Label: NUZYRA- omadacycline injection, powder, lyophilized, for solution
NUZYRA- omadacycline tablet, film coated



-
NDC Code(s):
71715-001-01,
71715-001-02,
71715-002-21,
71715-002-23, view more71715-002-24, 71715-002-27, 71715-002-28
- Packager: Paratek Pharmaceuticals, Inc.
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated March 31, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use NUZYRA ®safely and effectively. See full prescribing information for NUZYRA.
NUZYRA (omadacycline) for injection, for intravenous use
NUZYRA (omadacycline) tablets, for oral use
Initial U.S. Approval: 2018RECENT MAJOR CHANGES
Warnings and Precautions, Tetracycline Class Effects ( 5.6) 3/2025
INDICATIONS AND USAGE
NUZYRA is a tetracycline class antibacterial indicated for the treatment of adult patients with the following infections caused by susceptible microorganisms ( 1):
- Community-acquired bacterial pneumonia (CABP) ( 1.1)
- Acute bacterial skin and skin structure infections (ABSSSI) ( 1.2)
To reduce the development of drug-resistant bacteria and maintain the effectiveness of NUZYRA and other antibacterial drugs, NUZYRA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. ( 1.3)
DOSAGE AND ADMINISTRATION
Infection Loading Doses Maintenance Dose CABP NUZYRA Injection:
Day 1: 200 mg by intravenous infusion over 60 minutes OR100 mg by intravenous infusion over 30 minutes twice ( 2.2) OR
NUZYRA Tablets:
Day 1: 300 mg orally twice ( 2.2)NUZYRA Injection:
100 mg by intravenous infusion over 30 minutes once daily OR
NUZYRA Tablets:
300 mg orally once daily ( 2.2)ABSSSI NUZYRA Injection:
Day 1: 200 mg by intravenous infusion over 60 minutes OR100 mg by intravenous infusion over 30 minutes twice (2.3)OR
NUZYRA Tablets:
Day 1 and Day 2: 450 mg orally once daily ( 2.3)NUZYRA Injection:
100 mg by intravenous infusion over 30 minutes once daily OR
NUZYRA Tablets:
300 mg orally once daily ( 2.3)- CABP and ABSSSI: Treatment duration is 7 to 14 days. ( 2.2, 2.3)
- Fast for at least 4 hours and then take NUZYRA tablets with water. After oral dosing, no food or drink (except water) is to be consumed for 2 hours and no dairy products, antacids, or multivitamins for 4 hours. ( 2.1)
- See full prescribing information for the preparation of NUZYRA IV and other administration instructions. ( 2.1, 2.5)
DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
Known hypersensitivity to omadacycline, tetracycline class antibacterial drugs or any of the excipients in NUZYRA ( 4)
WARNINGS AND PRECAUTIONS
- Mortality Imbalance in Patients with CABP: In the CABP trial, mortality rate of 2% was observed in NUZYRA-treated patients compared to 1% in moxifloxacin-treated patients. The cause of the mortality imbalance has not been established. Closely monitor clinical response to therapy in CABP patients, particularly in those at higher risk for mortality. ( 5.1, 6.1)
- Tooth Discoloration and Enamel Hypoplasia: The use of NUZYRA during tooth development (last half of pregnancy, infancy and childhood to the age of 8 years) may cause permanent discoloration of the teeth (yellow-gray-brown) and enamel hypoplasia. ( 5.2, 8.1, 8.4)
- Inhibition of Bone Growth:The use of NUZYRA during the second and third trimester of pregnancy, infancy and childhood up to the age of 8 years may cause reversible inhibition of bone growth. ( 5.3, 8.1, 8.4)
- Clostridioides difficile-associated diarrhea : Evaluate if diarrhea occurs. ( 5.5)
ADVERSE REACTIONS
The most common adverse reactions (incidence ≥2%) are nausea, vomiting, infusion site reactions, alanine aminotransferase increased, aspartate aminotransferase increased, gamma-glutamyl transferase increased, hypertension, headache, diarrhea, insomnia, and constipation. ( 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Paratek Pharmaceuticals, Inc. at 1-833-727-2835 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- Patients who are on anticoagulant therapy may require downward adjustment of their anticoagulant dosage while taking NUZYRA. ( 7.1)
- Absorption of tetracyclines, including NUZYRA, is impaired by antacids containing aluminum, calcium, or magnesium, bismuth subsalicylate and iron containing preparations. ( 2.1, 7.2)
USE IN SPECIFIC POPULATIONS
Lactation: Breastfeeding is not recommended during treatment with NUZYRA. ( 8.2)
See 17 for PATIENT COUNSELING INFORMATION.
Revised: 3/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
1.1 Community-Acquired Bacterial Pneumonia (CABP)
1.2 Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
1.3 Usage
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
2.2 Dosage in Adults with Community-Acquired Bacterial Pneumonia (CABP)
2.3 Dosage in Adults with Acute Bacterial Skin Structure and Skin Infections (ABSSSI)
2.4 Dosage Adjustments in Patients with Renal or Hepatic Impairment
2.5 Preparation and Administration of NUZYRA for Injection Intravenous Solution
3 DOSAGE FORMS AND STRENGTHS
3.1 NUZYRA for Injection
3.2 NUZYRA Tablets
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Mortality Imbalance in Patients with Community-Acquired Bacterial Pneumonia
5.2 Tooth Discoloration and Enamel Hypoplasia
5.3 Inhibition of Bone Growth
5.4 Hypersensitivity Reactions
5.5 Clostridioides difficile-Associated Diarrhea
5.6 Tetracycline Class Effects
5.7 Development of Drug-Resistant Bacteria
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
7 DRUG INTERACTIONS
7.1 Anticoagulant Drugs
7.2 Antacids and Iron Preparations
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.3 Females and Males of Reproductive Potential
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.4 Microbiology
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.2 Animal Toxicology and/or Pharmacology
14 CLINICAL STUDIES
14.1 Community-Acquired Bacterial Pneumonia
14.2 Acute Bacterial Skin and Skin Structure Infections
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
16.2 Storage and Handling
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
1 INDICATIONS AND USAGE
1.1 Community-Acquired Bacterial Pneumonia (CABP)
NUZYRA is indicated for the treatment of adult patients with community-acquired bacterial pneumonia (CABP) caused by the following susceptible microorganisms: Streptococcus pneumoniae, Staphylococcus aureus(methicillin-susceptible isolates), Haemophilus influenzae, Haemophilus parainfluenzae, Klebsiella pneumoniae, Legionella pneumophila, Mycoplasma pneumoniae, and Chlamydophila pneumoniae.
1.2 Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
NUZYRA is indicated for the treatment of adult patients with acute bacterial skin and skin structure infections (ABSSSI) caused by the following susceptible microorganisms: Staphylococcus aureus(methicillin-susceptible and -resistant isolates), Staphylococcus lugdunensis, Streptococcus pyogenes, Streptococcus anginosus grp. (includes S. anginosus, S. intermedius, and S. constellatus), Enterococcus faecalis, Enterobacter cloacae,and Klebsiella pneumoniae.
1.3 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of NUZYRA and other antibacterial drugs, NUZYRA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
-
2 DOSAGE AND ADMINISTRATION
2.1 Important Administration Instructions
NUZYRA for Injection: Do NOT administer NUZYRA for injection with any solution containing multivalent cations, e.g., calcium and magnesium, through the same intravenous line [see Drug Interactions (7.2)] . Co-infusion with other medications has not been studied [see Dosage and Administration (2.5)] .
NUZYRA Tablets: Fast for at least 4 hours and then take with water. After oral dosing, no food or drink (except water) is to be consumed for 2 hours and no dairy products, antacids, or multivitamins for 4 hours [see Drug Interactions (7.2)and Clinical Pharmacology (12.3)].
2.2 Dosage in Adults with Community-Acquired Bacterial Pneumonia (CABP)
For treatment of adults with CABP the recommended dosage regimen (loading and maintenance) of NUZYRA is described in Table 1 below.
