Label: REGLAN- metoclopramide hydrochloride tablet

-
Contains inactivated NDC Code(s)
NDC Code(s): 16590-906-30, 16590-906-60, 16590-906-90 - Packager: STAT RX USA LLC
- This is a repackaged label.
- Source NDC Code(s): 68220-151
- Category: HUMAN PRESCRIPTION DRUG LABEL
Drug Label Information
Updated August 31, 2010
If you are a healthcare professional or from the pharmaceutical industry please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
- SPL UNCLASSIFIED SECTION
-
BOXED WARNING
(What is this?)
WARNING: TARDIVE DYSKINESIA
Treatment with metoclopramide can cause tardive dyskinesia, a serious movement disorder that is often irreversible. The risk of developing tardive dyskinesia increases with duration of treatment and total cumulative dose.
Metoclopramide therapy should be discontinued in patients who develop signs or symptoms of tardive dyskinesia. There is no known treatment for tardive dyskinesia. In some patients, symptoms may lessen or resolve after metoclopramide treatment is stopped.
Treatment with metoclopramide for longer than 12 weeks should be avoided in all but rare cases where therapeutic benefit is thought to outweigh the risk of developing tardive dyskinesia.
See WARNINGS
-
DESCRIPTION
For oral administration, reglan® tablets (metoclopramide tablets, USP) 10 mg are white, scored, capsule-shaped tablets engraved "REGLAN" on one side and "AP 10" on the opposite side.
Each tablet contains:
Metoclopramide base .................................................. 10 mg
(as the monohydrochloride monohydrate)Inactive Ingredients
Magnesium Stearate, Mannitol, Microcrystalline Cellulose, Stearic Acid.reglan® tablets (metoclopramide tablets, USP) 5 mg are green, elliptical-shaped tablets engraved "REGLAN" over "5" on one side and "AP" on the opposite side.
Each tablet contains:
Metoclopramide base .................................................... 5 mg
(as the monohydrochloride monohydrate)Inactive Ingredients
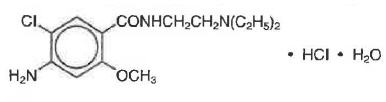
Corn starch, D&C Yellow 10 Aluminum Lake, FD&C Blue 1 Aluminum Lake, Lactose, Microcrystalline Cellulose, Silicon Dioxide, Stearic Acid.Metoclopramide hydrochloride is a white crystalline, odorless substance, freely soluble in water. Chemically, it is 4-amino-5-chloro-N-[2-(diethylamino)ethyl]-2-methoxy benzamide monohydrochloride monohydrate. Its molecular formula is C14H22ClN3O2•HCl•H2O. Its molecular weight is 354.3.
-
CLINICAL PHARMACOLOGY
Metoclopramide stimulates motility of the upper gastrointestinal tract without stimulating gastric, biliary, or pancreatic secretions. Its mode of action is unclear. It seems to sensitize tissues to the action of acetylcholine. The effect of metoclopramide on motility is not dependent on intact vagal innervation, but it can be abolished by anticholinergic drugs.
Metoclopramide increases the tone and amplitude of gastric (especially antral) contractions, relaxes the pyloric sphincter and the duodenal bulb, and increases peristalsis of the duodenum and jejunum resulting in accelerated gastric emptying and intestinal transit. It increases the resting tone of the lower esophageal sphincter. It has little, if any, effect on the motility of the colon or gallbladder.
In patients with gastroesophageal reflux and low LESP (lower esophageal sphincter pressure), single oral doses of metoclopramide produce dose-related increases in LESP. Effects begin at about 5 mg and increase through 20 mg (tile largest dose tested). The increase in LESP from a 5 mg dose lasts about 45 minutes and that of 20 mg lasts between 2 and 3 hours. Increased rate of stomach emptying has been observed with single oral doses of 10 mg.
The antiemetic properties of metoclopramide appear to be a result of its antagonism of central and peripheral dopamine receptors. Dopamine produces nausea and vomiting by stimulation of the medullary chemoreceptor trigger zone (CTZ), and metoclopramide blocks stimulation of the CTZ by agents like l-dopa or apomorphine which are known to increase dopamine levels or to possess dopamine-like effects. Metoclopramide also abolishes the slowing of gastric emptying caused by apomorphine.
Like the phenothiazines and related drugs, which are also dopamine antagonists, metoclopramide produces sedation and may produce extrapyramidal reactions, although these are comparatively rare (see WARNINGS). Metoclopramide inhibits the central and peripheral effects of apomorphine, induces release of prolactin and causes a transient increase in circulating aldosterone levels, which may be associated with transient fluid retention.
