Label: RELPAX- eletriptan hydrobromide tablet, film coated

-
Contains inactivated NDC Code(s)
NDC Code(s): 16590-201-06 - Packager: STAT Rx USA LLC
- This is a repackaged label.
- Source NDC Code(s): 0049-2340
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated August 13, 2012
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
DESCRIPTION
RELPAX® (eletriptan) Tablets contain eletriptan hydrobromide, which is a selective 5-hydroxytryptamine 1B/1D (5-HT1B/1D) receptor agonist. Eletriptan is chemically designated as (R)-3-[(1-Methyl-2-pyrrolidinyl)methyl]-5-[2-(phenylsulfonyl)ethyl]-1H-indole monohydrobromide, and it has the following chemical structure:
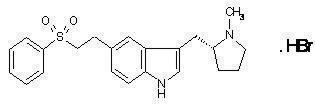
The empirical formula is C22H26N2O2S . HBr, representing a molecular weight of 463.40. Eletriptan hydrobromide is a white to light pale colored powder that is readily soluble in water.
Each RELPAX Tablet for oral administration contains 24.2 or 48.5 mg of eletriptan hydrobromide equivalent to 20 mg or 40 mg of eletriptan, respectively. Each tablet also contains the inactive ingredients microcrystalline cellulose NF, lactose NF, croscarmellose sodium NF, magnesium stearate NF, titanium dioxide USP, hypromellose, triacetin USP and FD&C Yellow No. 6 aluminum lake.
-
CLINICAL PHARMACOLOGY
Mechanism of Action
Eletriptan binds with high affinity to 5-HT1B, 5-HT1D and 5-HT1F receptors, has modest affinity for 5-HT1A, 5-HT1E, 5-HT2B and 5-HT7 receptors, and little or no affinity for 5-HT2A, 5-HT2C, 5-HT3, 5-HT4, 5-HT5A and 5-HT6 receptors. Eletriptan has no significant affinity or pharmacological activity at adrenergic alpha1, alpha2, or beta; dopaminergic D1 or D2; muscarinic; or opioid receptors.
Two theories have been proposed to explain the efficacy of 5-HT receptor agonists in migraine. One theory suggests that activation of 5-HT1 receptors located on intracranial blood vessels, including those on the arteriovenous anastomoses, leads to vasoconstriction, which is correlated with the relief of migraine headache. The other hypothesis suggests that activation of 5-HT1 receptors on sensory nerve endings in the trigeminal system results in the inhibition of pro-inflammatory neuropeptide release.
In the anesthetized dog, eletriptan has been shown to reduce carotid arterial blood flow, with only a small increase in arterial blood pressure at high doses. While the effect on blood flow was selective for the carotid arterial bed, decreases in coronary artery diameter were observed. Eletriptan has also been shown to inhibit trigeminal nerve activity in the rat.
Pharmacokinetics
Absorption
Eletriptan is well absorbed after oral administration with peak plasma levels occurring approximately 1.5 hours after dosing to healthy subjects. In patients with moderate to severe migraine the median Tmax is 2.0 hours. The mean absolute bioavailability of eletriptan is approximately 50%. The oral pharmacokinetics are slightly more than dose-proportional over the clinical dose range. The AUC and Cmax of eletriptan are increased by approximately 20 to 30% following oral administration with a high fat meal.
Distribution
The volume of distribution of eletriptan following IV administration is 138L. Plasma protein binding is moderate and approximately 85%.
Metabolism
The N-demethylated metabolite of eletriptan is the only known active metabolite. This metabolite causes vasoconstriction similar to eletriptan in animal models. Though the half-life of the metabolite is estimated to be about 13 hours, the plasma concentration of the N-demethylated metabolite is 10–20% of parent drug and is unlikely to contribute significantly to the overall effect of the parent compound.
In vitro studies indicate that eletriptan is primarily metabolized by cytochrome P-450 enzyme CYP3A4 (see WARNINGS, DOSAGE AND ADMINISTRATION and CLINICAL PHARMACOLOGY: Drug Interactions).
Special Populations
Age
The pharmacokinetics of eletriptan are generally unaffected by age.
Eletriptan has been given to only 50 patients over the age of 65. Blood pressure was increased to a greater extent in elderly subjects than in young subjects. The pharmacokinetic disposition of eletriptan in the elderly is similar to that seen in younger adults (see PRECAUTIONS).
There is a statistically significant increased half-life (from about 4.4 hours to 5.7 hours) between elderly (65 to 93 years of age) and younger adult subjects (18 to 45 years of age) (see PRECAUTIONS).
Race
A comparison of pharmacokinetic studies run in western countries with those run in Japan has indicated an approximate 35% reduction in the exposure of eletriptan in Japanese male volunteers compared to western males. Population pharmacokinetic analysis of two clinical studies indicates no evidence of pharmacokinetic differences between Caucasians and non-Caucasian patients.
Menstrual Cycle
In a study of 16 healthy females, the pharmacokinetics of eletriptan remained consistent throughout the phases of the menstrual cycle.
Renal Impairment
There was no significant change in clearance observed in subjects with mild, moderate or severe renal impairment, though blood pressure elevations were observed in this population (see WARNINGS).
Hepatic Impairment
The effects of severe hepatic impairment on eletriptan metabolism have not been evaluated. Subjects with mild or moderate hepatic impairment demonstrated an increase in both AUC (34%) and half-life. The Cmax was increased by 18% (see PRECAUTIONS and DOSAGE AND ADMINISTRATION).
Drug Interactions
CYP3A4 inhibitors
In vitro studies have shown that eletriptan is metabolized by the CYP3A4 enzyme. A clinical study demonstrated about a 3-fold increase in Cmax and about a 6-fold increase in the AUC of eletriptan when combined with ketoconazole. The half-life increased from 5 hours to 8 hours and the Tmax increased from 2.8 hours to 5.4 hours. Another clinical study demonstrated about a 2-fold increase in Cmax and about a 4-fold increase in AUC when erythromycin was co-administered with eletriptan. It has also been shown that co-administration of verapamil and eletriptan yields about a 2-fold increase in Cmax and about a 3-fold increase in AUC of eletriptan, and that co-administration of fluconazole and eletriptan yields about a 1.4-fold increase in Cmax and about a 2-fold increase in AUC of eletriptan.