Table 1: Dosage of NUZYRA in Adult CABP Patients Loading Doses Maintenance Dose Treatment Duration NUZYRA Injection:
200 mg by intravenous infusion over 60 minutes on day 1.
OR
100 mg by intravenous infusion over 30 minutes, twice on day 1.
ORNUZYRA Injection:
100 mg by intravenous infusion over 30 minutes once daily.
OR
NUZYRA Tablets:
300 mg orally once daily.7 to 14 Days NUZYRA Tablets:
300 mg orally twice on day 1.2.3 Dosage in Adults with Acute Bacterial Skin Structure and Skin Infections (ABSSSI)
For treatment of adults with ABSSSI, the recommended dosage regimen (loading and maintenance) of NUZYRA is described in Table 2 below.
Table 2: Dosage of NUZYRA in Adult ABSSSI Patients Loading Doses Maintenance Dose Treatment Duration NUZYRA Injection:
200 mg by intravenous infusion over 60 minutes on day 1.
OR
100 mg by intravenous infusion over 30 minutes, twice on day 1.
ORNUZYRA Injection:
100 mg by intravenous infusion over 30 minutes once daily.
OR
NUZYRA Tablets:
300 mg orally once daily.7 to 14 Days NUZYRA Tablets:
450 mg orally once a day on day 1 and day 2.2.4 Dosage Adjustments in Patients with Renal or Hepatic Impairment
No dosage adjustment is warranted in patients with renal or hepatic impairment [see Clinical Pharmacology (12.3)].
2.5 Preparation and Administration of NUZYRA for Injection Intravenous Solution
Reconstitution and Dilution:
- NUZYRA must be reconstituted and then further diluted under aseptic conditions. To prepare the required dose for intravenous infusion, reconstitute and dilute the appropriate number of vials, as determined from Table 3 below.
- Reconstitute each 100 mg vial of NUZYRA with 5 mL of Sterile Water, 0.9% Sodium Chloride Injection, USP, or 5% Dextrose Injection, USP, for Injection.
- Gently swirl the contents and let the vial stand until the cake has completely dissolved and any foam disperses. Do not shake the vial.
- The reconstituted NUZYRA solution should be yellow to dark orange in color; if not, the solution should be discarded. Visually inspect the reconstituted NUZYRA solution for particulate matter and discoloration prior to further dilution and administration. If necessary, invert the vial to dissolve any remaining powder and swirl gently to prevent foaming.
- Immediately (within 1 hour), withdraw 5 mL or 10 mL of the reconstituted solution and further dilute to a 100 mL (nominal volume) of 0.9% Sodium Chloride Injection, USP, or 5% Dextrose Injection, USP, bag for injection. The concentration of the final diluted infusion solution will either be 1 mg/mL or 2 mg/mL in accordance with Table 3 below. Discard any unused portion of the reconstituted solution.
- Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
Table 3: Preparation of NUZYRA Intravenous Infusion NUZYRA for Injection Dose Number of Vials to Reconstitute for Further Dilution Volume of Reconstituted Solution (5 mL/vial) to Withdraw for Further Dilution Final Infusion Concentration of NUZYRA 200 mg 2 Vials 10 mL 2 mg/mL 100 mg 1 Vial 5 mL 1 mg/mL Storage of the Diluted Infusion Solution
The NUZYRA diluted infusion solution may be used within 24 hours at room temperature (less than or equal to 25°C) or within 7 days when refrigerated (2°C to 8°C). Do not freeze. Allow the infusion bag to reach room temperature prior to use.
Administration
After reconstitution and dilution, administer NUZYRA by intravenous infusion, using a total infusion time of 60 minutes for a 200 mg dose, or a total infusion time of 30 minutes for a 100 mg dose [see Dosage and Administration (2.2, 2.3)] .
Administer NUZYRA intravenously through a dedicated line or through a Y-site. If the same intravenous line is used for sequential infusion of several drugs, the line should be flushed with 0.9% Sodium Chloride Injection, USP, or 5% Dextrose Injection, USP, before and after infusion of NUZYRA. The compatibility of NUZYRA with other drugs and infusion solutions other than 5% Dextrose Injection, USP or 0.9% Sodium Chloride Injection, USP has not been established.
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
NUZYRA is contraindicated in patients with known hypersensitivity to omadacycline or tetracycline class antibacterial drugs, or to any of the excipients [see Warnings and Precautions (5.3)and Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Mortality Imbalance in Patients with Community-Acquired Bacterial Pneumonia
Mortality imbalance was observed in the CABP clinical trial with eight deaths (2%) occurring in patients treated with NUZYRA compared to four deaths (1%) in patients treated with moxifloxacin. The cause of the mortality imbalance has not been established.
All deaths, in both treatment arms, occurred in patients >65 years of age; most patients had multiple comorbidities [see Use in Specific Populations (8.5)] . The causes of death varied and included worsening and/or complications of infection and underlying conditions. Closely monitor clinical response to therapy in CABP patients, particularly in those at higher risk for mortality [see Adverse Reactions (6.1)].
5.2 Tooth Discoloration and Enamel Hypoplasia
The use of NUZYRA during tooth development (last half of pregnancy, infancy, and childhood up to the age of 8 years) may cause permanent discoloration of the teeth (yellow-gray-brown). This adverse reaction is more common during long-term use of the tetracycline class drugs, but it has been observed following repeated short-term courses. Enamel hypoplasia has also been reported with tetracycline class drugs. Advise the patient of the potential risk to the fetus if NUZYRA is used during the second or third trimester of pregnancy [see Use in Specific Populations (8.1, 8.4)].
5.3 Inhibition of Bone Growth
The use of NUZYRA during the second and third trimester of pregnancy, infancy and childhood up to the age of 8 years may cause reversible inhibition of bone growth. All tetracyclines form a stable calcium complex in any bone-forming tissue. A decrease in fibula growth rate has been observed in premature infants given oral tetracycline in doses of 25 mg/kg every 6 hours. This reaction was shown to be reversible when the drug was discontinued. Advise the patient of the potential risk to the fetus if NUZYRA is used during the second or third trimester of pregnancy [see Use in Specific Populations (8.1, 8.4)] .
5.4 Hypersensitivity Reactions
Hypersensitivity reactions have been reported with NUZYRA [see Adverse Reactions (6.1)]. Life-threatening hypersensitivity (anaphylactic) reactions have been reported with other tetracycline class antibacterial drugs. NUZYRA is structurally similar to other tetracycline class antibacterial drugs and is contraindicated in patients with known hypersensitivity to tetracycline class antibacterial drugs [see Contraindications (4)] . Discontinue NUZYRA if an allergic reaction occurs.
5.5 Clostridioides difficile-Associated Diarrhea
Clostridioides difficile-associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial drug use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial drug use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial drug treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.6 Tetracycline Class Effects
NUZYRA is structurally similar to tetracycline class of antibacterial drugs and may have similar adverse reactions. Adverse reactions including photosensitivity, fixed drug eruption, pseudotumor cerebri, and anti-anabolic action which has led to increased BUN, azotemia, acidosis, hyperphosphatemia, pancreatitis, and abnormal liver function tests, have been reported for other tetracycline class antibacterial drugs, and may occur with NUZYRA. Discontinue NUZYRA if any of these adverse reactions are suspected.
5.7 Development of Drug-Resistant Bacteria
Prescribing NUZYRA in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria [see Indications and Usage (1.3)].
