Label: revex- nalmefene hydrochloride injection, solution
-
Contains inactivated NDC Code(s)
NDC Code(s): 10019-311-22, 10019-311-39, 10019-315-21, 10019-315-39 - Packager: Baxter Healthcare Corporation
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
Drug Label Information
Updated June 21, 2007
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
- N/A - Section Title Not Found In Database
-
DESCRIPTION
REVEX (nalmefene hydrochloride injection), an opioid antagonist, is a 6-methylene analogue of naltrexone. The chemical structure is shown below:
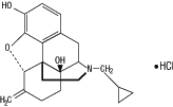
Molecular Formula: C 21H25NO3•HCl
Molecular Weight: 375.9, CAS # 58895-64-0
Chemical Name: 17-(Cyclopropylmethyl)-4,5α-epoxy-6-methylenemorphinan-3,14-diol, hydrochloride salt.
Nalmefene hydrochloride is a white to off-white crystalline powder which is freely soluble in water up to 130 mg/mL and slightly soluble in chloroform up to 0.13 mg/mL, with a pK a of 7.6.
REVEX is available as a sterile solution for intravenous, intramuscular, and subcutaneous administration in two concentrations, containing 100 µg or 1.0 mg of nalmefene free base per mL. The 100 µg/mL concentration contains 110.8 µg of nalmefene hydrochloride and the 1.0 mg/mL concentration contains 1.108 mg of nalmefene hydrochloride per mL. Both concentrations contain 9.0 mg of sodium chloride per mL and the pH is adjusted to 3.9 with hydrochloric acid.
Concentrations and dosages of REVEX are expressed as the free base equivalent of nalmefene.
-
CLINICAL PHARMACOLOGY
Pharmacodynamics
REVEX prevents or reverses the effects of opioids, including respiratory depression, sedation, and hypotension. Pharmacodynamic studies have shown that REVEX has a longer duration of action than naloxone at fully reversing doses. REVEX has no opioid agonist activity.
REVEX is not known to produce respiratory depression, psychotomimetic effects, or pupillary constriction. No pharmacological activity was observed when REVEX was administered in the absence of opioid agonists.
REVEX has not been shown to produce tolerance, physical dependence, or abuse potential.
REVEX can produce acute withdrawal symptoms in individuals who are opioid dependent.
Pharmacokinetics
Nalmefene exhibited dose proportional pharmacokinetics following intravenous administration of 0.5 mg to 2.0 mg. Pharmacokinetic parameters for nalmefene after a 1 mg intravenous administration in adult male volunteers are listed in Table 1.
Table 1: Mean (CV%) Nalmefene Pharmacokinetic Parameters
In Adult Males Following a 1 mg Intravenous DoseParameter Young, N=18 Elderly, N=11 Age 19-32 62-80 Cp at 5 min. (ng/mL) 3.7 (29) 5.8 (38) Vdss (L/kg) 8.6 (19) 8.6 (29) Vc (L/kg) 3.9 (29) 2.8 (41) AUC0-inf (ng-hr/mL) 16.6 (27) 17.3 (14) Terminal T½ (hr) 10.8 (48) 9.4 (49) Clplasma (L/hr/kg) 0.8 (23) 0.8 (18) Absorption
Nalmefene was completely bioavailable following intramuscular or subcutaneous administration in 12 male volunteers relative to intravenous nalmefene. The relative bioavailabilities of intramuscular and subcutaneous routes of administration were 101.5%± 8.1% (Mean ± SD) and 99.7%± 6.9%, respectively. Nalmefene will be administered primarily as an intravenous bolus, however, nalmefene can be given intra-muscularly (IM) or subcutaneously (SC) if venous access cannot be established. While the time to maximum plasma nalmefene concentration was 2.3 ± 1.1 hours following intramuscular and 1.5 ± 1.2 hours following subcutaneous administrations, therapeutic plasma concentrations are likely to be reached within 5-15 minutes after a 1 mg dose in an emergency. Because of the variability in the speed of absorption for IM& SC dosing, and the inability to titrate to effect, great care should be taken if repeated doses must be given by these routes.