Eletriptan should not be used within at least 72 hours of treatment with the following potent CYP3A4 inhibitors: ketoconazole, itraconazole, nefazodone, troleandomycin, clarithromycin, ritonavir and nelfinavir. Eletriptan should not be used within 72 hours with drugs that have demonstrated potent CYP3A4 inhibition and have this potent effect described in the CONTRAINDICATIONS, WARNINGS or PRECAUTIONS sections of their labeling (see WARNINGS and DOSAGE AND ADMINISTRATION).
Propranolol
The Cmax and AUC of eletriptan were increased by 10 and 33%, respectively, in the presence of propranolol. No interactive increases in blood pressure were observed. No dosage adjustment appears to be needed for patients taking propranolol (see PRECAUTIONS).
The effect of eletriptan on other drugs
The effect of eletriptan on enzymes other than cytochrome P-450 has not been investigated. In vitro human liver microsome studies suggest that eletriptan has little potential to inhibit CYP1A2, 2C9, 2E1 and 3A4 at concentrations up to 100µM. While eletriptan has an effect on CYP2D6 at high concentration, this effect should not interfere with metabolism of other drugs when eletriptan is used at recommended doses. There is no in vitro or in vivo evidence that clinical doses of eletriptan will induce drug metabolizing enzymes. Therefore, eletriptan is unlikely to cause clinically important drug interactions mediated by these enzymes.
-
CLINICAL STUDIES
The efficacy of RELPAX in the acute treatment of migraines was evaluated in eight randomized, double-blind placebo-controlled studies. All eight studies used 40 mg. Seven studies evaluated an 80 mg dose and two studies included a 20 mg dose.
In all eight studies, randomized patients treated their headaches as outpatients. Seven studies enrolled adults and one study enrolled adolescents (age 11 to 17). Patients treated in the seven adult studies were predominantly female (85%) and Caucasian (94%) with a mean age of 40 years (range 18 to 78). In all studies, patients were instructed to treat a moderate to severe headache. Headache response, defined as a reduction in headache severity from moderate or severe pain to mild or no pain, was assessed up to 2 hours after dosing. Associated symptoms such as nausea, vomiting, photophobia and phonophobia were also assessed.
Maintenance of response was assessed for up to 24 hours post dose. In the adult studies, a second dose of RELPAX Tablets or other medication was allowed 2 to 24 hours after the initial treatment for both persistent and recurrent headaches. The incidence and time to use of these additional treatments were also recorded.
In the seven adult studies, the percentage of patients achieving headache response 2 hours after treatment was significantly greater among patients receiving RELPAX Tablets at all doses compared to those who received placebo. The two-hour response rates from these controlled clinical studies are summarized in Table 1.
Table 1: Percentage of Patients with Headache Response (Mild or No Headache) 2 Hours Following Treatment Placebo RELPAX
20 mgRELPAX
40 mgRELPAX
80 mgNA - Not Applicable - *
- p value < 0.05 vs placebo
Study 1 23.8%
(n=126)54.3%*
(n=129)65.0%*
(n=117)77.1%*
(n=118)Study 2 19.0%
(n=232)NA 61.6%*
(n=430)64.6%*
(n=446)Study 3 21.7%
(n=276)47.3%*
(n=273)61.9%*
(n=281)58.6%*
(n=290)Study 4 39.5%
(n=86)NA 62.3%*
(n=175)70.0%*
(n=170)Study 5 20.6%
(n=102)NA 53.9%*
(n=206)67.9%*
(n=209)Study 6 31.3%
(n=80)NA 63.9%*
(n=169)66.9%*
(n=160)Study 7 29.5%
(n=122)NA 57.5%*
(n=492)NA Comparisons of the performance of different drugs based upon results obtained in different clinical trials are never reliable. Because studies are generally conducted at different times, with different samples of patients, by different investigators, employing different criteria and/or different interpretations of the same criteria, under different conditions (dose, dosing regimen, etc.), quantitative estimates of treatment response and the timing of response may be expected to vary considerably from study to study.
The estimated probability of achieving an initial headache response within 2 hours following treatment is depicted in Figure 1.
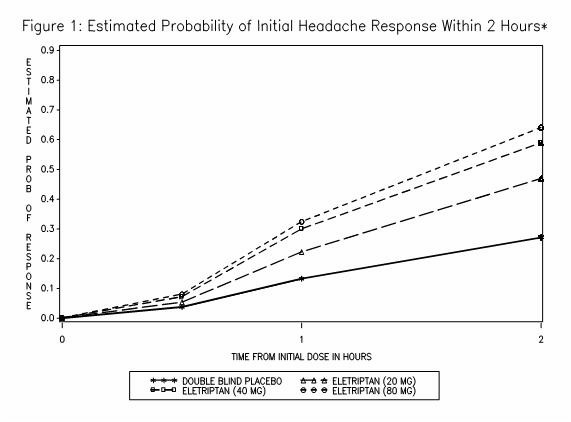
*Figure 1 shows the Kaplan-Meier plot of probability over time of obtaining headache response (no or mild pain) following treatment with eletriptan. The plot is based on 7 placebo-controlled, outpatient trials in adults providing evidence of efficacy (Studies 1 through 7). Patients not achieving headache response or taking additional treatment prior to 2 hours were censored at 2 hours.
For patients with migraine-associated photophobia, phonophobia, and nausea at baseline, there was a decreased incidence of these symptoms following administration of RELPAX as compared to placebo.
Two to 24 hours following the initial dose of study treatment, patients were allowed to use additional treatment for pain relief in the form of a second dose of study treatment or other medication. The estimated probability of taking a second dose or other medications for migraine over the 24 hours following the initial dose of study treatment is summarized in Figure 2.