-
6 ADVERSE REACTIONS
The following clinically significant adverse reactions are described in greater detail in the Warnings and Precautions section of labeling:
- Mortality Imbalance in Patients with Community-Acquired Bacterial Pneumonia [see Warnings and Precautions (5.1)]
- Tooth Development and Enamel Hypoplasia [see Warnings and Precautions (5.2)]
- Inhibition of Bone Growth [see Warnings and Precautions (5.3)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.4)]
- Tetracycline Class Effects [see Warnings and Precautions (5.6)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Overview of the Safety Evaluation of NUZYRA
NUZYRA was evaluated in three Phase 3 clinical trials (Trial 1, Trial 2 and Trial 3). These trials included a single Phase 3 trial in CABP patients (Trial 1) and two Phase 3 trials in ABSSSI patients (Trial 2 and Trial 3). Across all Phase 3 trials, a total of 1073 patients were treated with NUZYRA (382 patients in Trial 1 and 691 in Trials 2 and 3 of which 368 patients were treated with only oral NUZYRA).
Clinical Trial Experience in Patients with Community-Acquired Bacterial Pneumonia
Trial 1 was a Phase 3 CABP trial that enrolled 774 adult patients, 386 randomized to NUZYRA (382 received at least one dose of NUZYRA and 4 patients did not receive the study drug) and 388 randomized to moxifloxacin (all 388 received at least one dose of moxifloxacin). The mean age of patients treated with NUZYRA was 61 years (range 19 to 97 years) and 42% were greater than or equal to 65 years of age. Overall, patients treated with NUZYRA were predominantly male (53.7%), white (92.4%), and had a mean body mass index (BMI) of 27.3 kg/m 2. Approximately 47% of NUZYRA treated patients had CrCl <90 ml/min. Patients were administered an IV to oral switch dosage regimen of NUZYRA. The total treatment duration was 7 to 14 days. Mean duration of IV treatment was 5.7 days and mean total duration of treatment was 9.6 days in both treatment arms.
Imbalance in Mortality
In Trial 1, eight deaths (2%) occurred in 382 patients treated with NUZYRA as compared to four deaths (1%) in 388 patients treated with moxifloxacin. All deaths, in both treatment arms, occurred in patients >65 years of age. The causes of death varied and included worsening and/or complications of infection and underlying conditions. The cause of the mortality imbalance has not been established [see Warnings and Precautions (5.1)] .
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In Trial 1, a total of 23/382 (6.0%) patients treated with NUZYRA and 26/388 (6.7%) patients treated with moxifloxacin experienced serious adverse reactions.
Discontinuation of treatment due to any adverse reactions occurred in 21/382 (5.5%) patients treated with NUZYRA and 27/388 (7.0%) patients treated with moxifloxacin.
Most Common Adverse Reactions
Table 4 lists the most common adverse reactions occurring in ≥2% of patients receiving NUZYRA in Trial 1.
Table 4: Adverse Reactions Occurring in ≥2% of Patients Receiving NUZYRA in Trial 1 Adverse Reaction NUZYRA
(N = 382)Moxifloxacin
(N = 388)Alanine aminotransferase increased 3.7 4.6 Hypertension 3.4 2.8 Gamma-glutamyl transferase increased 2.6 2.1 Insomnia 2.6 2.1 Vomiting 2.6 1.5 Constipation 2.4 1.5 Nausea 2.4 5.4 Aspartate aminotransferase increased 2.1 3.6 Headache 2.1 1.3 Clinical Trials Experience in Patients with Acute Bacterial Skin and Skin Structure Infections
Trial 2 was a Phase 3 ABSSSI trial that enrolled 655 adult patients, 329 randomized to NUZYRA and 326 randomized to linezolid. Trial 3 was a Phase 3 ABSSSI trial that enrolled 735 adult patients, 368 randomized to NUZYRA and 367 randomized to linezolid.
In Trial 2 (IV to oral switch trial), the mean age of patients treated with NUZYRA was 47 years (range 19 to 88). Overall, patients treated with NUZYRA were predominantly male (62.8%), white (91.0%) and had a mean BMI of 28.1 kg/m 2.
In Trial 3 (oral only trial), the mean age of patients was 43 years (range 18 to 86). Patients treated with NUZYRA were predominantly male (65.8%), white (88.9%), and had a mean BMI of 27.9 kg/m 2.
In Trials 2 and 3, approximately 12% of NUZYRA treated patients had CrCl <90 ml/min. Overall, the mean and median calculated lesion area was similar across both trials. Trial 2 required at least 3 days of IV treatment followed by switch to oral regimen based on physician's discretion. Mean duration of IV treatment in Trial 2 was 4 days and mean total duration of treatment was 9 days in both treatment arms. In Trial 3, only oral therapy was administered, and mean total duration of treatment was 8 days in both treatment arms. The median days on treatment in the pooled ABSSSI trials was 9 days for both NUZYRA and linezolid.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In the pooled ABSSSI trials, serious adverse reactions occurred in 16/691 (2.3%) of patients treated with NUZYRA and 13/689 (1.9%) of patients treated with comparator. Discontinuation of treatment due to adverse events occurred in 12 (1.7%) NUZYRA treated patients, and 10 (1.5%) comparator treated patients. There was 1 death (0.1%) reported in NUZYRA treated patients and 3 deaths (0.4%) reported in linezolid patients in ABSSSI trials.
Most Common Adverse Reactions
Table 5 includes the most common adverse reactions occurring in ≥2% of patients receiving NUZYRA in Trials 2 and 3.
Table 5: Adverse Reactions Occurring in ≥2% of Patients Receiving NUZYRA in Pooled Trials 2 and 3 Adverse Reaction NUZYRA
(N = 691)Linezolid
(N = 689)- *
- In Trial 2, which included IV to oral dosing of NUZYRA, 40 (12%) patients experienced nausea and 17 (5%) patients experienced vomiting in NUZYRA treatment group as compared to 32 (10%) patients experienced nausea and 16 (5%) patients experienced vomiting in the comparator group. One patient (0.3%) in the NUZYRA group discontinued treatment due to nausea and vomiting. In Trial 3, which included the oral loading dose of NUZYRA, 111 (30%) patients experienced nausea and 62 (17%) patients experienced vomiting in NUZYRA treatment group as compared to 28 (8%) patients experienced nausea and 11 (3%) patients experienced vomiting in the linezolid group. One patient (0.3%) in the NUZYRA group discontinued treatment due to nausea and vomiting
- †
- Infusion site extravasation, pain, erythema, swelling, inflammation, irritation, peripheral swelling and skin induration.
Nausea * 21.9 8.7 Vomiting 11.4 3.9 Infusion site reactions † 5.2 3.6 Alanine aminotransferase increased 4.1 3.6 Aspartate aminotransferase increased 3.6 3.5 Headache 3.3 3.0 Diarrhea 3.2 2.9 Selected Adverse Reactions Occurring in Less Than 2% of Patients Receiving NUZYRA in Trials 1, 2 and 3
The following selected adverse reactions were reported in NUZYRA-treated patients at a rate of less than 2% in Trials 1, 2 and 3.
Cardiovascular System Disorders:tachycardia, atrial fibrillation
Blood and Lymphatic System Disorders:anemia, thrombocytosis
Ear and Labyrinth Disorders:vertigo
Gastrointestinal Disorders:abdominal pain, dyspepsia
General Disorders and Administration Site Conditions:fatigue
Immune System Disorders:hypersensitivity
Infections and Infestations:oral candidiasis, vulvovaginal mycotic infection
Investigations:creatinine phosphokinase increased, bilirubin increased, lipase increased, alkaline phosphatase increased
Nervous System Disorders:dysgeusia, lethargy
Respiratory, Thoracic, and Mediastinal disorders:oropharyngeal pain
Skin and Subcutaneous Tissue Disorders:pruritus, erythema, hyperhidrosis, urticaria
-
7 DRUG INTERACTIONS
7.1 Anticoagulant Drugs
Because tetracyclines have been shown to depress plasma prothrombin activity, patients who are on anticoagulant therapy may require downward adjustment of their anticoagulant dosage while also taking NUZYRA.
