88% of patients with a median duration of 7.5 days (range: 1 to 119 days). Of patients developing neurotoxicity, 83% also developed CRS.
The most common neurologic toxicities (≥ 5%) included encephalopathy, tremor, aphasia, delirium, and headache.
Monitor patients daily for at least 7 days following BREYANZI infusion for signs and symptoms of neurologic toxicities and assess for other causes of neurological symptoms. Continue to monitor patients for signs or symptoms of neurologic toxicities for at least 2 weeks after infusion and treat promptly. Manage neurologic toxicity with supportive care and/or corticosteroid as needed [see Dosage and Administration (2.2, 2.3)]. Advise patients to avoid driving for at least 2 weeks following infusion.
Counsel patients to seek immediate medical attention should signs or symptoms of neurologic toxicity occur at any time [see Patient Counseling Information (17)].
5.3 Hypersensitivity Reactions
Allergic reactions may occur with the infusion of BREYANZI. Serious hypersensitivity reactions, including anaphylaxis, may be due to dimethyl sulfoxide (DMSO).
5.4 Serious Infections
Severe infections, including life-threatening or fatal infections, have occurred in patients after BREYANZI infusion.
In clinical trials of BREYANZI, infections of any grade occurred in 33% of patients, with Grade 3 or higher infections occurred in 12% of all patients. Grade 3 or higher infections with an unspecified pathogen occurred in 7%, bacterial infections in 3.5%, viral infections in 2%, and fungal infections in 0.7% of patients. One patient with FL, who received four prior lines of therapy developed a fatal case of John Cunningham (JC) virus progressive multifocal leukoencephalopathy four months after treatment with BREYANZI. One patient with MCL, who received three prior lines of therapy, developed a fatal case of cryptococcal meningoencephalitis 35 days after treatment with BREYANZI.
Febrile neutropenia developed after BREYANZI infusion in 8% of patients. Febrile neutropenia may be concurrent with CRS. In the event of febrile neutropenia, evaluate for infection and manage with broad‑spectrum antibiotics, fluids, and other supportive care as medically indicated.
Monitor patients for signs and symptoms of infection before and after BREYANZI administration and treat appropriately. Administer prophylactic antimicrobials according to standard institutional guidelines.
Avoid administration of BREYANZI in patients with clinically significant, active systemic infections.
Viral Reactivation
Hepatitis B virus (HBV) reactivation, in some cases resulting in fulminant hepatitis, hepatic failure, and death, can occur in patients treated with drugs directed against B cells.
In clinical trials of BREYANZI, 35 of 38 patients with a prior history of HBV were treated with concurrent antiviral suppressive therapy. Perform screening for HBV, HCV, and HIV in accordance with clinical guidelines before collection of cells for manufacturing. In patients with prior history of HBV, consider concurrent antiviral suppressive therapy to prevent HBV reactivation per standard guidelines.
5.5 Prolonged Cytopenias
Patients may exhibit cytopenias not resolved for several weeks following lymphodepleting chemotherapy and BREYANZI infusion.
In clinical trials of BREYANZI, Grade 3 or higher cytopenias persisted at Day 29 following BREYANZI infusion in 35% of patients, and included thrombocytopenia in 25%, neutropenia in 22%, and anemia in 6% of patients.
Monitor complete blood counts prior to and after BREYANZI administration.
5.6 Hypogammaglobulinemia
B-cell aplasia and hypogammaglobulinemia can occur in patients receiving BREYANZI.
In clinical trials of BREYANZI, hypogammaglobulinemia was reported as an adverse reaction in 9% of patients. Hypogammaglobulinemia, either as an adverse reaction or laboratory IgG level below 500 mg/dL after infusion, was reported in 30% of patients.
Monitor immunoglobulin levels after treatment with BREYANZI and manage using infection precautions, antibiotic prophylaxis, and immunoglobulin replacement as clinically indicated.
Live Vaccines
The safety of immunization with live viral vaccines during or following BREYANZI treatment has not been studied. Vaccination with live virus vaccines is not recommended for at least 6 weeks prior to the start of lymphodepleting chemotherapy, during BREYANZI treatment, and until immune recovery following treatment with BREYANZI.
5.7 Secondary Malignancies
Patients treated with BREYANZI may develop secondary malignancies. T cell malignancies have occurred following treatment of hematologic malignancies with BCMA- and CD19-directed genetically modified autologous T cell immunotherapies, including BREYANZI. Mature T cell malignancies, including CAR-positive tumors, may present as soon as weeks following infusion, and may include fatal outcomes [see Boxed Warning, Adverse Reactions (6.2), Patient Counseling Information (17)]. Monitor lifelong for secondary malignancies. In the event that a secondary malignancy occurs, contact Bristol-Myers Squibb at 1-888-805-4555 for reporting and to obtain instructions on collection of patient samples for testing.
5.8 Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome (IEC-HS)
Immune Effector Cell-Associated Hemophagocytic Lymphohistiocytosis-Like Syndrome (IEC-HS), including fatal or life-threatening reactions, occurred following treatment with BREYANZI. Seven out of 769 (0.9%) patients with R/R NHL exposed to BREYANZI developed IEC-HS (1 LBCL; 3 CLL/SLL; 3 MZL). Time to onset of IEC-HS ranged from 7 to 32 days. Of the 7 patients, 3 patients developed IEC-HS with overlapping occurrence of CRS and neurotoxicity, 2 patients developed IEC-HS with overlapping occurrence of neurotoxicity, and 1 patient developed IEC-HS with overlapping occurrence of CRS. IEC-HS was fatal in 2 of 7 patients. One patient had fatal IEC-HS and one had ongoing IEC-HS at time of death.
IEC-HS is a life-threatening condition with a high mortality rate if not recognized and treated early. Treatment of IEC-HS should be administered per current practice guidelines.
-
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety data described in the WARNINGS and PRECAUTIONS and in this section reflects exposure to a single dose of BREYANZI in 769 patients in five clinical studies as described below.
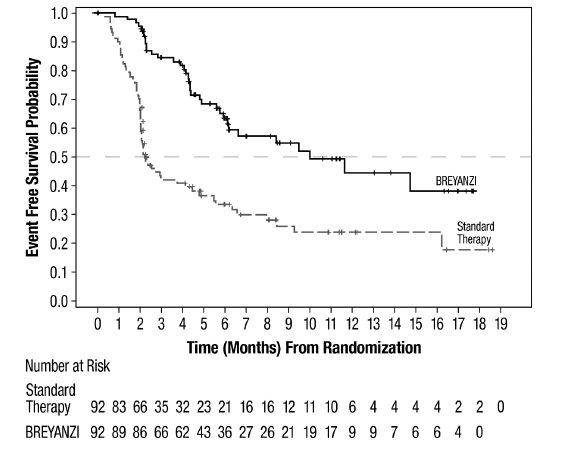
Study 1 (JCAR017-BCM-003; Relapsed or Refractory LBCL After One Line of Therapy)
Study 1 was a randomized, open-label, multicenter study, in which patients with primary refractory LBCL or relapse within 1 year of first-line chemoimmunotherapy received BREYANZI (N=89) or standard therapy (N=91) [see Clinical Studies (14.1)]. Patients had not yet received treatment for relapsed or refractory lymphoma and were potential candidates for autologous HSCT. The trial excluded patients who were ineligible for transplant or who had age > 75 years, Eastern Cooperative Oncology Group (ECOG) performance status >1, history of central nervous system (CNS) disorders (such as seizures or cerebrovascular ischemia), uncontrolled infection, CrCl < 45 mL/min, alanine aminotransferase (ALT) > 5 times the upper limit of normal (ULN), left ventricular ejection fraction (LVEF) < 40%, or absolute neutrophil count (ANC) < 1.0 × 109 cells/L or platelets < 50 × 109 cells/L in the absence of bone marrow involvement.
The planned dose of BREYANZI was 100 × 106 CAR-positive viable T cells. The median age of the BREYANZI-treated population was 59 years (range: 20 to 74 years); 47% were male; 58% were White, 11% were Asian, and 5% were Black.
Serious adverse reactions occurred in 38% of patients. The most common nonlaboratory serious adverse reactions (> 2%) were CRS, sepsis, fever, febrile neutropenia, headache, aphasia, COVID-19 infection, and pulmonary embolism.
Table 4 presents selected nonlaboratory adverse reactions in patients treated with BREYANZI, and Table 5 describes selected new or worsening Grade 3 or 4 laboratory abnormalities.
The most common nonlaboratory adverse reactions (≥ 20%) were fever, CRS, musculoskeletal pain, headache, fatigue, nausea, constipation, and dizziness.
Table 4: Adverse Reactions in ≥ 10% of Patients with Relapsed or Refractory LBCL Treated with BREYANZI in Study 1 (N=89) * Represents multiple related terms. a Edema includes edema peripheral, localized edema, pleural effusion swelling. b Dizziness includes dizziness, dizziness postural, syncope, vertigo. c Motor dysfunction includes fine motor skill dysfunction, muscle spasms, muscular weakness. d Tremor includes resting tremor, tremor, essential tremor. e Rash includes catheter site rash, dermatitis acneiform, dermatitis exfoliative generalized, erythema multiforme, rash, rash maculo-papular, rash pruritic. f Hemorrhage includes conjunctival hemorrhage, cystitis hemorrhagic, epistaxis, gastrointestinal hemorrhage, hematoma, hematuria, retinal hemorrhage, vaginal hemorrhage. Adverse Reaction
Any Grade (%)
Grade 3 or Higher (%)
Blood and lymphatic system disorders
Febrile neutropenia
10
10
Cardiac disorders
Tachycardia*
15
1.1
Gastrointestinal disorders
Nausea
24
0
Constipation
20
2.2
Diarrhea
18
0
Abdominal pain*
13
2.2
Vomiting
11
0
General disorders and administration site conditions
Fever
55
3.4
Fatigue*
28
1.1
Edemaa
13
0
Immune system disorders
Cytokine release syndrome
49
1.1
Infections and infestations
Bacterial infectious disorders*
12
6
Infections with pathogen unspecified*
12
6
Sepsis*
10
7
Metabolism and nutrition disorders
Decreased appetite
15
0
Musculoskeletal and connective tissue disorders
Musculoskeletal pain*
36
3.4
Nervous system disorders
Headache*
34
6
Dizzinessb
20
1.1
Motor dysfunctionc
12
3.4
Tremord
11
1.1
Psychiatric disorders
Insomnia*
15
0
Respiratory, thoracic, and mediastinal disorders
Cough*
11
0
Skin and subcutaneous tissue disorders
Rashe
12
1.1
Vascular disorders
Hypotension*
15
2.2
Hemorrhagef
12
0
Other clinically important adverse reactions in < 10% of patients treated with BREYANZI included the following:
- •
- Immune system disorders: Hemophagocytic lymphohistiocytosis (1.1%)
- •
- Infections and infestations: Viral infection (9%), fungal infection (4.5%), pneumonia (2.2%)
- •
- Nervous system disorders: Encephalopathy (8%), aphasia (4.5%), peripheral neuropathy (4.5%), ataxia (3.4%), paresis (1.1%)
- •
- Psychiatric disorders: Delirium (2.2%)
- •
- Renal and urinary disorders: Renal failure (3.4%)
- •
- Respiratory, thoracic, and mediastinal disorders: Dyspnea (8%)
- •
- Vascular disorders: Thrombosis (8%), hypertension (7%)
Table 5: Grade 3 or 4 Laboratory Abnormalities Occurring in ≥ 10% of Patients with Relapsed or Refractory LBCL Treated with BREYANZI in Study 1 a Baseline lab values were assessed prior to lymphodepleting chemotherapy. b Based on 88 evaluable patients, defined as those with both a baseline grade and at least one post-baseline grade for the particular lab. Laboratory Abnormalitya
Grade 3 or 4 (%)b
Lymphocyte count decreased
98
Neutrophil count decreased
89
Platelet count decreased
48
Hemoglobin decreased
32
Grade 4 laboratory abnormalities in ≥ 10% of patients were lymphocyte decrease (64%), neutrophil decrease (66%), and platelet decrease (34%).
Study 2 (017006; Relapsed or Refractory LBCL After One Line of Therapy)
Study 2 was a single-arm open-label study in transplant-ineligible patients with R/R LBCL after one line of chemoimmunotherapy [see Clinical Studies (14.1)]. The study enrolled patients who were ineligible for high-dose therapy and autologous HSCT due to organ function or age, but who had adequate organ function for CAR-T cell therapy. Patients with a history of relevant CNS disorders (such as seizures or cerebrovascular ischemia), ECOG performance status > 2, or uncontrolled infection were ineligible. The trial required LVEF ≥ 40%, adequate oxygen saturation on room air with ≤ Grade 1 dyspnea, AST, and ALT ≤ 5 x ULN, total bilirubin < 2.0 mg/dL, creatinine clearance > 30 mL/min, and adequate bone marrow function to receive lymphodepleting chemotherapy. The planned dose of BREYANZI was 100 × 106 CAR-positive viable T cells.
The median age was 74 years (range: 53 to 84 years), 90% were age ≥ 65 years, 61% were male. The ECOG performance status was 0 or 1 in 74% of patients and 2 in 26% of patients; 25% had CrCl < 60 ml/min; 20% had a baseline ANC < 1000/μL.
