Label: BRILINTA- ticagrelor tablet

- NDC Code(s): 55154-9618-0, 55154-9618-8
- Packager: Cardinal Health 107, LLC
- This is a repackaged label.
- Source NDC Code(s): 0186-0777
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated July 22, 2024
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Medication Guide: HTML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use BRILINTA safely and effectively. See full prescribing information for BRILINTA.
BRILINTA® (ticagrelor) tablets, for oral use
Initial U.S. Approval: 2011WARNING: BLEEDING RISK
See full prescribing information for complete boxed warning.
- •
-
BRILINTA, like other antiplatelet agents, can cause significant,
sometimes fatal bleeding. (5.1, 6.1)
- •
-
Do not use BRILINTA in patients with active pathological
bleeding or a history of intracranial hemorrhage. (4.1, 4.2)
- •
-
Do not start BRILINTA in patients undergoing urgent coronary
artery bypass graft surgery (CABG). (5.1, 6.1)
- •
-
If possible, manage bleeding without discontinuing BRILINTA.
Stopping BRILINTA increases the risk of subsequent
cardiovascular events. (5.2)
INDICATIONS AND USAGE
BRILINTA is a P2Y12 platelet inhibitor indicated
- •
- to reduce the risk of cardiovascular (CV) death, myocardial infarction (MI), and stroke in patients with acute coronary syndrome (ACS) or a history of MI. For at least the first 12 months following ACS, it is superior to clopidogrel.
BRILINTA also reduces the risk of stent thrombosis in patients who have been stented for treatment of ACS. (1.1) - •
- to reduce the risk of a first MI or stroke in patients with coronary artery disease (CAD) at high risk for such events. While use is not limited to this setting, the efficacy of BRILINTA was established in a population with type 2 diabetes mellitus (T2DM). (1.2)
- •
- to reduce the risk of stroke in patients with acute ischemic stroke (NIH Stroke Scale score ≤5) or high-risk transient ischemic attack (TIA). (1.3)
DOSAGE AND ADMINISTRATION
- •
-
ACS or History of MI
- o
- Initiate treatment with 180 mg oral loading dose of BRILINTA. Then administer 90 mg twice daily during the first year. After one year, administer 60 mg twice daily. (2.2)
- •
-
Patients with CAD and No Prior Stroke or MI
- o
- Administer 60 mg BRILINTA twice daily. (2.3)
- •
-
Acute Ischemic Stroke
- o
- Initiate treatment with a 180 mg loading dose of BRILINTA then continue with 90 mg twice daily for up to 30 days. (2.4)
Use BRILINTA with a daily maintenance dose of aspirin of 75-100 mg. (2) However, in patients who have undergone PCI, consider single antiplatelet therapy with BRILINTA based on the evolving risk for thrombotic versus bleeding events. (2.2)
DOSAGE FORMS AND STRENGTHS
- •
- 60 mg and 90 mg tablets (3)
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
- •
- Dyspnea was reported more frequently with BRILINTA than with control agents in clinical trials. Dyspnea from BRILINTA is self-limiting. (5.3)
- •
- Severe Hepatic Impairment: Likely increase in exposure to ticagrelor. (5.6)
- •
- Laboratory Test Interference: False negative platelet functional test results have been reported for Heparin Induced Thrombocytopenia (HIT). BRILINTA is not expected to impact PF4 antibody testing for HIT. (5.8)
ADVERSE REACTIONS
Most common adverse reactions (>5%) are bleeding and dyspnea. (5.1, 5.3, 6.1)
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
DRUG INTERACTIONS
- •
- Avoid use with strong CYP3A inhibitors or CYP3A inducers. (7.1, 7.2)
- •
- Opioids: Decreased exposure to ticagrelor. Consider use of parenteral anti-platelet agent. (7.3)
- •
- Patients receiving more than 40 mg per day of simvastatin or lovastatin may be at increased risk of statin-related adverse effects. (7.4)
- •
- Monitor digoxin levels with initiation of or any change in BRILINTA. (7.5)
USE IN SPECIFIC POPULATIONS
- •
- Lactation: Breastfeeding not recommended. (8.2)
See 17 for PATIENT COUNSELING INFORMATION and Medication Guide.
Revised: 3/2024
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
WARNING: BLEEDING RISK
1 INDICATIONS AND USAGE
1.1 Acute Coronary Syndrome or a History of Myocardial Infarction
1.2 Coronary Artery Disease but No Prior Stroke or Myocardial Infarction
1.3 Acute Ischemic Stroke or Transient Ischemic Attack (TIA)
2 DOSAGE AND ADMINISTRATION
2.1 General Instructions
2.2 Acute Coronary Syndrome or a History of Myocardial Infarction
2.3 Coronary Artery Disease but No Prior Stroke or Myocardial Infarction
2.4 Acute Ischemic Stroke or Transient Ischemic Attack (TIA)
3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
4.1 History of Intracranial Hemorrhage
4.2 Active Bleeding
4.3 Hypersensitivity
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Bleeding
5.2 Discontinuation of BRILINTA in Patients Treated for Coronary Artery Disease
5.3 Dyspnea
5.4 Bradyarrhythmias
5.5 Severe Hepatic Impairment
5.6 Central Sleep Apnea
5.7 Laboratory Test Interferences
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
7 DRUG INTERACTIONS
7.1 Strong CYP3A Inhibitors
7.2 Strong CYP3A Inducers
7.3 Opioids
7.4 Simvastatin, Lovastatin
7.5 Digoxin
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.6 Hepatic Impairment
8.7 Renal Impairment
10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.5 Pharmacogenomics
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
14 CLINICAL STUDIES
14.1 Acute Coronary Syndromes and Secondary Prevention after Myocardial Infarction
14.2 Coronary Artery Disease but No Prior Stroke or Myocardial Infarction
14.3 Acute Ischemic Stroke or Transient Ischemic Attack (TIA)
16 HOW SUPPLIED/STORAGE AND HANDLING
17 PATIENT COUNSELING INFORMATION
- *
- Sections or subsections omitted from the full prescribing information are not listed.
-
BOXED WARNING
(What is this?)
WARNING: BLEEDING RISK
- •
- BRILINTA, like other antiplatelet agents, can cause significant, sometimes fatal bleeding (5.1, 6.1).
- •
- Do not use BRILINTA in patients with active pathological bleeding or a history of intracranial hemorrhage
- (4.1, 4.2).
- •
- Do not start BRILINTA in patients undergoing urgent coronary artery bypass graft surgery (CABG) (5.1,
- 6.1).
- •
- If possible, manage bleeding without discontinuing BRILINTA. Stopping BRILINTA increases the risk of
- subsequent cardiovascular events (5.2).
-
1 INDICATIONS AND USAGE
1.1 Acute Coronary Syndrome or a History of Myocardial Infarction
BRILINTA is indicated to reduce the risk of cardiovascular (CV) death, myocardial infarction (MI), and stroke in patients with acute coronary syndrome (ACS) or a history of MI. For at least the first 12 months following ACS, it is superior to clopidogrel.
BRILINTA also reduces the risk of stent thrombosis in patients who have been stented for treatment of ACS [see Clinical Studies (14.1)].
1.2 Coronary Artery Disease but No Prior Stroke or Myocardial Infarction
BRILINTA is indicated to reduce the risk of a first MI or stroke in patients with coronary artery disease (CAD) at high risk for such events [see Clinical Studies (14.2)]. While use is not limited to this setting, the efficacy of BRILINTA was established in a population with type 2 diabetes mellitus (T2DM).
1.3 Acute Ischemic Stroke or Transient Ischemic Attack (TIA)
BRILINTA is indicated to reduce the risk of stroke in patients with acute ischemic stroke (NIH Stroke Scale score ≤5) or high-risk transient ischemic attack (TIA) [see Clinical Studies (14.3)].
-
2 DOSAGE AND ADMINISTRATION
2.1 General Instructions
Advise patients who miss a dose of BRILINTA to take their next dose at its scheduled time.
For patients who are unable to swallow tablets whole, BRILINTA tablets can be crushed, mixed with water, and drunk.
The mixture can also be administered via a nasogastric tube (CH8 or greater) [see Clinical Pharmacology (12.3)].
Do not administer BRILINTA with another oral P2Y12 platelet inhibitor.
Avoid aspirin at doses higher than recommended [see Clinical Studies (14.1)].
2.2 Acute Coronary Syndrome or a History of Myocardial Infarction
Initiate treatment with a 180 mg loading dose of BRILINTA. Administer the first 90 mg maintenance dose of
BRILINTA, 6 to 12 hours after the loading dose. Administer 90 mg of BRILINTA twice daily during the first year after
an ACS event. After one year, administer 60 mg of BRILINTA twice daily.
Initiate BRILINTA with a daily maintenance dose of aspirin of 75 mg to 100 mg. However, in patients who have
undergone percutaneous coronary intervention (PCI), consider single antiplatelet therapy with BRILINTA based on the
evolving risk for thrombotic versus bleeding events [see Warnings and Precautions (5.1) and Clinical Studies (14)].
2.3 Coronary Artery Disease but No Prior Stroke or Myocardial Infarction
Administer 60 mg of BRILINTA twice daily.
Generally, use BRILINTA with a daily maintenance dose of aspirin of 75 mg to 100 mg [see Clinical Studies (14)].
2.4 Acute Ischemic Stroke or Transient Ischemic Attack (TIA)
Initiate treatment with a 180 mg loading dose of BRILINTA and then continue with 90 mg twice daily for up to 30 days.
Administer the first maintenance dose 6 to 12 hours after the loading dose.
Use BRILINTA with a loading dose of aspirin (300 mg to 325 mg) and a daily maintenance dose of aspirin of 75 mg to
100 mg [see Clinical Studies (14)].
- 3 DOSAGE FORMS AND STRENGTHS
-
4 CONTRAINDICATIONS
4.1 History of Intracranial Hemorrhage
BRILINTA is contraindicated in patients with a history of intracranial hemorrhage (ICH) because of a high risk of recurrent ICH in this population [see Clinical Studies (14.1), (14.2)].
4.2 Active Bleeding
BRILINTA is contraindicated in patients with active pathological bleeding such as peptic ulcer or intracranial hemorrhage [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)].
-
5 WARNINGS AND PRECAUTIONS
5.1 Risk of Bleeding
Drugs that inhibit platelet function including BRILINTA increase the risk of bleeding [see Warnings and Precautions
(5.2) and Adverse Reactions (6.1)].
Patients treated for acute ischemic stroke or TIA
Patients at NIHSS >5 and patients receiving thrombolysis were excluded from THALES and use of BRILINTA in such patients is not recommended.
5.2 Discontinuation of BRILINTA in Patients Treated for Coronary Artery Disease
Discontinuation of BRILINTA will increase the risk of myocardial infarction, stroke, and death in patients being treated
for coronary artery disease. If BRILINTA must be temporarily discontinued (e.g., to treat bleeding or for significant
surgery), restart it as soon as possible. When possible, interrupt therapy with BRILINTA for five days prior to surgery that
has a major risk of bleeding. Resume BRILINTA as soon as hemostasis is achieved.
5.3 Dyspnea
In clinical trials, about 14% (PLATO and PEGASUS) to 21% (THEMIS) of patients treated with BRILINTA developed dyspnea. Dyspnea was usually mild to moderate in intensity and often resolved during continued treatment but led to study drug discontinuation in 0.9% (PLATO), 1.0% (THALES), 4.3% (PEGASUS), and 6.9% (THEMIS) of patients.
In a substudy of PLATO, 199 subjects underwent pulmonary function testing irrespective of whether they reported dyspnea. There was no indication of an adverse effect on pulmonary function assessed after one month or after at least 6 months of chronic treatment.
If a patient develops new, prolonged, or worsened dyspnea that is determined to be related to BRILINTA, no specific treatment is required; continue BRILINTA without interruption if possible. In the case of intolerable dyspnea requiring discontinuation of BRILINTA, consider prescribing another antiplatelet agent.