7.2 Antacids and Iron Preparations
Absorption of oral tetracyclines, including NUZYRA, is impaired by antacids containing aluminum, calcium, or magnesium, bismuth subsalicylate, and iron containing preparations [see Dosage and Administration (2.1)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
NUZYRA, like other tetracycline class antibacterial drugs, may cause discoloration of deciduous teeth and reversible inhibition of bone growth when administered during the second and third trimester of pregnancy [see Warnings and Precautions (5.2, 5.3), Data, Use in Specific Populations (8.4)].
The limited available data of NUZYRA use in pregnant women is insufficient to inform drug associated risk of major birth defects and miscarriages. Animal studies indicate that administration of omadacycline during the period of organogenesis resulted in fetal loss and/or congenital malformations in pregnant rats and rabbits at 7 times and 3 times the mean AUC exposure, respectively, of the clinical intravenous dose of 100 mg and the oral dose of 300 mg. Reductions in fetal weight occurred in rats at all administered doses (see Data). In a fertility study, administration to rats during mating and early pregnancy resulted in embryo loss at 20 mg/kg/day; systemic exposure based on AUC was approximately equal to the clinical exposure level [see Nonclinical Toxicology (13.1)] . Results of studies in rats with omadacycline have shown tooth discoloration.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15-20%.
Data
Animal Data
Intravenous infusion of omadacycline to pregnant rats during organogenesis (gestation days 6-17) at doses of 5 to 80 mg/kg/day resulted in maternal lethality at 80 mg/kg/day. Increased embryo-fetal lethality and fetal malformations (whole body edema) occurred at 60 mg/kg/day (7 times the clinical AUC), dose-dependent reductions in fetal body weight occurred at all doses, and delayed skeletal ossification occurred at doses as low as 10 mg/kg/day (Systemic exposure based on AUC at a similar dose in unmated female rats in a separate study was approximately half the clinical exposure). In pregnant rabbits, intravenous infusion of 5, 10 or 20 mg/kg/day during organogenesis (gestation days 7-18) resulted in maternal lethality and body weight loss at 20 mg/kg/day. Embryo-fetal lethality, congenital malformations of the skeleton, and reduced fetal weight also occurred at 20 mg/kg/day (7 times the clinical AUC). Cardiac and lung malformations were present in dose-related incidence at 10 and 20 mg/kg/day. The fetal no-adverse-effect-level in the rabbit embryo-fetal development study was 5 mg/kg/day, at approximately 1.2 times the clinical steady state AUC.
Intravenous infusion of omadacycline to pregnant and lactating rats at doses of 7.5, 15 and 30 mg/kg/day did not adversely affect survival, growth (other than lower pup body weights and/or gains at the high dose that were only statistically significant at sporadic intervals), postnatal development, behavior, or reproductive capability of offspring at maternal doses up to 30 mg/kg/day (approximately equivalent to 3 times the IV clinical dose of 100 mg/day, based on doses normalized for total body surface area), the highest dose tested, although dosing was discontinued early in a number of animals in this group due to injection site intolerance.
Results of animal studies indicate that tetracyclines cross the placenta, are found in fetal tissues, and can have toxic effects on the developing fetus (often related to retardation of skeletal development). Evidence of embryotoxicity also has been noted in animals treated early in pregnancy.
8.2 Lactation
Risk Summary
There is no information on the presence of omadacycline in human milk, the effects on the breastfed infant or the effects on milk production. Tetracyclines are excreted in human milk; however, the extent of absorption of tetracyclines, including omadacycline, by the breastfed infant is not known. Because there are other antibacterial drug options available to treat CABP and ABSSSI in lactating women and because of the potential for serious adverse reactions, including tooth discoloration and inhibition of bone growth, advise patients that breastfeeding is not recommended during treatment with NUZYRA and for 4 days (based on half-life) after the last dose.
8.3 Females and Males of Reproductive Potential
Contraception
Females
NUZYRA may produce embryonic or fetal harm [see Use in Specific Populations (8.1)]. Advise patients to use an acceptable form of contraception while taking NUZYRA.
Infertility
Males
In rat studies, injury to the testis and reduced sperm counts and motility occurred in male rats after treatment with omadacycline [see Nonclinical Toxicology (13.1)].
Females
In rat studies, omadacycline affected fertility parameters in female rats, resulting in reduced ovulation and increased embryonic loss at intended human exposures [see Nonclinical Toxicology (13.1)] .
8.4 Pediatric Use
Safety and effectiveness of NUZYRA in pediatric patients below the age of 18 years have not been established.
Due to the adverse effects of the tetracycline class of drugs, including NUZYRA on tooth development and bone growth, use of NUZYRA in pediatric patients less than 8 years of age is not recommended [see Warnings and Precautions (5.1, 5.2)].
8.5 Geriatric Use
Of the total number of patients who received NUZYRA in the Phase 3 clinical trials (n=1073), 200 patients were ≥65 years of age, including 92 patients who were ≥75 years of age. In Trial 1, numerically lower clinical success rates at early clinical response (ECR) timepoint for NUZYRA-treated and moxifloxacin-treated patients (75.5% and 78.7%, respectively) were observed in CABP patients ≥ 65 years of age as compared to patients <65 years of age (85.2% and 86.3%, respectively). Additionally, all deaths in the CABP trial occurred in patients >65 years of age [see Adverse Reactions (6.1)].
No significant difference in NUZYRA exposure was observed between healthy elderly subjects and younger subjects following a single 100 mg IV dose of NUZYRA [see Clinical Pharmacology (12.3)] .
8.6 Hepatic Impairment
No dose adjustment of NUZYRA is warranted in patients with mild, moderate, or severe hepatic insufficiency (Child-Pugh classes A, B, or C) [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dose adjustment of NUZYRA is warranted in patients with mild, moderate, or severe renal impairment, including patients with end stage renal disease who are receiving hemodialysis [see Clinical Pharmacology (12.3)].
- 10 OVERDOSAGE
-
11 DESCRIPTION
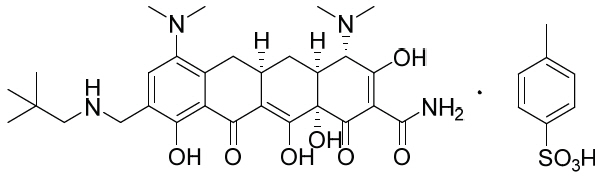
NUZYRA contains omadacycline tosylate, an aminomethylcycline which is a semisynthetic derivative of the tetracycline class of antibacterial drugs, for intravenous or oral administration. The chemical name of omadacycline tosylate is (4S,4aS,5aR,12aS)-4,7-bis(dimethylamino)-9-(2,2-dimethylpropylaminomethyl)-3,10,12,12a-tetrahydroxy-1,11-dioxo-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide, 4-methylbenzenesulfonate.
The molecular formula is C 36H 48N 4O 10S (monotosylate salt) and the molecular weight is 728.9 (monotosylate salt). The following represents the chemical structure of omadacycline tosylate:
NUZYRA (omadacycline) for injection is a yellow to dark orange sterile lyophilized powder. Each vial of NUZYRA for injection contains 100 mg of omadacycline (equivalent to 131 mg omadacycline tosylate). Inactive ingredients: Sucrose (100 mg); may include hydrochloric acid and/or sodium hydroxide for pH adjustment.
NUZYRA (omadacycline) tablets for oral administration are yellow film coated tablets containing 150 mg of omadacycline (equivalent to 196 mg omadacycline tosylate), and the following inactive ingredients: Colloidal silicon dioxide, crospovidone, glycerol monocaprylocaprate, iron oxide yellow, lactose monohydrate, microcrystalline cellulose, polyvinyl alcohol, sodium bisulfite, sodium lauryl sulfate, sodium stearyl fumarate, talc, and titanium dioxide.
-
12 CLINICAL PHARMACOLOGY
12.2 Pharmacodynamics
Cardiac Electrophysiology
Based on the nonclinical and clinical data, including electrocardiogram evaluation in the phase 3 clinical trials, one of which had moxifloxacin as a control group, no clinically relevant QTc prolongation was observed at the maximum recommended dose of omadacycline.