Serious adverse reactions occurred in 33% of patients. The most common nonlaboratory, serious adverse reactions (> 2%) were CRS, confusional state, gastrointestinal hemorrhage, muscular weakness, musculoskeletal pain, pulmonary embolism, and sepsis.
Table 6 presents selected nonlaboratory adverse reactions, and Table 7 describes selected new or worsening Grade 3 or 4 laboratory abnormalities.
The most common nonlaboratory adverse reactions (≥ 20%) were fatigue, CRS, fever, nausea, encephalopathy, hypotension, musculoskeletal pain, and edema.
Table 6: Adverse Reactions in ≥ 10% of Patients with Relapsed or Refractory LBCL Treated with BREYANZI in Study 2 (N=61) * Represents multiple related terms. a Encephalopathy includes amnesia, apraxia, cognitive disorder, confusional state, depressed level of consciousness, disturbance in attention, dyscalculia, encephalopathy, lethargy, memory impairment, mental status changes, somnolence. b Dizziness includes dizziness, dizziness postural, syncope, vertigo. c Tremor includes resting tremor, tremor. Adverse Reaction
Any Grade (%)
Grade 3 or Higher (%)
Cardiac disorders
Tachycardia*
10
0
Gastrointestinal disorders
Nausea
25
1.6
Diarrhea
15
0
Constipation
11
0
General disorders and administration site conditions
Fatigue*
44
1.6
Fever
38
1.6
Edema*
20
0
Immune system disorders
Cytokine release syndrome
39
1.6
Infections and infestations
Infections with pathogen unspecified*
13
4.9
Upper respiratory tract infection*
13
0
Bacterial infectious disorders*
10
3.3
Metabolism and nutrition disorders
Decreased appetite
13
1.6
Musculoskeletal and connective tissue disorders
Musculoskeletal pain*
23
4.9
Nervous system disorders
Encephalopathya
23
4.9
Dizzinessb
16
1.6
Tremorc
16
0
Headache
11
1.6
Psychiatric disorders
Insomnia
11
0
Respiratory, thoracic, and mediastinal disorders
Cough*
18
0
Dyspnea*
16
4.9
Vascular disorders
Hypotension*
23
1.6
Hypertension
10
4.9
Other clinically important adverse reactions in < 10% of patients included the following:
- •
- Blood and lymphatic system disorders: Febrile neutropenia (1.6%)
- •
- Eye disorders: Vision blurred (3.3%)
- •
- Gastrointestinal disorders: Vomiting (8%), abdominal pain (7%), gastrointestinal hemorrhage (4.9%)
- •
- Infections and infestations: Fungal infection (4.9%), sepsis (3.3%), viral infection (3.3%)
- •
- Nervous system disorders: Motor dysfunction (7%), aphasia (4.9%), ataxia (4.9%), peripheral neuropathy (4.9%)
- •
- Psychiatric disorders: Delirium (3.3%)
- •
- Renal and urinary disorders: Renal failure (7%)
- •
- Respiratory, thoracic, and mediastinal disorders: Hypoxia (4.9%)
- •
- Skin and subcutaneous tissue disorders: Rash (7%)
- •
- Vascular disorders: Thrombosis (7%)
Table 7: Grade 3 or 4 Laboratory Abnormalities Occurring in ≥ 10% of Patients with Relapsed or Refractory LBCL Treated with BREYANZI in Study 2 (N=61) a Baseline lab values were assessed prior to lymphodepleting chemotherapy. Laboratory Abnormalitya
Grade 3 or 4 (%)
Lymphocyte count decreased
97
Neutrophil count decreased
80
Hemoglobin decreased
30
Platelet count decreased
26
Grade 4 laboratory abnormalities in ≥ 10% of patients were lymphocyte decrease (95%), neutrophil decrease (57%), and platelet decrease (20%).
Study 3 (017001; Relapsed or Refractory LBCL After Two or More Lines of Therapy)
Study 3 was an open-label, single-arm study which evaluated 268 adult patients with R/R LBCL after 2 or more prior lines of therapy received a single dose of CAR-positive viable T cells [see Clinical Studies (14.1)]. Patients with a history of CNS disorders (such as seizures or cerebrovascular ischemia) or autoimmune disease requiring systemic immunosuppression were ineligible. The median age of the study population was 63 years (range: 18 to 86 years); 65% were male. The ECOG performance status at screening was 0 in 41% of patients, 1 in 58% of patients, and 2 in 1.5% of patients.
Serious adverse reactions occurred in 46% of patients. The most common nonlaboratory serious adverse reactions (> 2%) were CRS, encephalopathy, sepsis, febrile neutropenia, aphasia, pneumonia, fever, hypotension, dizziness, and delirium. Fatal adverse reactions occurred in 4% of patients.
Table 8 presents selected nonlaboratory adverse reactions reported in patients treated with BREYANZI, and Table 9 describes selected new or worsening Grade 3 or 4 laboratory abnormalities.
The most common nonlaboratory adverse reactions (≥ 20%) were fatigue, CRS, musculoskeletal pain, nausea, headache, encephalopathy, infections (pathogen unspecified), decreased appetite, diarrhea, hypotension, tachycardia, dizziness, cough, constipation, abdominal pain, vomiting, and edema.
Table 8: Adverse Reactions in ≥ 10% of Patients with Relapsed or Refractory LBCL Treated with BREYANZI in Study 3 (N=268) * Represents multiple related terms. a Edema includes edema, edema peripheral, fluid overload, fluid retention, generalized edema, hypervolemia, peripheral swelling, pulmonary congestion, pulmonary edema, swelling. b Encephalopathy includes amnesia, bradyphrenia, cognitive disorder, confusional state, depersonalization/derealization disorder, depressed level of consciousness, disturbance in attention, encephalopathy, flat affect, hypersomnia, incoherent, lethargy, leukoencephalopathy, loss of consciousness, memory impairment, mental impairment, mental status changes, somnolence. c Dizziness includes dizziness, presyncope, syncope, vertigo. d Tremor includes essential tremor, resting tremor, tremor. e Peripheral neuropathy includes hyperesthesia, hypoesthesia, meralgia paresthetica, neuralgia, neuropathy peripheral, paresthesia, peripheral sensory neuropathy, sciatica, sensory loss. f Aphasia includes aphasia, disorganized speech, dysarthria, dysphemia, dysphonia, slow speech, speech disorder. g Motor dysfunction includes eyelid ptosis, motor dysfunction, muscle rigidity, muscle spasms, muscle spasticity, muscle tightness, muscle twitching, muscular weakness, myoclonus, myopathy. h Delirium includes agitation, delirium, delusion, disorientation, hallucination, ‘hallucination, visual’, irritability, restlessness. i Rash includes erythema, dermatitis acneiform, perineal rash, rash, rash erythematous, rash macular, rash maculo-papular, rash morbilliform, rash papular, rash pruritic, rash pustular. j Hemorrhage includes catheter site hemorrhage, conjunctival hemorrhage, epistaxis, hematoma, hematuria, hemorrhage, hemorrhage intracranial, pulmonary hemorrhage, retinal hemorrhage, vaginal hemorrhage. Adverse Reaction
Any Grade (%)
Grade 3 or Higher (%)
Cardiac disorders
Tachycardia*
25
0
Gastrointestinal disorders
Nausea
33
1.5
Diarrhea
26
0.4
Constipation
23
0
Abdominal pain*
21
3.0
Vomiting
21
0.4
General disorders and administration site conditions
Fatigue*
48
3.4
Edemaa
21
1.1
Fever
16
0
Chills
12
0
Immune system disorders
Cytokine release syndrome
46
4.1
Infections and infestations*
Infection with pathogen unspecified*
29
16
Bacterial infection*
13
5
Upper respiratory tract infection*
13
0.7
Viral infection
10
1.5
Metabolism and nutrition disorders
Decreased appetite
28
2.6
Musculoskeletal and connective tissue disorders
Musculoskeletal pain*
37
2.2
Nervous system disorders
Headache*
30
1.1
Encephalopathyb
29
9
Dizzinessc
24
2.6
Tremord
16
0
Peripheral neuropathye
11
0
Aphasiaf
10
2.2
Motor dysfunctiong
10
1.1
Psychiatric disorders
Insomnia*
14
0.4
Anxiety*
10
0
Deliriumh
10
2.2
Renal and urinary disorders
Renal failure*
11
3.0
Respiratory, thoracic, and mediastinal disorders
Cough*
23
0
Dyspnea*
16
2.6
Skin and subcutaneous tissue disorders
Rashi
13
0.4
Vascular disorders
Hypotension*
26
3.4
Hypertension
14
4.5
Hemorrhagej
10
1.5
Other clinically important adverse reactions in < 10% of patients included the following:
- •
- Cardiac disorders: Arrhythmia (6%), cardiomyopathy (1.5%)
- •
- Gastrointestinal disorders: Gastrointestinal hemorrhage (4.1%)
- •
- Infections and infestations: Pneumonia (8%), fungal infection (8%), sepsis (4.5%), urinary tract infection (4.1%)
- •
- Metabolism and nutrition disorders: Tumor lysis syndrome (0.7%)
- •
- Nervous system disorders: Ataxia or gait disturbance (7%), visual disturbance (5%), paresis (2.6%), cerebrovascular events (1.9%), seizure (1.1%), brain edema (0.4%)
- •
- Procedural complications: Infusion-related reaction (1.9%)
- •
- Respiratory, thoracic, and mediastinal disorders: Pleural effusion (7%), hypoxia (6%)
- •
- Vascular disorder: Thrombosis (7%)
Table 9: Grade 3 or 4 Laboratory Abnormalities Occurring in ≥ 10% of Patients with Relapsed or Refractory LBCL Treated with BREYANZI in Study 3 a Baseline lab values were assessed prior to lymphodepleting chemotherapy. b The denominator varied from 239 to 268, based on the number of patients with a baseline value and at least one post-treatment value for the particular lab. Laboratory Abnormalitya
Grade 3 or 4 (%)b
Lymphocyte count decreased
95
Neutrophil count decreased
88
Platelet count decreased
41
Hemoglobin decreased
32
Phosphate decreased
16
Fibrinogen decreased
14
Study 4 (017004; Relapsed or Refractory CLL/SLL)
Study 4 was an open-label, single-arm study which evaluated 89 adult patients with R/R CLL/SLL who had received at least 2 prior lines of therapy including a BTK inhibitor and a BCL-2 inhibitor before receiving a single dose of CAR positive viable T cells [see Clinical Studies (14.2)]. Patients with a history of CNS disorders (such as seizures or cerebrovascular ischemia) or autoimmune disease requiring systemic immunosuppression, Richter’s transformation, ECOG performance status >1 were ineligible. The trial required LVEF ≥ 40%, adequate oxygen saturation on room air with ≤ Grade 1 dyspnea, ALT ≤ 5 x ULN, total bilirubin < 2.0 mg/dL, creatinine clearance > 30 mL/min, and adequate bone marrow function to receive lymphodepleting chemotherapy.
The median age of the study population was 66 years (range: 49 to 82 years); 69% were male, 84% were White, 3% were Black, 1% were Asian. Two percent were Hispanic, and 89% were non-Hispanic. The ECOG performance status at screening was 0 in 40% of patients, and 1 in 60% of patients.
Serious adverse reactions occurred in 60% of patients. The most common nonlaboratory serious adverse reactions (> 2%) were CRS, encephalopathy, febrile neutropenia, pneumonia, hemorrhage, fever, renal failure, aphasia, abdominal pain, delirium, tumor lysis syndrome, upper respiratory tract infection, and hemophagocytic lymphohistiocytosis [IEC-HS]. Fatal adverse reactions occurred in 1.1% of patients.
Table 10 presents selected nonlaboratory adverse reactions reported in patients treated with BREYANZI, and Table 11 describes selected new or worsening Grade 3 or 4 laboratory abnormalities.
The most common nonlaboratory adverse reactions (≥ 20%) were CRS, encephalopathy, fatigue, musculoskeletal pain, nausea, edema, diarrhea, dyspnea, headache, fever, decreased appetite, constipation, tremor, dizziness, Infection with pathogen unspecified, rash, tachycardia, cough, and delirium.