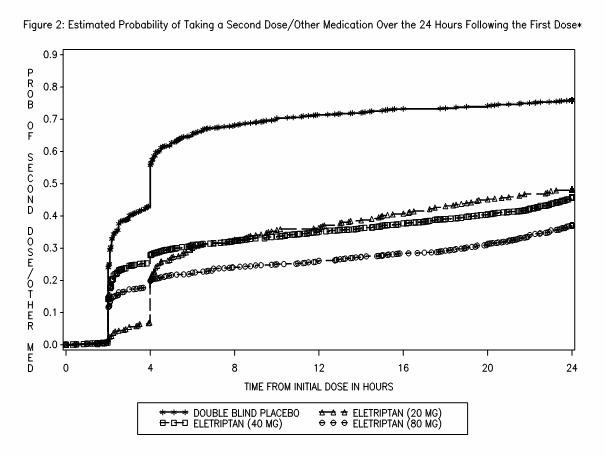
*This Kaplan-Meier plot is based on data obtained in 7 placebo-controlled trials in adults (Studies 1 through 7). Patients were instructed to take a second dose of study medication as follows: a) in the event of no response at 2 hours (studies 2 and 4–7) or at 4 hours (study 3); b) in the event of headache recurrence within 24 hours (studies 2–7). Patients not using additional treatments were censored at 24 hours. The plot includes both patients who had headache response at 2 hours and those who had no response to the initial dose. It should be noted that the protocols did not allow remedication within 2 hours post dose.
The efficacy of RELPAX was unaffected by the duration of attack; gender or age of the patient; relationship to menses; or concomitant use of estrogen replacement therapy/oral contraceptives or frequently used migraine prophylactic drugs.
In a single study in adolescents (n=274), there were no statistically significant differences between treatment groups. The headache response rate at 2 hours was 57% for both RELPAX 40 mg Tablets and placebo.
-
INDICATIONS AND USAGE
RELPAX is indicated for the acute treatment of migraine with or without aura in adults.
RELPAX is not intended for the prophylactic therapy of migraine or for use in the management of hemiplegic or basilar migraine (see CONTRAINDICATIONS). Safety and effectiveness of RELPAX Tablets have not been established for cluster headache, which is present in an older, predominantly male population.
-
CONTRAINDICATIONS
RELPAX Tablets should not be given to patients with ischemic heart disease (e.g., angina pectoris, history of myocardial infarction, or documented silent ischemia) or to patients who have symptoms, or findings consistent with ischemic heart disease, coronary artery vasospasm, including Prinzmetal's variant angina, or other significant underlying cardiovascular disease (see WARNINGS).
RELPAX Tablets should not be given to patients with cerebrovascular syndromes including (but not limited to) strokes of any type as well as transient ischemic attacks (see WARNINGS).
RELPAX Tablets should not be given to patients with peripheral vascular disease including (but not limited to) ischemic bowel disease (see WARNINGS).
Because RELPAX Tablets may increase blood pressure, it should not be given to patients with uncontrolled hypertension (see WARNINGS).
RELPAX Tablets should not be administered to patients with hemiplegic or basilar migraine.
RELPAX Tablets should not be used within 24 hours of treatment with another 5-HT1 agonist, an ergotamine-containing or ergot-type medication such as dihydroergotamine (DHE) or methysergide.
RELPAX Tablets should not be used in patients with known hypersensitivity to eletriptan or any of its inactive ingredients.
RELPAX Tablets should not be given to patients with severe hepatic impairment.
-
WARNINGS
RELPAX Tablets should only be used where a clear diagnosis of migraine has been established.
CYP3A4 Inhibitors
Eletriptan should not be used within at least 72 hours of treatment with the following potent CYP3A4 inhibitors: ketoconazole, itraconazole, nefazodone, troleandomycin, clarithromycin, ritonavir, and nelfinavir. Eletriptan should not be used within 72 hours with drugs that have demonstrated potent CYP3A4 inhibition and have this potent effect described in the CONTRAINDICATIONS, WARNINGS or PRECAUTIONS sections of their labeling (see CLINICAL PHARMACOLOGY: Drug Interactions and DOSAGE AND ADMINISTRATION).
In a coronary angiographic study of rapidly infused intravenous eletriptan to concentrations exceeding those achieved with 80 mg oral eletriptan in the presence of potent CYP3A4 inhibitors, a small dose-related decrease in coronary artery diameter similar to that seen with a 6 mg subcutaneous dose of sumatriptan was observed.
Risk of Myocardial Ischemia and/or Infarction and Other Cardiac Events
Because of the potential of 5-HT1 agonists to cause coronary vasospasm, eletriptan should not be given to patients with documented ischemic or vasospastic coronary artery disease (CAD) (see CONTRAINDICATIONS). It is strongly recommended that eletriptan not be given to patients in whom unrecognized CAD is predicted by the presence of risk factors (e.g., hypertension, hypercholesterolemia, smoker, obesity, diabetes, strong family history of CAD, female with surgical or physiological menopause, or male over 40 years of age) unless a cardiovascular evaluation provides satisfactory clinical evidence that the patient is reasonably free of coronary artery and ischemic myocardial disease or other significant underlying cardiovascular disease. The sensitivity of cardiac diagnostic procedures to detect cardiovascular disease or predisposition to coronary artery vasospasm is modest, at best. If, during the cardiovascular evaluation, the patient's medical history, electrocardiographic, or other investigations reveal findings indicative of, or consistent with coronary artery vasospasm or myocardial ischemia, eletriptan should not be administered (see CONTRAINDICATIONS).
For patients with risk factors predictive of CAD, who are determined to have a satisfactory cardiovascular evaluation, it is strongly recommended that administration of the first dose of eletriptan take place in the setting of a physician's office or similar medically staffed and equipped facility unless the patient has previously received eletriptan. Because cardiac ischemia can occur in the absence of clinical symptoms, consideration should be given to obtaining on the first occasion of use an electrocardiogram (ECG) during the interval immediately following administration of RELPAX Tablets, in these patients with risk factors.
It is recommended that patients who are intermittent long-term users of 5-HT1 agonists including RELPAX Tablets, and who have or acquire risk factors predictive of CAD, as described above, undergo periodic cardiovascular evaluation as they continue to use RELPAX Tablets.
The systematic approach described above is intended to reduce the likelihood that patients with unrecognized cardiovascular disease will be inadvertently exposed to eletriptan.
Cardiac Events and Fatalities
Serious adverse cardiac events, including acute myocardial infarction, life-threatening disturbances of cardiac rhythm, and death have been reported within a few hours following the administration of 5-HT1 agonists including RELPAX. Considering the extent of use of 5-HT1 agonists in patients with migraine, the incidence of these events is extremely low.