Cardiac Physiology-Increase in Heart Rate
In phase 1 studies conducted in healthy volunteers, reversible dose-dependent increases in heart rate have been observed following administration of single and multiple doses of omadacycline. The clinical implication of this finding is unknown [see Adverse Reactions (6.1)].
In a standard radiolabeled ligand binding assays, omadacycline was shown to inhibit binding of H-scopolamine to the M2 subtype of the muscarinic acetylcholine receptor. In the heart, muscarinic M2 receptors serve as mediators of the parasympathetic input that normally is received via the vagus nerve and stimulation of the receptor increases membrane potassium conductance through the acetylcholine-dependent channel, which slows depolarization and reduces pacemaker activity in the sinoatrial node.
12.3 Pharmacokinetics
The pharmacokinetic parameters of NUZYRA after single and multiple oral and intravenous doses are summarized in Table 6.
Table 6: Mean (SD) Pharmacokinetic Parameters of NUZYRA in Healthy Adult Subjects Dose and Route of Administration 100 mg IV 300 mg Oral 450 mg Oral C max= maximum plasma concentration, AUC = area under concentration-time curve, IV = intravenous, ND = not determined, T max= time to C max PK Parameters * C maxng/mL Single dose 1507 (582)
(n=63)548 (146)
(n=103)874 (232)
(n=24)Steady state 2116 (680)
(n=41)952 (420)
(n=43)1077 (269)
(n=24)AUCh*ng/mL Single dose † 9358 (2072)
(n=62)9399 (2559)
(n=102)13504 (3634)
(n=24)Steady state ‡ 12140 (3223)
(n=41)11156 (5010)
(n=43)13367 (3469)
(n=24)Accumulation Accumulation ratio 1.5 Absorption Bioavailability 34.5% following single 300 mg dose of NUZYRA T maxMedian (min, max) Single dose 0.6 (0.3, 0.7)
(n=63)2.5 (1, 4.1)
(n=103)2.5 (1.5, 3)
(n=24)Steady state 0.5 (0, 1)
(n=41)2.5 (0, 8)
(n=43)2.5 (1.5, 4)
(n=24)Distribution Plasma Protein Binding 20%; not concentration dependent Volume of DistributionL Single dose 256 (66)
(n=62)794 §(188)
(n=27)914 §(821.9)
(n=23)Steady state 190 (53)
(n=41)440 §(262)
(n=34)607 §(197.4)
(n=24)Elimination Elimination Half-Lifeh Single dose 16.4 (2.1)
(n=62)15.0 (2.5)
(n=81)13.45 (1.7)
(n=23)Steady state 16.0 (3.5)
(n=41)15.5 (1.7)
(n=21)16.83 (1.4)
(n=23)Systemic ClearanceL/h Single dose 11.24 (2.7)
(n=62)34.6 §(10.7)
(n=27)43.4 §(49.8)
(n=23)Steady state 8.8 (2.2)
(n=41)18.3 §(8.3)
(n=34)21.2 §(8.9)
(n=24)Renal ClearanceL/h 3.1 (0.69)
(n=8)Metabolism Omadacycline is not metabolized Excretion(% dose) Urine 27 (3.5)
(n=8)14.4 ¶(2.3)
(n=6)ND Feces ND 81.1 ¶(2.3)
(n=6)ND Absorption
The exposure to omadacycline is similar between a 300 mg oral dose and a 100 mg intravenous dose of NUZYRA in healthy fasted subjects.
Effect of Food
Ingestion of a standard high-fat nondairy meal (855 calories; 59% calories from fat) and standard high-fat meal including dairy (985 calories; 60% calories from fat) 2-hours before administration of a single 300 mg oral dose of NUZYRA decreased the rate (Cmax) and extent of absorption (AUC) by 40% and 42%, and 59% and 63%, respectively compared to administration of NUZYRA under fasting conditions. The rate and extent of absorption of NUZYRA were not substantially decreased when a high-fat nondairy meal (800-1000 calories; 50% calories from fat) was ingested 4 hours pre-dose.
Following ingestion of either a light non-fat (300-350 calories; ≤5% calories from fat), or a standard low-fat (800-1000 calories; 30% calories from fat), or a standard high fat (800-1000 calories; 50% calories from fat) meal 2 hours post-dose, the AUC and C maxwere not substantially altered, as compared to fasting conditions.
Distribution
Plasma protein binding of omadacycline is approximately 20% and is not concentration dependent. The mean (% CV) volume of distribution of omadacycline at steady-state following IV administration of NUZYRA in healthy subjects was 190 (27.7) L.
Excretion
Following a 100 mg IV dose of NUZYRA, 27% of the dose was recovered as unchanged omadacycline in the urine. In healthy male volunteers receiving 300 mg oral [ 14C] NUZYRA, 77.5% to 84.0% of the dose was recovered in the feces, approximately 14.4% (range 10.8% to 17.4%) in the urine, with 95.5% of the administered radioactive dose recovered after 7 days.
Lung Penetration
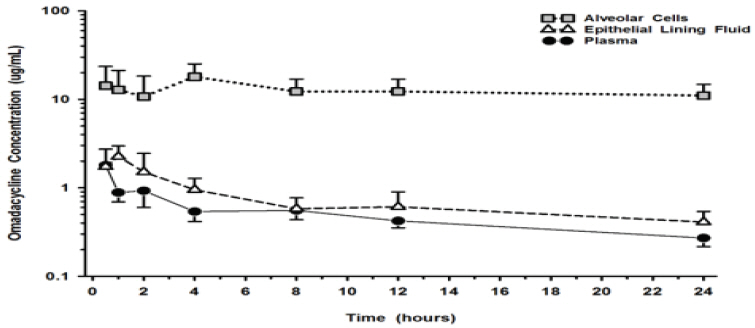
The mean omadacycline concentrations over time for alveolar cells (AC), epithelial lining fluid (ELF), and plasma following IV administration of multiple doses of 100 mg of NUZYRA to healthy volunteers are shown in Figure 1. The steady-state omadacycline AUC 0-24h(302.5 hr*mcg/mL) in AC was 25.8-fold higher than the plasma AUC 0-24h, and the AUC 0-24h(17.2 hr*mcg/mL) in ELF was 1.5-fold higher than the AUC 0-24hin plasma.
Figure 1: Mean (± SD) Concentrations of Omadacycline in Alveolar Cells, Epithelial Lining, and Plasma Following Multiple 100 mg IV Doses of NUZYRA to Healthy Subjects During Bronchoscopy Sampling Times
Specific Populations
No clinically significant differences in the pharmacokinetics of omadacycline were observed based on age, gender, race, weight, renal impairment or end-stage renal disease, and hepatic impairment.
Patients with Renal Impairment
A study was conducted to compare NUZYRA pharmacokinetics following 100 mg IV administration in 8 subjects with end-stage renal disease (ESRD) on stable hemodialysis, with and 8-matched healthy control subjects. In the ESRD subjects, NUZYRA was administered on two separate occasions; immediately prior to dialysis and after dialysis, and the AUC, C max, and CL of NUZYRA were comparable between the renally impaired subjects and the matching healthy subjects. During dialysis, 7.9% of omadacycline was recovered in the dialysate. Renal impairment did not impact NUZYRA elimination.
Patients with Hepatic Impairment
A study was conducted to compare NUZYRA pharmacokinetics following intravenous and oral dosing to 5 subjects with mild hepatic impairment (Child-Pugh Class A), 6 subjects with moderate hepatic impairment (Child-Pugh Class B), and 6 subjects with severe hepatic impairment (Child-Pugh Class C) as compared to 12 matched healthy control subjects. The AUC and C maxof NUZYRA were comparable between the hepatically impaired subjects and the matching healthy subjects, and similar clearance was observed across all cohorts. Hepatic impairment did not impact NUZYRA elimination.