Table 10: Adverse Reactions in ≥ 10% of Patients with Relapsed or Refractory CLL/SLL Treated with BREYANZI in Study 4 (N=89) * Represents multiple related terms. a Edema includes ascites, face edema, hypervolemia, edema peripheral, pleural effusion, pulmonary edema, scrotal edema. b Encephalopathy includes cognitive disorder, confusional state, disturbance in attention, encephalopathy, lethargy, memory impairment, mental status changes, somnolence. c Dizziness includes dizziness, presyncope, syncope, vertigo. d Motor dysfunction includes asterixis, muscle spasms, muscular weakness, myoclonus. ePeripheral neuropathy includes hyperesthesia, hypoesthesia, neuralgia, neuropathy peripheral, paresthesia, peripheral sensory neuropathy. f Delirium includes agitation, delirium, hallucination, hallucination visual, intensive care unit delirium, irritability, restlessness. g Rash includes dermatitis contact, erythema, petechiae, rash, rash macular, rash maculo-papular, scrotal erythema, seborrheic keratosis, urticaria. hHemorrhage includes epistaxis, hemorrhage intracranial, hematoma, hematuria, hemorrhoidal hemorrhage, intraventricular hemorrhage, lower gastrointestinal hemorrhage, traumatic hemothorax. Adverse Reaction
Any Grade (%)
Grade 3 or Higher (%)
Blood and lymphatic system disorders
Febrile neutropenia
12
12
Cardiac disorders
Tachycardia*
21
0
Gastrointestinal disorders
Nausea
35
0
Diarrhea*
30
1.1
Constipation
24
0
Abdominal pain*
18
0
Vomiting
15
0
General disorders and administration site conditions
Fatigue*
40
4.5
Edemaa
30
4.5
Fever*
27
1.1
Chills
17
1.1
Immune system disorders
Cytokine release syndrome
83
9
Infections and infestations
Infection with pathogen unspecified*
23
10
Upper respiratory tract infection*
19
1.1
Viral infection*
10
1.1
Metabolism and nutrition disorders
Decreased appetite
27
4.5
Tumor lysis syndrome
11
11
Musculoskeletal and connective tissue disorders
Musculoskeletal pain*
42
1.1
Nervous system disorders
Encephalopathyb
44
18
Headache*
28
1.1
Tremor
24
2.2
Dizzinessc
21
1.1
Motor dysfunctiond
14
2.2
Peripheral neuropathye
12
0
Taste disorder*
10
0
Psychiatric disorders
Deliriumf
20
3.4
Insomnia
16
1.1
Anxiety
12
1.1
Renal and urinary disorders
Renal failure*
15
3.4
Respiratory, thoracic, and mediastinal disorders
Dyspnea*
27
8
Cough*
20
0
Skin and subcutaneous tissue disorders
Rashg
23
2.2
Vascular disorders
Hypotension*
17
0
Hemorrhageh
16
1.1
Hypertension
10
4.5
Other clinically important adverse reactions in < 10% of patients included the following:
- •
- Cardiac disorders: Chest discomfort (4.5%), Arrhythmia (2.2%).
- •
- Eye disorders: Vision blurred (4.5%).
- •
- Gastrointestinal disorders: Dyspepsia (9%), abdominal distension (7%).
- •
- Immune system disorders: Hemophagocytic lymphohistiocytosis [IEC-HS] (3.4%).
- •
- Infections and infestations: Fungal infection (9%), pneumonia (7%), urinary tract infection (7%), bacterial infectious disorders (4.5%), sepsis (2.2%).
- •
- Injury, poisoning and procedural complications: Infusion related reaction (1.1%).
- •
- Nervous system disorders: Aphasia (8%), Ataxia (3.4%), Paresis (3.4%), Seizure (1.1%).
- •
- Psychiatric disorders: Affective disorder (7%).
- •
- Respiratory, thoracic, and mediastinal disorders: Oral pain (8%), hypoxia (8%).
- •
- Skin and subcutaneous tissue disorders: Ecchymosis (8%), xerosis (7%), pruritus (6%).
- •
- Vascular disorder: Thrombosis (6%).
Table 11: Grade 3 or 4 Laboratory Abnormalities Occurring in ≥ 10% of Patients with Relapsed or Refractory CLL/SLL Treated with BREYANZI in Study 4 a Baseline lab values were assessed prior to lymphodepleting chemotherapy. b The denominator ranged from 85 to 89 for other measurements, based on the number of patients with a baseline value and at least one post-treatment value for the particular lab. Laboratory Abnormalitya
Grade 3 or 4 (%)b
Neutrophil count decreased
94
Lymphocyte count decreased
87
White blood cell decreased
85
Platelet count decreased
53
Hemoglobin decreased
49
Hypophosphatemia
24
Hyponatremia
18
Hypocalcemia
11
Grade 4 laboratory abnormalities in ≥ 10% of patients were neutrophil count decreased (81%), lymphocyte count decreased (73%), white blood cell decreased (72%), and platelet count decreased (30%).
Study 5 (JCAR017-FOL-001; Relapsed or Refractory FL Cohort)
Study 5 was an open-label, single-arm study which evaluated 107 adult patients with relapsed or refractory FL after two or more prior lines of therapy received a single dose of BREYANZI [see Clinical Studies (14.3)]. Patients with a history of CNS disorders and active autoimmune disease requiring immunosuppressive therapy were ineligible. The median age was 62 years (range: 23 to 80 years), 38 % were female, and ECOG performance status was 0 in 61% and 1 in 39% of patients; 56% were White, 3% were Black, 9% were Asian; 5% were Hispanic and 69% were non-Hispanic.
Serious adverse reactions occurred in 26% of patients. The most common nonlaboratory serious adverse reactions (> 2%) were CRS, aphasia, febrile neutropenia, fever, and tremor.
Table 12 presents selected nonlaboratory adverse reactions reported in patients treated with BREYANZI, and Table 13 describes selected new or worsening Grade 3 or 4 laboratory abnormalities.
The most common nonlaboratory adverse reactions (≥ 20%) were CRS, headache, musculoskeletal pain, fatigue, constipation, and fever.
Table 12: Adverse Reactions** in ≥ 10% of Patients with Relapsed or Refractory FL Treated with BREYANZI in Study 5 (N=107) * Represents multiple related terms. ** Includes adverse reactions up to 90 days following treatment with BREYANZI. Adverse Reaction
Any Grade (%)
Grade 3 or Higher (%)
Gastrointestinal disorders
Constipation
21
0
Diarrhea
15
0
General disorders and administration site conditions
Fatigue*
23
0
Fever*
20
0
Immune system disorders
Cytokine release syndrome
59
0.9
Infections and infestations
Infection with pathogen unspecified*
16
4.7
Musculoskeletal and connective tissue disorders
Musculoskeletal pain*
28
0
Nervous system disorders
Headache
28
0
Tremor
15
0
Other clinically important adverse reactions in < 10% of patients included the following:
- •
- Blood and lymphatic system disorders: Febrile neutropenia (6%).
- •
- Cardiac disorders: Tachycardia (2.8%).
- •
- Eye disorders: Vision blurred (1.9%).
- •
- Gastrointestinal disorders: Nausea (9%), abdominal pain (4.7%), vomiting (3.7%).
- •
- General disorders and administration site conditions: Edema (4.7%), chills (3.7%).
- •
- Infections and infestations: Upper respiratory tract infection (8%), bacterial infectious disorders (6%), urinary tract infection (4.7%), viral infectious disorders (1.9%), pneumonia (1.9%), sepsis (0.9%).
- •
- Nervous system disorders: Encephalopathy (7%), aphasia (8%), dizziness (4.7%), motor dysfunction (3.7%), ataxia (3.7%), neuropathy peripheral (4.7%).
- •
- Psychiatric disorders: Insomnia (4.7%), delirium (4.7%), anxiety (1.9%).
- •
- Renal and urinary disorders: Acute kidney injury (0.9%).
- •
- Respiratory, thoracic and mediastinal disorders: Cough (7%), dyspnea (1.9%), hypoxia (1.9%).
- •
- Vascular disorders: Hypotension (8%), hypertension (6%), thrombosis (4.7%).
- •
- Skin and subcutaneous tissue disorders: Rash (7%).
Table 13: Grade 3 or 4 Laboratory Abnormalities* Occurring in ≥ 10% of Patients with Relapsed or Refractory FL Treated with BREYANZI in Study 5 (N=107) * Includes laboratory abnormalities up to 90 days following treatment with BREYANZI. a Baseline lab values were assessed prior to lymphodepleting chemotherapy. b Based on the number of patients with a baseline value and at least one post treatment value for the particular lab. Laboratory Abnormalitya
Grade 3 or 4 (%)b
Lymphocyte count decreased
94
Neutrophil count decreased
79
White blood cell decreased
74
Platelet count decreased
17
Grade 4 laboratory abnormalities in ≥ 10% of patients were lymphocyte count decreased (78%), neutrophil count decreased (61%), white blood cell decreased (41%), and platelet count decreased (11%).
Study 3 (017001; Relapsed or Refractory MCL Cohort)
Study 3 was an open-label, single-arm study which evaluated 88 adult patients with relapsed or refractory MCL received a single dose of CAR-positive viable T cells [see Clinical Studies (14.4)]. Patients with a history of CNS disorders (such as seizures or cerebrovascular ischemia) or autoimmune disease requiring systemic immunosuppression were ineligible. The trial required left ventricular ejection fraction ≥ 40%, adequate oxygen saturation on room air with ≤ Grade 1 dyspnea, ALT ≤ 5 x ULN, total bilirubin < 2.0 mg/dL, creatinine clearance > 30 mL/min, and adequate bone marrow function to receive lymphodepleting chemotherapy. The median age of the study population was 69 years (range: 36 to 86 years); 76% were male, 88% were White, 6% were Asian and 2.3% were Black. Four percent were Hispanic, and 92% were non-Hispanic. The ECOG performance status at screening was 0 in 54% of patients, and 1 in 46% of patients.
Serious adverse reactions occurred in 53% of patients. The most common nonlaboratory serious adverse reactions (> 2%) were CRS, confusional state, fever, encephalopathy, mental status changes, pleural effusion, upper respiratory tract infection, and decreased appetite. Fatal adverse reactions occurred in 4.5% of patients.
Table 14 presents selected nonlaboratory adverse reactions reported in patients treated with BREYANZI, and Table 15 describes selected new or worsening Grade 3 or 4 laboratory abnormalities.
The most common nonlaboratory adverse reactions (≥ 20%) were CRS, fatigue, musculoskeletal pain, encephalopathy, edema, headache, and decreased appetite.
Table 14: Adverse Reactions** in ≥ 10% of Patients with Relapsed or Refractory MCL Treated with BREYANZI in Study 3 (N=88) * Represents multiple related terms. ** Includes adverse reactions up to 90 days following treatment with BREYANZI. a Edema includes hypervolemia, localized edema, edema, edema peripheral, peripheral swelling, pleural effusion, pulmonary edema. b Encephalopathy includes confusional state, depressed level of consciousness, encephalopathy, lethargy, memory impairment, mental status changes, somnolence. c Dizziness includes dizziness, dizziness postural, syncope, vertigo. d Motor dysfunction includes fine motor skill dysfunction, muscle spasms, muscle tightness, muscular weakness. e Rash includes dermatitis contact, rash, rash erythematous, rash macular, rash maculo-papular, rash pruritic. f Hemorrhage includes catheter site hemorrhage, epistaxis, hematoma, hematuria, hemorrhage, hemorrhoidal hemorrhage, rectal hemorrhage. Adverse Reaction
Any Grade (%)
Grade 3 or Higher (%)
Cardiac disorders
Tachycardia*
17
3.4
Gastrointestinal disorders
Nausea
18
2.3
Diarrhea
17
0
Abdominal pain*
15
3.4
Constipation
14
0
General disorders and administration site conditions
Fatigue*
39
2.3
Edemaa
25
1.1
Fever*
17
0
Chills
11
0
Immune system disorders
Cytokine release syndrome
61
1.1
Infections and infestations
Infection with pathogen unspecified*
16
6
Upper respiratory tract infection*
13
2.3
Metabolism and nutrition disorders
Decreased appetite
21
4.5
Musculoskeletal and connective tissue disorders
Musculoskeletal pain*
38
2.3
Nervous system disorders
Encephalopathyb
30
9
Headache
23
0
Dizzinessc
11
2.3
Motor dysfunctiond
11
0
Tremor
11
0
Psychiatric disorders
Insomnia*
14
0
Anxiety
13
1.1
Renal and urinary disorders
Renal failure*
15
0
Respiratory, thoracic, and mediastinal disorders
Dyspnea*
11
0
Cough
10
0
Skin and subcutaneous tissue disorders
Rashe
11
1.1
Vascular disorders
Hypotension*
15
0
Hemorrhagef
10
0
Hypertension
10
3.4
Other clinically important adverse reactions in < 10% of patients included the following:
- •
- Blood and lymphatic system disorders: Febrile neutropenia (6%).
- •
- Cardiac disorders: Arrhythmia (2.3%).
- •
- Eye disorders: Vision blurred (3.4%).
- •
- Gastrointestinal disorders: Vomiting (6%).
- •
- Infections and infestations: Bacterial infection (9%), viral infection (9%), fungal infection (8%), urinary tract infection (6%), sepsis (3.4%), pneumonia (2.3%).
- •
- Injury, poisoning and procedural complications: Infusion related reaction (4.5%).
- •
- Metabolism and nutrition disorders: Tumor lysis syndrome (2.3%).
- •
- Nervous system disorders: Neuropathy peripheral (9%), aphasia (8%), ataxia (4.5%), cerebral infarction (1.1%), seizure (1.1%).
- •
- Psychiatric disorders: Delirium (7%).
- •
- Respiratory, thoracic, and mediastinal disorders: Hypoxia (3.4%).
- •
- Vascular disorder: Thrombosis (4.5%).