Distribution
Following a 1 mg parenteral dose, nalmefene was rapidly distributed. In a study of brain receptor occupancy, a 1 mg dose of nalmefene blocked over 80% of brain opioid receptors within 5 minutes after administration. The apparent volumes of distribution centrally (Vc) and at steady-state (Vdss) are 3.9 ± 1.1 L/kg and 8.6 ± 1.7 L/kg, respectively. Ultrafiltration studies of nalmefene have demonstrated that 45% (CV 4.1%) is bound to plasma proteins over a concentration range of 0.1 to 2µg/mL. An in vitro determination of the distribution of nalmefene in human blood demonstrated that nalmefene distributed 67% (CV 8.7%) into red blood cells and 39% (CV 6.4%) into plasma. The whole blood to plasma ratio was 1.3 (CV 6.6%) over the nominal concentration range in whole blood from 0.376 to 30 ng/mL.
Metabolism
Nalmefene is metabolized by the liver, primarily by glucuronide conjugation, and excreted in the urine. Nalmefene is also metabolized to trace amounts of an N-dealkylated metabolite. Nalmefene glucuronide is inactive and the N-dealkylated metabolite has minimal pharmacological activity. Less than 5% of nalmefene is excreted in the urine unchanged. Seventeen percent (17%) of the nalmefene dose is excreted in the feces. The plasma concentration-time profile in some subjects suggests that nalmefene undergoes enterohepatic recycling.
Elimination
After intravenous administration of 1 mg REVEX to normal males (ages 19-32), plasma concentrations declined biexponentially with a redistribution and a terminal elimination half-life of 41 ± 34 minutes and 10.8 ± 5.2 hours, respectively. The systemic clearance of nalmefene is 0.8 ± 0.2 L/hr/kg and the renal clearance is 0.08 ± 0.04 L/hr/kg.
Special Populations
Elderly
Dose proportionality was observed in nalmefene AUC0-inf following 0.5 to 2 mg intravenous administration to elderly male subjects. Following a 1 mg intravenous nalmefene dose, there were no significant differences between young (19-32 years) and elderly (62-80 years) adult male subjects with respect to plasma clearance, steady-state volume of distribution, or half-life. There was an apparent age-related decrease in the central volume of distribution (young: 3.9± 1.1 L/kg, elderly: 2.8 ± 1.1 L/kg) that resulted in a greater initial nalmefene concentration in the elderly group. While initial nalmefene plasma concentrations were transiently higher in the elderly, it would not be anticipated that this population would require dosing adjustment. No clinical adverse events were noted in the elderly following the 1 mg intravenous nalmefene dose.
Patients with Hepatic Impairment
Subjects with hepatic disease, when compared to matched normal controls, had a 28.3% decrease in plasma clearance of nalmefene (0.56 ± 0.21 L/hr/kg versus 0.78 ± 0.24 L/hr/kg, respectively). Elimination half-life increased from 10.2 ± 2.2 hours to 11.9 ± 2.0 hours in the hepatically impaired. No dosage adjustment is recommended since nalmefene will be administered as an acute course of therapy.
Patients with Renal Impairment
There was a statistically significant 27% decrease in plasma clearance of nalmefene in the end-stage renal disease (ESRD) population during interdialysis (0.57± 0.20 L/hr/kg) and a 25% decreased plasma clearance in the ESRD population during intradialysis (0.59 ± 0.18 L/hr/kg) compared to normals (0.79± 0.24 L/hr/kg). The elimination half-life was prolonged in ESRD patients from 10.2 ± 2.2 hours in normals to 26.1 ± 9.9 hours. (SeeDOSAGE AND ADMINISTRATION.)
-
CLINICAL TRIALS
REVEX has been administered to reverse the effects of opioids after general anesthesia and in the treatment of overdose. It has also been used to reverse the systemic effects of intrathecal opioids.
Reversal of Postoperative Opioid Depression
REVEX (nalmefene hydrochloride injection) (N=326) was studied in 5 controlled trials in patients who had received morphine or fentanyl intraoperatively. The primary efficacy criterion was the reversal of respiratory depression. A positive reversal was defined as both an increase in respiratory rate by 5 breaths per minute and a minimum respiratory rate of 12 breaths per minute. Five minutes after administration, initial single REVEX doses of 0.1, 0.25, 0.5, or 1.0 µg/kg had effectively reversed respiratory depression in a dose-dependent manner. Twenty minutes after initial administration, respiratory depression had been effectively reversed in most patients receiving cumulative doses within the recommended range (0.1 to 1.0 µg/kg). Total doses of REVEX above 1.0 µg/kg did not increase the therapeutic response. The postoperative administration of REVEX at the recommended doses did not prevent the analgesic response to subsequently administered opioids.