The onset of pharmacological action of metoclopramide is 1 to 3 minutes following an intravenous dose, 10 to 15 minutes following intramuscular administration, and 30 to 60 minutes following an oral dose; pharmacological effects persist for 1 to 2 hours.
Pharmacokinetics
Metoclopramide is rapidly and well absorbed. Relative to an intravenous dose of 20 mg, the absolute oral bioavailability of metoclopramide is 80% ± 15.5% as demonstrated in a crossover study of 18 subjects. Peak plasma concentrations occur at about 1 to 2 hr after a single oral dose. Similar time to peak is observed after individual doses at steady state.
In a single dose study of 12 subjects, the area under the drug concentration-time curve increases linearly with doses from 20 to 100 mg. Peak concentrations increase linearly with dose; time to peak concentrations remains the same; whole body clearance is unchanged; and the elimination rate remains the same. The average elimination half-life in individuals with normal renal function is 5 to 6 hr. Linear kinetic processes adequately describe the absorption and elimination of metoclopramide.
Approximately 85% of the radioactivity of an orally administered dose appears in the urine within 72 hr. Of the 85% eliminated in the urine, about half is present as free or conjugated metoclopramide.
The drug is not extensively bound to plasma proteins (about 30%). The whole body volume of distribution is high (about 3.5 L/kg) which suggests extensive distribution of drug to the tissues.
Renal impairment affects the clearance of metoclopramide. In a study with patients with varying degrees of renal impairment, a reduction in creatinine clearance was correlated with a reduction in plasma clearance, renal clearance, non-renal clearance, and increase in elimination half-life. The kinetics of metoclopramide in the presence of renal impairment remained linear however. The reduction in clearance as a result of renal impairment suggests that adjustment downward of maintenance dosage should be done to avoid drug accumulation.
Adult Pharmacokinetic Data Parameter Value Vd (Lkg) - 3.5 Plasma Protein Binding - 30% t1/2 (hr) 5 to 6 Oral Bioavailability 80% ± 15.5% In pediatric patients, the pharmacodynamics of metoclopramide following oral and intravenous administration are highly variable and a concentration-effect relationship has not been established.
There are insufficient reliable data to conclude whether the pharmacokinetics of metoclopramide in adults and the pediatric population are similar. Although there are insufficient data to support the efficacy of metoclopramide in pediatric patients with symptomatic gastroesophageal reflux (GER) or cancer chemotherapy-related nausea and vomiting, its pharmacokinetics have been studied in these patient populations.
In an open-label study, six pediatric patients (age range, 3.5 weeks to 5.4 months) with GER received metoclopramide 0.15 mg/kg oral solution every 6 hours for 10 doses. The mean peak plasma concentration of metoclopramide after the tenth dose was 2-fold (56.8 µg/L) higher compared to that observed after the first dose (29 µg/L) indicating drug accumulation with repeated dosing. After the tenth dose, the mean time to reach peak concentrations (2.2 hr), half-life (4.1 hr), clearance (0.67 L/h/kg), and volume of distribution (4.4 L/kg) of metoclopramide were similar to those observed after the first dose. In the youngest patient (age, 3.5 weeks), metoclopramide half-life after the first and the tenth dose (23.1 and 10.3 hr, respectively) was significantly longer compared to other infants due to reduced clearance. This may be attributed to immature hepatic and renal systems at birth.
Single intravenous doses of metoclopramide 0.22 to 0.46 mg/kg (mean, 0.35 mg/kg) were administered over 5 minutes to 9 pediatric cancer patients receiving chemotherapy (mean age, 11.7 years; range, 7 to 14 yr) for prophylaxis of cytotoxic-induced vomiting. The metoclopramide plasma concentrations extrapolated to time zero ranged from 65 to 395 µg/L (mean, 152 µg/L). The mean elimination half-life, clearance, and volume of distribution of metoclopramide were 4.4 hr (range, 1.7 to 8.3 hr), 0.56 L/h/kg (range, 0.12 to 1.20 L/h/kg), and 3.0 L/kg (range, 1.0 to 4.8 L/kg), respectively.
In another study, nine pediatric cancer patients (age range, 1 to 9 yr) received 4 to 5 intravenous infusions (over 30 minutes) of metoclopramide at a dose of 2 mg/kg to control emesis. After the last dose, the peak serum concentrations of metoclopramide ranged from 1060 to 5680 µg/L. The mean elimination half-life, clearance, and volume of distribution of metoclopramide were 4.5 hr (range, 2.0 to 12.5 hr), 0.37 L/h/kg (range, 0.10 to 1.24 L/h/kg), and 1.93 L/kg (range, 0.95 to 5.50 L/kg), respectively.