Table 15: Grade 3 or 4 Laboratory Abnormalities* Occurring in ≥ 10% of Patients with Relapsed or Refractory MCL Treated with BREYANZI in Study 3 * Includes lab abnormalities up to 90 days following treatment with BREYANZI. a Baseline lab values were assessed prior to lymphodepleting chemotherapy. b The denominator ranged from 87 to 88 for laboratory measurements, based on the number of patients with a baseline value and at least one post-treatment value for the particular lab. Laboratory Abnormalitya
Grade 3 or 4 (%)b
Lymphocyte count decreased
89
Neutrophil count decreased
85
White blood cell decreased
83
Platelet count decreased
39
Hemoglobin decreased
33
Uric acid increased
10
Sodium decreased
10
Grade 4 laboratory abnormalities in ≥ 10% of patients were lymphocyte count decreased (84%), neutrophil count decreased (52%), white blood cell decreased (52%), and platelet count decreased (22%).
Study 5 (JCAR017-FOL-001; Relapsed or Refractory MZL Cohort)
Study 5 was a an open-label, single-arm study which evaluated 67 adult patients with relapsed or refractory MZL after two or more prior lines of therapy or relapsed after hematopoietic stem cell transplant (HSCT) received a single dose of CAR-positive viable T cells [see Clinical Studies (14.5)]. Patients who had previously received CD19-directed therapy had to have biopsy proven CD19 positive lymphoma [see Clinical Studies (14)]. Patients with a history of CNS disorders (such as seizures or cerebrovascular ischemia) and active autoimmune disease requiring immunosuppressive therapy were ineligible. The median age was 62 years (range: 37 to 81 years), 58% were male, ECOG performance status was 0 in 55% and 1 in 45% of patients; 57% were White, 6% were Asian, 2% were Black; race was not reported in 36% of patients; and 2% were Hispanic.
Serious adverse reactions occurred in 39% of patients. The most common nonlaboratory serious adverse reactions (> 2%) were CRS, encephalopathy, aphasia, sepsis, tremor, delirium, dizziness, infusion related hypersensitivity reaction, and transient ischemic attack. Fatal adverse reactions occurred in 3% of patients.
Table 16 presents nonlaboratory adverse reactions reported in ≥ 10% of patients treated with BREYANZI, and Table 17 describes selected new or worsening Grade 3 or 4 laboratory abnormalities.
The most common nonlaboratory adverse reactions (≥ 20%) were CRS, diarrhea, fatigue, musculoskeletal pain, and headache.
Table 16: Adverse Reactions** in ≥ 10% of Patients with Relapsed or Refractory MZL Treated with BREYANZI in Study 5 (N=67) * Represents multiple related terms. ** Includes adverse reactions up to 90 days following treatment with BREYANZI. a Edema includes ascites, fluid retention, hypervolaemia, oedema peripheral, peripheral swelling, pleural effusion, pulmonary oedema. b Encephalopathy includes brain fog, cognitive disorder, confusional state, disturbance in attention, dyscalculia, dysgraphia, memory impairment, somnolence. c Delirium includes delirium, disorientation, hallucination, irritability, restlessness. Adverse Reaction
Any Grade (%)
Grade 3 or Higher (%)
Gastrointestinal disorders
Diarrhea
28
1.5
Nausea
18
1.5
Abdominal pain*
10
0
General disorders and administration site conditions
Fatigue*
27
3
Edemaa
18
3
Fever*
10
0
Immune system disorders
Cytokine release syndrome
76
4.5
Infections and infestations
Infection with pathogen unspecified*
16
6
Metabolism and nutrition disorders
Decreased appetite
10
3
Musculoskeletal and connective tissue disorders
Musculoskeletal pain*
22
0
Nervous system disorders
Headache
21
1.5
Tremor
21
0
Encephalopathyb
21
1.5
Dizziness
16
0
Aphasia
10
0
Psychiatric disorders
Deliriumc
10
3
Renal and urinary disorders
Renal failure*
10
1.5
Vascular disorders
Hypotension
10
0
Other clinically important adverse reactions in < 10% of patients included the following:
- •
- Blood and lymphatic system disorders: Febrile neutropenia (3%).
- •
- Cardiac disorders: Tachycardia (7.5%).
- •
- Gastrointestinal disorders: Vomiting (9%), constipation (3%), dyspepsia (3%).
- •
- General disorders and administration site conditions: Chills (7.5%), infusion related hypersensitivity reactions (3%).
- •
- Immune system disorders: Hemophagocytic lymphohistiocytosis (4.5%).
- •
- Infections and infestations: Bacterial infections (6%), viral infections (6%), fungal infections (3%).
- •
- Metabolism and nutrition disorders: Tumor lysis syndrome (1.5%).
- •
- Nervous system disorders: Motor dysfunction (4.5%), ataxia (3%), neuropathy peripheral (3%), transient ischaemic attack (3.0%).
- •
- Psychiatric disorders: Insomnia (9%), affective disorder (7.5%).
- •
- Respiratory, thoracic and mediastinal disorders: Dyspnea (6%), cough (4.5%), hypoxia (4.5%).
- •
- Skin and subcutaneous tissue disorders: Rash (4.5%).
- •
- Vascular disorders: Hemorrhage (9%), hypertension (6%), thrombosis (6%).
Table 17: Grade 3 or 4 Laboratory Abnormalities* Occurring in ≥ 10% of Patients with Relapsed or Refractory MZL Treated with BREYANZI in Study 5 (N=67) * Includes laboratory abnormalities up to 90 days following treatment with BREYANZI. a Baseline lab values were assessed prior to lymphodepleting chemotherapy. b Based on the number of patients with a baseline value and at least one post treatment value for the particular lab. Laboratory Abnormalitya
Grade 3 or 4 (%)b
Lymphocyte count decreased
99
Neutrophil count decreased
84
White blood cell decreased
84
Platelet count decreased
28
Hemoglobin decreased
25
Fibrinogen decreased
10
Grade 4 laboratory abnormalities in ≥ 10% of patients were lymphocyte count decreased (85%), neutrophil count decreased (64%), white blood cell decreased (49%), and platelet count decreased (19%).
6.2 Postmarketing Experience
Because adverse events to marketed products are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to product exposure.
The following adverse events have been identified during postmarketing use of BREYANZI.
Nervous System Disorder: Immune effector cell-associated neurotoxicity syndrome (ICANS).
Neoplasms: T cell malignancies
Eye disorders: Blindness
- 7 DRUG INTERACTIONS
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
There are no available data with BREYANZI use in pregnant women. No animal reproductive and developmental toxicity studies have been conducted with BREYANZI to assess whether it can cause fetal harm when administered to a pregnant woman.
It is not known if BREYANZI has the potential to be transferred to the fetus. Based on the mechanism of action, if the transduced cells cross the placenta, they may cause fetal toxicity, including B-cell lymphocytopenia and hypogammaglobulinemia. Therefore, BREYANZI is not recommended for women who are pregnant, and pregnancy after BREYANZI infusion should be discussed with the treating physician.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
8.2 Lactation
Risk Summary
There is no information regarding the presence of BREYANZI in human milk, the effect on the breastfed infant, and the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for BREYANZI and any potential adverse effects on the breastfed infant from BREYANZI or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Pregnancy Testing
Pregnancy status of females with reproductive potential should be verified. Sexually active females of reproductive potential should have a pregnancy test prior to starting treatment with BREYANZI.
Contraception
See the prescribing information for fludarabine and cyclophosphamide for information on the need for effective contraception in patients who receive lymphodepleting chemotherapy.
There are insufficient exposure data to provide a recommendation concerning duration of contraception following treatment with BREYANZI.
8.4 Pediatric Use
The safety and efficacy of BREYANZI have not been established in pediatric patients.
8.5 Geriatric Use
In clinical trials of BREYANZI, 111 (41%) of 268 patients with two or more prior lines of therapy for LBCL, and 89 (59%) of 150 patients with one prior line of therapy for LBCL, were 65 years of age or older; 27 (10%) and 28 (19%) were 75 years of age or older, respectively. In patients with CLL/SLL, 51 (57%) of 89 were 65 years of age or older, and 9 (10%) were 75 years of age or older. In patients with FL, 42 (39%) of 107 were 65 years of age or older, and 10 (9%) were 75 years of age or older. In patients with MCL, 64 (73%) of 88 patients were 65 years of age or older, and 18 (21%) were 75 years of age or older. In patients with MZL, 30 (45%) of 67 patients were 65 years of age or older, and 10 (15%) were 75 years of age or older. No clinically important differences in safety or effectiveness of BREYANZI were observed between patients aged ≥ 65 and younger patients.
-
11 DESCRIPTION
BREYANZI (lisocabtagene maraleucel) is a CD19-directed genetically modified autologous T cell immunotherapy administered as a defined composition of CAR-positive viable T cells (consisting of CD8 and CD4 components). The CAR is comprised of the FMC63 monoclonal antibody-derived single-chain variable fragment (scFv), IgG4 hinge region, CD28 transmembrane domain, 4-1BB (CD137) costimulatory domain, and CD3 zeta activation domain. In addition, BREYANZI includes a nonfunctional truncated epidermal growth factor receptor (EGFRt) that is co-expressed on the cell surface with the CD19-specific CAR.
BREYANZI is a T cell product. BREYANZI is prepared from the patient’s T cells, which are obtained via a standard leukapheresis procedure. The purified CD8-positive and CD4-positive T cells are separately activated and transduced with the replication-incompetent lentiviral vector containing the anti-CD19 CAR transgene. The transduced T cells are expanded in cell culture, washed, formulated into a suspension, and cryopreserved as separate CD8 and CD4 component vials that together constitute a single dose of BREYANZI. The product must pass a sterility test before release for shipping as a frozen suspension in patient-specific vials. The product is thawed prior to administration [see Dosage and Administration (2.2) and How Supplied/Storage and Handling (16)].
The BREYANZI formulation contains 75% (v/v) Cryostor® CS10 [containing 7.5% dimethylsulfoxide (v/v)], 24% (v/v) Multiple Electrolytes for Injection, Type 1, 1% (v/v) of 25% albumin (human).
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
BREYANZI is a CD19-directed genetically modified autologous cell immunotherapy administered as a defined composition to reduce variability in CD8-positive and CD4-positive T cell dose. The CAR is comprised of an FMC63 monoclonal antibody-derived single chain variable fragment (scFv), IgG4 hinge region, CD28 transmembrane domain, 4-1BB (CD137) costimulatory domain, and CD3 zeta activation domain. CD3 zeta signaling is critical for initiating activation and antitumor activity, while 4-1BB (CD137) signaling enhances the expansion and persistence of BREYANZI.
CAR binding to CD19 expressed on the cell surface of tumor and normal B cells induces activation and proliferation of CAR T cells, release of pro-inflammatory cytokines, and cytotoxic killing of target cells.
12.2 Pharmacodynamics
Following BREYANZI infusion, pharmacodynamic responses were evaluated over a 4-week period by measuring transient elevation of soluble biomarkers such as cytokines, chemokines, and other molecules. Peak elevation of soluble biomarkers was observed within the first 14 days after BREYANZI infusion and returned to baseline levels within 28 days.
B-cell aplasia, defined as CD19+ B cells comprising less than 3% of peripheral blood lymphocytes, is an on-target effect of BREYANZI. B-cell aplasia was observed in the majority of patients for up to 1 year following BREYANZI infusion.
12.3 Pharmacokinetics
Following infusion, BREYANZI exhibited an initial expansion followed by a bi-exponential decline.
Relapsed or Refractory LBCL
The median time of maximal expansion in peripheral blood occurred 10-12 days after infusion. BREYANZI was present in peripheral blood for an estimated median of 12.1 months (range: 0.1+ to 24.2+ months).
Among patients who received two or more prior lines of therapy for LBCL (Study 3 – LBCL Cohort), responders (N=135) had a 2.3-fold higher median Cmax than nonresponders (N=37) (35,335 vs. 15,527 copies/µg). Responders had a 1.8-fold higher median AUC0-28d than nonresponders (273,552 vs. 155,240 day*copies/µg).
Patients < 65 years old (N=142) had a 3.1-fold and 2.3-fold higher median Cmax and AUC0-28d, respectively, compared to patients ≥ 65 years old (N=96). Sex, race, ethnicity, and body weight did not show clear relationships to Cmax and AUC0-28d.
Relapsed or Refractory CLL/SLL
The median time of maximal expansion in peripheral blood occurred 14 days after infusion. BREYANZI was present in peripheral blood for an estimated median of 12.0 months (range: 0.1+ to 30.1+ months).
Among patients who received prior therapy for CLL/SLL (Study 4), responders (N=27) had a 2.0-fold higher median Cmax than nonresponders (N=25) (99,559 vs. 48,948 copies/µg). Responders had a 1.9-fold higher median AUC0-28d than nonresponders (793,893 vs. 408,307 day*copies/µg).
Relapsed or Refractory FL
The median time of maximal expansion in peripheral blood occurred 10 days after infusion. Median Cmax and AUC0-28d are 30,530 copies/µg and 253,400 day*copies/µg, respectively. BREYANZI was present in peripheral blood for an estimated median of 12.0 months (range: 0.3+ to 24.0+ months).
Relapsed or Refractory MCL
The median time of maximal expansion in peripheral blood occurred 10 days after infusion. Median Cmax and AUC0-28d were 30,968 copies/µg and 375,006 day*copies/µg, respectively. BREYANZI was present in peripheral blood for an estimated median of 17.9 months (range: 0.1+ to 24.2+ months).