Reversal of the Effect of Intrathecally Administered Opioids
Intravenous REVEX at doses of 0.5 and 1.0 µg/kg was administered to 47 patients given intrathecal morphine. One to 2 doses of 0.5 and 1.0 µg/kg REVEX reversed respiratory depression in most patients. The administration of REVEX at the recommended doses did not prevent the analgesic response to subsequently administered opioids.
Management of Known or Suspected Opioid Overdose
REVEX (N=284) at doses of 0.5 mg to 2.0 mg was studied in 4 trials of patients who were presumed to have taken an opioid overdose. REVEX doses of 0.5 mg to 1.0 mg effectively reversed respiratory depression within 2 to 5 minutes in most patients subsequently confirmed to have opioid overdose. A total dose greater than 1.5 mg did not increase the therapeutic response.
- INDICATIONS AND USAGE
- CONTRAINDICATIONS
-
WARNINGS
Use of REVEX in Emergencies
REVEX, like all drugs in this class, is not the primary treatment for ventilatory failure. In most emergency settings, treatment with REVEX should follow, not precede, the establishment of a patent airway, ventilatory assistance, administration of oxygen, and establishment of circulatory access.
Risk of Recurrent Respiratory Depression
Accidental overdose with long acting opioids [such as methadone and levo-alpha-acetylmethadol (LAAM)] may result in prolonged respiratory depression. Respiratory depression in both the postoperative and overdose setting may be complex and involve the effects of anesthetic agents, neuromuscular blockers, and other drugs. While REVEX has a longer duration of action than naloxone in fully reversing doses, the physician should be aware that a recurrence of respiratory depression is possible, even after an apparently adequate initial response to REVEX treatment.
Patients treated with REVEX should be observed until, in the opinion of the physician, there is no reasonable risk of recurrent respiratory depression.
-
PRECAUTIONS
General
Cardiovascular Risks with Narcotic Antagonists
Pulmonary edema, cardiovascular instability, hypotension, hypertension, ventricular tachycardia, and ventricular fibrillation have been reported in connection with opioid reversal in both postoperative and emergency department settings. In many cases, these effects appear to be the result of abrupt reversal of opioid effects.
Although REVEX has been used safely in patients with pre-existing cardiac disease, all drugs of this class should be used with caution in patients at high cardiovascular risk or who have received potentially cardiotoxic drugs. (See DOSAGE AND ADMINISTRATION.)
Risk of Precipitated Withdrawal
REVEX, like other opioid antagonists, is known to produce acute withdrawal symptoms and, therefore, should be used with extreme caution in patients with known physical dependence on opioids or following surgery involving high doses of opioids. Imprudent use or excessive doses of opioid antagonists in the postoperative setting has been associated with hypertension, tachycardia, and excessive mortality in patients at high risk for cardiovascular complications. (See PRECAUTIONS.)
Incomplete Reversal of Buprenorphine
Preclinical studies have shown that nalmefene at doses up to 10 mg/kg (437 times the maximum recommended human dose) produced incomplete reversal of buprenorphine-induced analgesia in animal models. This appears to be a consequence of a high affinity and slow displacement of buprenorphine from the opioid receptors. Hence, REVEX may not completely reverse buprenorphine-induced respiratory depression.
Drug Interactions
REVEX has been administered after benzodiazepines, inhalational anesthetics, muscle relaxants, and muscle relaxant antagonists administered in conjunction with general anesthesia. It also has been administered in outpatient settings, both in trials in conscious sedation and in the emergency management of overdose following a wide variety of agents. No deleterious interactions have been observed.
Preclinical studies have shown that both flumazenil and nalmefene can induce seizures in animals. The coadministration of both flumazenil and nalmefene produced fewer seizures than expected in a study in rodents, based on the expected effects of each drug alone. Based on these data, an adverse interaction from the coadministration of the two drugs is not expected, but physicians should remain aware of the potential risk of seizures from agents in these classes.