-
INDICATIONS AND USAGE
The use of reglan® tablets is recommended for adults only. Therapy should not exceed 12 weeks in duration.
Symptomatic Gastroesophageal Reflux
reglan® tablets are indicated as short-term (4 to 12 weeks) therapy for adults with symptomatic, documented gastroesophageal reflux who fail to respond to conventional therapy.
The principal effect of metoclopramide is on symptoms of postprandial and daytime heartburn with less observed effect on nocturnal symptoms. If symptoms are confined to particular situations, such as following the evening meal, use of metoclopramide as single doses prior to the provocative situation should be considered, rather than using the drug throughout the day. Healing of esophageal ulcers and erosions has been endoscopically demonstrated at the end of a 12-week trial using doses of 15 mg q.i.d. As there is no documented correlation between symptoms and healing of esophageal lesions, patients with documented lesions should be monitored endoscopically.
Diabetic Gastroparesis (Diabetic Gastric Stasis)
reglan® tablets (metoclopramide tablets, USP) is indicated for the relief of symptoms associated with acute and recurrent diabetic gastric stasis. The usual manifestations of delayed gastric emptying (e.g., nausea, vomiting, heartburn, persistent fullness after meals, and anorexia) appear to respond to reglan® within different time intervals. Significant relief of nausea occurs early and continues to improve over a three-week period. Relief of vomiting and anorexia may precede the relief of abdominal fullness by one week or more.
-
CONTRAINDICATIONS
Metoclopramide should not be used whenever stimulation of gastrointestinal motility might be dangerous, e.g., in the presence of gastrointestinal hemorrhage, mechanical obstruction, or perforation.
Metoclopramide is contraindicated in patients with pheochromocytoma because the drug may cause a hypertensive crisis, probably due to release of catecholamines from the tumor. Such hypertensive crises may be controlled by phentolamine.
Metoclopramide is contraindicated in patients with known sensitivity or intolerance to the drug.
Metoclopramide should not be used in epileptics or patients receiving other drugs which are likely to cause extrapyramidal reactions, since the frequency and severity of seizures or extrapyramidal reactions may be increased.
-
WARNINGS
Mental depression has occurred in patients with and without prior history of depression. Symptoms have ranged from mild to severe and have included suicidal ideation and suicide. Metoclopramide should be given to patients with a prior history of depression only if the expected benefits outweigh the potential risks.
Extrapyramidal symptoms, manifested primarily as acute dystonic reactions, occur in approximately 1 in 500 patients treated with the usual adult dosages of 30 to 40 mg/day of metoclopramide. These usually are seen during the first 24 to 48 hours of treatment with metoclopramide, occur more frequently in pediatric patients and adult patients less than 30 years of age and are even more frequent at higher doses. These symptoms may include involuntary movements of limbs and facial grimacing, torticollis, oculogyric crisis, rhythmic protrusion of tongue, bulbar type of speech, trismus, or dystonic reactions resembling tetanus. Rarely, dystonic reactions may present as stridor and dyspnea, possibly due to laryngospasm. If these symptoms should occur, inject 50 mg diphenhydramine hydrochloride intramuscularly, and they usually will subside. Benztropine mesylate, 1 to 2 mg intramuscularly, may also be used to reverse these reactions.
Parkinsonian-like symptoms have occurred, more commonly within the first 6 months after beginning treatment with metoclopramide, but occasionally after longer periods. These symptoms generally subside within 2 to 3 months following discontinuance of metoclopramide. Patients with preexisting Parkinson's disease should be given metoclopramide cautiously, if at all, since such patients may experience exacerbation of parkinsonian symptoms when taking metoclopramide.
Tardive Dyskinesia
(see Boxed Warnings)
Treatment with metoclopramide can cause tardive dyskinesia (TD), a potentially irreversible and disfiguring disorder characterized by involuntary movements of the face, tongue, or extremities. Although the risk of TD with metoclopramide has not been extensively studied, one published study reported a TD prevalence of 20% among patients treated for at least 12 weeks. Treatment with metoclopramide for longer than 12 weeks should be avoided in all but rare cases where therapeutic benefit is thought to outweigh the risk of developing TD.
Although the risk of developing TD in the general population may be increased among the elderly, women, and diabetics, it is not possible to predict which patients will develop metoclopramide-induced TD. Both the risk of developing TD and the likelihood that TD will become irreversible increase with duration of treatment and total cumulative dose.
Metoclopramide should be discontinued in patients who develop signs or symptoms of TD. There is no known effective treatment for established cases of TD, although in some patients, TD may remit, partially or completely, within several weeks to months after metoclopramide is withdrawn.
Metoclopramide itself may suppress, or partially suppress, the signs of TD, thereby masking the underlying disease process. The effect of this symptomatic suppression upon the long-term course of TD is unknown. Therefore, metoclopramide should not be used for the symptomatic control of TD.