Drug Interaction Studies
Clinical Studies
Administration of oral verapamil (P-gp inhibitor) two hours prior to a single 300 mg oral dose of NUZYRA increased omadacycline AUC by approximately 25% and C max by approximately 9%.
In vitro Studies
In vitro studies in human liver microsomes indicate that omadacycline does not inhibit nor induce metabolism mediated by CYP 1A1, 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4/5, or UGT1A1. Therefore, NUZYRA is not expected to alter the pharmacokinetics of drugs metabolized by the above stated human hepatic enzymes.
Omadacycline is not an inhibitor of P-gp and organic anion transporting polypeptide (OATP) 1B1 and OATP1B3. Omadacycline is a substrate of P-gp (see Clinical Studies above). Omadacycline is not a substrate or inhibitor of the major organic anion transporters (OAT-1 and 3), breast cancer resistance protein (BCRP), or multidrug resistance-associated protein 2 (MRP2). Omadacycline was not an OATP1B1 or OATP1B3 substrate at supra-therapeutic concentrations (5-13 fold higher than clinically relevant concentrations).
12.4 Microbiology
Mechanism of Action
Omadacycline is an aminomethylcycline antibacterial within the tetracycline class of antibacterial drugs. Omadacycline binds to the 30S ribosomal subunit and blocks protein synthesis. In general, omadacycline is considered bacteriostatic; however, omadacycline has demonstrated bactericidal activity against some isolates of S. pneumoniae and H. influenzae.
Resistance
The following in vitro data are available, but their clinical significance is unknown. Omadacycline was active in vitro against Gram-positive bacteria expressing ribosomal protection proteins (TetM) and tetracycline resistance active efflux pumps (TetK and TetL), and in Enterobacteriaceae expressing the TetB efflux pump. Additionally, omadacycline was active against some S. aureus, S. pneumoniae, and H. influenzae strains carrying macrolide resistance genes ( ermA,Band/or C), or ciprofloxacin resistance genes ( gyrAand parC) and beta-lactamase positive H. influenzae.
Interaction with Other Antimicrobials
In vitro studies have not demonstrated antagonism between omadacycline and other commonly used antibacterials (ampicillin, ceftazidime, ceftriaxone, imipenem, piperacillin/tazobactam, gentamicin, vancomycin, daptomycin, linezolid).
Antimicrobial Activity
Omadacycline has been shown to be active against most isolates of the following bacteria, both in vitro and in clinical infections [see Indications and Usage (1.1, 1.2)] .
Community-Acquired Bacterial Pneumonia (CABP)
-
Gram-positive bacteria
Streptococcus pneumoniae
Staphylococcus aureus (methicillin-susceptible isolates)
-
Gram-negative bacteria
Haemophilus influenzae
Haemophilus parainfluenzae
Klebsiella pneumoniae
-
Other microorganisms
Chlamydophila pneumoniae
Legionella pneumophila
Mycoplasma pneumoniae
Acute Bacterial Skin and Skin Structure Infections (ABSSSI)
-
Gram-positive bacteria
Enterococcus faecalis
Staphylococcus aureus (methicillin-susceptible and -resistant isolates)
Staphylococcus lugdunensis
Streptococcus anginosus grp. (includes S. anginosus, S. intermedius, and S. constellatus)
Streptococcus pyogenes
-
Gram-negative bacteria
Enterobacter cloacae
Klebsiella pneumoniae
The following in vitro data are available, but their clinical significance is unknown. At least 90% of isolates of the following bacteria exhibit an in-vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for NUZYRA against isolates of similar genus or organism group. However, the efficacy of NUZYRA in treating clinical infections due to these bacteria has not been established in adequate and well controlled clinical trials.
-
Gram-positive bacteria
Enterococcus faecium(vancomycin-susceptible and -resistant isolates)
Streptococcus agalactiae
-
Gram-negative bacteria
Enterobacter aerogenes
Escherichia coli
Citrobacter freundii
Citrobacter koseri
Klebsiella oxytoca
Moraxella catarrhalis
-
Gram-positive bacteria
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies with omadacycline have not been conducted. However, there has been evidence of oncogenic activity in rats in studies with the related antibacterial drugs, oxytetracycline (adrenal and pituitary tumors), and minocycline (thyroid tumors).
Mutagenesis
Omadacycline was positive for clastogenicity and aneugenicity in an in vitro chromosome aberration assay in Chinese hamster ovary (CHO) cells and for mutagenicity in an in vitro forward mutation assay in mouse lymphoma cells. These effects were seen in the presence of metabolizing enzymes.
Omadacycline was negative in a chromosomal aberration test in Chinese hamster V79 cells and in vivo micronucleus assays administered intraperitoneally to ICR mice or intravenously to HanRcc: WIST rats.
Impairment of Fertility
Omadacycline administration to male rats in a fertility study caused reduced sperm counts and sperm motility at 20-mg/kg/day (approximately 1.3 times clinical systemic exposure, based on AUC in a separate study in rats at a similar dose), but had no effect on male fertility parameters. In general toxicity studies, inhibition of spermatogenesis occurred after administration of 45-mg/kg/day omadacycline (6 to 8 times the clinical AUC exposure) for 37 days or longer, but not at lower doses (15-mg/kg/day, ≤ 2 times clinical AUC exposure) or shorter treatment periods (4 weeks or less). In female rats, fertility was reduced at the 20-mg/kg/day dose (approximately equivalent to human exposures in a separate study in unmated females), characterized by reduced ovulation and increased embryonic loss when treatment occurred from before mating through early pregnancy.
13.2 Animal Toxicology and/or Pharmacology
Hyperpigmentation of the thyroid has been produced by members of the tetracycline class in the following species: in rats by omadacycline, oxytetracycline, doxycycline, tetracycline PO4, and methacycline; in minipigs by doxycycline, minocycline, tetracycline PO4, and methacycline; in dogs by doxycycline and minocycline; in monkeys by omadacycline and minocycline.
Minocycline, tetracycline PO4, methacycline, doxycycline, tetracycline base, oxytetracycline HCl, and tetracycline HCl were goitrogenic in rats fed a low iodine diet. This goitrogenic effect was accompanied by high radioactive iodine uptake. Administration of minocycline also produced a large goiter with high radioiodine uptake in rats fed a relatively high iodine diet.
Treatment of various animal species with this class of drugs has also resulted in the induction of thyroid hyperplasia in the following: in rats and dogs (minocycline); in chickens (chlortetracycline); and in rats and mice (oxytetracycline). Adrenal gland hyperplasia has been observed in goats and rats treated with oxytetracycline.
-
14 CLINICAL STUDIES
14.1 Community-Acquired Bacterial Pneumonia
A total of 774 adults with CABP were randomized in a multinational, double-blind, double-dummy trial (Trial 1, NCT #02531438) comparing NUZYRA to moxifloxacin. NUZYRA was administered 100 mg intravenously every 12 hours for two doses on Day 1, followed by 100 mg intravenously daily, or 300 mg orally, daily. Moxifloxacin 400 mg was administered intravenously or orally daily. Total treatment duration was 7-14 days. All enrolled patients were expected to require a minimum of at least 3 days of intravenous treatment. Efficacy and safety of an oral loading dose was not evaluated in CABP.
A total of 386 patients were randomized to NUZYRA and 388 patients were randomized to moxifloxacin. Patient demographic and baseline characteristics were balanced between the treatment groups. Patients were predominantly male (55%) and white (92%). Approximately 60% of patients in each group belonged to PORT Risk Class III, 26% were PORT Risk Class IV and 14.5% were PORT Risk Class II. The median age was 62 years, mean BMI was 27.34 kg/m 2, and approximately 47% of NUZYRA treated patients had CrCl <90 ml/min. Among NUZYRA-treated patients, common comorbid conditions included hypertension (49.5%), diabetes mellitus (16.3%), chronic lung disease (21.2%), atrial fibrillation (10.1%), and coronary artery disease (9.1%). The majority of sites were in Eastern Europe, which accounted for 82% of enrollment; 3 patients were enrolled in the US.