Carcinogenesis and Mutagenesis and Impairment of Fertility
Nalmefene did not have mutagenic activity in the Ames test with five bacterial strains or the mouse lymphoma assay. Clastogenic activity was not observed in the mouse micronucleus test or in the cytogenic bone marrow assay in rats. However, nalmefene did exhibit a weak but significant clastogenic activity in the human lymphocyte metaphase assay in the absence but not in the presence of exogenous metabolic activation. Oral administration of nalmefene up to 1200 mg/m 2/day did not affect fertility, reproductive performance, and offspring survival in rats.
Use in Pregnancy
Pregnancy Category B
Reproduction studies have been performed in rats (up to 1200 mg/m 2/day) and rabbits (up to 2400 mg/m2/day) by oral administration of nalmefene and in rabbits by intravenous administration up to 96 mg/m2/day (114 times the human dose). There was no evidence of impaired fertility or harm to the fetus. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, this drug should be used during pregnancy only if clearly needed.
Nursing Mothers
Nalmefene and its metabolites were secreted into rat milk, reaching concentrations approximately three times those in plasma at one hour and decreasing to about half the corresponding plasma concentrations by 24 hours following bolus administration. As no clinical information is available, caution should be exercised when REVEX is administered to a nursing woman.
Use in Pediatric Patients
Safety and effectiveness of REVEX in pediatric patients have not been established.
Use in Neonates
The safety and effectiveness of REVEX in neonates have not been established in clinical studies. In a preclinical study, nalmefene was administered by subcutaneous injection to rat pups at doses up to 205 mg/m 2/day throughout maternal lactation without producing adverse effects. A preclinical study evaluating the irritancy of the dosage form following arterial and venous administration in animals showed no vascular irritancy.
REVEX (nalmefene hydrochloride injection) should only be used in the resuscitation of the newborn when, in the opinion of the treating physician, the expected benefits outweigh the risks.
Geriatric Use
Clinical studies of REVEX (nalmefene hydrochloride injection) did not include sufficient number of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
-
ADVERSE REACTIONS
Adverse event information was obtained following administration of REVEX to 152 normal volunteers and in controlled clinical trials to 1127 patients for the treatment of opioid overdose or for postoperative opioid reversal.
Nalmefene was well tolerated and showed no serious toxicity during experimental administration to healthy individuals, even when given at 15 times the highest recommended dose. In a small number of subjects, at doses exceeding the recommended REVEX dose, nalmefene produced symptoms suggestive of reversal of endogenous opioids, such as have been reported for other narcotic antagonist drugs. These symptoms (nausea, chills, myalgia, dysphoria, abdominal cramps, and joint pain) were usually transient and occurred at very low frequency.
Such symptoms of precipitated opioid withdrawal at the recommended clinical doses were seen in both postoperative and overdose patients who were later found to have had histories of covert opioid use. Symptoms of precipitated withdrawal were similar to those seen with other opioid antagonists, were transient following the lower doses used in the postoperative setting, and more prolonged following the administration of the larger doses used in the treatment of overdose.
Tachycardia and nausea following the use of nalmefene in the postoperative setting were reported at the same frequencies as for naloxone at equivalent doses. The risk of both these adverse events was low at doses giving partial opioid reversal and increased with increases in dose. Thus, total doses larger than 1.0 µg/kg in the postoperative setting and 1.5 mg/70 kg in the treatment of overdose are not recommended.
Relative Frequencies of Common Adverse Reactions
With an Incidence Greater than 1%
(all patients, all clinical settings)Adverse Event Nalmefene Naloxone Placebo N=1127 N=369 N=77 Nausea 18% 18% 6% Vomiting 9% 7% 4% Tachycardia 5% 8% - Hypertension 5% 7% - Postoperative pain 4% 4% N/A Fever 3% 4% - Dizziness 3% 4% 1% Headache 1% 1% 4% Chills 1% 1% - Hypotension 1% 1% - Vasodilatation 1% 1% - Incidence less than 1%
CARDIOVASCULAR: Bradycardia, arrhythmia
DIGESTIVE: Diarrhea, dry mouth
NERVOUS SYSTEM: Somnolence, depression, agitation, nervousness, tremor, confusion, withdrawal syndrome, myoclonus
The incidence of adverse events was highest in patients who received more than the recommended dose of REVEX.