Relapsed or Refractory MZL
The median time of maximal expansion in peripheral blood occurred 10 days after infusion. Median Cmax and AUC0-28d were 81,390 copies/µg and 657,799 day*copies/µg, respectively. BREYANZI was present in peripheral blood for an estimated median of 18.0 months (range: 0.6 to 42.1 months).
Patients receiving medications for CRS and/or neurologic toxicities
In general, patients with higher CAR-T cell expansion tended to have higher rates of CRS and neurologic toxicities. Some patients required tocilizumab and/or corticosteroids for the management of CRS or neurologic toxicities [see Dosage and Administration (2.3)]. BREYANZI continued to expand and persist in patients who received tocilizumab and/or corticosteroids treatment. In Study 3 - LBCL Cohort, patients treated with tocilizumab (N=49) had a 3.6-fold and 3.7-fold higher median Cmax and AUC0-28d of BREYANZI, respectively, compared to patients who did not receive tocilizumab (N=189). Similarly, patients who received corticosteroids (N=50) had a 3.8-fold and 3.7-fold higher median Cmax and AUC0‑28d of BREYANZI, respectively, compared to patients who did not receive corticosteroids (N=188).
12.6 Immunogenicity
The observed incidence of anti-product antibodies is highly dependent on the sensitivity and specificity of the assay. Differences in assay methods preclude meaningful comparisons of the incidence of anti-product antibodies in the studies described below with the incidence of anti-product antibodies in other studies, including those of BREYANZI or of other similar products.
BREYANZI has the potential to induce anti-product antibodies. The immunogenicity of BREYANZI has been evaluated using an electrochemiluminescence (ECL) immunoassay for the detection of binding antibodies against the extracellular CD19-binding domain of BREYANZI. Pre-existing anti-product antibodies were detected in 11% (28/261) in Study 3 - LBCL Cohort, 1% (1/89) in Study 1, 0% (0/51) in Study 2, 2% (2/86) in Study 4, 2% (2/102) in Study 5 - FL Cohort, 13% (11/87) in Study 3 - MCL Cohort, and 0% (0/66) in Study 5 - MZL Cohort of patients. Treatment-induced or treatment-boosted anti-product antibodies were detected in 11% (27/257) in Study 3 - LBCL Cohort, 1% (1/89) in Study 1, 2% (1/49) in Study 2, 7% (6/84) in Study 4, 18% (18/100) in Study 5 - FL Cohort, 18% (15/85) in Study 3 - MCL Cohort, and 20% (13/66) in Study 5 - MZL Cohort of patients. Due to the small number of patients who had anti-product antibodies, the relationship between anti-product antibody status and efficacy, safety, or pharmacokinetics was not conclusive.
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
No carcinogenicity or genotoxicity studies have been conducted with BREYANZI. No studies have been conducted to evaluate the effects of BREYANZI on fertility. In vitro studies with BREYANZI manufactured from healthy donors and patients showed no evidence for transformation and/or immortalization and no preferential integration near genes associated with oncogenic transformation.
-
14 CLINICAL STUDIES
14.1 Relapsed or Refractory Large B-Cell Lymphoma
Relapsed or Refractory LBCL After One Line of Therapy
Study 1
A randomized, open-label, multicenter trial evaluated the efficacy of BREYANZI in adult patients with relapsed or refractory LBCL after first-line chemoimmunotherapy (Study 1: JCAR017-BCM-003; NCT03575351). Patients had not yet received treatment for relapsed or refractory lymphoma, were potential candidates for autologous HSCT, and were required to have primary refractory disease or relapse within 12 months from complete response (CR) to initial chemoimmunotherapy. Eligibility criteria required adequate organ function and blood counts for HSCT.
In total, 184 patients were randomized in a 1:1 ratio to receive a single infusion of BREYANZI (planned dose, 100 × 106 CAR-positive viable T cells) or to receive standard therapy consisting of 3 cycles of chemoimmunotherapy followed by high-dose therapy and autologous HSCT in patients who attained CR or PR. All patients underwent leukapheresis prior to randomization.
Patients randomized to BREYANZI were to receive lymphodepleting chemotherapy consisting of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day concurrently for 3 days followed by BREYANZI infusion 2 to 7 days after completion of lymphodepleting chemotherapy. Bridging chemotherapy was permitted between leukapheresis and the start of lymphodepleting chemotherapy. BREYANZI was administered in the inpatient (79%) and outpatient (21%) setting.
In the overall study population, the median age was 59 years (range: 20 to 75 years), 57% were male, 59% were White, 10% were Asian, and 4% were Black. Diagnoses included de novo DLBCL NOS (55%), high-grade B-cell lymphoma (23%), primary mediastinal large B-cell lymphoma (10%), and DLBCL arising from indolent lymphoma (8%). Of these patients, 73% had primary refractory disease to last therapy and 27% had relapsed disease within 12 months of achieving CR to first-line therapy.
Of 92 patients randomized to receive BREYANZI, 89 (97%) received BREYANZI. The median time from leukapheresis to product availability was 26 days (range: 19 to 84 days), and the median time from leukapheresis to product infusion was 36 days (range: 25 to 91 days). Fifty-eight (63%) patients received bridging therapy. One patient received a nonconforming product (manufacturing failure; 1.1%).
Of the 92 patients randomized to receive standard therapy, 91 started treatment and 43 (47%) received high-dose therapy and HSCT. The most common reason for not receiving HSCT was lack of efficacy of the salvage chemotherapy.
The primary efficacy measure was event-free survival (EFS) as determined by an independent review committee (IRC). Other efficacy measures included progression-free survival. Efficacy is summarized in Table 18 and Figure 1. The estimated 1-year EFS was 45% [95% CI: 29, 59] in the BREYANZI arm and 24% [95% CI: 14, 35] in the standard therapy arm.
Of the 92 patients in the BREYANZI arm, the estimated median duration of response (DOR) was not reached (95% CI: 7.9 months, NR) in patients who achieved CR (N=61) and 2.3 months (95% CI: 2.1, NR) in patients who achieved a best response of PR (N=18).
An interim analysis of overall survival was conducted at the time of the primary EFS analysis. The interim analysis of overall survival did not meet the criteria for statistical significance. Forty-six patients randomized to the standard therapy arm (50%) subsequently received BREYANZI on protocol.
Table 18: Efficacy Results in Patients with Relapsed or Refractory LBCL (Study 1) NR=not reached; CI=confidence interval. a Per the Lugano criteria, as assessed by an IRC. b EFS is defined as time from randomization to the earliest date of disease progression or relapse, death from any cause, failure to achieve CR or PR by 9 weeks post-randomization, or start of new lymphoma therapy due to efficacy concerns. c Kaplan-Meier estimate. d Based on a stratified Cox proportional hazards model. For all stratified analyses, stratification was based on response to first-line therapy (primary refractory vs relapsed) and second-line age-adjusted International Prognostic Index. e Cochran-Mantel-Haenszel test. f p-value is compared with 0.012 of the allocated alpha for this pre-specified interim analysis. Outcomea
BREYANZI Arm
(N=92)
Standard Therapy Arm
(N=92)
Event-Free Survivalb
Number of events, n (%)
35 (38)
63 (69)
Median, months [95% CI]c
10.1 [6.1, NR]
2.3 [2.2, 4.3]
Hazard ratio [95% CI]d
0.34 [0.22, 0.52]
One-sided p-value
<0.0001f
Complete Response Rate, % [95% CI]
66 [56, 76]
39 [29, 50]
Difference in CR rate, % [95% CI]
27 [12, 41]
One-sided p-valuee
<0.0001f
Progression-Free Survival
Number of events, n (%)
28 (30)
43 (47)
Median, months [95% CI]c
14.8 [6.6, NR]
5.7 [3.9, 9.4]
Hazard ratio [95% CI]d
0.41 [0.25, 0.66]
One-sided p-value
0.0001f
Figure 1: Kaplan-Meier Plot of IRC-Assessed Event-Free Survival (Intention-to- Treat Analysis)
Study 2
The efficacy of BREYANZI was evaluated in a single-arm, open-label, multicenter trial (Study 2: 017006; NCT03483103) in transplant-ineligible patients with relapsed or refractory LBCL after one line of chemoimmunotherapy. The study enrolled patients who were not eligible for high-dose therapy and autologous HSCT due to organ function or age, while also having adequate organ function for CAR-T cell therapy. The study required at least one of the following criteria: age ≥ 70 years, adjusted diffusing capacity of the lung for carbon monoxide (DLCO) ≤ 60%; LVEF < 50%; creatinine clearance < 60 mL/min; AST or ALT greater than 2 × ULN, or ECOG performance status of 2. The planned dose of BREYANZI was 100 × 106 CAR-positive viable T cells. Bridging therapy for disease control was permitted between leukapheresis and the start of lymphodepleting chemotherapy. Of the 61 patients treated with BREYANZI, 32 (53%) received bridging therapy.
BREYANZI was administered 2 to 7 days following completion of lymphodepleting chemotherapy. The lymphodepleting chemotherapy regimen consisted of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day concurrently for 3 days. BREYANZI was administered in the inpatient (67%) and outpatient (33%) setting.
Of 74 patients who underwent leukapheresis, 61 (82%) received BREYANZI and comprise the main efficacy population; 1 (1.4%) received CAR-positive T cells that did not meet the product specifications for BREYANZI (manufacturing failure); and 12 (16%) did not receive CAR-positive T cells for other reasons.
Of the 61 patients who received BREYANZI, the median age was 74 years (range: 53 to 84 years), 61% were male, 89% were White, 3% were Asian, and 2% were Black. Diagnoses included de novo DLBCL NOS (51%), high-grade B-cell lymphoma (33%), and DLBCL arising from follicular lymphoma (15%). Of these patients, 53% had primary refractory disease, 23% had relapse within 12 months of completing first-line therapy, and 25% had relapse >12 months after first-line therapy.
Efficacy was based on CR rate and DOR, as determined by an independent review committee (IRC) using 2014 Lugano criteria (Tables 19 and 20). The median time to CR was 1 month (range 0.8 to 6.9 months).
Table 19: Response Rate in Relapsed or Refractory LBCL (Study 2) CI=confidence interval; NR=Not reached. a Per the Lugano criteria, as assessed by an IRC. b 2-sided 95% exact Clopper-Pearson confidence intervals. Outcomea
BREYANZI-Treated
(N=61)
All Leukapheresed
(N=74)
Overall Response, n (%)
[95% CI]b
49 (80%)
[68%, 89%]
50 (68%)
[56%, 78%]
Complete Response
[95% CI]
33 (54%)
[41%, 67%]
34 (46%)
[34%, 58%]
Partial Response
[95% CI]
16 (26%)
[16%, 39%]
16 (22%)
[13%, 33%]
Table 20: Duration of Response in Study 2 DOR=duration of response; CI=confidence interval; CR=complete response; PR=partial response; NR=not reached. a Per the Lugano criteria, as assessed by an IRC. b Kaplan-Meier method is used to obtain 2-sided 95% confidence intervals. c Kaplan-Meier estimate. + Indicates a censored value. Outcomea
BREYANZI-Treated
(N=61)
Number of Responders
49
DOR
Median [95% CI], months b
Range, months
11.2 [5.1, NR]
0.0+ to 22.8+
DOR if Best Response is CR
Median [95% CI], months
Range, months
Rate at 6 months [95% CI] c
Rate at 12 months [95% CI] c
NR [11.2, NR]
2.0+ to 22.8+
83% [64, 93]
68% [45, 83]
DOR if Best Response is PR
Median [95% CI], months
2.1 [1.4, 2.3]
Range, months
0.0+ to 7.9
Rate at 6 months [95% CI] c
8.2% [0.5, 30.5]
Relapsed or Refractory LBCL After Two or More Lines of Therapy
The efficacy of BREYANZI was evaluated in an open-label, multicenter, single-arm trial (Study 3 - LBCL Cohort: 017001; NCT02631044) in adult patients with relapsed or refractory large B-cell non-Hodgkin lymphoma after at least 2 lines of therapy. The study included patients with ECOG performance status ≤ 2, prior autologous and/or allogeneic HSCT, and secondary CNS lymphoma involvement. The study excluded patients with a creatinine clearance of less than 30 mL/min, ALT > 5 times the ULN, or LVEF < 40%. There was no prespecified threshold for blood counts; patients were eligible to enroll if they were assessed by the investigator to have adequate bone marrow function to receive lymphodepleting chemotherapy. Bridging therapy for disease control was permitted between apheresis and the start of lymphodepleting chemotherapy, including intrathecal chemotherapy or radiation therapy for treatment of CNS involvement with lymphoma.
BREYANZI was administered 2 to 7 days following completion of lymphodepleting chemotherapy. The lymphodepleting chemotherapy regimen consisted of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day concurrently for 3 days. BREYANZI was administered in the inpatient and outpatient setting.
Of 299 patients who underwent leukapheresis for whom BREYANZI was manufactured in the dose range of 50 to 110 × 106 CAR-positive viable T cells:
- •
- 44 (15%) did not receive CAR-positive T cells either due to manufacturing failures (n=2), death (n=29), disease complications (n=6), or other reasons (n=7).
