Label: THIOLA- tiopronin tablet, sugar coated


- NDC Code(s): 0178-0900-01
- Packager: Mission Pharmacal Company
- Category: HUMAN PRESCRIPTION DRUG LABEL
- DEA Schedule: None
- Marketing Status: New Drug Application
Drug Label Information
Updated September 25, 2025
If you are a consumer or patient please visit this version.
- Download DRUG LABEL INFO: PDF XML
- Official Label (Printer Friendly)
-
HIGHLIGHTS OF PRESCRIBING INFORMATION
HIGHLIGHTS OF PRESCRIBING INFORMATION
These highlights do not include all the information needed to use THIOLA® safely and effectively.
See full prescribing information for THIOLA.
THIOLA (tiopronin) tablets, for oral use
Initial U.S. Approval: 1988INDICATIONS AND USAGE
(1)
(1)
(1)
--------------------------------------------INDICATIONS AND USAGE----------------------------------------- (1)
THIOLA is a reducing and complexing thiol indicated, in combination with high fluid intake, alkali, and diet
modification, for the prevention of cystine stone formation in adults and pediatric patients 20 kg and greater
with severe homozygous cystinuria, who are not responsive to these measures alone. (1) (1)DOSAGE AND ADMINISTRATION
------------------------------------------DOSAGE AND ADMINISTRATION-------------------------------------
• The recommended initial dosage in adult patients is 800 mg/day. In clinical studies, the average dosage was
about 1,000 mg/day. (2.1)
• The recommended initial dosage in pediatric patients 20 kg and greater is 15 mg/kg/day. Avoid dosages greater
than 50 mg/kg per day in pediatric patients. (2.1, 5.1, 8.4)
• Administer THIOLA in 3 divided doses at the same times each day at least one hour before or 2 hours after meals.
(2.1)
• Measure urinary cystine 1 month after initiation of THIOLA and every 3 months thereafter. (2.2) (2)DOSAGE FORMS AND STRENGTHS
CONTRAINDICATIONS
WARNINGS AND PRECAUTIONS
------------------------------------------WARNINGS AND PRECAUTIONS------------------------------------- (5)
• Proteinuria, including nephrotic syndrome, and membranous nephropathy, has been reported with tiopronin use.
Pediatric patients receiving greater than 50 mg/kg of tiopronin per day may be at increased risk for proteinuria.
(2.1, 5.1, 8.4)
• Hypersensitivity reactions have been reported during tiopronin treatment. (4, 5.2) (5)ADVERSE REACTIONS
---------------------------------------------ADVERSE REACTIONS------------------------------------------- (6)
Most common adverse reactions (≥10%) are nausea, diarrhea or soft stools, oral ulcers, rash, fatigue, fever,
arthralgia, proteinuria, and emesis. (6)
To report SUSPECTED ADVERSE REACTIONS, contact Mission Pharmacal Company at toll-free phone #
1-800-298-1087 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch. (6)USE IN SPECIFIC POPULATIONS
-----------------------------------------USE IN SPECIFIC POPULATIONS-------------------------------------- (7)
• Lactation: Breastfeeding is not recommended. (8.2)
• Geriatric: Choose dose carefully and monitor renal function in the elderly. (8.5) (7)(7)
(7)
See 17 for PATIENT COUNSELING INFORMATION. (7)
Revised: 01/2021 (7)
Revised: 9/2025
-
Table of Contents
FULL PRESCRIBING INFORMATION: CONTENTS*
1 INDICATIONS AND USAGE
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Monitoring3 DOSAGE FORMS AND STRENGTHS
4 CONTRAINDICATIONS
5 WARNINGS AND PRECAUTIONS
5.1 Proteinuria
5.2 Hypersensitivity Reactions6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use10 OVERDOSAGE
11 DESCRIPTION
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility16 HOW SUPPLIED/STORAGE AND HANDLING
- *
- Sections or subsections omitted from the full prescribing information are not listed.