Premarketing experience with eletriptan among the 7,143 unique individuals who received eletriptan during premarketing clinical trials: In a clinical pharmacology study, in subjects undergoing diagnostic coronary angiography, a subject with a history of angina, hypertension and hypercholesterolemia, receiving intravenous eletriptan (Cmax of 127 ng/mL equivalent to 60 mg oral eletriptan), reported chest tightness and experienced angiographically documented coronary vasospasm with no ECG changes of ischemia.
There was also one report of atrial fibrillation in a patient with a past history of atrial fibrillation.
Postmarketing experience with eletriptan: Serious cardiovascular events, some resulting in death, have been reported in association with the use of RELPAX. In very rare cases, these events have occurred in the absence of known cardiovascular diseases. The uncontrolled nature of postmarketing surveillance, however, makes it impossible to determine definitively if the cases were actually caused by eletriptan or to reliably assess causation in individual cases.
Cerebrovascular Events and Fatalities Associated With 5-HT1 Agonists
Cerebral hemorrhage, subarachnoid hemorrhage, stroke, and other cerebrovascular events have been reported in patients treated with 5-HT1 agonists, and some have resulted in fatalities. In a number of cases, it appears possible that the cerebrovascular events were primary, the agonist having been administered in the incorrect belief that the symptoms experienced were a consequence of migraine, when they were not. It should be noted that patients with migraine may be at increased risk of certain cerebrovascular events (e.g., stroke, hemorrhage, and transient ischemic attack).
Other Vasospasm-Related Events
5-HT1 agonists may cause vasospastic reactions other than coronary artery vasospasm. Both peripheral vascular ischemia and colonic ischemia with abdominal pain and bloody diarrhea have been reported with 5-HT1 agonists.
Serotonin Syndrome
The development of a potentially life-threatening serotonin syndrome may occur with triptans, including Relpax treatment, particularly during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs). If concomitant treatment with Relpax and an SSRI (e.g., fluoxetine, paroxetine, sertraline, fluvoxamine, citalopram, escitalopram) or SNRI (e.g., venlafaxine, duloxetine) is clinically warranted, careful observation of the patient is advised, particularly during treatment initiation and dose increases. Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, coma), autonomic instability (e.g., tachycardia, labile blood pressure, hyperthermia), neuromuscular aberrations (e.g., hyperreflexia, incoordination) and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea). (See PRECAUTIONS—DRUG INTERACTIONS).
Increase in Blood Pressure
Significant elevation in blood pressure, including hypertensive crisis, has been reported on rare occasions in patients receiving 5-HT1 agonists with and without a history of hypertension. In clinical pharmacology studies, oral eletriptan (at doses of 60 mg or more) was shown to cause small, transient dose-related increases in blood pressure, predominantly diastolic, consistent with its mechanism of action and with other 5-HT1B/1D agonists. The effect was more pronounced in renally impaired and elderly subjects. A single patient with hepatic cirrhosis received eletriptan 80 mg and experienced a blood pressure of 220/96 mm Hg five hours after dosing. The treatment-related event persisted for seven hours.
Eletriptan is contraindicated in patients with uncontrolled hypertension (see CONTRAINDICATIONS).
An 18% increase in mean pulmonary artery pressure was seen following dosing with another 5-HT1 agonist in a study evaluating subjects undergoing cardiac catheterization.
-
PRECAUTIONS
General
As with other 5-HT1 agonists, sensations of tightness, pain, pressure and heaviness have been reported after treatment with eletriptan in the precordium, throat, and jaw. Events that are localized to the chest, throat, neck and jaw have not been associated with arrhythmias or ischemic ECG changes in clinical trials; in a clinical pharmacology study of subjects undergoing diagnostic coronary angiography, one subject with a history of angina, hypertension and hypercholesterolemia, receiving intravenous eletriptan, reported chest tightness and experienced angiographically documented coronary vasospasm with no ECG changes of ischemia. Because 5-HT1 agonists may cause coronary artery vasospasm, patients who experience signs or symptoms suggestive of angina following dosing should be evaluated for the presence of CAD or a predisposition to Prinzmetal's variant angina before receiving additional doses of medication, and should be monitored electrocardiographically if dosing is resumed and similar symptoms recur. Similarly, patients who experience other symptoms or signs suggestive of decreased arterial flow, such as ischemic bowel syndrome or Raynaud's syndrome following the use of any 5-HT1 agonist are candidates for further evaluation (see CONTRAINDICATIONS and WARNINGS).
Overuse
Excessive use of any anti-migraine medicinal product can lead to daily chronic headaches. Overuse of all triptans has been reported primarily in patients with chronic daily headache.
Hepatically Impaired Patients
The effects of severe hepatic impairment on eletriptan metabolism was not evaluated. Subjects with mild or moderate hepatic impairment demonstrated an increase in both AUC (34%) and half-life. The Cmax was increased by 18%. Eletriptan should not be used in patients with severe hepatic impairment. No dose adjustment is necessary in mild to moderate impairment (see DOSAGE AND ADMINISTRATION).
Binding to Melanin-Containing Tissues
In rats treated with a single intravenous (3 mg/kg) dose of radio-labeled eletriptan, elimination of radioactivity from the retina was prolonged, suggesting that eletriptan and/or its metabolites may bind to the melanin of the eye. Because there could be accumulation in melanin-rich tissues over time, this raises the possibility that eletriptan could cause toxicity in these tissues after extended use. Although no systematic monitoring of ophthalmologic function was undertaken in clinical trials, and no specific recommendations for ophthalmologic monitoring are offered, prescribers should be aware of the possibility of long-term ophthalmologic effects.
Corneal Opacities
Transient corneal opacities were seen in dogs receiving oral eletriptan at 5 mg/kg and above. They were observed during the first week of treatment, but were not present thereafter despite continued treatment. Exposure at the no-effect dose level of 2.5 mg/kg was approximately equal to that achieved in humans at the maximum recommended daily dose.
Information for Patients
See PATIENT INFORMATION at the end of this labeling for the text of the separate leaflet provided for patients.
Patients should be cautioned about the risk of serotonin syndrome with the use of RELPAX or other triptans, especially during combined use with selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs).
Drug Interactions
Ergot-containing drugs
Ergot-containing drugs have been reported to cause prolonged vasospastic reactions. Because these effects may be additive, use of ergotamine-containing or ergot-type medications (like dihydroergotamine [DHE] or methysergide) and eletriptan within 24 hours of each other is not recommended (see CONTRAINDICATIONS).