- •
- 204 (68%) received BREYANZI in the intended dose range, of whom 192 were evaluable for efficacy (main efficacy population); 12 were not evaluable due to absence of PET positive disease at study baseline or after bridging therapy.
- •
- 51 (17%) either received BREYANZI outside of the intended dose range (n=26) or received CAR-positive T cells that did not meet the product specifications for BREYANZI (manufacturing failures; n=25).
Of the 192 patients in the main efficacy population, the median age was 63 years (range: 18 to 86 years), 69% were male, 84% were White, 6% were Black, and 4.7% were Asian. The median number of prior therapies was 3 (range: 1 to 8). Diagnoses were de novo DLBCL (53%), DLBCL transformed from indolent lymphoma (25%), high-grade B-cell lymphoma (14%), primary mediastinal large B-cell lymphoma (7%), follicular lymphoma, grade 3B (1.0%). Of these patients, 64% had disease refractory to last therapy, 53% had primary refractory disease, 37% had prior HSCT and 2.6% had CNS involvement.
Efficacy was based on CR rate and DOR, as determined by an IRC using 2014 Lugano criteria (Tables 21 and 22). The median time to first response (CR or partial response [PR]) was 1.0 month (range: 0.7 to 8.9 months). The median time to first CR was 1.0 month (range 0.8 to 12.5 months). Of the 104 patients who achieved CR, 23 initially had stable disease (6 patients) or PR (17 patients), with a median time to improvement of 2.2 months (range: 0.7 to 11.6 months).
Table 21: Response Rate in Main Efficacy Population (Study 3 - LBCL Cohort) CI=confidence interval. a Per the Lugano criteria, as assessed by an IRC. BREYANZI-Treated
N=192Overall Response Ratea, n
[95% CI]141 (73%)
[67%, 80%]Complete Response, n
[95% CI]104 (54%)
[47%, 61%]Partial Response, n
[95% CI]37 (19%)
[14%, 26%]Table 22: Duration of Response in Study 3 - LBCL Cohort DOR=duration of response; CI=confidence interval; CR=complete response; PR=partial response; NR=not reached. a Evaluable for efficacy. b KM method was used to obtain 2-sided 95% confidence intervals. + Indicates a censored value. BREYANZI-Treateda
N=192Number of Responders
141
DOR (Months)
Median [95% CI]b
Range
16.7 [5.3, NR]
0.0+ to 23.5+
DOR if Best Response is CR (Months)
Median [95% CI]
Range
NR [16.7, NR]
0.7+ to 23.5+
DOR if Best Response is PR (Months)
Median [95% CI]
Range
1.4 [1.1, 2.2]
0.0+ to 22.8+
Response durations were longer in patients who achieved a CR, as compared to patients with a best response of PR (Table 22). Of the 104 patients who achieved CR, 68 (65%) had remission lasting at least 6 months and 64 (62%) had remission lasting at least 9 months.
Of the 287 patients who underwent leukapheresis and had radiographically evaluable disease, 27 additional patients achieved a response, apart from the responses noted in Table 22. The IRC-assessed overall response rate in the leukapheresed population (n=287) was 59% (95% CI: 53, 64), with a CR rate of 43% (95% CI: 37, 49) and PR rate of 15% (95% CI: 11, 20). These efficacy results include responses that may have been contributed solely by bridging therapy, responses after receipt of product outside of the intended dose range, and responses to product that did not meet release specifications.
14.2 Relapsed or Refractory Chronic Lymphocytic Lymphoma or Small Lymphocytic Lymphoma
The efficacy of BREYANZI was evaluated in a phase 1/2, open-label, multicenter, single-arm trial (Study 4: 017004; NCT03331198) in adult patients with R/R CLL or SLL who had received at least 2 prior lines of therapy including a BTK inhibitor and a BCL-2 inhibitor. Patients with del(17p), complex karyotype, and unmutated immunoglobulin heavy chain variable region (IGHV) were included in the study. The study enrolled patients with ECOG performance status of ≤ 1. The study excluded patients with a creatinine clearance of less than 30 mL/min, ALT > 5 times the ULN (except for subjects with leukemic infiltration of the liver), or LVEF < 40%. There was no prespecified threshold for blood counts; patients were eligible to enroll if they were assessed by the investigator to have adequate bone marrow function to receive lymphodepleting chemotherapy. The planned dose of BREYANZI was 100 × 106 CAR-positive viable T cells. Bridging therapy for disease control was permitted between apheresis and the start of lymphodepleting chemotherapy.
Of the 89 patients, BREYANZI was administered 2 to 11 days following completion of lymphodepleting chemotherapy. The lymphodepleting chemotherapy regimen consisted of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day concurrently for 3 days. BREYANZI was administered in the inpatient (88%) and outpatient (12%) settings.
Of 113 patients who underwent leukapheresis, 94 received BREYANZI. Of the 94, 84 received BREYANZI at 90 to 111 × 106 CAR-positive T cells and 10 received a cell-dose outside of this dose range; 3 received CAR-positive T cells that did not meet the product specifications for BREYANZI (manufacturing failure); and 16 other patients did not receive BREYANZI for other reasons.
The median time from leukapheresis to product availability was 24 days (range: 19 to 84 days), and the median time from leukapheresis to product infusion was 36 days (range: 28 to 384 days). Of the 89 patients treated with BREYANZI, 69 (78%) received bridging therapy.
Patients had measurable disease present before BREYANZI administration based on IRC assessment. Nineteen of 84 patients were not evaluable for efficacy (13 patients did not have baseline disease assessments performed after completion of bridging therapy, 1 patient lacked measurable disease, and 5 had Richter’s transformation). Of the 65 efficacy-evaluable patients, the median age was 66 years (range: 49 to 82 years), 68% were male, 80% were White, 3% were Black, 1% were Asian. Two-percent were Hispanic and 89% were non-Hispanic. Eighty-three percent of patients had at least one high risk genetic attribute including 43% del(17p), 45% TP53 mutation, 45% unmutated IGHV, and 62% with complex karyotype. Fifty-one percent of the patients had bulky disease. The median number of prior therapies was 5 (range: 2 to 12). All 65 patients were exposed to a BTK inhibitor, of which 88% were refractory, 1.5% were relapsed, and 11% were intolerant. Of 65 patients who received a BCL-2 inhibitor, 92% were refractory, none relapsed, and 6% were intolerant. A total of 83% had disease refractory to last therapy.
Efficacy was based on ORR (including CR and PR) and DOR as determined by an IRC using 2018 International Workshop CLL (iwCLL) criteria (Tables 23 and 24). The median time to first response (CR or PR) was 1.1 months (range: 0.8 to 17.4 months). The median time to first CR was 3.0 months (range 1.1 to 17.9 months).
Table 23: Response Rate in Relapsed or Refractory CLL/SLL (Study 4) CI=confidence interval; iwCLL=international workshop on chronic lymphocytic leukemia; IRC=independent review committee. a Per the 2018 iwCLL criteria, as assessed by IRC. b 2-sided 95% exact Clopper-Pearson CIs. Outcome
BREYANZI Treated
(N=65)
All Leukapheresed
(N=113)
Overall Response Ratea, n (%)
29 (45)
42 (37)
[95% CI]b
[32.3, 57.5]
[28.3, 46.8]
Complete Responsea, n (%)
13 (20)
16 (14)
[95% CI]b
[11.1, 31.8]
[8.3, 22.0]
Partial Responsea, n (%)
16 (25)
26 (23)
[95% CI]b
[14.8, 36.9]
[15.6, 31.9]
Table 24: Duration of Response in Study 4 CI=confidence interval; NR=not reached. a Per the 2018 iwCLL criteria, as assessed by IRC. b Kaplan-Meier method was used to obtain 2-sided 95% CIs. + Indicates a censored value. BREYANZI-Treated
N=65
Number of Responders
29
DORa (months)
Median [95% CI]b
35.3 [12.4, NR]
Range
2.0+, 35.3
DOR if Best Response is CRa(months)
N=13
Median [95% CI]b
NR [15.0, NR]
Range
2.0+, 30.0+
Rate at 12 months (%) [95% CI]b
100 [NR, NR]
Rate at 18 months (%) [95% CI]b
87.5 [38.7, 98.1]
DOR if Best response is PRa(months)
N=16
Median [95% CI]b
12.4 [8.9, NR]
Range
4.1, 35.3
Rate at 12 months (%) [95% CI]b
60.3 [32.0, 79.8]
Rate at 18 months (%) [95% CI]b
46.9 [21.4, 68.9]
Minimal residual disease (MRD) negative status was defined as less than one CLL cell per 104 leukocytes using ClonoSEQ, a next generation sequencing assay (NGS) at any time post infusion (MRD‑negativity rate). There was no statistical testing of MRD-negativity rate. In patients who achieved CR, the MRD-negativity rate was 100% (13/13, 95% CI: 75.3, 100) in peripheral blood and 92.3% (12/13, 95% CI: 64, 99.8) in the bone marrow.
14.3 Relapsed or Refractory Follicular Lymphoma
The efficacy of BREYANZI was evaluated in a Phase 2, open-label, multicenter, single-arm trial (Study 5 ‑ FL Cohort: JCAR017-FOL-001; NCT04245839) in adult patients with relapsed or refractory FL after two or more lines of systemic therapy (including an anti-CD20 antibody and an alkylating agent). The study enrolled patients with ECOG performance status of ≤ 1. The study excluded patients with a creatinine clearance of ≤ 30 mL/min, ALT > 5 times the ULN, or LVEF < 40%. There was no prespecified threshold for blood counts; patients were eligible to enroll if they were assessed by the investigator to have adequate bone marrow function to receive lymphodepleting chemotherapy. The planned dose of BREYANZI was 100 × 106 CAR-positive viable T cells. Bridging therapy for disease control was permitted between apheresis and the start of lymphodepleting chemotherapy.
A single dose of BREYANZI was administered 2 to 7 days following completion of lymphodepleting chemotherapy. The lymphodepleting chemotherapy regimen consisted of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day concurrently for 3 days. Six percent of patients were treated with BREYANZI in outpatient setting.
Of 114 patients who underwent leukapheresis, 107 received BREYANZI and the median dose administered was 100.02 x 106 CAR-positive viable T-cells (range: 93.4 to 109.2 x 106 CAR-positive viable T cells).The primary efficacy analysis included 94 patients who had PET-positive disease at study baseline or confirmation of PET-positive disease after bridging therapy, received conforming product in intended dose range and had at least 9 months of follow up from the date of first response.
The median time from leukapheresis to product availability was 29 days (range: 20 to 55 days), and the median time from leukapheresis to product infusion was 50 days (range: 31 to 313 days). Of the 107 patients treated with BREYANZI, 40 (40%) received bridging therapy.
The median age was 63 years (range: 23 to 80 years), 62% were male, ECOG performance status was 0 in 63% and 1 in 37% of patients; 55% were White, 3% were Black, 9% were Asian; 5% were Hispanic and 69% were non-Hispanic. The median number of prior systemic therapies was 3 (range: 2 to 10), with 46% receiving 2 prior lines, 22% receiving 3 prior lines and 32% receiving ≥ 4 prior lines. Twenty percent had received rituximab with lenalidomide, 25% had received a PI3K inhibitor, and 29% of patients had prior autologous HSCT. Eighty-nine percentage patients had Stage III-IV disease at study entry, 29% had bulky disease, 64% had progression within 6 months of the most recent regimen, and 50% had progression within 24 months of initial diagnosis (POD24).
Efficacy was based on ORR, defined as the percentage of patients with a best overall response (BOR) of CR or PR after BREYANZI infusion and duration of response as determined by an IRC (Tables 25 and 26). The median time to first response (CR or PR) was 1.0 month (range: 0.6 to 3.3 months). The median time to first CR was 3.0 months (range 0.6 to 18.0 months).