5.4 Bradyarrhythmias
BRILINTA can cause ventricular pauses [see Adverse Reactions (6.1)]. Bradyarrhythmias including AV block have been
reported in the postmarketing setting. Patients with a history of sick sinus syndrome, 2nd or 3rd degree AV block or
bradycardia-related syncope not protected by a pacemaker were excluded from clinical studies and may be at increased
risk of developing bradyarrhythmias with ticagrelor.
5.5 Severe Hepatic Impairment
Avoid use of BRILINTA in patients with severe hepatic impairment. Severe hepatic impairment is likely to increase
serum concentration of ticagrelor. There are no studies of BRILINTA patients with severe hepatic impairment [see
5.6 Central Sleep Apnea
Central sleep apnea (CSA) including Cheyne-Stokes respiration (CSR) has been reported in the post-marketing setting in
patients taking ticagrelor, including recurrence or worsening of CSA/CSR following rechallenge. If central sleep apnea is
suspected, consider further clinical assessment.
5.7 Laboratory Test Interferences
False negative functional tests for Heparin Induced Thrombocytopenia (HIT)
BRILINTA has been reported to cause false negative results in platelet functional tests (including the heparin-induced
platelet aggregation (HIPA) assay) for patients with Heparin Induced Thrombocytopenia (HIT). This is related to
inhibition of the P2Y12-receptor on the healthy donor platelets in the test by ticagrelor in the affected patient’s
serum/plasma. Information on concomitant treatment with BRILINTA is required for interpretation of HIT functional
tests. Based on the mechanism of BRILINTA interference, BRILINTA is not expected to impact PF4 antibody testing for
HIT.
-
6 ADVERSE REACTIONS
The following adverse reactions are also discussed elsewhere in the labeling:
- •
- Bleeding [see Warnings and Precautions (5.1)]
- •
- Dyspnea [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
BRILINTA has been evaluated for safety in more than 58,000 patients.
Bleeding in PLATO (Reduction in risk of thrombotic events in ACS)
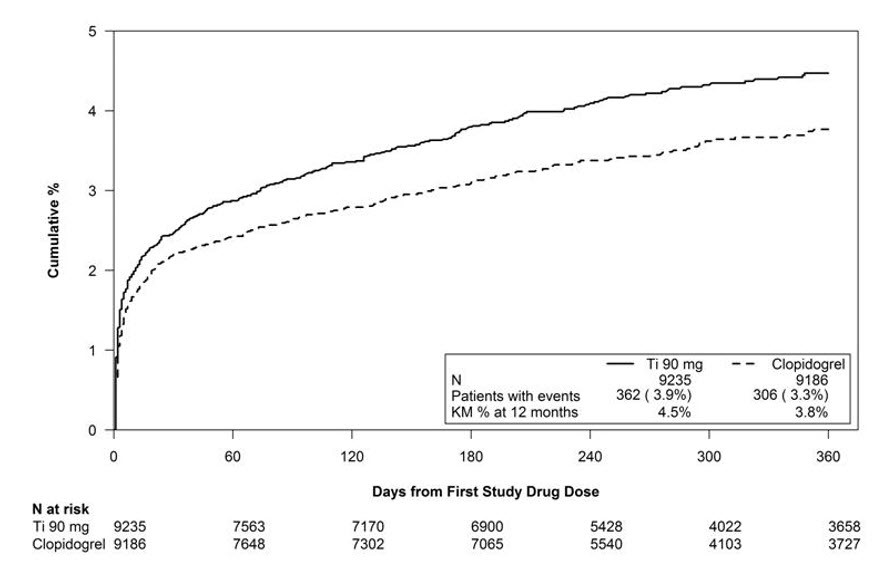
Figure 1 is a plot of time to the first non-CABG major bleeding event.
Figure 1 - Kaplan-Meier estimate of time to first non-CABG PLATO-defined major bleeding event (PLATO)
Frequency of bleeding in PLATO is summarized in Tables 1 and 2. About half of the non-CABG major bleeding events were in the first 30 days.
Table 1 - Non-CABG related bleeds (PLATO) - *
- 90 mg BID
BRILINTA*
N=9235Clopidogrel
N=9186n (%) patients
with event
n (%) patients
with event
PLATO Major + Minor
713 (7.7)
567 (6.2)
Major
362 (3.9)
306 (3.3)
Fatal/Life-threatening
171 (1.9)
151 (1.6)
Fatal
15 (0.2)
16 (0.2)
Intracranial hemorrhage (Fatal/Life-threatening)
26 (0.3)
15 (0.2)
PLATO Minor bleed: requires medical intervention to stop or treat bleeding.
PLATO Major bleed: any one of the following: fatal; intracranial; intrapericardial with cardiac tamponade; hypovolemic shock or severe hypotension requiring intervention; significantly disabling (e.g., intraocular with permanent vision loss); associated with a decrease in Hb of at least 3 g/dL (or a fall in hematocrit (Hct) of at least 9%); transfusion of 2 or more units.
PLATO Major bleed, fatal/life-threatening: any major bleed as described above and associated with a decrease in Hb of more than 5 g/dL (or a fall in hematocrit (Hct) of at least 15%); transfusion of 4 or more units.
Fatal: A bleeding event that directly led to death within 7 days.
No baseline demographic factor altered the relative risk of bleeding with BRILINTA compared to clopidogrel.
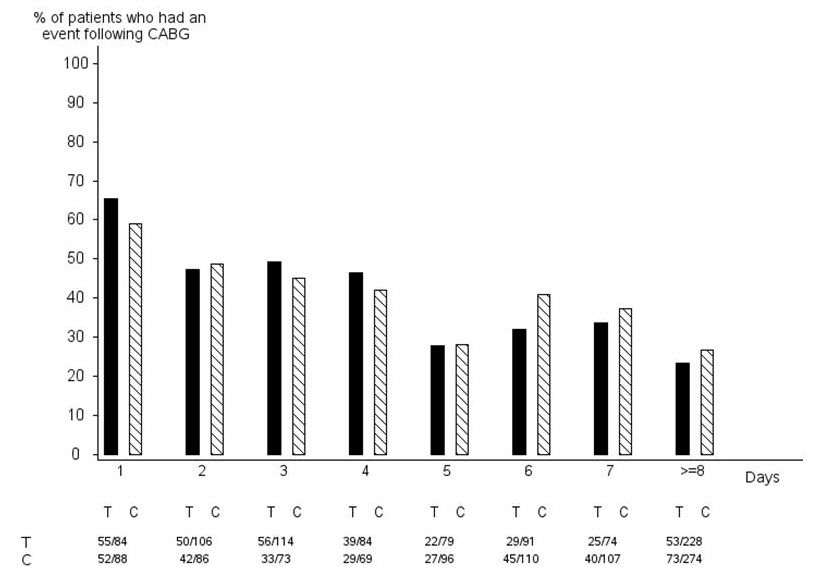
In PLATO, 1584 patients underwent CABG surgery. The percentages of those patients who bled are shown in Figure 2 and Table 2.
Figure 2 - ‘Major fatal/life-threatening’ CABG-related bleeding by days from last dose of study drug to CABG procedure (PLATO)
X-axis is days from last dose of study drug prior to CABG.
The PLATO protocol recommended a procedure for withholding study drug prior to CABG or other major surgery without unblinding. If surgery was elective or non-urgent, study drug was interrupted temporarily, as follows: If local practice was to allow antiplatelet effects to dissipate before surgery, capsules (blinded clopidogrel) were withheld 5 days before surgery and tablets (blinded ticagrelor) were withheld for a minimum of 24 hours and a maximum of 72 hours before surgery. If local practice was to perform surgery without waiting for dissipation of antiplatelet effects capsules and tablets were withheld 24 hours prior to surgery and use of aprotinin or other hemostatic agents was allowed. If local practice was to use IPA monitoring to determine when surgery could be performed both the capsules and tablets were withheld at the same time and the usual monitoring procedures followed.
T Ticagrelor; C Clopidogrel.
Table 2 - CABG-related bleeding (PLATO) - *
- 90 mg BID
BRILINTA*
N=770
Clopidogrel
N=814
n (%) patients
with event
n (%) patients
with event
PLATO Total Major
626 (81.3)
666 (81.8)
Fatal/Life-threatening
337 (43.8)
350 (43.0)
Fatal
6 (0.8)
7 (0.9)
PLATO Major bleed: any one of the following: fatal; intracranial; intrapericardial with cardiac tamponade; hypovolemic shock or severe hypotension requiring intervention; significantly disabling (e.g., intraocular with permanent vision loss); associated with a decrease in Hb of at least 3 g/dL (or a fall in hematocrit (Hct) of at least 9%); transfusion of 2 or more units.
PLATO Major bleed, fatal/life-threatening: any major bleed as described above and associated with a decrease in Hb of more than 5 g/dL (or a fall in hematocrit (Hct) of at least 15%); transfusion of 4 or more units.
When antiplatelet therapy was stopped 5 days before CABG, major bleeding occurred in 75% of BRILINTA treated patients and 79% on clopidogrel.
Other Adverse Reactions in PLATO
Adverse reactions that occurred at a rate of 4% or more in PLATO are shown in Table 3.
Table 3 - Percentage of patients reporting non-hemorrhagic adverse reactions at least 4% or more in either group and more frequently on BRILINTA (PLATO) - *
- 90 mg BID
BRILINTA*
N=9235Clopidogrel
N=9186Dyspnea
13.8
7.8
Dizziness
4.5
3.9
Nausea
4.3
3.8
Bleeding in PEGASUS (Secondary Prevention in Patients with a History of Myocardial Infarction)
Overall outcome of bleeding events in the PEGASUS study are shown in Table 4.
Table 4 - Bleeding events (PEGASUS) - *
- 60 mg BID
BRILINTA*
N=6958
Placebo
N=6996
Events / 1000 patient years
Events / 1000 patient years
TIMI Major
8
3
Fatal
1
1
Intracranial hemorrhage
2
1
TIMI Major or Minor
11
5
TIMI Major: Fatal bleeding, OR any intracranial bleeding, OR clinically overt signs of hemorrhage associated with a drop in hemoglobin (Hgb) of ≥5 g/dL, or a fall in hematocrit (Hct) of ≥15%.
Fatal: A bleeding event that directly led to death within 7 days.
TIMI Minor: Clinically apparent with 3-5 g/dL decrease in hemoglobin.
The bleeding profile of BRILINTA 60 mg compared to aspirin alone was consistent across multiple pre-defined subgroups (e.g., by age, gender, weight, race, geographic region, concurrent conditions, concomitant therapy, stent, and medical history) for TIMI Major and TIMI Major or Minor bleeding events.
Other Adverse Reactions in PEGASUS
Adverse reactions that occurred in PEGASUS at rates of 3% or more are shown in Table 5.