CYP3A4 Inhibitors
Eletriptan is metabolized primarily by CYP3A4 (see WARNINGS regarding use with potent CYP3A4 inhibitors).
Monoamine Oxidase Inhibitors
Eletriptan is not a substrate for monoamine oxidase (MAO) enzymes; therefore, there is no expectation of an interaction between eletriptan and MAO inhibitors.
Propranolol
The Cmax and AUC of eletriptan were increased by 10 and 33% respectively in the presence of propranolol. No interactive increases in blood pressure were observed. No dosage adjustment appears to be needed for patients taking propranolol (see CLINICAL PHARMACOLOGY).
Selective Serotonin Reuptake Inhibitors/Serotonin Norepinephrine Reuptake Inhibitors and Serotonin Syndrome
Cases of life-threatening serotonin syndrome have been reported during combined use of selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs) and triptans (See WARNINGS).
Other 5-HT1 agonists
Concomitant use of other 5-HT1 agonists within 24 hours of RELPAX treatment is not recommended (see CONTRAINDICATIONS).
Drug/Laboratory Test Interactions
RELPAX Tablets are not known to interfere with commonly employed clinical laboratory tests.
Carcinogenesis
Lifetime carcinogenicity studies, 104 weeks in duration, were carried out in mice and rats by administering eletriptan in the diet. In rats, the incidence of testicular interstitial cell adenomas was increased at the high dose of 75 mg/kg/day. The estimated exposure (AUC) to parent drug at that dose was approximately 6 times that achieved in humans receiving the maximum recommended daily dose (MRDD) of 80 mg, and at the no-effect dose of 15 mg/kg/day it was approximately 2 times the human exposure at the MRDD. In mice, the incidence of hepatocellular adenomas was increased at the high dose of 400 mg/kg/day. The exposure to parent drug (AUC) at that dose was approximately 18 times that achieved in humans receiving the MRDD, and the AUC at the no-effect dose of 90 mg/kg/day was approximately 7 times the human exposure at the MRDD.
Mutagenesis
Eletriptan was not mutagenic in bacterial or mammalian cell assays in vitro, testing negative in the Ames reverse mutation test and the hypoxanthine-guanine phosphoribosyl transferase (HGPRT) mutation test in Chinese hamster ovary cells. It was not clastogenic in two in vivo mouse micronucleus assays. Results were equivocal in in vitro human lymphocyte clastogenicity tests, in which the incidence of polyploidy was increased in the absence of metabolic activation (-S9 conditions), but not in the presence of metabolic activation.
Impairment of Fertility
In a rat fertility and early embryonic development study, doses tested were 50, 100 and 200 mg/kg/day, resulting in systemic exposures to parent drug in rats, based on AUC, that were 4, 8 and 16 times MRDD, respectively, in males and 7, 14 and 28 times MRDD, respectively, in females. There was a prolongation of the estrous cycle at the 200 mg/kg/day dose due to an increase in duration of estrus, based on vaginal smears. There were also dose-related, statistically significant decreases in mean numbers of corpora lutea per dam at all 3 doses, resulting in decreases in mean numbers of implants and viable fetuses per dam. This suggests a partial inhibition of ovulation by eletriptan. There was no effect on fertility of males and no other effect on fertility of females.
Pregnancy
Pregnancy Category C
In reproductive toxicity studies in rats and rabbits, oral administration of eletriptan was associated with developmental toxicity (decreased fetal and pup weights and an increased incidence of fetal structural abnormalities). Effects on fetal and pup weights were observed at doses that were, on a mg/m2 basis, 6 to 12 times greater than the clinical maximum recommended daily dose (MRDD) of 80 mg. The increase in structural alterations occurred in the rat and rabbit at doses that, on a mg/m2 basis, were 12 times greater than (rat) and approximately equal to (rabbit) the MRDD.
When pregnant rats were administered eletriptan during the period of organogenesis at doses of 10, 30 or 100 mg/kg/day, fetal weights were decreased and the incidences of vertebral and sternebral variations were increased at 100 mg/kg/day (approximately 12 times the MRDD on a mg/m2 basis). The 100 mg/kg dose was also maternally toxic, as evidenced by decreased maternal body weight gain during gestation. The no-effect dose for developmental toxicity in rats exposed during organogenesis was 30 mg/kg, which is approximately 4 times the MRDD on a mg/m2 basis.
When doses of 5, 10 or 50 mg/kg/day were given to New Zealand White rabbits throughout organogenesis, fetal weights were decreased at 50 mg/kg, which is approximately 12 times the MRDD on a mg/m2 basis. The incidences of fused sternebrae and vena cava deviations were increased in all treated groups. Maternal toxicity was not produced at any dose. A no-effect dose for developmental toxicity in rabbits exposed during organogenesis was not established, and the 5 mg/kg dose is approximately equal to the MRDD on a mg/m2 basis.
There are no adequate and well-controlled studies in pregnant women; therefore, eletriptan should be used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Nursing Mothers
Eletriptan is excreted in human breast milk. In one study of 8 women given a single dose of 80 mg, the mean total amount of eletriptan in breast milk over 24 hours in this group was approximately 0.02% of the administered dose. The ratio of eletriptan mean concentration in breast milk to plasma was 1:4, but there was great variability. The resulting eletriptan concentration-time profile was similar to that seen in the plasma over 24 hours, with very low concentrations of drug (mean 1.7 ng/mL) still present in the milk 18–24 hours post dose. The N-desmethyl active metabolite was not measured in the breast milk. Caution should be exercised when RELPAX is administered to nursing women.
Pediatric Use
Safety and effectiveness of RELPAX Tablets in pediatric patients have not been established; therefore, RELPAX is not recommended for use in patients under 18 years of age.
The efficacy of RELPAX Tablets (40 mg) in patients 11–17 was not established in a randomized, placebo-controlled trial of 274 adolescent migraineurs (see CLINICAL STUDIES). Adverse events observed were similar in nature to those reported in clinical trials in adults. Postmarketing experience with other triptans includes a limited number of reports that describe pediatric patients who have experienced clinically serious adverse events that are similar in nature to those reported rarely in adults. Long-term safety of eletriptan was studied in 76 adolescent patients who received treatment for up to one year. A similar profile of adverse events to that of adults was observed. The long-term safety of eletriptan in pediatric patients has not been established.