Laboratory findings
Transient increases in CPK were reported as adverse events in 0.5% of the postoperative patients studied. These increases were believed to be related to surgery and not believed to be related to the administration of REVEX. Increases in AST were reported as adverse events in 0.3% of the patients receiving either nalmefene or naloxone. The clinical significance of this finding is unknown. No cases of hepatitis or hepatic injury due to either nalmefene or naloxone were observed in the clinical trials.
- DRUG ABUSE AND DEPENDENCE
-
OVERDOSAGE
Intravenous doses of up to 24 mg of nalmefene, administered to healthy volunteers in the absence of opioid agonists, produced no serious adverse reactions, severe signs or symptoms, or clinically significant laboratory abnormalities. As with all opioid antagonists, use in patients physically dependent on opioids can result in precipitated withdrawal reactions that may result in symptoms that require medical attention. Treatment of such cases should be symptomatic and supportive. Administration of large amounts of opioids to patients receiving opioid antagonists in an attempt to overcome a full blockade has resulted in adverse respiratory and circulatory reactions.
-
DOSAGE AND ADMINISTRATION
Important Information - Dosage Forms
REVEX is supplied in two concentrations that can be identified by their color coded container labels: a concentration suitable for postoperative use (100 µg/mL) in a blue labeled ampul containing ONE (1) mL and a concentration suitable for the management of overdose (1 mg/mL, 10 times as concentrated, 20 times as much drug) in a green labeled ampul containing TWO (2) mL. Proper steps should be taken to prevent use of the incorrect concentration.
General Principles
REVEX should be titrated to reverse the undesired effects of opioids. Once adequate reversal has been established, additional administration is not required and may actually be harmful due to unwanted reversal of analgesia or precipitated withdrawal.
Duration of Action
The duration of action of REVEX is as long as most opioid analgesics. The apparent duration of action of REVEX will vary, however, depending on the half-life and plasma concentration of the narcotic being reversed, the presence or absence of other drugs affecting the brain or muscles of respiration, and the dose of REVEX administered. Partially reversing doses of REVEX (1 µg/kg) lose their effect as the drug is redistributed through the body, and the effects of these low doses may not last more than 30-60 minutes in the presence of persistent opioid effects. Fully reversing doses (1 mg/70 kg) have been shown to last many hours in both experimental and clinical studies, but may complicate the management of patients who are in pain, at high cardiovascular risk, or who are physically dependent on opioids.
The recommended doses represent a compromise between a desirable controlled reversal and the need for prompt response and adequate duration of action. Using higher dosages or shorter intervals between incremental doses is likely to increase the incidence and severity of symptoms related to acute withdrawal such as nausea, vomiting, elevated blood pressure, and anxiety.
Patients Tolerant to or Physically Dependent on Opioids
REVEX may cause acute withdrawal symptoms in individuals who have some degree of tolerance to and dependence on opioids. These patients should be closely observed for symptoms of withdrawal following administration of the initial and subsequent injections of REVEX. Subsequent doses should be administered with intervals of at least 2-5 minutes between doses to allow the full effect of each incremental dose of REVEX to be reached.
Recommended Doses for Reversal of Postoperative Opioid Depression
Use 100µg/mL dosage strength (blue label) and see Table 2 for initial doses.
The goal of treatment with REVEX in the postoperative setting is to achieve reversal of excessive opioid effects without inducing a complete reversal and acute pain. This is best accomplished with an initial dose of 0.25 µg/kg followed by 0.25µg/kg incremental doses at 2-5 minute intervals, stopping as soon as the desired degree of opioid reversal is obtained. A cumulative total dose above 1.0 µg/kg does not provide additional therapeutic effect.
Table 2: Reversal of Postoperative Opioid Depression Body Weight mL of REVEX
100µg/mL Solution50 kg 0.125 60 kg 0.150 70 kg 0.175 80 kg 0.200 90 kg 0.225 100 kg 0.250 In cases where the patient is known to be at increased cardiovascular risk, it may be desirable to dilute REVEX 1:1 with saline or sterile water and use smaller initial and incremental doses of 0.1 µg/kg.