Table 5 - Non-hemorrhagic adverse reactions reported in >3.0% of patients in the ticagrelor 60 mg treatment group (PEGASUS) - *
- 60 mg BID
BRILINTA*
N=6958Placebo
N=6996Dyspnea
14.2%
5.5%
Dizziness
4.5%
4.1%
Diarrhea
3.3%
2.5%
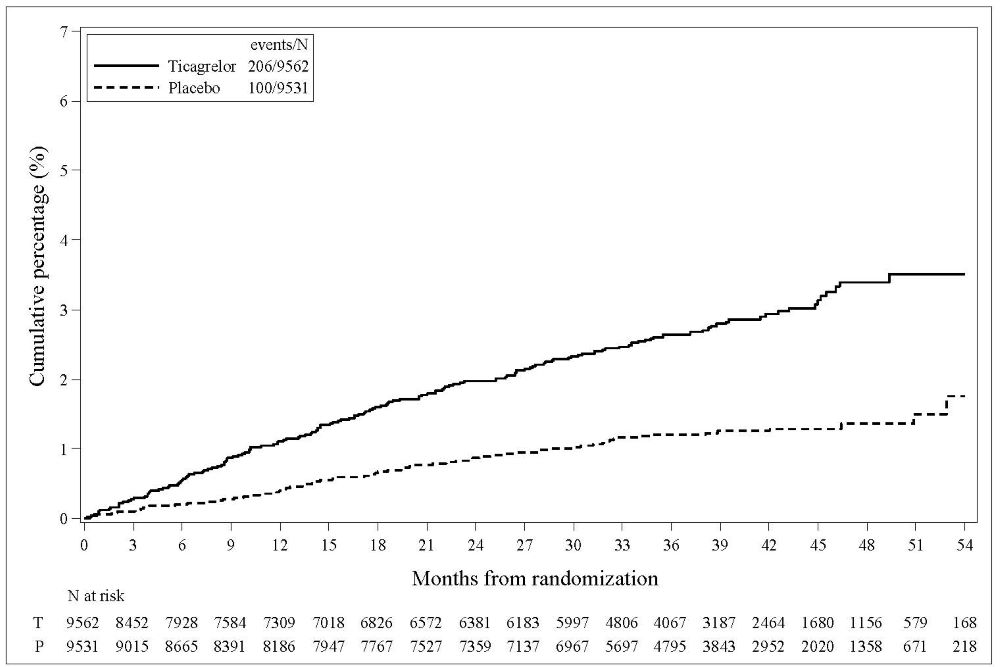
Bleeding in THEMIS (Prevention of major CV events in patients with CAD and Type 2 Diabetes Mellitus)
The Kaplan-Meier curve of time to first TIMI Major bleeding event is presented in Figure 3.
Figure 3 – Time to first TIMI Major bleeding event (THEMIS)
T = Ticagrelor; P = Placebo; N = Number of patients
The bleeding events in THEMIS are shown below in Table 6.
Table 6 – Bleeding events (THEMIS) BRILINTA
N=9562
Placebo
N=9531
Events / 1000 patient years
Events / 1000 patient years
TIMI Major
9
4
TIMI Major or Minor
12
5
TIMI Major or Minor or Requiring medical attention
46
18
Fatal bleeding
1
0
Intracranial hemorrhage
3
2
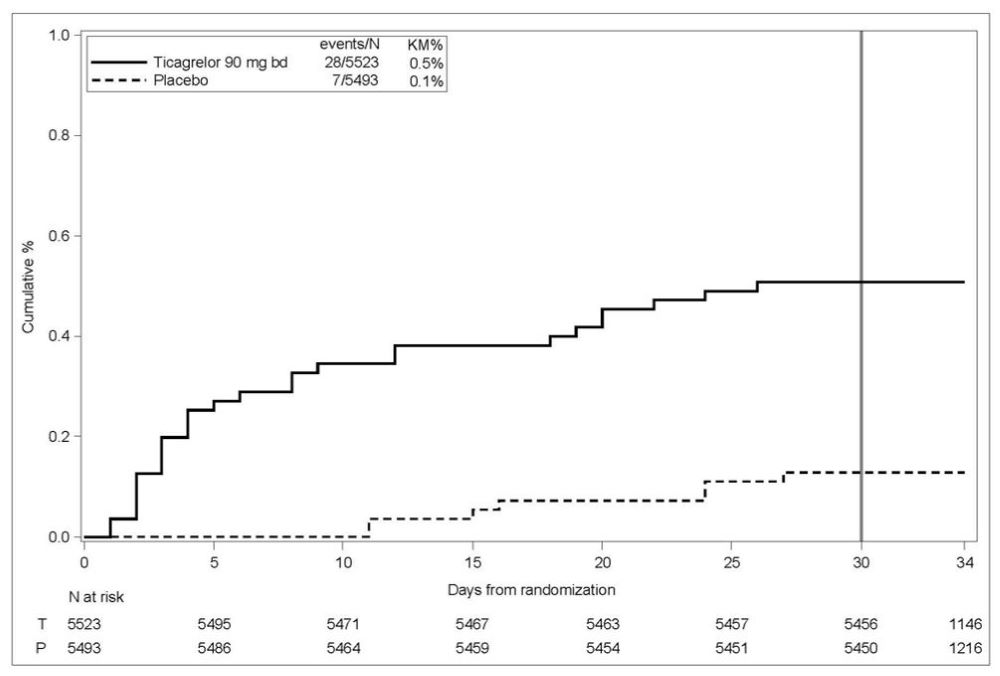
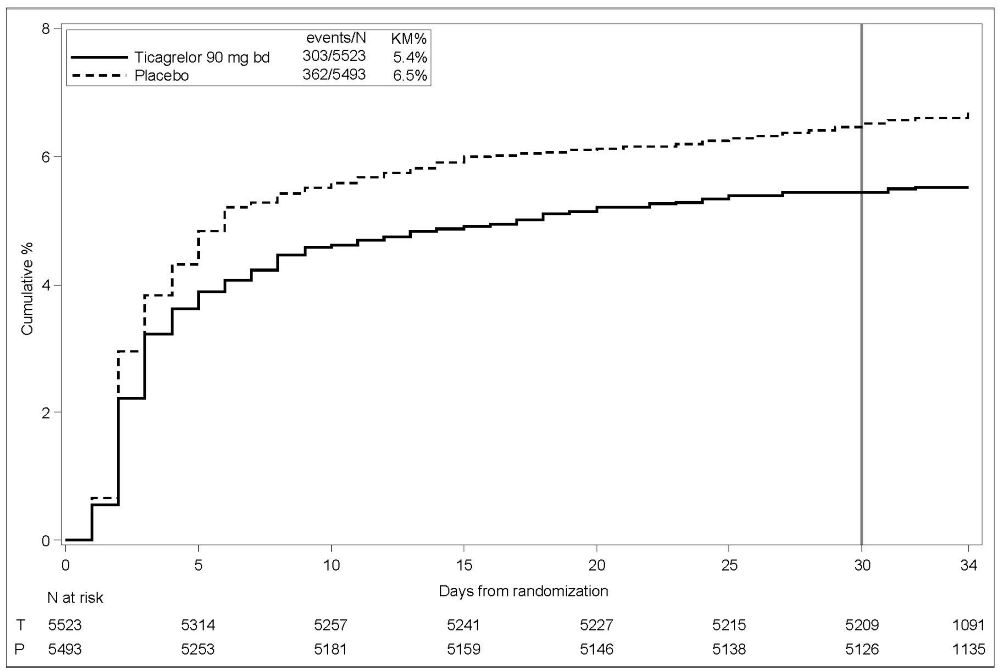
Bleeding in THALES (Reduction in risk of stroke in patients with acute ischemic stroke or TIA)
The Kaplan-Meier curve of time course of GUSTO severe bleeding events is presented in Figure 4.
Figure 4 – Time course of GUSTO severe bleeding events
KM%: Kaplan-Meier percentage evaluated at Day 30; T = Ticagrelor; P = placebo; N = Number of patients
GUSTO Severe: Any one of the following: fatal bleeding, intracranial bleeding (excluding asymptomatic hemorrhagic transformations of ischemic brain infarctions and excluding microhemorrhages < 10 mm evident only on gradient-echo magnetic resonance imaging), bleeding that caused hemodynamic compromise requiring intervention (eg, systolic blood pressure <90 mmg Hg that required blood or fluid replacement, or vasopressor/inotropic support, or surgical intervention).
Intracranial bleeding and fatal bleeding in THALES: In total, there were 21 intracranial hemorrhages (ICHs) for BRILINTA and 6 ICHs for placebo. Fatal bleedings, almost all ICH, occurred in 11 for BRILINTA and in 2 for placebo.
Bradycardia
In a Holter substudy of about 3000 patients in PLATO, more patients had ventricular pauses with BRILINTA (6.0%) than with clopidogrel (3.5%) in the acute phase; rates were 2.2% and 1.6%, respectively, after 1 month. PLATO, PEGASUS, THEMIS and THALES excluded patients at increased risk of bradycardic events (e.g., patients who have sick sinus syndrome, 2nd or 3rd degree AV block, or bradycardic-related syncope and not protected with a pacemaker).
Lab abnormalities
Serum Uric Acid:
In PLATO, serum uric acid levels increased approximately 0.6 mg/dL from baseline on BRILINTA 90 mg and approximately 0.2 mg/dL on clopidogrel. The difference disappeared within 30 days of discontinuing treatment. Reports of gout did not differ between treatment groups in PLATO (0.6% in each group).
In PEGASUS, serum uric acid levels increased approximately 0.2 mg/dL from baseline on BRILINTA 60 mg and no elevation was observed on aspirin alone. Gout occurred more commonly in patients on BRILINTA than in patients on aspirin alone (1.5%, 1.1%). Mean serum uric acid concentrations decreased after treatment was stopped.
Serum Creatinine:
In PLATO, a >50% increase in serum creatinine levels was observed in 7.4% of patients receiving BRILINTA 90 mg compared to 5.9% of patients receiving clopidogrel. The increases typically did not progress with ongoing treatment and often decreased with continued therapy. Evidence of reversibility upon discontinuation was observed even in those with the greatest on treatment increases. Treatment groups in PLATO did not differ for renal-related serious adverse events such as acute renal failure, chronic renal failure, toxic nephropathy, or oliguria.
In PEGASUS, serum creatinine concentration increased by >50% in approximately 4% of patients receiving BRILINTA 60 mg, similar to aspirin alone. The frequency of renal related adverse events was similar for ticagrelor and aspirin alone regardless of age and baseline renal function.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of BRILINTA. Because these reactions are reported voluntarily from a population of an unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and lymphatic system disorders: Thrombotic Thrombocytopenic Purpura (TTP) has been rarely reported with the use of BRILINTA. TTP is a serious condition which can occur after a brief exposure (<2 weeks) and requires prompt treatment.
Immune system disorders: Hypersensitivity reactions including angioedema [see Contraindications (4.3)].
Respiratory Disorders: Central sleep apnea, Cheyne-Stokes respiration
Skin and subcutaneous tissue disorders: Rash
-
7 DRUG INTERACTIONS
7.1 Strong CYP3A Inhibitors
Strong CYP3A inhibitors substantially increase ticagrelor exposure and so increase the risk of dyspnea, bleeding, and other adverse events. Avoid use of strong inhibitors of CYP3A (e.g., ketoconazole, itraconazole, voriconazole, clarithromycin, nefazodone, ritonavir, saquinavir, nelfinavir, indinavir, atazanavir and telithromycin) [see Clinical Pharmacology (12.3)].
7.2 Strong CYP3A Inducers
Strong CYP3A inducers substantially reduce ticagrelor exposure and so decrease the efficacy of ticagrelor. Avoid use with strong inducers of CYP3A (e.g., rifampin, phenytoin, carbamazepine and phenobarbital) [see Clinical Pharmacology (12.3)].
7.3 Opioids
As with other oral P2Y12 inhibitors, co-administration of opioid agonists delay and reduce the absorption of ticagrelor and its active metabolite presumably because of slowed gastric emptying [see Clinical Pharmacology (12.3)]. Consider the use of a parenteral anti-platelet agent in acute coronary syndrome patients requiring co-administration of morphine or other opioid agonists.
7.4 Simvastatin, Lovastatin
BRILINTA increases serum concentrations of simvastatin and lovastatin because these drugs are metabolized by CYP3A4. Avoid simvastatin and lovastatin doses greater than 40 mg [see Clinical Pharmacology (12.3)].
7.5 Digoxin
BRILINTA inhibits the P-glycoprotein transporter; monitor digoxin levels with initiation of or change in BRILINTA therapy [see Clinical Pharmacology (12.3)].