Geriatric Use
Eletriptan has been given to only 50 patients over the age of 65. Blood pressure was increased to a greater extent in elderly subjects than in young subjects. The pharmacokinetic disposition of eletriptan in the elderly is similar to that seen in younger adults (see CLINICAL PHARMACOLOGY). In clinical trials, there were no apparent differences in efficacy or the incidence of adverse events between patients under 65 years of age and those 65 and above (n=50).
There is a statistically significantly increased half-life (from about 4.4 hours to 5.7 hours) between elderly (65 to 93 years of age) and younger adult subjects (18 to 45 years of age) (see CLINICAL PHARMACOLOGY).
-
ADVERSE REACTIONS
Serious cardiac events, including some that have been fatal, have occurred following the use of 5-HT1 agonists including RELPAX. These events are extremely rare and most have been reported in patients with risk factors predictive of CAD. Events reported have included coronary artery vasospasm, transient myocardial ischemia, myocardial infarction, ventricular tachycardia, and ventricular fibrillation (see CONTRAINDICATIONS, WARNINGS and PRECAUTIONS).
Incidence in Controlled Clinical Trials
Among 4,597 patients who treated the first migraine headache with RELPAX in short-term placebo-controlled trials, the most common adverse events reported with treatment with RELPAX were asthenia, nausea, dizziness, and somnolence. These events appear to be dose-related.
In long-term open-label studies where patients were allowed to treat multiple migraine attacks for up to 1 year, 128 (8.3%) out of 1,544 patients discontinued treatment due to adverse events.
Table 2 lists adverse events that occurred in the subset of 5,125 migraineurs who received eletriptan doses of 20 mg, 40 mg and 80 mg or placebo in worldwide placebo-controlled clinical trials. The events cited reflect experience gained under closely monitored conditions of clinical trials in a highly selected patient population. In actual clinical practice or in other clinical trials, those frequency estimates may not apply, as the conditions of use, reporting behavior, and the kinds of patients treated may differ.
Only adverse events that were more frequent in a RELPAX treatment group compared to the placebo group with an incidence greater than or equal to 2% are included in Table 2.
Table 2: Adverse Experience Incidence in Placebo-Controlled Migraine Clinical Trials: Events Reported by ≥ 2% Patients Treated with RELPAX and More Than Placebo Adverse Event Type Placebo
(n=988)RELPAX
20 mg
(n=431)RELPAX
40 mg
(n=1774)RELPAX
80 mg
(n=1932)ATYPICAL SENSATIONS Paresthesia 2% 3% 3% 4% Flushing/feeling of warmth 2% 2% 2% 2% PAIN AND PRESSURE SENSATIONS Chest –
tightness/pain/pressure1% 1% 2% 4% Abdominal –
pain/discomfort/stomach pain/cramps/pressure1% 1% 2% 2% DIGESTIVE Dry mouth 2% 2% 3% 4% Dyspepsia 1% 1% 2% 2% Dysphagia –
throat tightness/difficulty swallowing0.2% 1% 2% 2% Nausea 5% 4% 5% 8% NEUROLOGICAL Dizziness 3% 3% 6% 7% Somnolence 4% 3% 6% 7% Headache 3% 4% 3% 4% OTHER Asthenia 3% 4% 5% 10% RELPAX is generally well tolerated. Across all doses, most adverse reactions were mild and transient. The frequency of adverse events in clinical trials did not increase when up to 2 doses of RELPAX were taken within 24 hours. The incidence of adverse events in controlled clinical trials was not affected by gender, age, or race of the patients. Adverse event frequencies were also unchanged by concomitant use of drugs commonly taken for migraine prophylaxis (e.g., SSRIs, beta blockers, calcium channel blockers, tricyclic antidepressants), estrogen replacement therapy and oral contraceptives.
Other Events Observed in Association With the Administration of RELPAX Tablets
In the paragraphs that follow, the frequencies of less commonly reported adverse clinical events are presented. Because the reports include events observed in open studies, the role of RELPAX Tablets in their causation cannot be reliably determined. Furthermore, variability associated with adverse event reporting, the terminology used to describe adverse events, etc., limit the value of the quantitative frequency estimates provided. Event frequencies are calculated as the number of patients reporting an event divided by the total number of patients (N=4,719) exposed to RELPAX. All reported events are included except those already listed in Table 2, those too general to be informative, and those not reasonably associated with the use of the drug. Events are further classified within body system categories and enumerated in order of decreasing frequency using the following definitions: frequent adverse events are those occurring in at least 1/100 patients, infrequent adverse events are those occurring in 1/100 to 1/1000 patients and rare adverse events are those occurring in fewer than 1/1000 patients.
General: Frequent were back pain, chills and pain. Infrequent were face edema and malaise. Rare were abdomen enlarged, abscess, accidental injury, allergic reaction, fever, flu syndrome, halitosis, hernia, hypothermia, lab test abnormal, moniliasis, rheumatoid arthritis and shock.
Cardiovascular: Frequent was palpitation. Infrequent were hypertension, migraine, peripheral vascular disorder and tachycardia. Rare were angina pectoris, arrhythmia, atrial fibrillation, AV block, bradycardia, hypotension, syncope, thrombophlebitis, cerebrovascular disorder, vasospasm and ventricular arrhythmia.
Digestive: Infrequent were anorexia, constipation, diarrhea, eructation, esophagitis, flatulence, gastritis, gastrointestinal disorder, glossitis, increased salivation and liver function tests abnormal. Rare were gingivitis, hematemesis, increased appetite, rectal disorder, stomatitis, tongue disorder, tongue edema and tooth disorder.
Endocrine: Rare were goiter, thyroid adenoma and thyroiditis.
Hemic and Lymphatic: Rare were anemia, cyanosis, leukopenia, lymphadenopathy, monocytosis and purpura.
Immune System Disorders: Allergic reactions, some of which may be serious, including angioedema.
Metabolic: Infrequent were creatine phosphokinase increased, edema, peripheral edema and thirst. Rare were alkaline phosphatase increased, bilirubinemia, hyperglycemia, weight gain and weight loss.