Table 25: Response Rate in Patients with Relapsed or Refractory FL CI=confidence interval. a Evaluable for efficacy. b Per the Lugano criteria, as assessed by an IRC. c Two-sided 95% confidence interval based on exact Clopper-Pearson method. d CR required a negative bone marrow biopsy after treatment in patients who did not have a negative bone marrow biopsy between their most recent disease progression and prior to initiation of lymphodepleting chemotherapy. BREYANZI‑Treateda
(N=94)
All Leukapheresed
(N=114)
Overall Response Rateb, n (%)
90 (95.7)
105 (92.1)
[95% CI]c
[89.5, 98.8)
[85.5, 96.3]
Complete Responsed, n (%)
69 (73.4)
78 (68.4)
[95% CI]
[63.3, 82.0]
[59.1, 76.8)
Partial Response, n (%)
21 (22.3)
27 (23.7)
[95% CI]
[14.4, 32.1]
[16.2, 32.6]
Table 26: Duration of Responsea in Patients with Relapsed or Refractory FL DOR= duration of response; CI=confidence interval; CR=complete response; PR=partial response; NR=not reached. a Median follow-up for DOR is 16.8 months (95% CI: 16.3 to 17.0). b Evaluable for efficacy. c Median [95% CI] are estimated from KM estimates. d KM estimate of probability of continued response at the specified month. + Indicates a censored value. BREYANZI Treatedb
N = 94
Number of Responders
90
DOR (months)
Median [95% CI]c
NR (18.0, NR)
Range
1.9, 23.1+
Rate at 12 months, (%) [95% CI]d
80.9 (71.0, 87.7)
Rate at 18 months, (%) [95% CI]d
77.1 (65.9, 85.0)
DOR if best response is CR (months)
N=69
Median [95% CI]c
NR [NR, NR]
Range
2.8, 23.1+
Rate at 12 months, (%) [95% CI]d
88.2 (77.7, 93.9)
Rate at 18 months, (%) [95% CI]d
83.1 (70.0, 90.9)
DOR if best response is PR (months)
N=21
Median [95% CI]c
18.04 [4.17, NR]
Range
1.9, 22.4+
Rate at 12 months, (%) [95% CI]d
56.7 (33.3, 74.7)
Rate at 18 months, (%) [95% CI]d
56.7 (33.3, 74.7)
14.4 Relapsed or Refractory Mantle Cell Lymphoma
The efficacy of BREYANZI was evaluated in an open-label, multicenter, single-arm trial (Study 3 - MCL Cohort: 017001; NCT02631044) in adult patients with relapsed or refractory MCL who had received at least two prior lines of therapy including a BTK inhibitor, an alkylating agent, and an anti-CD20 agent. The study included patients with ECOG performance status of ≤ 1, prior autologous and/or allogeneic HSCT, and secondary CNS lymphoma involvement. The study excluded patients with a creatinine clearance ≤ 30 mL/min, alanine aminotransferase > 5 times the upper limit of normal or left ventricular ejection fraction (LVEF) < 40%. There was no prespecified threshold for blood counts; patients were eligible to enroll if they were assessed by the investigator to have adequate bone marrow function to receive lymphodepleting chemotherapy. The planned dose of BREYANZI was 100 × 106 CAR-positive viable T cells. Bridging therapy for disease control was permitted between apheresis and the start of lymphodepleting chemotherapy.
BREYANZI was administered 2 to 7 days following completion of lymphodepleting chemotherapy. The lymphodepleting chemotherapy regimen consisted of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day concurrently for 3 days. BREYANZI was administered in the inpatient (85%) and outpatient (15%) setting.
Of 89 patients who underwent leukapheresis, 71 received BREYANZI and the median dose administered was 99.8 x 106 CAR-positive viable T cells (range: 90 to 103 x 106 CAR-positive viable T cells). The primary efficacy analysis included a total of 68 patients with MCL who received at least 2 prior lines of therapy including a BTK inhibitor, had PET-positive disease at study baseline or after bridging therapy, received conforming product in the intended dose range, and had at least 6 months of follow up from the date of first response.
The median time from leukapheresis to product availability was 24 days (range: 17 to 80 days), and the median time from leukapheresis to product infusion was 36 days (range: 28 to 489 days). Of the 68 patients in the main efficacy population, 44 (65%) received bridging therapy.
The median age was 69 years (range: 36 to 86 years); 75% were male, ECOG performance status was 0 in 57% and 1 in 43% of patients; 87% were White, 1.5% were Black, 6% were Asian; 6% were Hispanic and 91% were non-Hispanic. The median number of prior therapies was 3 (range: 2 to 11) and 32% had received prior HSCT. Twenty-nine percent of patients had blastoid morphology and 10% had CNS lymphoma involvement at baseline. Sixty-nine percent of patients had disease refractory to their last therapy for MCL, defined as best response of partial response (PR), stable disease (SD), or progressive disease (PD) to last systemic or HSCT treatment with curative intent. All 68 patients were exposed to BTK inhibitor, of which 56% were refractory, defined as any response to prior BTK inhibitor that was less than PR.
Efficacy was based on ORR, defined as the percentage of patients with BOR of either CR or PR after BREYANZI infusion, as determined by an IRC using 2014 Lugano classification. Other efficacy measures included complete response rate and DOR, as determined by IRC (Tables 27 and Table 28).
Both the median time to first response (CR or PR) and the median time to first CR were 1 month (range: 0.7 to 3 months).
Table 27: Response Rate in Relapsed or Refractory MCL (Study 3 - MCL Cohort) CI=confidence interval. a Evaluable for efficacy. b Per the 2014 Lugano classification (including bone marrow biopsy assessments), as assessed by IRC. c 2-sided 95% exact Clopper-Pearson CIs. Outcome
BREYANZI-Treateda
(N=68)
All Leukapheresed
(N=89)
Overall Response Rateb, n (%)
[95% CI]c
58 (85.3)
(74.6, 92.7)
65 (73.0)
(62.6, 81.9)
Complete Response, n (%)
[95% CI]
46 (67.6)
(55.2, 78.5)
51 (57.3)
(46.4, 67.7)
Partial Response, n (%)
[95% CI]
12 (17.6)
(9.5, 28.8)
14 (15.7)
(8.9, 25.0)
Table 28: Duration of Responsea in Study 3 - MCL Cohort CI=confidence interval; NE=not evaluable; NR=not reached.
a Median follow-up for DOR is 22.2 months (95% CI: 16.7 to 22.8).
b Per the 2014 Lugano classification (including bone marrow biopsy assessments), as assessed by IRC.
c Kaplan-Meier method was used to obtain 2-sided 95% CIs.
d KM estimate of probability of continued response at the specified month.
+ Indicates a censored value.BREYANZI-Treated
(N=68)
Number of Responders
58
DORb (months)
Median [95% CI]c
Range
13.3 [6.0, 23.3]
0.0+, 23.3+
Rate at 12 months (%) [95% CI]d
51.4 [37.5, 63.7]
Rate at 18 months (%) [95% CI]d
38.8 [25.0, 52.4]
DOR if Best Response is CRb (months)
Median [95% CI]c
Range
N=46
17.5 [7.5, NR]
0.6, 23.3+
Rate at 12 months (%) [95% CI]d
57.8 [41.9, 70.7]
Rate at 18 months (%) [95% CI]d
48.0 [31.6, 62.6]
DOR if Best Response is PRb (months)
Median [95% CI]c
Range
N=12
2.2 [1.8, 13.3]
0.0+, 14.5
Rate at 12 months (%) [95% CI]d
27.3 [6.5, 53.9]
Rate at 18 months (%) [95% CI]d
0.0 [NE, NE]
14.5 Relapsed or Refractory Marginal Zone Lymphoma
The efficacy of BREYANZI was evaluated in an open-label, multicenter, single-arm study (Study 5 – MZL Cohort: JCAR017-FOL-001; NCT04245839) in adult patients with relapsed or refractory MZL. The study enrolled patients who had received at least two or more lines of systemic therapy (including an anti-CD20 antibody and an alkylating agent) or who relapsed after hematopoietic stem cell transplant (HSCT). Confirmation of the presence of CD19-positive lymphoma on biopsy was required for patients with prior CD19-directed therapy. The indication for systemic treatment was based on investigator’s assessment. The study included patients with ECOG performance status of ≤ 1 and excluded patients with a creatinine clearance of ≤ 30 mL/min, ALT > 5 times the ULN, or LVEF < 40%. The study also excluded patients with transformed lymphoma, active or serious infections, active autoimmune disease requiring immunosuppressive therapy, or a history of a central nervous system disorder. There was no prespecified threshold for blood counts; patients were eligible to enroll if they were assessed by the investigator to have adequate bone marrow function to receive lymphodepleting chemotherapy.
The planned dose of BREYANZI was 100 × 106 CAR-positive viable T cells. Bridging therapy for disease control was permitted between apheresis and the start of lymphodepleting chemotherapy.
A single dose of BREYANZI was administered 2 to 7 days following completion of lymphodepleting chemotherapy. The lymphodepleting chemotherapy regimen consisted of fludarabine 30 mg/m2/day and cyclophosphamide 300 mg/m2/day concurrently for 3 days. BREYANZI was administered in the inpatient (79%) and outpatient (21%) setting.
Of 77 patients who underwent leukapheresis, 69 received lymphodepleting chemotherapy, 67 received BREYANZI and 2 patients received nonconforming product. Thirty-four (44%) of the 77 leukapheresed patients received bridging therapy. The most commonly used agents (>= 5%) included: anti-CD20 directed therapy (26%), steroids (17%), alkylating agents (18%), vincristine (9%), platinum chemotherapy (7%), Bruton tyrosine kinase inhibitors (10%), anthracycline (8%), and gemcitabine (5%). Eight patients did not receive lymphodepleting chemotherapy following leukapheresis due to progressive disease in 4 patients, failure to meet treatment criteria in 3 patients, and suicide in 1 patient.
The population demographics and disease characteristics of the 77 patients who were leukapheresed were as follows: median age was 64 years (range: 37 to 81 years), 61% were male, ECOG performance status was 0 in 53% and 1 in 47% of patients; 56% were White, 5% were Asian, 3% were Black; and 4% were Hispanic. Thirty-seven patients (48%) had nodal MZL, 19 (25%) patients had extranodal MZL/mucosa-associated lymphoid tissue (MALT), and 21 (27%) patients had splenic MZL. The median number of prior systemic therapies was 3 (range: 2 to 12), with 43% receiving 2 prior lines, 23% receiving 3 prior lines, and 34% receiving ≥ 4 prior lines. Seventy-nine percent had received bendamustine, 20% had received rituximab with lenalidomide, 9% had received prior zanubrutinib, and 14% of patients had prior autologous HSCT. Eighty-seven percent of patients had Stage III-IV disease at study entry, 23% had bulky disease, 35% had progression within 6 months of the most recent regimen, and 33% had progression within 24 months of initiation of first line chemoimmunotherapy with anti-CD20 and alkylating agent (POD24).
The median dose administered was 100.2 × 106 CAR-positive viable T-cells (range: 97.3 to 102.8 × 106 CAR-positive viable T cells). The primary efficacy analysis included 66 patients who had confirmed measurable disease by CT at study baseline or after bridging therapy and received conforming product in intended dose range and had at least 9 months of follow up from the date of first response.
The median time from leukapheresis to product availability was 29 days (range: 21 to 44 days), and the median time from leukapheresis to product infusion was 50 days (range: 30 to 121 days).
The main efficacy outcome measure was based on overall response rate (ORR), defined as the percentage of patients with the best overall response (BOR) of CR or PR after BREYANZI infusion, and duration of response as determined by an independent review committee (IRC). In the BREYANZI-treated patients, the median time to first response (CR or PR) was 0.95 months (range: 0.8 to 29.7 months). The median time to first CR was 5.55 months (range 0.9 to 29.7 months).
The efficacy results are summarized in Table 29 and Table 30 below.
Table 29: Response Rate in Patients with Relapsed or Refractory MZL CI=confidence interval. a Evaluable for efficacy. b Per the Lugano criteria (based on CT only), as assessed by an IRC. c Two-sided 95% confidence interval based on exact Clopper-Pearson method. d CR required a negative bone marrow biopsy after treatment in patients who did not have a negative bone marrow biopsy between their most recent disease progression and prior to initiation of lymphodepleting chemotherapy. e Leukapheresed patients includes patients who were leukapheresed but did not receive BREYANZI for the following reasons: Progressive disease (n=4); Failure to meet treatment criteria (n=3); Received nonconforming product (n=2); and Suicide (n=1). BREYANZI‑Treateda
(N=66)
All Leukapheresede
(N=77)
Overall Response Rateb, n (%)
[95% CI]c
63 (95.5)
65 (84.4)
[87.3, 99.1]
[74.4, 91.7]
Complete Responsed, n (%)
[95% CI]
41 (62.1)
[49.3, 73.8]
43 (55.8)
[44.1, 67.2]
Partial Response, n (%)
[95% CI]
22 (33.3)
[22.2, 46.0]
22 (28.6)
[18.8, 40.0]
Table 30: Duration of Responsea in Patients with Relapsed or Refractory MZL DOR= duration of response; CI=confidence interval; CR=complete response; PR=partial response; NR=not reached. a Median follow-up for DOR is 21.59 months (95% CI: 17.28 to 22.77) for treated set and 22.31 months (95% CI: 17.28, 22.97) for Leukapheresed Set. b Evaluable for efficacy. c Median [95% CI] are estimated from KM estimates. + Indicates a censored value. d KM estimate of probability of continued response at the specified month. BREYANZI-Treatedb
(N = 66)
Number of Responders
63
DOR (months)
Median [95% CI]c
NR [25.59, NR]
Range
0.0+, 35.3+
Rate at 12 months, (%) [95% CI]d
96.7 (87.3, 99.2)
Rate at 24 months, (%) [95% CI]d
90.1 (73.1, 96.6)
DOR if best response is CR (months)
N=41
Median [95% CI]c
NR [24.48, NR]
Range
0.0+, 35.3+
Rate at 12 months, (%) [95% CI]d
94.9 (81.2, 98.7)
Rate at 24 months, (%) [95% CI]d
89.0 (66.6, 96.7)
DOR if best response is PR (months)
N=22
Median [95% CI]c
25.59 [25.59, NR]
Range
2.0+, 32.8+
Rate at 12 months, (%) [95% CI]d
100 (100.0, 100.0)
Rate at 24 months, (%) [95% CI]d
93.3 (61.3, 99.0)
In subgroup analysis, overall response rate per IRC was 94.1% (95% CR: 71.3, 99.9) with a CR rate of 47.1% (95% CI: 23.0, 72.2) among 17 patients with extranodal marginal zone lymphoma; 96.9% (95% CI: 83.8, 99.9) with a CR rate of 62.5% (95% CI: 43.7, 78.9) among 32 patients with nodal marginal zone lymphoma; and 94.1% (95% CI 71.3, 99.9) with a CR rate of 76.5% (95% CI: 50.1, 93.2) among 17 patients with splenic marginal zone lymphoma.