Neuroleptic Malignant Syndrome (NMS)
There have been rare reports of an uncommon but potentially fatal symptom complex sometimes referred to as Neuroleptic Malignant Syndrome (NMS) associated with metoclopramide. Clinical manifestations of NMS include hyperthermia, muscle rigidity, altered consciousness, and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis and cardiac arrhythmias).
The diagnostic evaluation of patients with this syndrome is complicated. In arriving at a diagnosis, it is important to identify cases where the clinical presentation includes both serious medical illness (e.g., pneumonia, systemic infection, etc.) and untreated or inadequately treated extrapyramidal signs and symptoms (EPS). Other important considerations in the differential diagnosis include central anticholinergic toxicity, heat stroke, malignant hyperthermia, drug fever and primary central nervous system (CNS) pathology.
The management of NMS should include 1) immediate discontinuation of metoclopramide and other drugs not essential to concurrent therapy, 2) intensive symptomatic treatment and medical monitoring, and 3) treatment of any concomitant serious medical problems for which specific treatments are available. Bromocriptine and dantrolene sodium have been used in treatment of NMS, but their effectiveness have not been established (see ADVERSE REACTIONS).
-
PRECAUTIONS
General
In one study in hypertensive patients, intravenously administered metoclopramide was shown to release catecholamines; hence, caution should be exercised when metoclopramide is used in patients with hypertension.
Because metoclopramide produces a transient increase in plasma aldosterone, certain patients, especially those with cirrhosis or congestive heart failure, may be at risk of developing fluid retention and volume overload. If these side effects occur at any time during metoclopramide therapy, the drug should be discontinued.
Adverse reactions, especially those involving the nervous system, may occur after stopping the use of reglan®. A small number of patients may experience a withdrawal period after stopping reglan® that could include dizziness, nervousness, and/or headaches.
Information for Patients
The use of reglan® is recommended for adults only. Metoclopramide may impair the mental and/or physical abilities required for the performance of hazardous tasks such as operating machinery or driving a motor vehicle. The ambulatory patient should be cautioned accordingly.
For additional information, patients should be instructed to see the Medication Guide for reglan® tablets.
Drug Interactions
The effects of metoclopramide on gastrointestinal motility are antagonized by anticholinergic drugs and narcotic analgesics. Additive sedative effects can occur when metoclopramide is given with alcohol, sedatives, hypnotics, narcotics, or tranquilizers.
The finding that metoclopramide releases catecholamines in patients with essential hypertension suggests that it should be used cautiously, if at all, in patients receiving monoamine oxidase inhibitors.
Absorption of drugs from the stomach may be diminished (e.g., digoxin) by metoclopramide, whereas the rate and/or extent of absorption of drugs from the small bowel may be increased (e.g., acetaminophen, tetracycline, levodopa, ethanol, cyclosporine).
Gastroparesis (gastric stasis) may be responsible for poor diabetic control in some patients. Exogenously administered insulin may begin to act before food has left the stomach and lead to hypoglycemia. Because the action of metoclopramide will influence the delivery of food to the intestines and thus the rate of absorption, insulin dosage or timing of dosage may require adjustment.
Carcinogenesis, Mutagenesis, Impairment of Fertility
A 77-week study was conducted in rats with oral doses up to about 40 times the maximum recommended human daily dose. Metoclopramide elevates prolactin levels and the elevation persists during chronic administration. Tissue culture experiments indicate that approximately one-third of human breast cancers are prolactin-dependent in vitro, a factor of potential importance if the prescription of metoclopramide is contemplated in a patient with previously detected breast cancer. Although disturbances such as galactorrhea, amenorrhea, gynecomastia, and impotence have been reported with prolactin-elevating drugs, the clinical significance of elevated serum prolactin levels is unknown for most patients. An increase in mammary neoplasms has been found in rodents after chronic administration of prolactin-stimulating neuroleptic drugs and metoclopramide. Neither clinical studies nor epidemiologic studies conducted to date, however, have shown an association between chronic administration of these drugs and mammary tumorigenesis; the available evidence is too limited to be conclusive at this time.
An Ames mutagenicity test performed on metoclopramide was negative.
Pregnancy Category B
Reproduction studies performed in rats, mice and rabbits by the IV., I.M., S.C., and oral routes at maximum levels ranging from 12 to 250 times the human dose have demonstrated no impairment of fertility or significant harm to the fetus due to metoclopramide. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Metoclopramide is excreted in human milk. Caution should be exercised when metoclopramide is administered to a nursing mother.
Pediatric Use
Safety and effectiveness in pediatric patients have not been established (see OVERDOSAGE).