Musculoskeletal: Infrequent were arthralgia, arthritis, arthrosis, bone pain, myalgia and myasthenia. Rare were bone neoplasm, joint disorder, myopathy and tenosynovitis.
Neurological: Frequent were hypertonia, hypesthesia and vertigo. Infrequent were abnormal dreams, agitation, anxiety, apathy, ataxia, confusion, depersonalization, depression, emotional lability, euphoria, hyperesthesia, hyperkinesia, incoordination, insomnia, nervousness, speech disorder, stupor, thinking abnormal and tremor. Rare were abnormal gait, amnesia, aphasia, catatonic reaction, dementia, diplopia, dystonia, hallucinations, hemiplegia, hyperalgesia, hypokinesia, hysteria, manic reaction, neuropathy, neurosis, oculogyric crisis, paralysis, psychotic depression, sleep disorder and twitching.
Respiratory: Frequent was pharyngitis. Infrequent were asthma, dyspnea, respiratory disorder, respiratory tract infection, rhinitis, voice alteration and yawn. Rare were bronchitis, choking sensation, cough increased, epistaxis, hiccup, hyperventilation, laryngitis, sinusitis and sputum increased.
Skin and Appendages: Frequent was sweating. Infrequent were pruritus, rash and skin disorder. Rare were alopecia, dry skin, eczema, exfoliative dermatitis, maculopapular rash, psoriasis, skin discoloration, skin hypertrophy and urticaria.
Special Senses: Infrequent was abnormal vision, conjunctivitis, ear pain, eye pain, lacrimation disorder, photophobia, taste perversion and tinnitus. Rare were abnormality of accommodation, dry eyes, ear disorder, eye hemorrhage, otitis media, parosmia and ptosis.
Urogenital: Infrequent were impotence, polyuria, urinary frequency and urinary tract disorder. Rare were breast pain, kidney pain, leukorrhea, menorrhagia, menstrual disorder and vaginitis.
Other Events Observed During Post-Marketing Use
The following adverse reaction(s) have been identified during postapproval use of RELPAX. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Neurological: seizure
Digestive: vomiting
- DRUG ABUSE AND DEPENDENCE
-
OVERDOSAGE
No significant overdoses in premarketing clinical trials have been reported. Volunteers (N=21) have received single doses of 120 mg without significant adverse effects. Daily doses of 160 mg were commonly employed in Phase III trials. Based on the pharmacology of the 5-HT1B/1D agonists, hypertension or other more serious cardiovascular symptoms could occur on overdose.
The elimination half-life of eletriptan is about 4 hours (see CLINICAL PHARMACOLOGY) and therefore monitoring of patients after overdose with eletriptan should continue for at least 20 hours, or longer should symptoms or signs persist.
There is no specific antidote to eletriptan. In cases of severe intoxication, intensive care procedures are recommended, including establishing and maintaining a patent airway, ensuring adequate oxygenation and ventilation, and monitoring and support of the cardiovascular system.
It is unknown what effect hemodialysis or peritoneal dialysis has on the serum concentration of eletriptan.
-
DOSAGE AND ADMINISTRATION
In controlled clinical trials, single doses of 20 mg and 40 mg were effective for the acute treatment of migraine in adults. A greater proportion of patients had a response following a 40 mg dose than following a 20 mg dose (see CLINICAL STUDIES). Individuals may vary in response to doses of RELPAX Tablets. The choice of dose should therefore be made on an individual basis. An 80 mg dose, although also effective, was associated with an increased incidence of adverse events. Therefore, the maximum recommended single dose is 40 mg.
If after the initial dose, headache improves but then returns, a repeat dose may be beneficial. If a second dose is required, it should be taken at least 2 hours after the initial dose. If the initial dose is ineffective, controlled clinical trials have not shown a benefit of a second dose to treat the same attack. The maximum daily dose should not exceed 80 mg.
The safety of treating an average of more than 3 headaches in a 30-day period has not been established.
CYP3A4 Inhibitors
Eletriptan is metabolized by the CYP3A4 enzyme. Eletriptan should not be used within at least 72 hours of treatment with the following potent CYP3A4 inhibitors: ketoconazole, itraconazole, nefazodone, troleandomycin, clarithromycin, ritonavir and nelfinavir. Eletriptan should not be used within 72 hours with drugs that have demonstrated potent CYP3A4 inhibition and have this potent effect described in the CONTRAINDICATIONS, WARNINGS or PRECAUTIONS sections of their labeling (see WARNINGS and CLINICAL PHARMACOLOGY: Drug Interactions).
Hepatic Impairment
The drug should not be given to patients with severe hepatic impairment since the effect of severe hepatic impairment on eletriptan metabolism was not evaluated. No dose adjustment is necessary in mild to moderate impairment (see CLINICAL PHARMACOLOGY, CONTRAINDICATIONS and PRECAUTIONS).
- HOW SUPPLIED
- SPL UNCLASSIFIED SECTION
-
PATIENT SUMMARY OF INFORMATIONRELPAX®
(eletriptan hydrobromide)
Please read this information before you start taking RELPAX and each time you renew your prescription. Remember, this summary does not take the place of discussions with your doctor. You and your doctor should discuss RELPAX when you start taking your medication and at regular checkups.
What is RELPAX?
RELPAX is a prescription medicine used to treat migraine headaches in adults. RELPAX is not for other types of headaches.
What is a Migraine Headache?
Migraine is an intense, throbbing headache. You may have pain on one or both sides of your head. You may have nausea and vomiting, and be sensitive to light and noise. The pain and symptoms of a migraine headache can be worse than a common headache. Some women get migraines around the time of their menstrual period. Some people have visual symptoms before the headache, such as flashing lights or wavy lines, called an aura.
How Does RELPAX Work?
Treatment with RELPAX reduces swelling of blood vessels surrounding the brain. This swelling is associated with the headache pain of a migraine attack. RELPAX blocks the release of substances from nerve endings that cause more pain and other symptoms like nausea, and sensitivity to light and sound.
It is thought that these actions contribute to relief of your symptoms by RELPAX.
Who should not take RELPAX?
Do not take RELPAX if you:
- have uncontrolled high blood pressure.
- have heart disease or a history of heart disease.
- have hemiplegic or basilar migraine (if you are not sure about this, ask your doctor).
- have or had a stroke or problems with your blood circulation.
- have serious liver problems.