- 15 REFERENCES
-
16 HOW SUPPLIED/STORAGE AND HANDLING
BREYANZI consists of genetically modified autologous T cells, supplied in vials as separate frozen suspensions of each CD8 component (NDC 73153-901-08) and CD4 component (NDC 73153-902-04). Each CD8 or CD4 component is packed in a carton containing up to 4 vials, depending upon the concentration of the cryopreserved drug product CAR-positive viable T cells. The cartons for each CD8 component and CD4 component are in an outer carton (NDC 73153-900-01). BREYANZI is shipped directly to the cell lab or clinical pharmacy associated with the infusion center in the vapor phase of a liquid nitrogen shipper. A Release for Infusion (RFI) Certificate for each component and patient-specific syringe labels are affixed inside the shipper.
- •
- Confirm patient identity upon receipt.
- •
- Store vials in the vapor phase of liquid nitrogen (less than or equal to minus 130°C) in a temperature-monitored system.
- •
- Thaw BREYANZI prior to infusion [see Dosage and Administration (2.2)].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Ensure that patients understand the risk (11%) of manufacturing failure. In case of a manufacturing failure, a second manufacturing of BREYANZI may be attempted. While the patient awaits the product, additional bridging therapy (not the lymphodepletion) may be necessary. This bridging therapy may be associated with adverse events during the pre-infusion period, which could delay or prevent the administration of BREYANZI.
Advise patients that they will be monitored daily for at least 7 days following the BREYANZI infusion and instruct patients to remain close to a healthcare facility for at least 2 weeks following the infusion.
Prior to infusion, advise patients of the following risks:
- •
- Cytokine Release Syndrome (CRS) – Signs and symptoms of CRS (fever, chills, hypotension, tachycardia, hypoxia, and fatigue). Counsel patients to seek immediate medical attention should signs or symptoms of CRS occur at any time [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
- •
- Neurologic Toxicities – Signs or symptoms associated with neurologic events including encephalopathy, confusion, decreased consciousness, speech disorders, tremor, and seizures. Counsel patients to seek immediate medical attention should signs or symptoms of neurologic toxicity occur at any time [see Warnings and Precautions (5.2) and Adverse Reactions (6.1)].
- •
- Serious Infections – Signs or symptoms associated with infection [see Warnings and Precautions (5.4) and Adverse Reactions (6.1)].
- •
- Prolonged Cytopenias – Signs or symptoms associated with bone marrow suppression including neutropenia, anemia, thrombocytopenia, or febrile neutropenia [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
- •
- Secondary Malignancies: Secondary malignancies, including T cell malignancies, have occurred [see Boxed Warning, Warnings and Precautions (5.7), Adverse Reactions (6.2)].
Advise patients of the need to:
- •
- Contact Bristol-Myers Squibb at 1-888-805-4555 if they are diagnosed with a secondary malignancy [see Warnings and Precautions (5.7)].
- •
- Avoid driving for at least 2 weeks after BREYANZI administration.
-
SPL UNCLASSIFIED SECTION
Manufactured by:
Juno Therapeutics Inc., a Bristol-Myers Squibb Company, Bothell, WA 98021. US License No.: 2156.
Celgene Corporation, a Bristol-Myers Squibb Company, Summit, NJ 07901. US License No.: 2252.
Bristol-Myers Squibb Company, Devens, MA 01434. US License No.: 1713.
BREYANZI® is a trademark of Juno Therapeutics, Inc., a Bristol-Myers Squibb Company.
Pat. https://www.bms.com/patient-and-caregivers/our-medicines.html
BREPI.010/MG.009
-
MEDICATION GUIDE
MEDICATION GUIDE
BREYANZI® (pronounced braye an' zee)
(lisocabtagene maraleucel)This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: December 2025 Read this Medication Guide before you start your BREYANZI treatment. The more you know about your treatment, the more active you can be in your care. Talk with your healthcare provider if you have questions about your health condition or treatment. Reading this Medication Guide does not take the place of talking with your healthcare provider about your treatment.
What is the most important information I should know about BREYANZI?
BREYANZI may cause side effects that are life-threatening and can lead to death. Call your healthcare provider or get emergency help right away if you get any of the following:- •
- difficulty breathing
- •
- fever (100.4°F/38°C or higher)
- •
- chills/shaking chills
- •
- confusion
- •
- severe nausea, vomiting, diarrhea
- •
- fast or irregular heartbeat
- •
- dizziness/lightheadedness
- •
- severe fatigue or weakness
It is important that you tell your healthcare providers that you have received BREYANZI and to show them your BREYANZI Patient Wallet Card. Your healthcare provider may give you other medicines to treat your side effects.
What is BREYANZI?
BREYANZI is a prescription medicine used to treat five types of non-Hodgkin lymphoma:- •
- Large B cell lymphoma, when:
- o
- your first treatment has not worked or your cancer returned within a year of your first treatment OR
- o
- your first treatment has not worked or your cancer returned after the first treatment, and you are not eligible for hematopoietic stem cell transplantation because of medical conditions or age OR
- o
- two or more kinds of treatment have not worked or stopped working.
- •
- Chronic lymphocytic leukemia or small lymphocytic lymphoma when two or more kinds of treatment have not worked or stopped working.
- •
- Follicular lymphoma, when two or more kinds of treatment have not worked or stopped working.
- •
- Mantle cell lymphoma when two or more kinds of treatment have not worked or stopped working, including a prior Bruton tyrosine kinase (BTK) inhibitor medicine.
- •
- Marginal zone lymphoma when two or more kinds of treatment have not worked or stopped working.
BREYANZI is different than other cancer medicines because it is made from your own white blood cells, which have been genetically modified to recognize and attack your lymphoma cells.
Before getting BREYANZI, tell your healthcare provider about all your medical problems, including if you have or have had:
- •
- Neurologic problems (such as seizures, stroke, or memory loss)
- •
- Lung or breathing problems
- •
- Heart problems
- •
- Liver problems
- •
- Kidney problems
- •
- A recent or active infection
Tell your healthcare provider about all the medications you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How will I receive BREYANZI?
- •
- BREYANZI is made from your own white blood cells, so your blood will be collected by a process called “leukapheresis” (LOO-kuh-feh-REE-sis).
- •
- It takes about 3-4 weeks from the time your cells are received at the manufacturing site and are available to be shipped back to your healthcare provider, but the time may vary.
- •
- Before you get BREYANZI, you will get 3 days of chemotherapy to prepare your body.
- •
- When your BREYANZI is ready, your healthcare provider will give it to you through a catheter (tube) placed into your vein (intravenous infusion). BREYANZI is given as infusions of 2 different cell types.
- o
- You will receive infusions of one cell type, immediately followed by the other cell type.
- o
- The time for infusion will vary but will usually be less than 15 minutes for each of the 2 cell types.
- •
- During the first week, you will be monitored daily.
- •
- You should plan to stay close to a healthcare facility for at least 2 weeks after getting BREYANZI. Your healthcare provider will check to see that your treatment is working and help you with any side effects that may occur.
- •
- You may be hospitalized for side effects and your healthcare provider will discharge you if your side effects are under control, and it is safe for you to leave the hospital.
- •
- Your healthcare provider will want to do blood tests to follow your progress. It is important that you do have your blood tested. If you miss an appointment, call your healthcare provider as soon as possible to reschedule.
What should I avoid after receiving BREYANZI?
- •
- Avoid driving for at least 2 weeks after you get BREYANZI.
- •
- Do not donate blood, organs, tissues, or cells for transplantation.
What are the possible or reasonably likely side effects of BREYANZI?
The most common side effects of BREYANZI are:
- •
- fatigue
- •
- difficulty breathing
- •
- fever (100.4°F/38°C or higher)
- •
- chills/shaking chills
- •
- confusion
- •
- difficulty speaking or slurred speech
- •
- severe nausea, vomiting, diarrhea
- •
- headache
- •
- dizziness/lightheadedness
- •
- fast or irregular heartbeat
- •
- swelling
- •
- low blood pressure
- •
- muscle pain
BREYANZI can increase the risk of life-threatening infections that may lead to death. Tell your healthcare provider right away if you develop fever, chills, or any signs or symptoms of an infection.
BREYANZI can lower one or more types of your blood cells (red blood cells, white blood cells, or platelets). After treatment, your healthcare provider will test your blood to check for this. Tell your healthcare provider right away if you get a fever, are feeling tired, or have bruising or bleeding.BREYANZI may increase your risk of getting cancers including certain types of blood cancers. Your healthcare provider should monitor you for this.
Having BREYANZI in your blood may cause a false-positive HIV test result by some commercial tests.
These are not all the possible side effects of BREYANZI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.General information about the safe and effective use of BREYANZI
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. If you would like more information about BREYANZI, talk with your healthcare provider. You can ask your healthcare provider for information about BREYANZI that is written for health professionals.
For more information, go to BREYANZI.com or call 1-888-805-4555.
Manufactured by:Juno Therapeutics Inc., a Bristol-Myers Squibb Company, Bothell, WA 98021. US License No.: 2156.
Celgene Corporation, a Bristol-Myers Squibb Company, Summit, NJ 07901. US License No.: 2252.
Bristol-Myers Squibb Company, Devens, MA 01434. US License No.: 1713.
BREYANZI® is a trademark of Juno Therapeutics, Inc., a Bristol-Myers Squibb Company.
Pat. https://www.bms.com/patient-and-caregivers/our-medicines.html
BREMG.009 12/2025 -
PRINCIPAL DISPLAY PANEL - Kit Carton
lisocabtagene maraleucel
Breyanzi™lisocabtagene maraleucel
Breyanzi®NDC 73153-900-01
Rx onlyFOR AUTOLOGOUS & INTRAVENOUS USE ONLY
Dispense with Medication Guide
Dosage: See Release for Infusion Certificate (inside shipper).
Contains DMSO 7.5% v/v.
1 to 4 single-dose vials/component. Each vial
contains up to 70 x 106 CAR-positive viable T cells/mL.
4.6 mL per vial.Store at ≤ -130°C; vapor phase of liquid nitrogen.
Do not filter or irradiate.
Not evaluated for infectious substances.
Discard unused portion.Manufactured by:
Juno Therapeutics, Inc., a Bristol-Myers Squibb Company
Bothell, WA 98021
Phone: 1-888-805-4555
US License #: 2156304029
NDC 73153-900-01
VERIFY
PATIENT
IDFirst: FIRST NAME
Last: LAST NAME
Date of birth: DD-MMM-YYYY
JOIN: XXXX-XXXXX
Aph ID/DIN: XXX XXX XXX XXXCD8
componentCD4
componentLot
XXXX-XXXXXY
XXXX-XXXXXYExpiration
DD-MMM-YYYY
DD-MMM-YYYYSTOP
Confirm patient ID
prior to infusion -
INGREDIENTS AND APPEARANCE
BREYANZI
lisocabtagene maraleucel kitProduct Information Product Type CELLULAR THERAPY Item Code (Source) NDC:73153-900 Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:73153-900-01 1 in 1 CARTON; Type 0: Not a Combination Product Quantity of Parts Part # Package Quantity Total Product Quantity Part 1 1 VIAL, SINGLE-DOSE 5 mL Part 2 1 VIAL, SINGLE-DOSE 5 mL Part 1 of 2 BREYANZI (CD4)
lisocabtagene maraleucel injection, suspensionProduct Information Item Code (Source) NDC:73153-902 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LISOCABTAGENE MARALEUCEL (UNII: 7K2YOJ14X0) (LISOCABTAGENE MARALEUCEL - UNII:7K2YOJ14X0) LISOCABTAGENE MARALEUCEL 50000000 in 5 mL Inactive Ingredients Ingredient Name Strength DIMETHYL SULFOXIDE (UNII: YOW8V9698H) ALBUMIN HUMAN (UNII: ZIF514RVZR) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:73153-902-04 5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125714 02/05/2021 Part 2 of 2 BREYANZI (CD8)
lisocabtagene maraleucel injection, suspensionProduct Information Item Code (Source) NDC:73153-901 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength LISOCABTAGENE MARALEUCEL (UNII: 7K2YOJ14X0) (LISOCABTAGENE MARALEUCEL - UNII:7K2YOJ14X0) LISOCABTAGENE MARALEUCEL 50000000 in 5 mL Inactive Ingredients Ingredient Name Strength DIMETHYL SULFOXIDE (UNII: YOW8V9698H) ALBUMIN HUMAN (UNII: ZIF514RVZR) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:73153-901-08 5 mL in 1 VIAL, SINGLE-DOSE; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125714 02/05/2021 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date BLA BLA125714 02/05/2021 Labeler - Juno Therapeutics, Inc. (079290042)