Care should be exercised in administering metoclopramide to neonates since prolonged clearance may produce excessive serum concentrations (see CLINICAL PHARMACOLOGY - Pharmacokinetics). In addition, neonates have reduced levels of NADH-cytochrome b5 reductase which, in combination with the aforementioned pharmacokinetic factors, make neonates more susceptible to methemoglobinemia (see OVERDOSAGE).
The safety profile of metoclopramide in adults cannot be extrapolated to pediatric patients. Dystonias and other extrapyramidal reactions associated with metoclopramlde are more common in the pediatric population than in adults. (See WARNINGS and ADVERSE REACTIONS - Extrapyramidal Reactions.)
Geriatric Use
Clinical studies of reglan® did not include sufficient numbers of subjects aged 65 and over to determine whether elderly subjects respond differently from younger subjects.
The risk of developing parkinsonian-like side effects increases with ascending dose. Geriatric patients should receive the lowest dose of reglan® that is effective. If parkinsonian-like symptoms develop in a geriatric patient receiving reglan®, reglan® should generally be discontinued before initiating any specific anti-parkinsonian agents (see WARNINGS and DOSAGE AND ADMINISTRATION - For the Relief of Symptomatic Gastroesophageal Reflux).
The elderly may be at greater risk for tardive dyskinesia (see WARNINGS - Tardive Dyskinesia).
Sedation has been reported in reglan® users. Sedation may cause confusion and manifest as oversedation in the elderly (see CLINICAL PHARMACOLOGY, PRECAUTIONS -Information for Patients and ADVERSE REACTIONS - CNS Effects).
reglan® is known to be substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in patients with impaired renal function (see DOSAGE AND ADMINISTRATION - Use in Patients with Renal or Hepatic Impairment).
For these reasons, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased renal function, concomitant disease, or other drug therapy in the elderly (see DOSAGE AND ADMINISTRATION - For the Relief of Symptomatic Gastroesophageal Reflux and Use in Patients with Renal or Hepatic Impairment).
Other Special Populations
Patients with NADH-cytochrome b5 reductase deficiency are at an increased risk of developing methemoglobinemia and/or sulfhemoglobinemia when metoclopramide is administered. In patients with G6PD deficiency who experience metoclopramide- induced methemoglobinemia, methylene blue treatment is not recommended (see OVERDOSAGE).
-
ADVERSE REACTIONS
In general, the incidence of adverse reactions correlates with the dose and duration of metoclopramide administration. The following reactions have been reported, although in most instances, data do not permit an estimate of frequency:
CNS Effects
Restlessness, drowsiness, fatigue, and lassitude occur in approximately 10% of patients receiving the most commonly prescribed dosage of 10 mg q.i.d. (see PRECAUTIONS). Insomnia, headache, confusion, dizziness, or mental depression with suicidal ideation (see WARNINGS) occur less frequently. The incidence of drowsiness is greater at higher doses. There are isolated reports of convulsive seizures without clearcut relationship to metoclopramide. Rarely, hallucinations have been reported.
Extrapyramidal Reactions (EPS)
Acute dystonic reactions, the most common type of EPS associated with metoclopramide, occur in approximately 0.2% of patients (1 in 500) treated with 30 to 40 mg of metoclopramide per day. Symptoms include involuntary movements of limbs, facial grimacing, torticollis, oculogyric crisis, rhythmic protrusion of tongue, bulbar type of speech, trismus, opisthotonus (tetanus-like reactions), and, rarely, stridor and dyspnea possibly due to laryngospasm; ordinarily these symptoms are readily reversed by diphenhydramine (see WARNINGS).
Parkinsonian-like symptoms may include bradykinesia, tremor, cogwheel rigidity, mask-like facies (see WARNINGS).
Tardive dyskinesia most frequently is characterized by involuntary movements of the tongue, face, mouth, or jaw, and sometimes by involuntary movements of the trunk and/or extremities; movements may be choreoathetotic in appearance (see WARNINGS).
Motor restlessness (akathisia) may consist of feelings of anxiety, agitation, jitteriness, and insomnia, as well as inability to sit still, pacing, foot tapping. These symptoms may disappear spontaneously or respond to a reduction in dosage.
Neuroleptic Malignant Syndrome
Rare occurrences of neuroleptic malignant syndrome (NMS) have been reported. This potentially fatal syndrome is comprised of the symptom complex of hyperthermia, altered consciousness, muscular rigidity, and autonomic dysfunction (see WARNINGS).
Endocrine Disturbances
Galactorrhea, amenorrhea, gynecomastia, impotence secondary to hyperprolactlnemia (see PRECAUTIONS). Fluid retention secondary to transient elevation of aldosterone (see CLINICAL PHARMACOLOGY).