- have taken any of the following medicines in the last 24 hours: other "triptans" or triptan combination products such as almotriptan (Axert®), frovatriptan (Frova™), naratriptan (Amerge®), rizatriptan (Maxalt®), sumatriptan (Imitrex®) sumatriptan and naproxen sodium, (Treximet™), zolmitriptan (Zomig®); ergotamines like Bellergal-S®, Cafergot®, Ergomar®, Wigraine®; dihydroergotamine like D.H.E. 45® or Migranal®; or methysergide (Sansert®). These medicines have side effects similar to RELPAX.1
- have taken the following medicines within at least 72 hours: ketoconazole (Nizoral®), itraconazole (Sporanox®), nefazodone (Serzone®), troleandomycin (TAO®), clarithromycin (Biaxin®), ritonavir (Norvir®), and nelfinavir (Viracept®). These medicines may cause an increase in the amount of RELPAX in the blood.1
- are allergic to RELPAX or any of its ingredients. The active ingredient is eletriptan. The inactive ingredients are listed at the end of this leaflet.
Tell your doctor about all the medicines you take or plan to take, including prescription and non-prescription medicines, supplements, and herbal remedies. Your doctor will decide if you can take RELPAX with your other medicines.
Some medicines used in treating depression such as the selective serotonin reuptake inhibitors (SSRIs) and serotonin norepinephrine reuptake inhibitors (SNRIs) may cause a condition called serotonin syndrome especially during combined use with certain migraine medications. Your doctor needs to know if you are taking any of these medicines, when taking Relpax.
- selective serotonin reuptake inhibitors (SSRIs) or serotonin norepinephrine reuptake inhibitors (SNRIs), two types of drugs for depression or other disorders. Common SSRIs are CELEXA® (citalopram HBr), LEXAPRO® (escitalopram oxalate), PAXIL® (paroxetine), PROZAC®/SARAFEM® (fluoxetine), SYMBYAX® (olanzapine/fluoxetine), ZOLOFT® (sertraline), and fluvoxamine. Common SNRIs are CYMBALTA® (duloxetine) and EFFEXOR® (venlafaxine).
Tell your doctor if you know that you have any of the following: risk factors for heart disease like high cholesterol, diabetes, smoking, obesity, menopause, or a family history of heart disease or stroke.
How should I take RELPAX?
RELPAX comes in 20 mg and 40 mg tablets. When you have a migraine headache, take your medicine as directed by your doctor.
- Take one RELPAX Tablet as soon as you feel a migraine coming on.
- If your headache improves and then comes back after 2 hours, you can take a second tablet.
- If the first tablet did not help your headache at all, do not take a second tablet without talking with your doctor.
- Do not take more than two RELPAX Tablets in any 24-hour period.
What are the possible side effects of RELPAX?
RELPAX is generally well tolerated. As with any medicine, people taking RELPAX may have side effects. The side effects are usually mild and do not last long.
The most common side effects of RELPAX are:
- dizziness
- nausea
- weakness
- tiredness
- pain or pressure sensation (e.g., in the chest or throat)
In very rare cases, patients taking triptans, such as RELPAX, may experience serious side effects, including heart attacks. Call your doctor right away if you have:
- severe chest pains
- shortness of breath
Some patients taking triptans may have a reaction called serotonin syndrome particularly during combined use with certain types of antidepressants, SSRIs or SNRIs. Symptoms may include confusion, hallucinations, fast heart beat, feeling faint, fever, sweating, muscle spasm, difficulty walking and/or diarrhea. Call your doctor right away if you have any of these symptoms after taking RELPAX.
Overuse of anti-migraine drugs like RELPAX can lead to daily headaches. Talk to your doctor if you have persistent non-migraine headaches.
This is not a complete list of side effects. Talk to your doctor if you develop any symptoms that concern you.
What to do in case of an overdose?
Call your doctor or poison control center or go to the ER.
General advice about RELPAX
Medicines are sometimes prescribed for conditions that are not mentioned in patient information leaflets. Do not use RELPAX for a condition for which it was not prescribed. Do not give RELPAX to other people, even if they have the same symptoms you have.
This leaflet summarizes the most important information about RELPAX. If you would like more information about RELPAX, talk with your doctor. You can ask your doctor or pharmacist for information on RELPAX that is written for health professionals. You can also call 1-866-4RELPAX (1-866-473-5729) or visit our web site at www.RELPAX.com.
What are the ingredients in RELPAX?
Active ingredient: eletriptan hydrobromide
Inactive ingredients: microcrystalline cellulose, lactose, croscarmellose sodium, magnesium stearate, titanium oxide, hypromellose, triacetin, and FD&C Yellow No. 6 aluminum lake.
Store RELPAX Tablets at room temperature 15–30°C (59–86°F).

LAB 0077-9.0
January 2012
- 1
- The brands listed are the trademarks of their respective owners and are not trademarks of Pfizer Inc.
- SPL UNCLASSIFIED SECTION
- PACKAGE LABEL - RELPAX 40 MG TABLETS
-
INGREDIENTS AND APPEARANCE
RELPAX
eletriptan hydrobromide tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:16590-201(NDC:0049-2340) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength eletriptan hydrobromide (UNII: M41W832TA3) (eletriptan - UNII:22QOO9B8KI) eletriptan 40 mg Inactive Ingredients Ingredient Name Strength cellulose, microcrystalline (UNII: OP1R32D61U) lactose (UNII: J2B2A4N98G) croscarmellose sodium (UNII: M28OL1HH48) magnesium stearate (UNII: 70097M6I30) titanium dioxide (UNII: 15FIX9V2JP) hypromelloses (UNII: 3NXW29V3WO) triacetin (UNII: XHX3C3X673) FD&C YELLOW NO. 6 (UNII: H77VEI93A8) Aluminum Oxide (UNII: LMI26O6933) Product Characteristics Color ORANGE Score no score Shape ROUND Size 8mm Flavor Imprint Code REP40;Pfizer Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:16590-201-06 6 in 1 BLISTER PACK Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA021016 12/26/2002 Labeler - STAT Rx USA LLC (786036330) Registrant - PSS World Medical Inc (101822682) Establishment Name Address ID/FEI Business Operations STAT Rx USA LLC 786036330 relabel(16590-201) , repack(16590-201)