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
Risk Summary
Available data from case reports with BRILINTA use in pregnant women have not identified a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Ticagrelor given to pregnant rats and pregnant rabbits during organogenesis caused structural abnormalities in the offspring at maternal doses about 5 to 7 times the maximum recommended human dose (MRHD) based on body surface area. When ticagrelor was given to rats during late gestation and lactation, pup death and effects on pup growth were seen at approximately 10 times the MRHD (see DATA).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
In reproductive toxicology studies, pregnant rats received ticagrelor during organogenesis at doses from 20 to 300 mg/kg/day. 20 mg/kg/day is approximately the same as the MRHD of 90 mg twice daily for a 60 kg human on a mg/m2 basis. Adverse outcomes in offspring occurred at doses of 300 mg/kg/day (16.5 times the MRHD on a mg/m2 basis) and included supernumerary liver lobe and ribs, incomplete ossification of sternebrae, displaced articulation of pelvis, and misshapen/misaligned sternebrae. At the mid-dose of 100 mg/kg/day (5.5 times the MRHD on a mg/m2 basis), delayed development of liver and skeleton was seen. When pregnant rabbits received ticagrelor during organogenesis at doses from 21 to 63 mg/kg/day, fetuses exposed to the highest maternal dose of 63 mg/kg/day (6.8 times the MRHD on a mg/m2 basis) had delayed gall bladder development and incomplete ossification of the hyoid, pubis and sternebrae occurred.
In a prenatal/postnatal study, pregnant rats received ticagrelor at doses of 10 to 180 mg/kg/day during late gestation and lactation. Pup death and effects on pup growth were observed at 180 mg/kg/day (approximately 10 times the MRHD on a mg/m2 basis). Relatively minor effects such as delays in pinna unfolding and eye opening occurred at doses of 10 and 60 mg/kg (approximately one-half and 3.2 times the MRHD on a mg/m2 basis).
8.2 Lactation
Risk Summary
There are no data on the presence of ticagrelor or its metabolites in human milk, the effects on the breastfed infant, or the effects on milk production. Ticagrelor and its metabolites were present in rat milk at higher concentrations than in maternal plasma. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Breastfeeding is not recommended during treatment with BRILINTA.
8.4 Pediatric Use
The safety and effectiveness of BRILINTA have not been established in pediatric patients. Effectiveness was not demonstrated in an adequate and well-controlled study conducted in 101 BRILINTA-treated pediatric patients, aged 2 to <18 for reducing the rate of vaso-occlusive crises in sickle cell disease.
8.5 Geriatric Use
About half of the patients in PLATO, PEGASUS, THEMIS, and THALES were ≥65 years of age and at least 15% were ≥75 years of age. No overall differences in safety or effectiveness were observed between elderly and younger patients.
8.6 Hepatic Impairment
Ticagrelor is metabolized by the liver and impaired hepatic function can increase risks for bleeding and other adverse events. Avoid use of BRILINTA in patients with severe hepatic impairment. There is limited experience with BRILINTA in patients with moderate hepatic impairment; consider the risks and benefits of treatment, noting the probable increase in exposure to ticagrelor. No dosage adjustment is needed in patients with mild hepatic impairment [see Warnings and Precautions (5.5) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustment is needed in patients with renal impairment [see Clinical Pharmacology (12.3)].
Patients with End-Stage Renal Disease on dialysis
Clinical efficacy and safety studies with BRILINTA did not enroll patients with end-stage renal disease (ESRD) on dialysis. In patients with ESRD maintained on intermittent hemodialysis, no clinically significant difference in concentrations of ticagrelor and its metabolite and platelet inhibition are expected compared to those observed in patients with normal renal function [see Clinical Pharmacology (12.3)]. It is not known whether these concentrations will lead to similar efficacy and safety in patients with ESRD on dialysis as were seen in PLATO, PEGASUS, THEMIS and THALES.
-
10 OVERDOSAGE
There is currently no known treatment to reverse the effects of BRILINTA, and ticagrelor is not dialyzable. Treatment of overdose should follow local standard medical practice. Bleeding is the expected pharmacologic effect of overdosing. If bleeding occurs, appropriate supportive measures should be taken.
Platelet transfusion did not reverse the antiplatelet effect of BRILINTA in healthy volunteers and is unlikely to be of clinical benefit in patients with bleeding.
Other effects of overdose may include gastrointestinal effects (nausea, vomiting, diarrhea) or ventricular pauses. Monitor the ECG.
-
11 DESCRIPTION
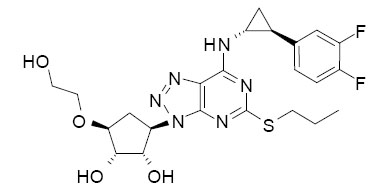
BRILINTA contains ticagrelor, a cyclopentyltriazolopyrimidine, inhibitor of platelet activation and aggregation mediated by the P2Y12 ADP-receptor. Chemically it is (1S,2S,3R,5S)-3-[7-{[(1R,2S)-2-(3,4-difluorophenyl)cyclopropyl]amino}-5-(propylthio)-3H-[1,2,3]-triazolo[4,5-d]pyrimidin-3-yl]-5-(2-hydroxyethoxy)cyclopentane-1,2-diol. The empirical formula of ticagrelor is C23H28F2N6O4S and its molecular weight is 522.57. The chemical structure of ticagrelor is:
Ticagrelor is a crystalline powder with an aqueous solubility of approximately 10 μg/mL at room temperature.
BRILINTA 90 mg tablets for oral administration contain 90 mg of ticagrelor and the following ingredients: mannitol, dibasic calcium phosphate, sodium starch glycolate, hydroxypropyl cellulose, magnesium stearate, hydroxypropyl methylcellulose, titanium dioxide, talc, polyethylene glycol 400, and ferric oxide yellow.
BRILINTA 60 mg tablets for oral administration contain 60 mg of ticagrelor and the following ingredients: mannitol, dibasic calcium phosphate, sodium starch glycolate, hydroxypropyl cellulose, magnesium stearate, hydroxypropyl methylcellulose, titanium dioxide, polyethylene glycol 400, ferric oxide black, and ferric oxide red.
-
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
Ticagrelor and its major metabolite reversibly interact with the platelet P2Y12 ADP-receptor to prevent signal transduction and platelet activation. Ticagrelor and its active metabolite are approximately equipotent.
12.2 Pharmacodynamics
The inhibition of platelet aggregation (IPA) by ticagrelor and clopidogrel was compared in a 6-week study examining both acute and chronic platelet inhibition effects in response to 20 μM ADP as the platelet aggregation agonist.
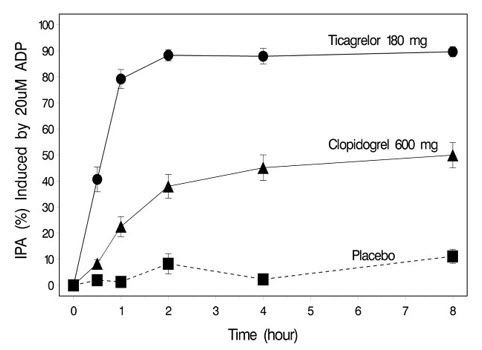
The onset of IPA was evaluated on Day 1 of the study following loading doses of 180 mg ticagrelor or 600 mg clopidogrel. As shown in Figure 5, IPA was higher in the ticagrelor group at all time points. The maximum IPA effect of ticagrelor was reached at around 2 hours, and was maintained for at least 8 hours.
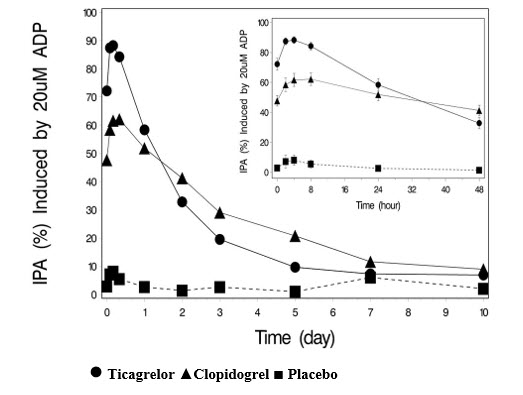
The offset of IPA was examined after 6 weeks on ticagrelor 90 mg twice daily or clopidogrel 75 mg daily, again in response to 20 μM ADP.
As shown in Figure 6, mean maximum IPA following the last dose of ticagrelor was 88% and 62% for clopidogrel. The insert in Figure 6 shows that after 24 hours, IPA in the ticagrelor group (58%) was similar to IPA in clopidogrel group (52%), indicating that patients who miss a dose of ticagrelor would still maintain IPA similar to the trough IPA of patients treated with clopidogrel. After 5 days, IPA in the ticagrelor group was similar to IPA in the placebo group. It is not known how either bleeding risk or thrombotic risk track with IPA, for either ticagrelor or clopidogrel.
Figure 5 - Mean inhibition of platelet aggregation (±SE) following single oral doses of placebo, 180 mg ticagrelor or 600 mg clopidogrel
Figure 6 - Mean inhibition of platelet aggregation (IPA) following 6 weeks on placebo, ticagrelor 90 mg twice daily, or clopidogrel 75 mg daily
Transitioning from clopidogrel to BRILINTA resulted in an absolute IPA increase of 26.4% and from BRILINTA to clopidogrel resulted in an absolute IPA decrease of 24.5%. Patients can be transitioned from clopidogrel to BRILINTA without interruption of antiplatelet effect [see Dosage and Administration (2)].
12.3 Pharmacokinetics
Ticagrelor demonstrates dose proportional pharmacokinetics, which are similar in patients and healthy volunteers.
Absorption
BRILINTA can be taken with or without food. Absorption of ticagrelor occurs with a median tmax of 1.5 h (range 1.0–4.0). The formation of the major circulating metabolite AR-C124910XX (active) from ticagrelor occurs with a median tmax of 2.5 h (range 1.5-5.0).
The mean absolute bioavailability of ticagrelor is about 36% (range 30%-42%). Ingestion of a high-fat meal had no effect on ticagrelor Cmax, but resulted in a 21% increase in AUC. The Cmax of its major metabolite was decreased by 22% with no change in AUC.
BRILINTA as crushed tablets mixed in water, given orally or administered through a nasogastric tube into the stomach, is bioequivalent to whole tablets (AUC and Cmax within 80-125% for ticagrelor and AR-C124910XX) with a median tmax of 1.0 hour (range 1.0 – 4.0) for ticagrelor and 2.0 hours (range 1.0 –8.0) for AR-C124910XX.
Distribution
The steady state volume of distribution of ticagrelor is 88 L. Ticagrelor and the active metabolite are extensively bound to human plasma proteins (>99%).
Metabolism
CYP3A4 is the major enzyme responsible for ticagrelor metabolism and the formation of its major active metabolite. Ticagrelor and its major active metabolite are weak P-glycoprotein substrates and inhibitors. The systemic exposure to the active metabolite is approximately 30-40% of the exposure of ticagrelor.
Excretion
The primary route of ticagrelor elimination is hepatic metabolism. When radiolabeled ticagrelor is administered, the mean recovery of radioactivity is approximately 84% (58% in feces, 26% in urine). Recoveries of ticagrelor and the active metabolite in urine were both less than 1% of the dose. The primary route of elimination for the major metabolite of ticagrelor is most likely to be biliary secretion. The mean t1/2 is approximately 7 hours for ticagrelor and 9 hours for the active metabolite.