Cardiovascular
Hypotension, hypertension, supraventricular tachycardia, bradycardia, fluid retention, acute congestive heart failure and possible AV block (see CONTRAINDICATIONS and PRECAUTIONS).
Gastrointestinal
Nausea and bowel disturbances, primarily diarrhea.
Hepatic
Rarely, cases of hepatotoxicity, characterized by such findings as jaundice and altered liver function tests, when metoclopramide was administered with other drugs with known hepatotoxic potential.
Renal
Urinary frequency and incontinence.
Hematologic
A few cases of neutropenia, leukopenia, or agranulocytosis, generally without clearcut relationship to metoclopramide. Methemoglobinemia, in adults and especially with overdosage in neonates (see OVERDOSAGE). Sulfhemoglobinemia in adults.
Allergic Reactions
A few cases of rash, urticaria, or bronchospasm, especially in patients with a history of asthma. Rarely, angioneurotic edema, including glossal or laryngeal edema.
Miscellaneous
Visual disturbances. Porphyria.
-
OVERDOSAGE
Symptoms of overdosage may include drowsiness, disorientation and extrapyramidal reactions. Anticholinergic or antiparkinson drugs or antihistamines with anticholinergic properties may be helpful in controlling the extrapyramidal reactions. Symptoms are self-limiting and usually disappear within 24 hours.
Hemodialysis removes relatively little metoclopramide, probably because of the small amount of the drug in blood relative to tissues. Similarly, continuous ambulatory peritoneal dialysis does not remove significant amounts of drug. It is unlikely that dosage would need to be adjusted to compensate for losses through dialysis. Dialysis is not likely to be an effective method of drug removal in overdose situations.
Unintentional overdose due to misadministration has been reported in infants and children with the use of metoclopramide oral solution. While there was no consistent pattern to the reports associated with these overdoses, events included seizures, extrapyramidal reactions, and lethargy.
Methemoglobinemia has occurred in premature and full-term neonates who were given overdoses of metoclopramide (1 to 4 mg/kg/day orally, intramuscularly or intravenously for 1 to 3 or more days). Methemoglobinemia can be reversed by the intravenous administration of methylene blue. However, methylene blue may cause hemolytic anemia in patients with G6PD deficiency, which may be fatal (see PRECAUTIONS-Other Special Populations).
-
DOSAGE AND ADMINISTRATION
Therapy with reglan® tablets should not exceed 12 weeks in duration.
For the Relief of Symptomatic Gastroesophageal Reflux
Administer from 10 mg to 15 mg reglan® (metoclopramide hydrochloride, USP) orally up to q.i.d. 30 minutes before each meal and at bedtime, depending upon symptoms being treated and clinical response (see CLINICAL PHARMACOLOGY and INDICATIONS AND USAGE). If symptoms occur only intermittently or at specific times of the day, use of metoclopramide in single doses up to 20 mg prior to the provoking situation may be preferred rather than continuous treatment. Occasionally, patients (such as elderly patients) who are more sensitive to the therapeutic or adverse effects of metoclopramide will require only 5 mg per dose.
Experience with esophageal erosions and ulcerations is limited, but healing has thus far been documented in one controlled trial using q.i.d. therapy at 15 mg/dose, and this regimen should be used when lesions are present, so long as it is tolerated (see ADVERSE REACTIONS). Because of the poor correlation between symptoms and endoscopic appearance of the esophagus, therapy directed at esophageal lesions is best guided by endoscopic evaluation.
Therapy longer than 12 weeks has not been evaluated and cannot be recommended.
For the Relief of Symptoms Associated with Diabetic Gastroparesis (Diabetic Gastric Stasis)
Administer 10 mg of metoclopramide 30 minutes before each meal and at bedtime for two to eight weeks, depending upon response and the likelihood of continued well-being upon drug discontinuation.
The initial route of administration should be determined by the severity of the presenting symptoms. If only the earliest manifestations of diabetic gastric stasis are present, oral administration of reglan® may be initiated. However, if severe symptoms are present, therapy should begin with metoclopramide injection (consult labeling of the injection prior to initiating parenteral administration).
Administration of metoclopramide injection up to 10 days may be required before symptoms subside, at which time oral administration may be instituted. Since diabetic gastric stasis is frequently recurrent, reglan® therapy should be reinstituted at the earliest manifestation.
Use in Patients with Renal or Hepatic Impairment
Since metoclopramide is excreted principally through the kidneys, in those patients whose creatinine clearance is below 40 mL/min, therapy should be initiated at approximately one-half the recommended dosage. Depending upon clinical efficacy and safety considerations, the dosage may be increased or decreased as appropriate.