- 1 INDICATIONS AND USAGE
-
2 DOSAGE AND ADMINISTRATION
2.1 Recommended Dosage
2.2 Monitoring
2.1 Recommended Dosage
Adults: The recommended initial dosage in adult patients is 800 mg/day. In clinical studies, the average
dosage was about 1,000 mg/day.
Pediatrics: The recommended initial dosage in pediatric patients weighing 20 kg and greater is 15 mg/kg/day.
Avoid dosages greater than 50 mg/kg per day in pediatric patients [see Warnings and Precautions (5.1), Use in
Specific Populations (8.4)].Administer THIOLA in 3 divided doses at the same times each day at least one hour before or 2 hours after
meals.Consider starting THIOLA at a lower dosage in patients with history of severe toxicity to d-penicillamine.
2.2 Monitoring
Measure urinary cystine 1 month after starting THIOLA and every 3 months thereafter. Adjust THIOLA dosage
to maintain urinary cystine concentration less than 250 mg/L.Assess for proteinuria before treatment and every 3 to 6 months during treatment [see Warnings and
Precautions (5.1)].Discontinue THIOLA in patients who develop proteinuria, and monitor urinary protein and renal function.
Consider restarting THIOLA treatment at a lower dosage after resolution of proteinuria. - 3 DOSAGE FORMS AND STRENGTHS
- 4 CONTRAINDICATIONS
-
5 WARNINGS AND PRECAUTIONS
5.1 Proteinuria
5.2 Hypersensitivity Reactions
5.1 Proteinuria
Proteinuria, including nephrotic syndrome, and membranous nephropathy, have been reported with tiopronin
use. Pediatric patients receiving greater than 50 mg/kg of tiopronin per day may be at increased risk for
proteinuria [see Dosage and Administration (2.2), Adverse Reactions (6.1, 6.2), Use in Specific Populations
(8.4)]. Monitor patients for the development of proteinuria and discontinue therapy in patients who develop
proteinuria [see Dosage and Administration (2.2)].
5.2 Hypersensitivity Reactions
Hypersensitivity reactions (drug fever, rash, fever, arthralgia and lymphadenopathy) have been reported [see
Contraindications (4)]. -
6 ADVERSE REACTIONS
6.1 Clinical Trials Experience
6.2 Postmarketing Experience
The following adverse reactions are discussed in greater detail in other sections of the labeling:
• Proteinuria [see Warnings and Precautions (5.1)]
• Hypersensitivity [see Warnings and Precautions (5.2)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed in
the clinical trials of the drug cannot be directly compared to rates in the clinical trials of another drug and may
not reflect the rates observed in practice.Adverse reactions occurring at an incidence of ≥5% in an uncontrolled trial in 66 patients with cystinuria
age 9 to 68 years are shown in the table below. Patients in group 1 had previously been treated with
d-penicillamine; those in group 2 had not. Of those patients who had stopped taking d-penicillamine due to
toxicity (34 out of 49 patients in group 1), 22 were able to continue treatment with THIOLA. In those without
prior history of d-penicillamine treatment, 6% developed reactions of sufficient severity to require THIOLA
withdrawal.
Table 1 presents adverse reactions ≥5% in either treatment group occurring in this trial.
Taste Disturbance
A reduction in taste perception may develop. It is believed to be the result of chelation of trace metals by
tiopronin. Hypogeusia is often self-limited.6.2 Postmarketing Experience
Adverse reactions have been reported from the literature, as well as during post-approval use of THIOLA.
Because the post-approval reactions are reported voluntarily from a population of uncertain size, it is not
always possible to reliably estimate their frequency or establish a causal relationship to THIOLA exposure.
Adverse reactions reported during the postmarketing use of THIOLA are listed by body system in Table 2.