Specific Populations
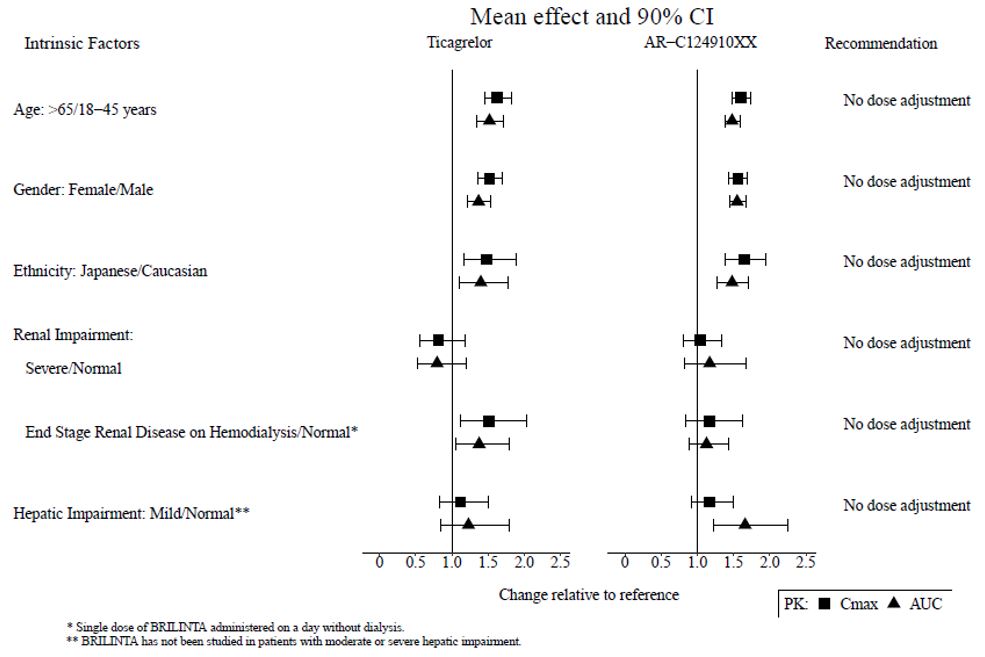
The effects of age, gender, ethnicity, renal impairment and mild hepatic impairment on the pharmacokinetics of ticagrelor are presented in Figure 7. Effects are modest and do not require dose adjustment.
Patients with End-Stage Renal Disease on Hemodialysis
In patients with end stage renal disease on hemodialysis AUC and Cmax of BRILINTA 90 mg administered on a day without dialysis were 38% and 51% higher respectively, compared to subjects with normal renal function. A similar increase in exposure was observed when BRILINTA was administered immediately prior to dialysis showing that BRILINTA is not dialyzable. Exposure of the active metabolite increased to a lesser extent. The IPA effect of BRILINTA was independent of dialysis in patients with end stage renal disease and similar to healthy adults with normal renal function.
Figure 7 - Impact of intrinsic factors on the pharmacokinetics of ticagrelor
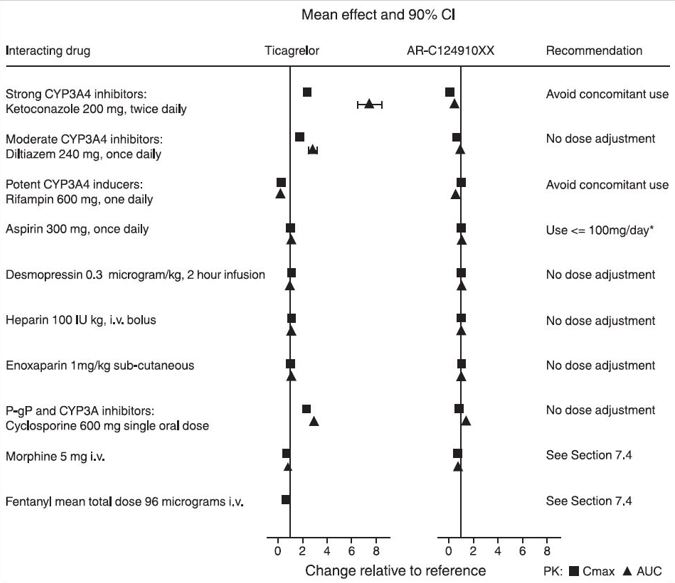
Effects of Other Drugs on BRILINTA
CYP3A4 is the major enzyme responsible for ticagrelor metabolism and the formation of its major active metabolite. The effects of other drugs on the pharmacokinetics of ticagrelor are presented in Figure 8 as change relative to ticagrelor given alone (test/reference). Strong CYP3A inhibitors (e.g., ketoconazole, itraconazole, and clarithromycin) substantially increase ticagrelor exposure. Moderate CYP3A inhibitors have lesser effects (e.g., diltiazem). CYP3A inducers (e.g., rifampin) substantially reduce ticagrelor blood levels. P-gp inhibitors (e.g., cyclosporine) increase ticagrelor exposure.
Co-administration of 5 mg intravenous morphine with 180 mg loading dose of ticagrelor decreased observed mean ticagrelor exposure by up to 25% in healthy adults and up to 36% in ACS patients undergoing PCI. Tmax was delayed by 1-2 hours. Exposure of the active metabolite decreased to a similar extent. Morphine co-administration did not delay or decrease platelet inhibition in healthy adults. Mean platelet aggregation was higher up to 3 hours post loading dose in ACS patients co-administered with morphine.
Co-administration of intravenous fentanyl with 180 mg loading dose of ticagrelor in ACS patients undergoing PCI resulted in similar effects on ticagrelor exposure and platelet inhibition.
Figure 8 - Effect of co-administered drugs on the pharmacokinetics of ticagrelor
*See Dosage and Administration (2)
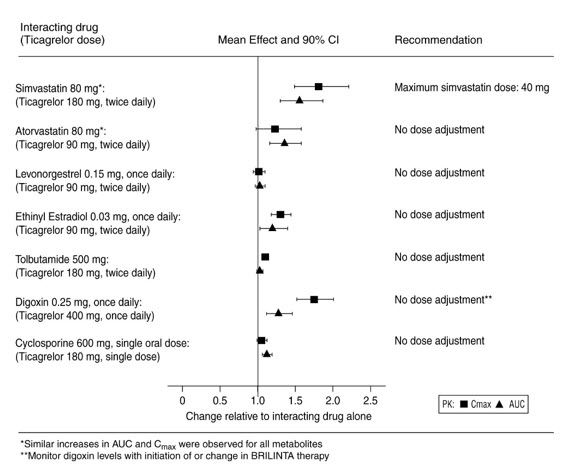
Effects of BRILINTA on Other Drugs
In vitro metabolism studies demonstrate that ticagrelor and its major active metabolite are weak inhibitors of CYP3A4, potential activators of CYP3A5 and inhibitors of the P-gp transporter. Ticagrelor and AR-C124910XX were shown to have no inhibitory effect on human CYP1A2, CYP2C19, and CYP2E1 activity. For specific in vivo effects on the pharmacokinetics of simvastatin, atorvastatin, ethinyl estradiol, levonorgesterol, tolbutamide, digoxin and cyclosporine, see Figure 9.
Figure 9 - Impact of BRILINTA on the pharmacokinetics of co-administered drugs
-
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Ticagrelor was not carcinogenic in the mouse at doses up to 250 mg/kg/day or in the male rat at doses up to 120 mg/kg/day (19 and 15 times the MRHD of 90 mg twice daily on the basis of AUC, respectively). Uterine carcinomas, uterine adenocarcinomas and hepatocellular adenomas were seen in female rats at doses of 180 mg/kg/day (29‑fold the maximally recommended dose of 90 mg twice daily on the basis of AUC), whereas 60 mg/kg/day (8‑fold the MRHD based on AUC) was not carcinogenic in female rats.
Mutagenesis
Ticagrelor did not demonstrate genotoxicity when tested in the Ames bacterial mutagenicity test, mouse lymphoma assay and the rat micronucleus test. The active O-demethylated metabolite did not demonstrate genotoxicity in the Ames assay and mouse lymphoma assay.
Impairment of Fertility
Ticagrelor had no effect on male fertility at doses up to 180 mg/kg/day or on female fertility at doses up to 200 mg/kg/day (>15-fold the MRHD on the basis of AUC). Doses of ≥10 mg/kg/day given to female rats caused an increased incidence of irregular duration estrus cycles (1.5-fold the MRHD based on AUC).
-
14 CLINICAL STUDIES
14.1 Acute Coronary Syndromes and Secondary Prevention after Myocardial Infarction
PLATO
PLATO (NCT00391872) was a randomized double-blind study comparing BRILINTA (N=9333) to clopidogrel (N=9291), both given in combination with aspirin and other standard therapy, in patients with acute coronary syndromes (ACS), who presented within 24 hours of onset of the most recent episode of chest pain or symptoms. The study’s primary endpoint was the composite of first occurrence of cardiovascular death, non-fatal MI (excluding silent MI), or non-fatal stroke.
Patients who had already been treated with clopidogrel could be enrolled and randomized to either study treatment. Patients with previous intracranial hemorrhage, gastrointestinal bleeding within the past 6 months, or with known bleeding diathesis or coagulation disorder were excluded. Patients taking anticoagulants were excluded from participating and patients who developed an indication for anticoagulation during the trial were discontinued from study drug. Patients could be included whether there was intent to manage the ACS medically or invasively, but patient randomization was not stratified by this intent.
All patients randomized to BRILINTA received a loading dose of 180 mg followed by a maintenance dose of 90 mg twice daily. Patients in the clopidogrel arm were treated with an initial loading dose of clopidogrel 300 mg, if clopidogrel therapy had not already been given. Patients undergoing PCI could receive an additional 300 mg of clopidogrel at investigator discretion. A daily maintenance dose of aspirin 75-100 mg was recommended, but higher maintenance doses of aspirin were allowed according to local judgment. Patients were treated for at least 6 months and for up to 12 months.
PLATO patients were predominantly male (72%) and Caucasian (92%). About 43% of patients were >65 years and 15% were >75 years. Median exposure to study drug was 276 days. About half of the patients received pre-study clopidogrel and about 99% of the patients received aspirin at some time during PLATO. About 35% of patients were receiving a statin at baseline and 93% received a statin sometime during PLATO.
Table 7 shows the study results for the primary composite endpoint and the contribution of each component to the primary endpoint. Separate secondary endpoint analyses are shown for the overall occurrence of CV death, MI, and stroke and overall mortality.
Table 7 - Patients with outcome events (PLATO) BRILINTA*
N=9333
Clopidogrel
N=9291
Hazard Ratio
(95% CI)
p-value
Events / 1000 patient years
Events / 1000 patient years
Composite of CV death, MI, or stroke
111
131
0.84 (0.77, 0.92)
0.0003
CV death
32
43
0.74
Non-fatal MI
64
76
0.84
Non-fatal stroke
15
12
1.24
Secondary endpoints†
CV death
45
57
0.79 (0.69, 0.91)
0.0013
MI‡
65
76
0.84 (0.75, 0.95)
0.0045
Stroke‡
16
14
1.17 (0.91, 1.52)
0.22
All-cause mortality
51
65
0.78 (0.69, 0.89)
0.0003
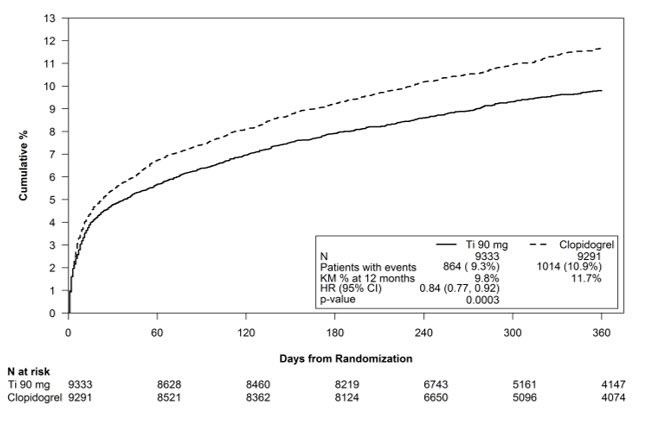
The Kaplan-Meier curve (Figure 10) shows time to first occurrence of the primary composite endpoint of CV death, non-fatal MI or non-fatal stroke in the overall study.