Management of Known or Suspected Opioid Overdose
Use 1.0 mg/mL dosage strength (green label).
The recommended initial dose of REVEX for non-opioid dependent patients is 0.5 mg/70 kg. If needed, this may be followed by a second dose of 1.0 mg/70 kg, 2-5 minutes later. If a total dose of 1.5 mg /70 kg has been administered without clinical response, additional REVEX (nalmefene hydrochloride injection) is unlikely to have an effect. Patients should not be given more REVEX than is required to restore the respiratory rate to normal, thus minimizing the likelihood of cardiovascular stress and precipitated withdrawal syndrome.
If there is a reasonable suspicion of opioid dependency, a challenge dose of REVEX 0.1 mg/70 kg should be administered initially. If there is no evidence of withdrawal in 2 minutes, the recommended dosing should be followed.
REVEX had no effect in cases where opioids were not responsible for sedation and hypoventilation. Therefore, patients should only be treated with REVEX when the likelihood of an opioid overdose is high, based on a history of opioid overdose or the clinical presentation of respiratory depression with concurrent pupillary constriction.
Repeated Dosing
REVEX is the longest acting of the currently available parenteral opioid antagonists. If recurrence of respiratory depression does occur, the dose should again be titrated to clinical effect using incremental doses to avoid over-reversal.
Hepatic and Renal Disease
Hepatic disease and renal failure substantially reduce the clearance of nalmefene (see Pharmacokinetics). For single episodes of opioid antagonism, adjustment of REVEX dosage is not required. However, in patients with renal failure, the incremental doses should be delivered slowly (over 60 seconds) to minimize the hypertension and dizziness reported following the abrupt administration of nalmefene to such patients.
Loss of Intravenous Access
Should intravenous access be lost or not readily obtainable, a pharmacokinetic study has shown that a single dose of REVEX should be effective within 5-15 minutes after intramuscular or subcutaneous doses of 1.0 mg. (See Pharmacokinetics.)
-
SAFETY AND HANDLING
REVEX is distributed in sealed ampuls which represent no known risk to health care workers. As with all parenterals, care should be taken to prevent the generation and inhalation of aerosols during preparation and use. Dermal absorption of spilled REVEX should be prevented by prompt removal of contaminated clothing and rinsing the skin thoroughly with cool water.
Parenteral drug products should be inspected visually for particulate matter and discoloration prior to administration, whenever solution and container permit.
-
HOW SUPPLIED
REVEX (nalmefene hydrochloride injection) is available in the following presentations:
An ampul containing 1 mL of 100 µg/mL nalmefene base (Blue Label) Box of 10 (NDC 10019-315-21)
An ampul containing 2 mL of 1 mg/mL nalmefene base (Green Label) Box of 10 (NDC 10019-311-22)
Store at controlled room temperature.
REVEX is a registered trademark of Ivax Laboratories, Inc.
Baxter is a registered trademark of Baxter International Inc.
-
INGREDIENTS AND APPEARANCE
REVEX
nalmefene hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:10019-311 Route of Administration INTRAVENOUS, INTRAMUSCULAR, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength nalmefene hydrochloride (UNII: K7K69QC05X) (nalmefene - UNII:TOV02TDP9I) 1 mg in 1 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) 9 mg in 1 mL hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:10019-311-22 10 in 1 BOX 1 NDC:10019-311-39 2 mL in 1 AMPULE REVEX
nalmefene hydrochloride injection, solutionProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:10019-315 Route of Administration INTRAVENOUS, INTRAMUSCULAR, SUBCUTANEOUS Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength nalmefene hydrochloride (UNII: K7K69QC05X) (nalmefene - UNII:TOV02TDP9I) 100 ug in 1 mL Inactive Ingredients Ingredient Name Strength sodium chloride (UNII: 451W47IQ8X) 9 mg in 1 mL hydrochloric acid (UNII: QTT17582CB) Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:10019-315-21 10 in 1 BOX 1 NDC:10019-315-39 1 mL in 1 AMPULE Labeler - Baxter Healthcare Corporation