-
8 USE IN SPECIFIC POPULATIONS
8.1 Pregnancy
8.2 Lactation
8.4 Pediatric Use
8.5 Geriatric Use
8.1 Pregnancy
Risk Summary
Available published case report data with tiopronin have not identified a drug-associated risk for major birth
defects, miscarriage, or adverse maternal or fetal outcomes. Renal stones in pregnancy may result in adverse
pregnancy outcomes (see Clinical Considerations). In animal reproduction studies, there were no adverse
developmental outcomes with oral administration of tiopronin to pregnant mice and rats during organogenesis
at doses up to 2 times a 2 grams/day human dose (based on mg/m 2). The estimated background risk of major
birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background
risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background
risk of major birth defects and miscarriage in clinically recognized pregnancies are 2% to 4% and 15% to
20%, respectively.
Clinical Considerations
Disease-associated maternal and/or embryo/fetal risk
Renal stones in pregnancy may increase the risk of adverse pregnancy outcomes, such as preterm birth and
low birth weight.
Data
Animal Data
No findings of fetal malformations could be attributed to the drug in reproduction studies in mice and rats at
doses up to 2 times the highest recommended human dose of 2 grams/day (based on mg/m 2).
8.2 Lactation
Risk Summary
There are no data on the presence of tiopronin in either human or animal milk or on the effects of the
breastfed child. A published study suggests that tiopronin may suppress milk production. Because of the
potential for serious adverse reactions, including nephrotic syndrome, advise patients that breastfeeding is not
recommended during treatment with THIOLA.
8.4 Pediatric Use
THIOLA is indicated in pediatric patients weighing 20 kg or more with severe homozygous cystinuria, in
combination with high fluid intake, alkali, and diet modification, for the prevention of cystine stone formation
who are not responsive to these measures alone. This indication is based on safety and efficacy data from a
trial in patients 9 years to 68 years of age and clinical experience. Proteinuria, including nephrotic syndrome,
has been reported in pediatric patients. Pediatric patients receiving greater than 50 mg/kg tiopronin per day
may be at greater risk [see Dosage and Administration (2.1, 2.2), Warnings and Precautions (5.1) and Adverse
Reactions (6.1)].
THIOLA tablets are not approved for use in pediatric patients weighing less than 20 kg or in pediatric patients
unable to swallow tablets [see Recommended Dosage (2.1)].
8.5 Geriatric Use
This drug is known to be substantially excreted by the kidney, and the risk of adverse reactions to this drug
may be greater in patients with impaired renal function. Because elderly patients are more likely to have
decreased renal function, care should be taken in dose selection, and it may be useful to monitor renal
function. - 10 OVERDOSAGE
-
11 DESCRIPTION
THIOLA (tiopronin) immediate-release tablets are a reducing and cystine-binding thiol drug (CBTD) for oral
use. Tiopronin is N-(2-Mercaptopropionyl) glycine and has the following structure: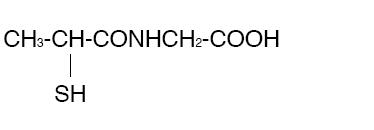
Tiopronin has the empirical formula C 5H 9NO 3S and a molecular weight of 163.20. In this drug product tiopronin
exists as a dl racemic mixture.Tiopronin is a white crystalline powder, which is freely soluble in water.
Each THIOLA tablet contains 100 mg of tiopronin. The inactive ingredients in THIOLA tablets include calcium
carbonate, carnauba wax, ethyl cellulose, dimethylaminoethyl methacrylate: butyl methacrylate: methyl
methacrylate copolymer (Eudragit E 100), hydroxy-propyl cellulose, lactose monohydrate, magnesium
stearate, povidone, sugar, talc, titanium dioxide. -
12 CLINICAL PHARMACOLOGY
12.1 Mechanism of Action
12.2 Pharmacodynamics
12.3 Pharmacokinetics
12.1 Mechanism of Action
The goal of therapy is to reduce urinary cystine concentration below its solubility limit. Tiopronin is an active
reducing agent which undergoes thiol-disulfide exchange with cystine to form a mixed disulfide of tiopronincysteine.
From this reaction, a water-soluble mixed disulfide is formed and the amount of sparingly soluble
cystine is reduced.