Figure 10 - Time to first occurrence of CV death, MI, or stroke (PLATO)
The curves separate by 30 days [relative risk reduction (RRR) 12%] and continue to diverge throughout the 12‑month treatment period (RRR 16%).
Among 11,289 patients with PCI receiving any stent during PLATO, there was a lower risk of stent thrombosis (1.3% for adjudicated “definite”) than with clopidogrel (1.9%) (HR 0.67, 95% CI 0.50-0.91; p=0.009). The results were similar for drug-eluting and bare metal stents.
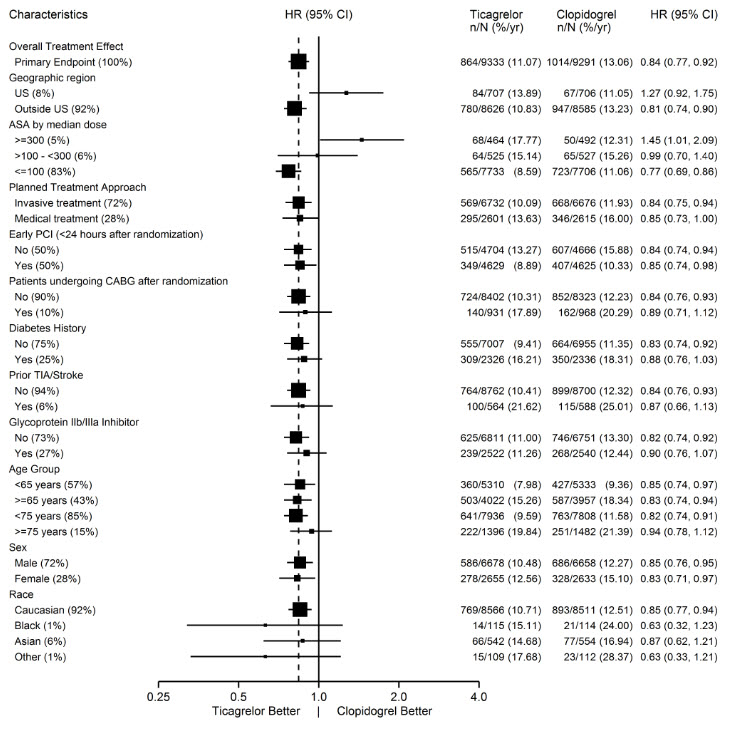
A wide range of demographic, concurrent baseline medications, and other treatment differences were examined for their influence on outcome. Some of these are shown in Figure 11. Such analyses must be interpreted cautiously, as differences can reflect the play of chance among a large number of analyses. Most of the analyses show effects consistent with the overall results, but there are two exceptions: a finding of heterogeneity by region and a strong influence of the maintenance dose of aspirin. These are considered further below.
Most of the characteristics shown are baseline characteristics, but some reflect post-randomization determinations (e.g., aspirin maintenance dose, use of PCI).
Figure 11 – Subgroup analyses of (PLATO)
Note: The figure above presents effects in various subgroups most of which are baseline characteristics and most of which were pre-specified. The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
Regional Differences
Results in the rest of the world compared to effects in North America (US and Canada) show a smaller effect in North America, numerically inferior to the control and driven by the US subset. The statistical test for the US/non-US comparison is statistically significant (p=0.009), and the same trend is present for both CV death and non-fatal MI. The individual results and nominal p-values, like all subset analyses, need cautious interpretation, and they could represent chance findings. The consistency of the differences in both the CV mortality and non-fatal MI components, however, supports the possibility that the finding is reliable.
A wide variety of baseline and procedural differences between the US and non-US (including intended invasive vs. planned medical management, use of GPIIb/IIIa inhibitors, use of drug eluting vs. bare-metal stents) were examined to see if they could account for regional differences, but with one exception, aspirin maintenance dose, these differences did not appear to lead to differences in outcome.
Aspirin Dose
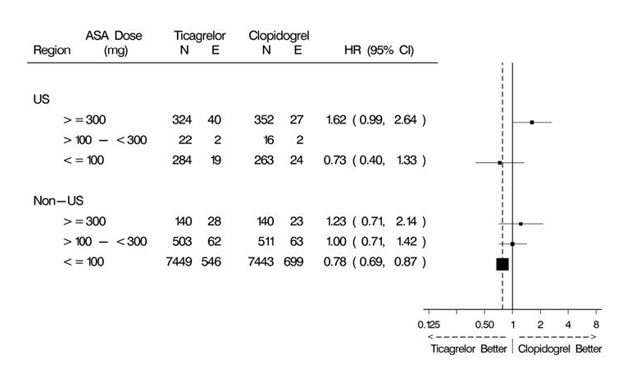
The PLATO protocol left the choice of aspirin maintenance dose up to the investigator and use patterns were different in US sites from sites outside of the US. About 8% of non-US investigators administered aspirin doses above 100 mg, and about 2% administered doses above 300 mg. In the US, 57% of patients received doses above 100 mg and 54% received doses above 300 mg. Overall results favored BRILINTA when used with low maintenance doses (≤100 mg) of aspirin, and results analyzed by aspirin dose were similar in the US and elsewhere. Figure 10 shows overall results by median aspirin dose. Figure 12 shows results by region and dose.
Figure 12 – CV death, MI, stroke by maintenance aspirin dose in the US and outside the US (PLATO)
Like any unplanned subset analysis, especially one where the characteristic is not a true baseline characteristic (but may be determined by usual investigator practice), the above analyses must be treated with caution. It is notable, however, that aspirin dose predicts outcome in both regions with a similar pattern, and that the pattern is similar for the two major components of the primary endpoint, CV death and non-fatal MI.
Despite the need to treat such results cautiously, there appears to be good reason to restrict aspirin maintenance dosage accompanying ticagrelor to 100 mg. Higher doses do not have an established benefit in the ACS setting, and there is a strong suggestion that use of such doses reduces the effectiveness of BRILINTA.
PEGASUS
The PEGASUS TIMI-54 study (NCT01225562) was a 21,162-patient, randomized, double-blind, placebo-controlled, parallel-group study. Two doses of ticagrelor, either 90 mg twice daily or 60 mg twice daily, co-administered with 75-150 mg of aspirin, were compared to aspirin therapy alone in patients with history of MI. The primary endpoint was the composite of first occurrence of CV death, non-fatal MI and non-fatal stroke. CV death and all-cause mortality were assessed as secondary endpoints.
Patients were eligible to participate if they were ≥50 years old, with a history of MI 1 to 3 years prior to randomization, and had at least one of the following risk factors for thrombotic cardiovascular events: age ≥65 years, diabetes mellitus requiring medication, at least one other prior MI, evidence of multivessel coronary artery disease, or creatinine clearance <60 mL/min. Patients could be randomized regardless of their prior ADP receptor blocker therapy or a lapse in therapy. Patients requiring or who were expected to require renal dialysis during the study were excluded. Patients with any previous intracranial hemorrhage, gastrointestinal bleeding within the past 6 months, or with known bleeding diathesis or coagulation disorder were excluded. Patients taking anticoagulants were excluded from participating and patients who developed an indication for anticoagulation during the trial were discontinued from study drug. A small number of patients with a history of stroke were included. Based on information external to PEGASUS, 102 patients with a history of stroke (90 of whom received study drug) were terminated early and no further such patients were enrolled.
Patients were treated for at least 12 months and up to 48 months with a median follow up time of 33 months.
Patients were predominantly male (76%) Caucasian (87%) with a mean age of 65 years, and 99.8% of patients received prior aspirin therapy.
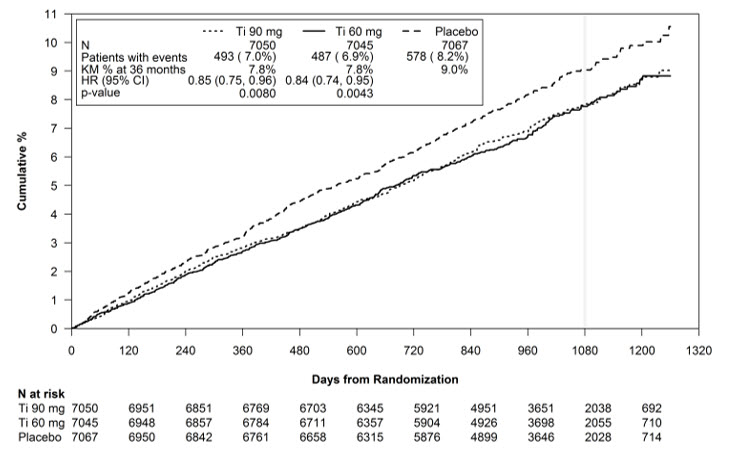
The Kaplan-Meier curve (Figure 13) shows time to first occurrence of the primary composite endpoint of CV death, non-fatal MI or non-fatal stroke.
Figure 13 – Time to First Occurrence of CV death, MI or Stroke (PEGASUS)
Ti = Ticagrelor BID, CI = Confidence interval; HR = Hazard ratio; KM = Kaplan-Meier; N = Number of patients.
Both the 60 mg and 90 mg regimens of BRILINTA in combination with aspirin were superior to aspirin alone in reducing the incidence of CV death, MI or stroke. The absolute risk reductions for BRILINTA plus aspirin vs. aspirin alone were 1.27% and 1.19% for the 60 and 90 mg regimens, respectively. Although the efficacy profiles of the two regimens were similar, the lower dose had lower risks of bleeding and dyspnea.
Table 8 shows the results for the 60 mg plus aspirin regimen vs. aspirin alone.
Table 8 - Incidences of the primary composite endpoint, primary composite endpoint components, and secondary endpoints (PEGASUS) BRILINTA*
N=7045Placebo
N=7067HR (95% CI)
p-value
Events / 1000 patient years
Events / 1000 patient years
Time to first CV death, MI, or stroke†
26
31
0.84 (0.74, 0.95)
0.0043
9
11
0.83 (0.68, 1.01)
Myocardial infarction§
15
18
0.84 (0.72, 0.98)
Stroke§
5
7
0.75 (0.57, 0.98)
All-cause mortality‡
16
18
0.89 (0.76, 1.04)
CI = Confidence interval; CV = Cardiovascular; HR = Hazard ratio; MI = Myocardial infarction; N = Number of patients.
In PEGASUS, the relative risk reduction (RRR) for the composite endpoint from 1 to 360 days (17% RRR) and from 361 days and onwards (16% RRR) were similar.
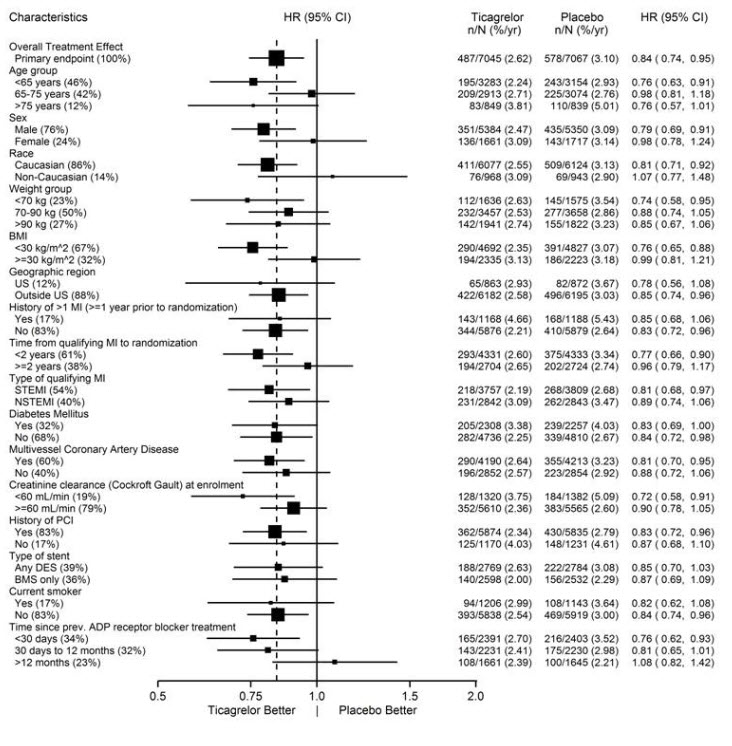
The treatment effect of BRILINTA 60 mg over aspirin appeared similar across most pre-defined subgroups, see Figure 14.