See OVERDOSAGE section for information regarding dialysis.
Metoclopramide undergoes minimal hepatic metabolism, except for simple conjugation. Its safe use has been described in patients with advanced liver disease whose renal function was normal.
-
HOW SUPPLIED
Each white, capsule-shaped, scored reglan® tablet (metoclopramide tablets, USP) contains 10 mg metoclopramide base (as the monohydrochloride monohydrate). Available in:
Bottles of 100 tablets (NDC 68220-151-10)
Each green, elliptical-shaped reglan® tablet (metoclopramide tablets, USP) contains 5 mg metoclopramide base (as the monohydrochloride monohydrate). Available in:
Bottles of 100 tablets (NDC 68220-150-10)
- SPL UNCLASSIFIED SECTION
-
Medication Guide
REGLAN (REG-lan) Tablets
(metoclopramide tablets)Read the Medication Guide that comes with REGLAN before you start taking it and each time you get a refill. There may be new information. If you take another product that contains metoclopramide (such as REGLAN injection, REGLAN ODT, or metoclopramide oral syrup), you should read the Medication Guide that comes with that product. Some of the information may be different. This Medication Guide does not take the place of talking with your doctor about your medical condition or your treatment.
What is the most important information I should know about REGLAN?
REGLAN can cause serious side effects, including:
Abnormal muscle movements called tardive dyskinesia (TD). These movements happen mostly in the face muscles. You can not control these movements. They may not go away even after stopping REGLAN. There is no treatment for TD, but symptoms may lessen or go away over time after you stop taking REGLAN.
Your chances for getting TD go up:
- the longer you take REGLAN and the more REGLAN you take. You should not take REGLAN for more than 12 weeks.
- if you are older, especially if you are a woman
- if you have diabetes
It is not possible for your doctor to know if you will get TD if you take REGLAN.
Call your doctor right away if you get movements you can not stop or control, such as:
- lip smacking, chewing, or puckering up your mouth
- frowning or scowling
- sticking out your tongue
- blinking and moving your eyes
- shaking of your arms and legs
See the section "What are the possible side effects of REGLAN?" for more information about side effects.
What is REGLAN?
REGLAN is a prescription medicine used:
- in adults for 4 to 12 weeks to relieve heartburn symptoms with gastroesophageal reflux disease (GERD) when certain other treatments do not work. REGLAN relieves daytime heartburn and heartburn after meals. It also helps ulcers in the esophagus to heal.
- to relieve symptoms of slow stomach emptying in people with diabetes. REGLAN helps treat symptoms such as nausea, vomiting, heartburn, feeling full long after a meal, and loss of appetite. Not all these symptoms get better at the same time.
It is not known if REGLAN is safe and works in children.
Who should not take REGLAN?
Do not take REGLAN if you:
- have stomach or intestine problems that could get worse with REGLAN, such as bleeding, blockage or a tear in the stomach or bowel wall
- have an adrenal gland tumor called a pheochromocytoma
- are allergic to REGLAN or anything in it. See the end of this Medication Guide for a list of ingredients in REGLAN.
- take medicines that can cause uncontrolled movements, such as medicines for mental illness
- have seizures
What should I tell my doctor before taking REGLAN?
Tell your doctor about all your medical conditions, including if you have:
- depression
- Parkinson's disease
- high blood pressure
- kidney problems. Your doctor may start with a lower dose.
- liver problems or heart failure. REGLAN may cause your body to hold fluids.
- diabetes. Your dose of insulin may need to be changed.
- breast cancer
- you are pregnant or plan to become pregnant. It is not known if REGLAN will harm your unborn baby.
- you are breast-feeding. REGLAN can pass into breast milk and may harm your baby. Talk with your doctor about the best way to feed your baby if you take REGLAN.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements. REGLAN and some other medicines may interact with each other and may not work as well, or cause possible side effects. Do not start any new medicines while taking REGLAN until you talk with your doctor.
Especially tell your doctor if you take:
- another medicine that contains metoclopramide, such as REGLAN ODT, or metoclopramide oral syrup
- a blood pressure medicine
- a medicine for depression, especially an Monoamine Oxidase Inhibitor (MAOI)
- insulin
- a medicine that can make you sleepy, such as anti-anxiety medicine, sleep medicines, and narcotics.
If you are not sure if your medicine is one listed above, ask your doctor or pharmacist.
Know the medicines you take. Keep a list of them and show it to your doctor and pharmacist when you get a new medicine.
How should I take REGLAN?
- REGLAN comes as a tablet you take by mouth.
- Take REGLAN exactly as your doctor tells you. Do not change your dose unless your doctor tells you.
- You should not take REGLAN for more than 12 weeks.