12.2 Pharmacodynamics
The decrement in urinary cystine produced by tiopronin is generally proportional to the dose. A reduction in
urinary cystine of 250-350 mg/day at tiopronin dosage of 1 g/day, and a decline of approximately 500 mg/day
at a dosage of 2 g/day, might be expected. Tiopronin has a rapid onset and offset of action, showing a fall in
cystine excretion on the first day of administration and a rise on the first day of drug withdrawal.
12.3 Pharmacokinetics
Absorption
THIOLA Tablets
When THIOLA single doses were given to fasted healthy subjects (n = 39), the median time to peak plasma
level (T max) was 1 (range: 0.5 to 2.1) hours.
EliminationExcretion
When tiopronin is given orally, up to 48% of dose appears in urine during the first 4 hours and up to 78% by
72 hours. -
13 NONCLINICAL TOXICOLOGY
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term carcinogenicity studies in animals have not been performed.
Mutagenesis
Tiopronin was not genotoxic in the chromosomal aberration, sister chromatid exchange, and in vivo
micronucleus assays.
Impairment of Fertility
High doses of tiopronin in experimental animals have been shown to interfere with maintenance of pregnancy
and viability of the fetus. In 2 published male fertility studies in rats, tiopronin at 20 mg/kg/day intramuscular
(IM) for 60 days induced reductions in testis, epididymis, vas deferens, and accessory sex glands weights and
in the count and motility of cauda epididymal sperm. - 16 HOW SUPPLIED/STORAGE AND HANDLING
- 17 PATIENT COUNSELING INFORMATION
- PRINCIPAL DISPLAY PANEL
-
INGREDIENTS AND APPEARANCE
THIOLA
tiopronin tablet, sugar coatedProduct Information Product Type HUMAN PRESCRIPTION DRUG Item Code (Source) NDC:0178-0900 Route of Administration ORAL Active Ingredient/Active Moiety Ingredient Name Basis of Strength Strength TIOPRONIN (UNII: C5W04GO61S) (TIOPRONIN - UNII:C5W04GO61S) TIOPRONIN 100 mg Inactive Ingredients Ingredient Name Strength SUCROSE (UNII: C151H8M554) 123 mg CALCIUM CARBONATE (UNII: H0G9379FGK) 42.7 mg LACTOSE (UNII: J2B2A4N98G) 39 mg DIMETHYLAMINOETHYL METHACRYLATE - BUTYL METHACRYLATE - METHYL METHACRYLATE COPOLYMER (UNII: 905HNO1SIH) 28.8 mg ETHYLCELLULOSES (UNII: 7Z8S9VYZ4B) 20 mg MAGNESIUM SILICATE (UNII: 9B9691B2N9) 14.4 mg MAGNESIUM STEARATE (UNII: 70097M6I30) 5.4 mg TITANIUM DIOXIDE (UNII: 15FIX9V2JP) 1.7 mg CARNAUBA WAX (UNII: R12CBM0EIZ) 0.05 mg FD&C RED NO. 40 (UNII: WZB9127XOA) ALUMINUM OXIDE (UNII: LMI26O6933) POVIDONE, UNSPECIFIED (UNII: FZ989GH94E) HYDROXYPROPYL CELLULOSE (70000 WAMW) (UNII: 66O7AQV0RT) Product Characteristics Color white Score no score Shape ROUND Size 8mm Flavor Imprint Code M Contains Packaging # Item Code Package Description Marketing Start Date Marketing End Date 1 NDC:0178-0900-01 100 in 1 BOTTLE; Type 0: Not a Combination Product 08/11/1988 Marketing Information Marketing Category Application Number or Monograph Citation Marketing Start Date Marketing End Date NDA NDA019569 08/11/1988 Labeler - Mission Pharmacal Company (008117095) Registrant - Mission Pharmacal Company (927726893) Establishment Name Address ID/FEI Business Operations Mission Pharmacal Company 927726893 manufacture(0178-0900)