Clinical success at the early clinical response (ECR) timepoint, 72 to 120 hours after the first dose, was defined as survival with improvement in at least two of four symptoms (cough, sputum production, chest pain, dyspnea) without deterioration in any of these four symptoms in the intent to treat population (ITT), which consisted of all randomized patients.
Table 7presents the clinical success rates at the ECR timepoint (ITT population).
Table 7: Clinical Success at the ECR Timepoint in Trial 1 (ITT Population) Endpoint NUZYRA (%) Moxifloxacin (%) Treatment Difference
(95% CI *)* Clinical Success at the early clinical response (ECR) timepoint, 72 to 120 hours after the first dose, was defined as survival with improvement in at least two of four symptoms (cough, sputum production, chest pain, dyspnea) from baseline without deterioration in any of these symptoms, with no receipt of antibacterial treatment either as a rescue for CABP or as a treatment for other infections that may be effective for CABP, and no discontinuation of study treatment due to AE. - *
- 95% confidence interval for the treatment difference
Clinical Success 81.1 82.7 -1.6 (-7.1, 3.8) Clinical response was also assessed by the investigator at the post therapy evaluation visit (PTE), 5 to 10 days after last dose of study drug and defined as survival and improvement in signs and symptoms of CABP, based on the clinician's judgment, to the extent that further antibacterial therapy is not necessary. Table 8 presents the results of clinical response at the PTE visit for both the ITT population and the Clinically Evaluable (CE) population, which consisted of all ITT patients who had a diagnosis of CABP, received a minimum number of expected doses of study drug, did not have any protocol deviations that would affect the assessment of efficacy, and had investigator assessment at the PTE visit. Clinical response rates by most common baseline pathogen in the microbiological ITT (micro-ITT) population, defined as all randomized patients with a baseline pathogen are presented in Table 9.
Table 8: Investigator's Overall Assessment of Clinical Response at PTE *in Trial 1 (ITT and CE Population) Endpoint Population NUZYRA n/N (%) Moxifloxacin n/N (%) Treatment Difference (95% CI †) - *
- Investigator's overall assessment of clinical response at PTE was defined as survival and improvement in signs and symptoms of CABP, based on the clinician's judgment, to the extent that further antibacterial therapy is not necessary in the ITT and CE populations.
- †
- 95% confidence interval for the treatment difference.
Clinical Success at PTE ITT 338/386 (87.6) 330/388 (85.1) 2.5 (-2.4, 7.4) Clinical Success at PTE CE 316/340 (92.9) 312/345 (90.4) 2.5 (-1.7, 6.8) Table 9: Investigator's Overall Assessment of Clinical Response at PTE by Baseline Pathogen in Trial 1 (micro-ITT population) Pathogen NUZYRA n/N (%) Moxifloxacin n/N (%) Streptococcus pneumoniae 37/43 (86.0) 31/34 (91.2) Methicillin-susceptible Staphylococcus aureus (MSSA ) 8/11 (72.7) 8/10 (80.0) Haemophilus influenzae 26/32 (81.3) 16/16 (100) Haemophilus parainfluenzae 15/18 (83.3) 13/17 (76.5) Klebsiella pneumoniae 10/13 (76.9) 11/13 (84.6) Legionella pneumophila 27/29 (93.1) 27/28 (96.4) Mycoplasma pneumoniae 31/35 (88.6) 25/29 (86.2) Chlamydophila pneumoniae 14/15 (93.3) 13/14 (92.9) 14.2 Acute Bacterial Skin and Skin Structure Infections
A total of 1390 adults with ABSSSI were randomized in two multicenter, multinational, double-blind, double-dummy trials (Trial 2 NCT #02378480 and Trial 3 NCT #02877927). Both trials compared 7 to 14 days of NUZYRA to linezolid. Patients with cellulitis, major abscess, or wound infection were enrolled in the trials.
In Trial 2, 329 patients were randomized to NUZYRA (100 mg intravenously every 12 hours for 2 doses followed by 100 mg intravenously every 24 hours, with the option to switch to 300 mg orally every 24 hours) and 326 patients were randomized to linezolid (600 mg intravenously every 12 hours, with the option to switch to 600 mg orally every 12 hours). Patients in the trial had the following infections: cellulitis (38%), wound infection (33%), and major abscess (29%). The mean surface area of the infected lesion was 455 cm 2in NUZYRA-treated patients and 498 cm 2in linezolid-treated patients. The mean age of patients was 47 years. Subjects were predominantly male (65%) and white (92%), and mean BMI was 28.1 kg/m 2. Among NUZYRA-treated patients, common comorbid conditions included drug abuse (53.9%), hepatitis C (29.1%), hypertension (20.4%), anxiety (19.5%), and depression (15.5%). Trial 2 was conducted globally including approximately 60% of patients enrolled in the United States.
In Trial 3, 368 patients were randomized to NUZYRA (450 mg oral once a day on Days 1 and 2, followed by 300 mg orally once a day) and 367 were randomized to linezolid (600 mg orally every 12 hours). All patients were enrolled in the United States. Patients in the trial had the following infections: wound infections (58%), cellulitis (24%), and major abscess (18%). The mean surface area of the infected lesion was 424 cm 2in NUZYRA-treated patients and 399 cm 2in linezolid-treated patients. The mean age of patients was 44 years. Subjects were predominantly male (63%) and white (91%) and mean BMI was 27.9 kg/m 2. The most common comorbid conditions included drug abuse (72.8%), tobacco use (12.0%), and chronic hepatitis C infection (31.5%).
In Trials 2 and 3, approximately 12% of NUZYRA-treated patients had CrCl <90 ml/min.
In both trials, efficacy was determined by the successful early clinical response at 48 to 72 hours after the first dose in the mITT population and was defined as a 20% or greater decrease in lesion size. Table 10 summarizes the clinical response rates in the two trials. The mITT population was defined as all randomized subjects without a sole Gram-negative causative pathogen at screening.
Table 10: Clinical Success *at the ECR Timepoint in the mITT Population in Trial 2 and Trial 3 Study NUZYRA (%) Linezolid (%) Treatment Difference
(Two-Sided 95% CI) †- *
- Clinical success at early clinical response (ECR) at 48 to 72 hours after the first dose, was defined as a 20% or greater decrease in lesion size without any reasons for failure (less than 20% reduction in lesion size, administration of rescue antibacterial therapy, use of another antibacterial or surgical procedure to treat for lack of efficacy, or death).
- †
- 95% confidence interval for the treatment difference.
Trial 2 84.8 85.5 -0.7 (-6.3, 4.9) Trial 3 87.3 82.2 +5.1 (-0.2, 10.5) Clinical response at the post therapy evaluation (PTE, 7 to 14 days after last dose) visit in the mITT and clinically evaluable (CE) populations was defined as survival after completion of study treatment without receiving any alternative antibacterial therapy other than NUZYRA, without unplanned major surgical intervention, and sufficient resolution of infection such that further antibacterial therapy is not needed (see Table 11). Clinical response rates at PTE by most common pathogen in the microbiological-mITT population, defined as all patients in the mITT population, who had at least 1 Gram- positive causative pathogen identified at baseline are provided in Table 12. The CE population consisted of all mITT patients who had a diagnosis of ABSSSI, received a minimum number of expected doses of study drug, did not have any protocol deviations that would affect the assessment of efficacy, and had investigator assessment at the PTE Visit.
Table 11: Investigator's Overall Assessment of Clinical Response at PTE in mITT and CE Population in Trial 2 and Trial 3 Study Population NUZYRA n/N (%) Linezolid n/N (%) Treatment Difference
(Two-Sided 95% CI) *- *
- 95% confidence interval for the treatment difference.