Figure 14 – Subgroup analyses of ticagrelor 60 mg (PEGASUS)
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and most of which were pre-specified. The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
14.2 Coronary Artery Disease but No Prior Stroke or Myocardial Infarction
THEMIS
The THEMIS study (NCT01991795) was a double-blind, parallel group, study in which 19,220 patients with CAD and Type 2 Diabetes Mellitus (T2DM) but no history of MI or stroke were randomized to twice daily BRILINTA or placebo, on a background of 75-150 mg of aspirin. The primary endpoint was the composite of first occurrence of CV death, MI, and stroke. CV death, MI, ischemic stroke, and all-cause death were assessed as secondary endpoints.
Patients were eligible to participate if they were ≥ 50 years old with CAD, defined as a history of PCI or CABG, or angiographic evidence of ≥ 50% lumen stenosis of at least 1 coronary artery and T2DM treated for at least 6 months with glucose-lowering medication. Patients with previous intracerebral hemorrhage, gastrointestinal bleeding within the past 6 months, known bleeding diathesis, and coagulation disorder were excluded. Patients taking anticoagulants or ADP receptor antagonists were excluded from participating, and patients who developed an indication for those medications during the trial were discontinued from study drug.
Patients were treated for a median of 33 months and up to 58 months.
Patients were predominantly male (69%) with a mean age of 66 years. At baseline, 80% had a history of coronary artery revascularization; 58% had undergone PCI, 29% had undergone a CABG and 7% had undergone both. The proportion of patients studied in the US was 12%. Patients in THEMIS had established CAD and other risk factors that put them at higher cardiovascular risk.
BRILINTA was superior to placebo in reducing the incidence of CV death, MI, or stroke. The effect on the composite endpoint was driven by the individual components MI and stroke; see Table 9.
Table 9 - Primary composite endpoint, primary endpoint components, and secondary endpoints (THEMIS) BRILINTA
N=9619Placebo
N=9601HR (95% CI)
p-value
Events / 1000 patient years
Events / 1000 patient years
Time to first CV death, MI, or stroke*
24
27
0.90 (0.81, 0.99)
0.04
CV death†
12
11
1.02 (0.88, 1.18)
Myocardial infarction†
9
11
0.84 (0.71, 0.98)
Stroke†
6
7
0.82 (0.67, 0.99)
Secondary endpoints
CV death
12
11
1.02 (0.88, 1.18)
Myocardial infarction
9
11
0.84 (0.71, 0.98)
Ischemic stroke
5
6
0.80 (0.64, 0.99)
All-cause death
18
19
0.98 (0.87, 1.10)
CI = Confidence interval; CV = Cardiovascular; HR = Hazard ratio; MI = Myocardial infarction.
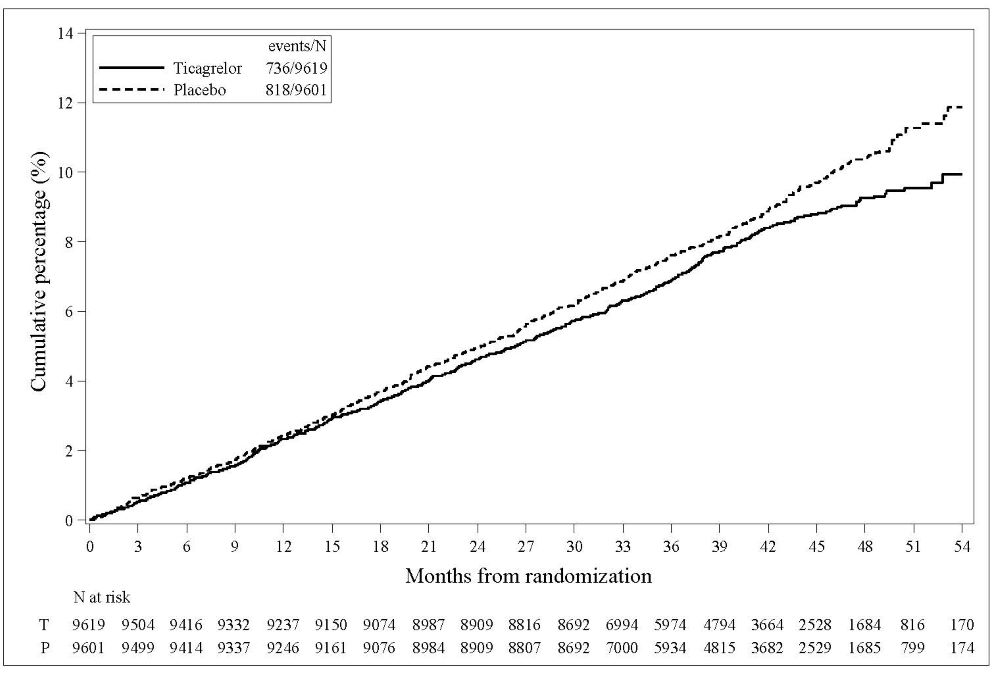
The Kaplan-Meier curve (Figure 15) shows time to first occurrence of the primary composite endpoint of CV death, MI, or stroke.
Figure 15 - Time to First Occurrence of CV death, MI or Stroke (THEMIS)
T = Ticagrelor; P = Placebo; N = Number of patients.
The treatment effect of BRILINTA appeared similar across patient subgroups, see Figure 16.
Figure 16 – Subgroup analyses of ticagrelor (THEMIS)
Note: The figure above presents effects in various subgroups all of which are baseline characteristics. The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
14.3 Acute Ischemic Stroke or Transient Ischemic Attack (TIA)
THALES
The THALES study (NCT03354429) was a 11016-patient, randomized, double-blind, parallel-group study of BRILINTA 90 mg twice daily versus placebo in patients with acute ischemic stroke or transient ischemic attack (TIA). The primary endpoint was the first occurrence of the composite of stroke and death up to 30 days. Ischemic stroke was assessed as one of the secondary endpoints.
Patients were eligible to participate if they were ≥40 years old, with non-cardioembolic acute ischemic stroke (NIHSS score ≤5) or high-risk TIA (defined as ABCD2 score ≥6 or ipsilateral atherosclerotic stenosis ≥50% in the internal carotid or an intracranial artery). Patients who received thrombolysis or thrombectomy within 24 hours prior to randomization were not eligible.
Patients were randomized within 24 hours of onset of an acute ischemic stroke or TIA to receive 30 days of either BRILINTA (90 mg twice daily, with an initial loading dose of 180 mg) or placebo, on a background of aspirin initially 300-325 mg then 75-100 mg daily. The median treatment duration was 31 days.
BRILINTA was superior to placebo in reducing the rate of the primary endpoint (composite of stroke and death), corresponding to a relative risk reduction (RRR) of 17% and an absolute risk reduction (ARR) of 1.1% (Table 10). The effect was driven primarily by a significant reduction in the stroke component of the primary endpoint (19% RRR, 1.1% ARR).
Table 10 - Incidences of the primary composite endpoint, primary composite endpoint components, and secondary endpoint (THALES) - *
- The number of patients with the event of interest. In the time to first stroke, patients who died are censored at the time of death.
BRILINTA
N=5523
Placebo
N=5493
HR (95% CI)
p-value
n (patients with event)
KM%
n (patients with event)
KM%
Time to first Stroke or Death
303
5.4%
362
6.5%
0.83 (0.71, 0.96)
0.015
Time to first Stroke*
284
5.1%
347
6.3%
0.81 (0.69, 0.95)
Time to Death*
36
0.6%
27
0.5%
1.33 (0.81, 2.19)
Secondary Endpoint
Time to first Ischemic Stroke
276
5.0%
345
6.2%
0.79 (0.68, 0.93)
0.004
CI = Confidence interval; HR = Hazard ratio; KM = Kaplan-Meier percentage calculated at 30 days; N = Number of patients
The Kaplan-Meier curve (Figure 17) shows the time to first occurrence of the primary composite endpoint of stroke and death.
Figure 17 – Time to First Occurrence of Stroke or Death (THALES)
KM%: Kaplan-Meier percentage evaluated at Day 30; T=Ticagrelor; P=placebo; N=Number of patients
BRILINTA’s treatment effect on stroke and on death accrued over the first 10 days and was sustained at 30 days. Although not studied, this suggests that shorter treatment could result in similar benefit and reduced bleeding risk.
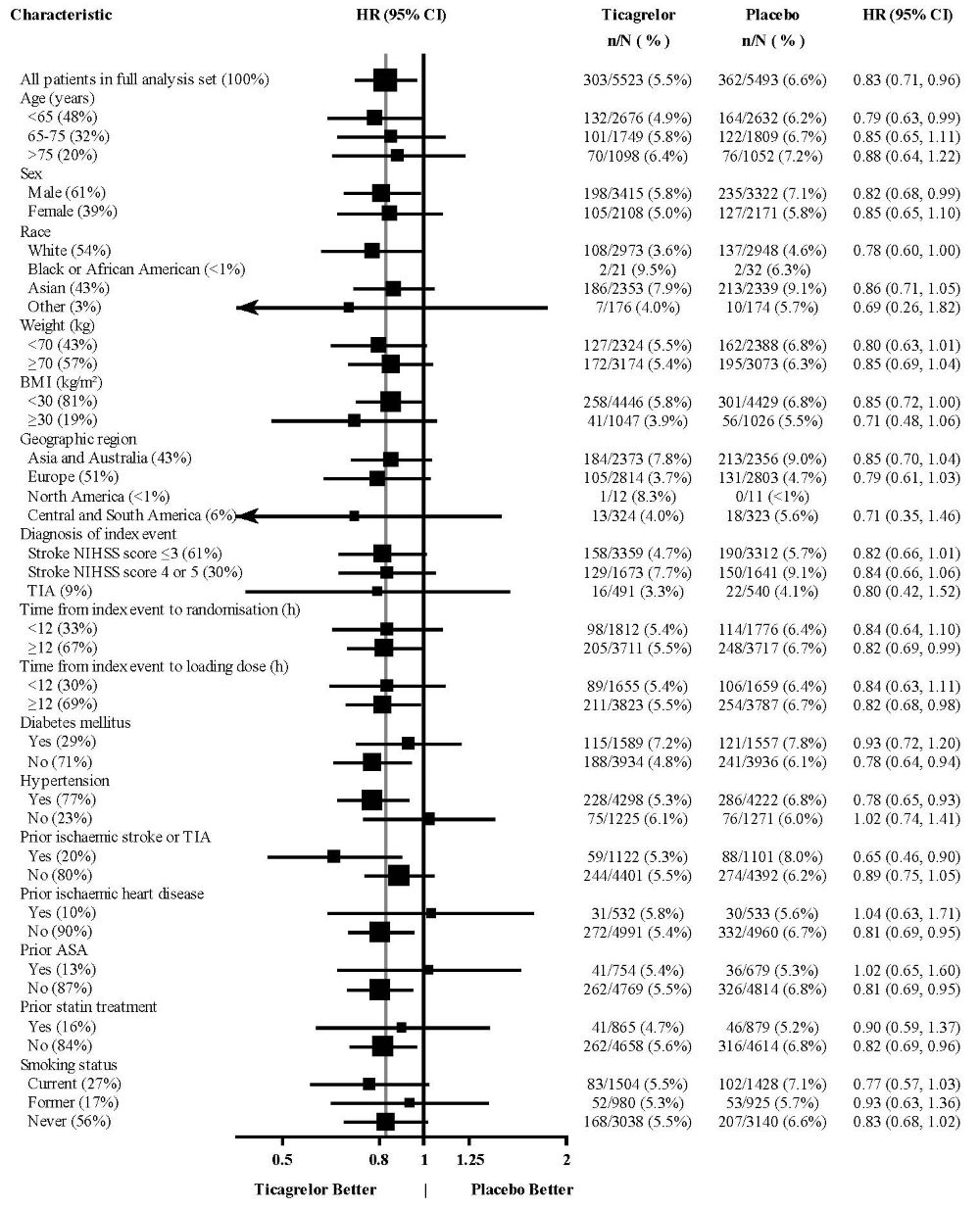
The treatment effect of BRILINTA was generally consistent across pre-defined subgroups (Figure 18).