- If you take too much REGLAN, call your doctor or Poison Control Center right away.
What should I avoid while taking REGLAN?
- Do not drink alcohol while taking REGLAN. Alcohol may make some side effects of REGLAN worse, such as feeling sleepy.
- Do not drive, work with machines, or do dangerous tasks until you know how REGLAN affects you. REGLAN may cause sleepiness.
What are the possible side effects of REGLAN?
Reglan can cause serious side effects, including:
- Abnormal muscle movements. See "What is the most important information I need to know about REGLAN?"
- Uncontrolled spasms of your face and neck muscles, or muscles of your body, arms, and legs (dystonia). These muscle spasms can cause abnormal movements and body positions. These spasms usually start within the first 2 days of treatment. These spasms happen more often in children and adults under age 30.
- Depression, thoughts about suicide, and suicide. Some people who take REGLAN become depressed. You may have thoughts about hurting or killing yourself. Some people who take Reglan have ended their own lives (suicide).
- Neuroleptic Malignant Syndrome (NMS). NMS is a very rare but very serious condition that can happen with Reglan. NMS can cause death and must be treated in a hospital. Symptoms of NMS include: high fever, stiff muscles, problems thinking, very fast or uneven heartbeat, and increased sweating.
- Parkinsonism. Symptoms include slight shaking, body stiffness, trouble moving or keeping your balance. If you already have Parkinson's disease, your symptoms may become worse while you are receiving REGLAN.
Call your doctor and get medical help right away if you:
- feel depressed or have thoughts about hurting or killing yourself
- have high fever, stiff muscles, problems thinking, very fast or uneven heartbeat, and increased sweating
- have muscle movements you cannot stop or control
- have muscle movements that are new or unusual
Common side effects of Reglan include:
- feeling restless, sleepy, tired, dizzy, or exhausted
- headache
- confusion
- trouble sleeping
You may have more side effects the longer you take REGLAN and the more REGLAN you take. You may still have side effects after stopping REGLAN. You may have symptoms from stopping (withdrawal) REGLAN such as headaches, and feeling dizzy or nervous.
Tell your doctor about any side effects that bother you or do not go away. These are not all the possible side effects of REGLAN.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store REGLAN?
- Keep REGLAN at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep REGLAN in the bottle it comes in. Keep the bottle closed tightly.
Keep REGLAN and all medicines out of the reach of children.
General information about REGLAN
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use REGLAN for a condition for which it was not prescribed. Do not give REGLAN to other people, even if they have the same symptoms that you have. It may harm them.
This Medication Guide summarizes the most important information about REGLAN. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about REGLAN that is written for health professionals. For more information, go to www.alavenpharm.com or call 1-888-317-0001.
What are the ingredients in REGLAN?
Active ingredient: metoclopramide
Inactive ingredients:
- REGLAN 10 mg tablets: magnesium stearate, mannitol, microcrystalline cellulose, stearic acid
- REGLAN 5 mg tablets: corn starch, D&C yellow 10 aluminum lake, FD&C blue 1 aluminum lake, lactose, microcrystalline cellulose, silicon dioxide, stearic acid
Manufactured for:
ALAVEN®
PHARMACEUTICAL LLC
Marietta, GA 30062For Medical Inquiries, call toll-free 1-888-317-0001
www.alavenpharm.comRev. 07/2009
PCS 9360
6-151-10-16-1009Revised July 2009
This Medication Guide has been approved by the U.S. Food and Drug Administration.
- PACKAGE LABEL
-
INGREDIENTS AND APPEARANCE
REGLAN
metoclopramide hydrochloride tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:16590-906(NDC:68220-151) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength METOCLOPRAMIDE HYDROCHLORIDE (UNII: W1792A2RVD) (METOCLOPRAMIDE - UNII:L4YEB44I46) METOCLOPRAMIDE HYDROCHLORIDE 10 mg Inactive Ingredients Ingredient Name Strength MAGNESIUM STEARATE (UNII: 70097M6I30) CELLULOSE, MICROCRYSTALLINE (UNII: OP1R32D61U) MANNITOL (UNII: 3OWL53L36A) STEARIC ACID (UNII: 4ELV7Z65AP) Product Characteristics Color white Score 2 pieces Shape OVAL Size 11mm Flavor Imprint Code REGLAN;AP;10 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:16590-906-30 30 in 1 BOTTLE 2 NDC:16590-906-60 60 in 1 BOTTLE 3 NDC:16590-906-90 90 in 1 BOTTLE Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA017854 01/15/2010 Labeler - STAT RX USA LLC (786036330) Establishment Name Address ID/FEI Business Operations STAT RX USA LLC 786036330 repack, relabel