Trial 2 mITT 272/316 (86.1) 260/311 (83.6) +2.5 (-3.2, 8.2) CE 259/269 (96.3) 243/260 (93.5) +2.8 (-1.0, 6.9) Trial 3 mITT 296/353 (83.9) 284/353 (80.5) +3.4 (-2.3, 9.1) CE 272/278 (97.8) 272/285 (95.4) +2.4 (-0.6, 5.8) Table 12: Investigator's Overall Assessment of Clinical Response at PTE by Baseline Pathogen in Trials 2 and 3 (micro-mITT population) Pathogen NUZYRA
n/N (%)Linezolid
n/N (%)Staphylococcus aureus 305/369 (82.7) 306/378 (81.0) Methicillin-susceptible Staphylococcus aureus(MSSA) 164/201 (81.6) 181/226 (80.1) Methicillin-resistant Staphylococcus aureus(MRSA) 146/173 (84.4) 128/157 (81.5) Staphylococcus lugdunensis 10/11 (90.9) 2/3 (66.7) Streptococcus anginosusgroup 84/104 (80.8) 59/82 (72.0) Streptococcus pyogenes 28/40 (70.0) 25/34 (73.5) Enterococcus faecalis 17/18 (94.4) 21/25 (84.0) Enterobacter cloacae 11/14 (78.6) 9/11 (81.8) Klebsiella pneumoniae 8/11 (72.7) 6/11 (54.5) -
16 HOW SUPPLIED/STORAGE AND HANDLING
16.1 How Supplied
NUZYRA for Injection
NUZYRA for Injection is supplied as a sterile lyophilized powder in a single-dose colorless glass vial, with each vial containing 100 mg of NUZYRA (equivalent to 131 mg omadacycline tosylate).
They are supplied as follows: 100 mg single-dose vial (NDC 71715-001-02), packaged in cartons of 10.
NUZYRA Tablets
NUZYRA Tablets contains 150 mg of omadacycline (equivalent to 196 mg omadacycline tosylate) in yellow, diamond-shaped, film-coated tablets debossed with OMC on one side and 150 on the other side.
They are supplied as follows:
Blister package of 6 (NDC 71715-002-21)
Blister package of 30 (5 blister cards of 6 tablets each) (NDC 71715-002-27)
16.2 Storage and Handling
NUZYRA for Injection and NUZYRA Tablets should be stored at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature] [see Dosage and Administration (2.5)]. Do not freeze.
-
17 PATIENT COUNSELING INFORMATION
Nausea and Vomiting
Advise patients that nausea and vomiting can be an adverse reaction to NUZYRA. Advise patients that a greater proportion of patients who received the oral loading dose of NUZYRA for treatment of ABSSSI experienced nausea and vomiting.
Allergic Reactions
Advise patients that allergic reactions, including serious allergic reactions, could occur and that serious allergic reactions require immediate treatment. Ask the patient about any previous hypersensitivity reactions to NUZYRA, or other tetracycline class antibacterials [see Warnings and Precautions (5.4)] .
Administration with Food
Instruct patients to fast 4 hours before and 2 hours after taking NUZYRA tablets and not to consume dairy products, antacids, or multivitamins for 4 hours after taking NUZYRA tablets [see Dosage and Administration (2.1)and Clinical Pharmacology (12.3)] .
Tooth Discoloration and Inhibition of Bone Growth
Advise patients that NUZYRA, like other tetracycline class drugs, may cause permanent tooth discoloration of deciduous teeth and reversible inhibition of bone growth when administered during the second and third trimesters of pregnancy. Tell your healthcare provider right away if you become pregnant during treatment [see Warnings and Precautions (5.1, 5.2)and Use in Specific Populations (8.1, 8.4)].
Lactation
Advise women not to breastfeed during treatment with NUZYRA and for 4 days after the last dose [see Use in Specific Populations (8.2)].
Diarrhea
Advise patients that diarrhea is a common problem caused by antibacterial drugs, including NUZYRA, which usually ends when the antibacterial drugs is discontinued. Sometimes after starting treatment with antibacterial drugs, patients can develop watery or bloody stools (with or without stomach cramps and fever). If this occurs, patients should contact their physician as soon as possible.
Tetracycline Class Adverse Reactions
Inform patients that NUZYRA is similar to tetracycline class antibacterial drugs and may have similar adverse reactions [see Warnings and Precautions (5.6)] .
Antibacterial Resistance
Advise patients that antibacterial drugs including NUZYRA should only be used to treat bacterial infections. They do not treat viral infections ( e.g., the common cold). When NUZYRA is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by NUZYRA or other antibacterial drugs in the future.
-
SPL UNCLASSIFIED SECTION
Distributed by:
Paratek Pharmaceuticals, Inc.
Boston, MA, USAPARATEK ®and the hexagon logo are registered trademarks of Paratek Pharmaceuticals, Inc.
NUZYRA ®and its design logo are registered trademarks of Paratek Pharmaceuticals, Inc.
For patent information: www.paratekpharma.com/products/patent.
© 2025 Paratek Pharmaceuticals, Inc. All rights reserved. - PRINCIPAL DISPLAY PANEL - 6 Tablet Blister Pack Carton
- PRINCIPAL DISPLAY PANEL - 10 Vial Carton
- PRINCIPAL DISPLAY PANEL - 30 Tablet Blister Pack Carton
-
INGREDIENTS AND APPEARANCE
NUZYRA
omadacycline injection, powder, lyophilized, for solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71715-001 Route of Administration INTRAVENOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMADACYCLINE (UNII: 090IP5RV8F) (OMADACYCLINE - UNII:090IP5RV8F) OMADACYCLINE 100 mg in 5 mL Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) HYDROCHLORIC ACID (UNII: QTT17582CB) SODIUM HYDROXIDE (UNII: 55X04QC32I) WATER (UNII: 059QF0KO0R) Product Characteristics Color yellow (Yellow to Dark Orange) Score Shape Size Flavor Imprint Code Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71715-001-02 10 in 1 CARTON 02/01/2019 1 NDC:71715-001-01 10 mL in 1 VIAL, GLASS; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209817 02/01/2019 NUZYRA
omadacycline tablet, film coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:71715-002 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength OMADACYCLINE (UNII: 090IP5RV8F) (OMADACYCLINE - UNII:090IP5RV8F) OMADACYCLINE 150 mg Inactive Ingredients Ingredient Name Strength SILICON DIOXIDE (UNII: ETJ7Z6XBU4) CROSPOVIDONE (UNII: 2S7830E561) GLYCERYL MONOCAPRYLOCAPRATE (UNII: G7515SW10N) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) LACTOSE MONOHYDRATE (UNII: EWQ57Q8I5X) MICROCRYSTALLINE CELLULOSE (UNII: OP1R32D61U) POLYVINYL ALCOHOL, UNSPECIFIED (UNII: 532B59J990) SODIUM BISULFITE (UNII: TZX5469Z6I) SODIUM LAURYL SULFATE (UNII: 368GB5141J) SODIUM STEARYL FUMARATE (UNII: 7CV7WJK4UI) TALC (UNII: 7SEV7J4R1U) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) Product Characteristics Color yellow Score no score Shape DIAMOND Size 19mm Flavor Imprint Code OMC;150 Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:71715-002-24 16 in 1 BLISTER PACK; Type 0: Not a Combination Product 02/01/2019 02/01/2020 2 NDC:71715-002-23 14 in 1 BLISTER PACK; Type 0: Not a Combination Product 02/01/2019 02/01/2020 3 NDC:71715-002-21 1 in 1 CARTON 02/01/2020 3 NDC:71715-002-28 6 in 1 BLISTER PACK; Type 0: Not a Combination Product 4 NDC:71715-002-27 5 in 1 CARTON 02/01/2020 4 NDC:71715-002-28 6 in 1 BLISTER PACK; Type 0: Not a Combination Product Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA209816 02/01/2019 Labeler - Paratek Pharmaceuticals, Inc. (076333934)