Figure 18 - Subgroup analyses of ticagrelor 90 mg (THALES)
Note: The figure above presents effects in various subgroups all of which are baseline characteristics and were pre-specified. The 95% confidence limits that are shown do not take into account how many comparisons were made, nor do they reflect the effect of a particular factor after adjustment for all other factors. Apparent homogeneity or heterogeneity among groups should not be over-interpreted.
At Day 30, there was an absolute reduction of 1.2% (95% CI: -2.1%, -0.3%) in the incidence of non-hemorrhagic stroke and death (excluding fatal bleed) favoring ticagrelor (294 events: 5.3%) over placebo (359 events: 6.5%) in the intention-to-treat population. In the same population, there was an absolute increase of 0.4% (95% CI: 0.2%, 0.6%) in the incidence of GUSTO severe bleeding unfavorable to ticagrelor arm (28 events: 0.5%) compared to the placebo arm (7 events: 0.1%).
-
16 HOW SUPPLIED/STORAGE AND HANDLING
BRILINTA (ticagrelor) 90 mg is supplied as a round, biconvex, yellow, film-coated tablet with a “90” above “T” on one side:
Overbagged with 10 tablets per bag, NDC 55154-9618-0
Approximately 1440 tablets per bottle, NDC 55154-9618-8
Storage and Handling
Store at 25°C (77°F); excursions permitted to 15 to 30°C (59 to 86°F) [see USP controlled room temperature].
-
17 PATIENT COUNSELING INFORMATION
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Advise patients daily doses of aspirin should not exceed 100 mg and to avoid taking any other medications that contain aspirin.
Advise patients that they:
- •
- Will bleed and bruise more easily
- •
- Will take longer than usual to stop bleeding
- •
- Should report any unanticipated, prolonged or excessive bleeding, or blood in their stool or urine.
Advise patients to contact their doctor if they experience unexpected shortness of breath, especially if severe.
Advise patients to inform physicians and dentists that they are taking BRILINTA before any surgery or dental procedure.
Advise women that breastfeeding is not recommended during treatment with BRILINTA [see Use in Specific Populations (8.2)].
BRILINTA® is a trademark of the AstraZeneca group of companies.
Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
Distributed By:
Cardinal Health
Dublin, OH 43017
L50533760124
ER7198S Rev. B
© AstraZeneca 2024
-
MEDICATION GUIDE
MEDICATION GUIDE
BRILINTA® (brih-LIN-tah)
(ticagrelor) Tablets
What is the most important information I should know about BRILINTA?
BRILINTA is used to lower your chance of having, or dying from, a heart attack or stroke. BRILINTA (and similar drugs) can cause bleeding that can be serious and sometimes lead to death. In cases of serious bleeding, such as internal bleeding, the bleeding may result in the need for blood transfusions or surgery. While you take BRILINTA:
- •
- you may bruise and bleed more easily
- •
- you are more likely to have nose bleeds
- •
- it will take longer than usual for any bleeding to stop
Call your healthcare provider right away, if you have any of these signs or symptoms of bleeding while taking BRILINTA:
- •
- bleeding that is severe or that you cannot control
- •
- pink, red or brown urine
- •
- vomiting blood or your vomit looks like “coffee grounds”
- •
- red or black stools (looks like tar)
- •
- coughing up blood or blood clots
Do not stop taking BRILINTA without talking to the healthcare provider who prescribes it for you. People who are treated with a stent, and stop taking BRILINTA too soon, have a higher risk of getting a blood clot in the stent, having a heart attack, or dying. If you stop BRILINTA because of bleeding, or for other reasons, your risk of a heart attack or stroke may increase.
Your healthcare provider may instruct you to stop taking BRILINTA 5 days before surgery. This will help to decrease your risk of bleeding with your surgery or procedure. Your healthcare provider should tell you when to start taking BRILINTA again, as soon as possible after surgery.
Taking BRILINTA with aspirin
BRILINTA is taken with aspirin, unless your healthcare provider specifically tells you otherwise. Talk to your healthcare provider about the dose of aspirin that you should take with BRILINTA. In most cases, you should not take a dose of aspirin higher than 100 mg daily. Do not take doses of aspirin higher than what your healthcare provider tells you to take. Tell your healthcare provider if you take other medicines that contain aspirin, and do not take new over-the-counter medicines with aspirin in them.
What is BRILINTA?
BRILINTA is a prescription medicine used to:
- •
- decrease your risk of death, heart attack, and stroke in people with a blockage of blood flow to the heart (acute coronary syndrome or ACS) or a history of a heart attack. BRILINTA can also decrease your risk of blood clots in your stent in people who have received stents for the treatment of ACS.
- •
- decrease your risk of a first heart attack or stroke in people who have a condition where the blood flow to the heart is decreased (coronary artery disease or CAD) who are at high risk for having a heart attack or stroke.
- •
- decrease your risk of stroke in people who are having a stroke (acute ischemic stroke) or mini-stroke (transient ischemic attack or TIA).
It is not known if BRILINTA is safe and effective in children.
Do not take BRILINTA if you:
- •
- have a history of bleeding in the brain
- •
- are bleeding now
- •
- are allergic to ticagrelor or any of the ingredients in BRILINTA. See the end of this Medication Guide for a complete list of ingredients in BRILINTA.
Before taking BRILINTA, tell your healthcare provider about all of your medical conditions, if you:
- •
- have had bleeding problems in the past
- •
- have had any recent serious injury or surgery
- •
- plan to have surgery or a dental procedure. See “What is the most important information I should know about BRILINTA?”
- •
- have a history of stomach ulcers or colon polyps
- •
- have lung problems, such as COPD or asthma
- •
- have liver problems
- •
- have a history of stroke
- •
- are pregnant or plan to become pregnant. It is not known if BRILINTA will harm your unborn baby. You and your healthcare provider should decide if you will take BRILINTA.
- •
- are breastfeeding or plan to breastfeed. It is not known if BRILINTA passes into your breast milk. You should not breastfeed during treatment with BRILINTA. Talk to your healthcare provider about the best way to feed your baby during treatment with BRILINTA.
Tell all of your healthcare providers and dentists that you are taking BRILINTA. They should talk to the healthcare provider who prescribed BRILINTA for you before you have any surgery or procedure.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. BRILINTA may affect the way other medicines work, and other medicines may affect how BRILINTA works. Certain medicines may increase your risk of bleeding.
Know the medicines you take. Keep a list of them to show your healthcare provider and pharmacist when you get a new medicine.
How should I take BRILINTA?
- •
- Take BRILINTA exactly as prescribed by your healthcare provider.
- •
- Your healthcare provider will tell you how many BRILINTA tablets to take and when to take them.
- •
- Take BRILINTA with aspirin, unless your healthcare provider specifically tells you otherwise. See “What is the most important information I should know about BRILINTA?”
- •
- You may take BRILINTA with or without food.
- •
- Take BRILINTA two times each day, around the same times each day.
- •
- If you miss your scheduled dose of BRILINTA, take your next dose at its scheduled time. Do not take 2 doses at the same time unless your healthcare provider tells you to.
- •
- If you take too much BRILINTA, call your healthcare provider or local poison control center or go to the nearest emergency room right away.
- If you are unable to swallow the tablet(s) whole, you may crush the BRILINTA tablet(s) and mix it with water. Drink all the water right away. Refill the glass with water, stir, and drink all the water.
- BRILINTA may also be given through certain nasogastric (NG) tubes. Ask your healthcare provider for instructions on how to take BRILINTA through a NG tube.
What are the possible side effects of BRILINTA?
BRILINTA can cause serious side effects, including:
- •
- See “What is the most important information I should know about BRILINTA?”
- •
- Shortness of breath. Tell your healthcare provider if you have new, worsening or unexpected shortness of breath when you are at rest, at night, or when you are doing any activity.
- Slow or irregular heartbeat.
- •
- Irregular breathing. Tell your healthcare provider if you develop irregular breathing patterns when asleep or awake such as speeding up, slowing down or short pauses in breathing. Your healthcare provider will decide if you need further evaluation.
These are not all of the possible side effects of BRILINTA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store BRILINTA?
- •
- Store BRILINTA at room temperature between 68°F to 77°F (20°C to 25°C).
Keep BRILINTA and all medicines out of the reach of children.
General information about the safe and effective use of BRILINTA.
Medicines are sometimes prescribed for purposes other than those listed in a Medication Guide. Do not use BRILINTA for a condition for which it was not prescribed. Do not give BRILINTA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about BRILINTA that is written for health professionals.
What are the ingredients in BRILINTA?
Active ingredient: ticagrelor
90 mg tablets:
Inactive ingredients: mannitol, dibasic calcium phosphate, sodium starch glycolate, hydroxypropyl cellulose, magnesium stearate, hydroxypropyl methylcellulose, titanium dioxide, talc, polyethylene glycol 400, and ferric oxide yellow.60 mg tablets:
Inactive ingredients: mannitol, dibasic calcium phosphate, sodium starch glycolate, hydroxypropyl cellulose, magnesium stearate, hydroxypropyl methylcellulose, titanium dioxide, polyethylene glycol 400, ferric oxide black and ferric oxide red.Distributed by: AstraZeneca Pharmaceuticals LP, Wilmington, DE 19850
Distributed By:
Cardinal Health
Dublin, OH 43017
L50533760124
ER7198S Rev. B
For more information call 1-800-236-9933 or go to www.Brilinta.com.
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 03/2024
- Package/Label Display Panel
-
INGREDIENTS AND APPEARANCE
BRILINTA
ticagrelor tabletProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:55154-9618(NDC:0186-0777) Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TICAGRELOR (UNII: GLH0314RVC) (TICAGRELOR - UNII:GLH0314RVC) TICAGRELOR 90 mg Inactive Ingredients Ingredient Name Strength MANNITOL (UNII: 3OWL53L36A) DIBASIC CALCIUM PHOSPHATE DIHYDRATE (UNII: O7TSZ97GEP) SODIUM STARCH GLYCOLATE TYPE A POTATO (UNII: 5856J3G2A2) HYDROXYPROPYL CELLULOSE (1600000 WAMW) (UNII: RFW2ET671P) MAGNESIUM STEARATE (UNII: 70097M6I30) HYPROMELLOSE, UNSPECIFIED (UNII: 3NXW29V3WO) TITANIUM DIOXIDE (UNII: 15FIX9V2JP) TALC (UNII: 7SEV7J4R1U) POLYETHYLENE GLYCOL 400 (UNII: B697894SGQ) FERRIC OXIDE YELLOW (UNII: EX438O2MRT) Product Characteristics Color YELLOW Score no score Shape ROUND (biconvex) Size 9mm Flavor Imprint Code 90;T Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:55154-9618-0 10 in 1 BAG 08/05/2011 1 1 in 1 BLISTER PACK; Type 0: Not a Combination Product 2 NDC:55154-9618-8 1440 in 1 BOTTLE, PLASTIC; Type 0: Not a Combination Product 08/05/2011 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA022433 08/05/2011 Labeler - Cardinal Health 107, LLC (118546603)